

Abstract
Retinitis pigmentosa (RP) is a group of inherited blinding diseases caused by mutations in multiple genes including RDS. RDS encodes rds/peripherin (rds), a 36-kDa glycoprotein in the rims of rod and cone outer-segment (OS) discs. Rom1 is related to rds with similar membrane topology and the identical distribution in OS. In contrast to RDS, no mutations in ROM1 alone have been associated with retinal disease. However, an unusual digenic form of RP has been described. Affected individuals in several families were doubly heterozygous for a mutation in RDS causing a leucine 185 to proline substitution in rds (L185P) and a null mutation in ROM1. Neither mutation alone caused clinical abnormalities. Here, we generated transgenic/knockout mice that duplicate the amino acid substitutions and predicted levels of rds and rom1 in patients with RDS-mediated digenic and dominant RP. Photoreceptor degeneration in the mouse model of digenic RP was faster than in the wild-type and monogenic controls by histological, electroretinographic, and biochemical analysis. We observed a positive correlation between the rate of photoreceptor loss and the extent of OS disorganization in mice of several genotypes. Photoreceptor degeneration in RDS-mediated RP appears to be caused by a simple deficiency of rds and rom1. The critical threshold for the combined abundance of rds and rom1 is ≈60% of wild type. Below this value, the extent of OS disorganization results in clinically significant photoreceptor degeneration.
Retinitis pigmentosa (RP) is a family of inherited retinal diseases characterized by progressive night blindness and loss of peripheral vision (1). Pathologically, RP is associated with degeneration of rod photoreceptors. Heterozygous mutations in the RDS gene are a common cause of autosomal dominant RP (2, 3). An example is the RDS P216L-allele (4). RDS encodes rds/peripherin (rds), a 36-kDa glycoprotein in the rims of rod and cone outer-segment (OS) discs (5, 6). These stacked membranous structures are the sites of photon-capture and reactions of visual transduction. Rom1 is a related disk-rim protein with 37% overall identity and similar membrane topology to rds (7). Mice homozygous for a knockout mutation in the rom1 gene displayed mild OS dysplasia (8), in contrast to complete absence of OS in rds−/− mice (9–11). Unlike RDS, no mutations in the ROM1 gene alone have been convincingly associated with human retinal disease (12, 13). However, an unusual digenic form of RP has been described. Affected individuals in four pedigrees were doubly heterozygous for a mutation in RDS causing a leucine 185 to proline substitution in rds (L185P) and a second presumptive null mutation in the unlinked ROM1 gene (13, 14). Neither mutation alone caused significant abnormalities.
The phenotype in rds−/− mutant mice (9, 10) indicates a critical role for rds in the formation of OS. Expression of a chimeric protein in transgenic mice on an rds−/− genetic-background established that rds is 2.5-fold more abundant than rom1, and that the rds–rom1 interaction involves the large, intradiscal D2 loop (15). In the current study, we sought to address three questions concerning the biochemical etiology of RDS-mediated retinal degeneration. First, can the clinical observation of digenic RP in humans be corroborated in an animal model? Second, how do the pathogenic L185P and P216L D2-loop substitutions affect the abundance of rds? Third, what is the relationship between OS disorganization and photoreceptor degeneration in the different mutant RDS alleles? To address these questions, we generated complex transgenic/mutant mice in which both the specific substitutions and levels of rds and rom1 closely matched those predicted for the corresponding human diseases.
Materials and Methods
Generation of Transgenic Mice.
We assembled a DNA construct containing a rhodopsin promoter upstream of the mouse rds coding region. We introduced a T to C transition into codon 185, resulting in L185P. The construct was otherwise identical to a previously described transgene encoding normal rds (16). Fertilized oocytes of hybrid strain B6 × DBA mice were microinjected with this construct. Of several L185P lines generated, line 1708 was selected for study based on its level of expression. P216L- and S231A-transgenic mice were generated as described (17, 18). The L185P, P216L, and S231A transgenes were crossed onto rds+/−, rds−/− (9, 10), rom1+/−, and rom1−/− (8) mutant backgrounds. Mice were analyzed for presence of the transgenes as described (17). Mice were maintained on a 12-hour light/dark cycle (25–30 lx). For all studies except electroretinogram (ERG) analysis, animals were killed between 4 and 6 h after light-onset.
Nuclease Protection Analysis of Retinal RNAs.
Total RNA was extracted from individual eyecups and hybridized to 32P-labeled cRNA probes of 1,041 nt for rds or 687 nt for rom1. After digestion with S1 nuclease, protected fragments were separated by electrophoresis through an 8% polyacrylamide gel containing 8 M urea. Bands were quantitated with reference to a standard curve of in vitro-transcribed sense rds-mRNA on a Molecular Dynamics model 425F PhosphorImager.
Electroretinography.
Full-field ERGs were obtained from mice of the genotypes indicated in Fig. 4. After overnight dark-adaptation, mice were anesthetized with ketamine (200 mg/kg) plus xylazine (10 mg/kg) and the pupils were dilated with topical 1.0% atropine sulfate. ERGs were obtained in a Ganzfeld dome, using a gold coil wire overlaid with 1% methylcellulose on the corneal surface, a similar reference electrode in the mouth, and a needle ground-electrode in the tail. A high-intensity flash unit (Novatron, Dallas, TX) provided short-wavelength flashes (Kodak Wratten 47B) at intensities 1 to 3.4 log scot td-sec in 0.3 log unit steps. The leading edge of the a-waves was fit (as an ensemble; ref. 19) by the Lamb and Pugh model for the activation phase of the phototransduction cascade (20). The maximal response (Rmp3) and the amplification constant (S) were calculated from this model.
Figure 4.

Electroretinographic analysis. ERGs were performed on five to 7-month-old mice of the genotypes: wild-type (n = 13), rom1+/− (n = 11), L185P rds+/− (n = 3), digenic (n = 7), and nontransgenic rds+/− (n = 8). Average values of Rmp3 for mice of the indicated genotypes are plotted in microvolts (μV ± SEM). An asterisk above the data bar denotes a significant difference from the wild-type value of Rmp3 (Student's t test; P < 0.01).
Preparation of Samples for Light and Electron Microscopy.
Mice were anesthetized with Nembutal (75 mg/kg) and fixed by vascular perfusion for 5 min with 1% formaldehyde and 2% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.2). Eyes were removed and immersed in the same fixative overnight at 4°C, then fixed for an additional hour in 1% osmium tetroxide. Tissues were dehydrated and embedded in Araldite 502 (Ted Pella, Redding, CA). Sections (0.5 μm) were cut on glass knives and stained with toluidine blue for light microscopy. Ultrathin sections were cut with a diamond knife and stained with uranium and lead salts for electron microscopy.
Immunoblot Analysis.
Antisera against residues 296–346 from the carboxy terminus of rds (rds C-term Ab) and residues 296–351 from the carboxy terminus of rom1 (rom1 C-term Ab) were prepared as described (15). For the quantitative immunoblotting, retinal homogenates were separated by SDS/PAGE and transferred to Immobilon P-SQ (Millipore) in buffer containing 0.05% SDS. After reacting with the primary antibody, blots were labeled by using 125I-protein A (ICN) in a 6-hr incubation at a concentration of 0.1 μCi/ml (1 Ci = 37 GBq). After five 8-min washes, the radioactive bands were visualized and quantitated on a Molecular Dynamics model 425F PhosphorImager. In a control experiment, the radioactive signal was shown to increase linearly with loaded protein up to 4% retina per lane. Quantitation was performed with 2% retina loaded per lane. Radioactive signals for rds and rom1 were determined as a fraction of the wild-type signal on each blot. The data presented in Table 1 represent the averages of these fractions.
Table 1.
Expected and observed levels of rds and rom1 monomers in mouse retinas
| Genotype | rds expected (% w/t) | rds observed (% w/t ± SEM) | rom1 expected (% w/t) | rom1 observed (% w/t ± SEM) |
|---|---|---|---|---|
| Wild-type | 100 | (100) | 100 | (100) |
| rom1+/− | 100 | 106 ± 4 | 50 | 58 ± 2 |
| L185Prds+/− | 100 | 62 ± 2 | 100 | 103 ± 2 |
| Digenic | 100 | 51 ± 2 | 50 | 42 ± 2 |
| rds+/− | 50 | 26 ± 2 | 100 | 67 ± 2 |
| P216L rds+/− | 100 | 8 ± 1 | 100 | 25 ± 3 |
| rom1−/− | 100 | 100 ± 4 | 0 | 0 |
| S231A rds+/− | 50 | 45 ± 4 | 100 | 99 ± 10 |
Expected levels of rds and rom1 monomers relative to wild-type (w/t) were based on measurements of the respective endogenous and transgenic mRNAs by nuclease protection analysis without considering the effects of reduced protein stability or outer-segment dysplasia. Observed levels of rds and rom1 were obtained by quantitative immunoblotting, as represented in Fig. 5a. Values are expressed as an average percent of the wild-type signal on each blot ± SEM. For S231A rds+/− mice, contributions of the nonglycosylated protein (lower rds band in Fig. 5a) were not included.
Immunoprecipitation Analysis.
Before immunoprecipitation, the rom1 C-term Ab was coupled to Affi-Gel Hz beads (Bio-Rad). Dissected retinas from mice of the indicated genotypes were homogenized in Triton buffer (50 mM Tris⋅HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.05% SDS, 2.5% glycerol, and protease inhibitors). Homogenates were cleared by a 2-min spin at 4°C and incubated with the coupled Ab at 4°C for 2 h. Beads were washed and the bound proteins were eluted with 2× sample buffer for immunoblot analysis. Approximately 50% of total rds in each extract was precipitated with the rom1 C-term Ab.
Results
Levels of the rds and rom1 mRNAs in Transgenic Retinas.
To determine expression of the rds and rom1 genes, we performed nuclease protection analysis on retinal RNA prepared from mice of the relevant genotypes (Fig. 1). The spontaneous rds mutation in mice results from insertion of an extraneous repetitive element into exon 2 (21). Although the disrupted gene is transcribed in rds−/− photoreceptors (protected band visible in Fig. 1), no protein product is made (16, 21). Thus, the spontaneous rds allele is null. The mRNA product of the L185P transgene in line 1708 mice was present at ≈50% the level of the endogenous transcript by quantitative nuclease-protection analysis (Fig. 1). No mRNA is transcribed from the disrupted gene in rom1 knockout mice (ref. 8; Fig. 1). The L185P transgene was crossed onto rds (9, 10) and rom1 (8) mutant backgrounds. Previously, we generated mice with a similar transgene encoding P216L-substituted rds (17). Mice from P216L line 1376 express the transgene in retina at ≈60% the level of the normal endogenous rds mRNA (17). We also generated transgenic mice that express an mRNA encoding S231A-substituted (nonglycosylated) rds at ≈80% the level of the normal endogenous rds mRNA (18).
Figure 1.

Nuclease protection analysis of rds and rom1 mRNAs. RNA was prepared from 3-week-old L185P-transgenic and nontransgenic mouse retinas of the indicated genotypes. (Upper) The three fragments protected with a 1,041-nt rds cRNA-probe: a band of 1,011 nt corresponding to the spontaneous rds-mutant mRNA (null), 899 nt corresponding to the normal endogenous rds mRNA (endo), and 843 nt corresponding to the mRNA product of the L185P-transgene (L185P). (Lower) The single 659-nt band protected by a 687-nt rom1 cRNA-probe corresponding to the normal rom1 mRNA (rom1).
Slow Photoreceptor Degeneration in a Mouse Model of Digenic RP.
We examined sections of retina from mice of the relevant genotypes by light microscopy. Fig. 2 shows a representative set of light micrographs from 9-month-old mice. The extent of photoreceptor loss can be estimated by observing the thickness of the outer nuclear layer (ONL) in retinal sections. The ONL thickness was reduced in L185P rds+/− and digenic compared with wild-type mice (Fig. 2). No difference in ONL thickness was apparent in rom1+/− compared with wild-type mice. These data indicate photoreceptor loss in L185P rds+/− and digenic mice.
Figure 2.

Light microscopy of mouse retinas. Outer retinas from 9-month-old mice of the indicated genotypes are shown. The OS, inner segment (IS), and ONL are indicated. Note the reduced ONL thickness in L185P rds+/− and digenic mice compared with the wild-type control. (×430.)
Expression of L185P-Substituted, but Not P216L-Substituted rds Corrects the OS Disorganization in rds+/− Retinas.
To estimate the degree of OS dysplasia in mice of the different genotypes, we examined retinas of 9-month-old mice by electron microscopy. The morphology of OS in rom1+/− mice was indistinguishable from wild type (Fig. 3). Mild dysplasia was noted in L185P-transgenic rds+/− mice, with slight shortening and widening of OS and misalignment of discs. The dysplasia was more severe in the digenic mice, with profound shortening of OS and disorganization of discs. As described (22), gross disorganization of OS was seen in rds+/− mice with replacement of the discs by large whorl structures. This dysplasia was not corrected by expression of P216L-substituted rds, as shown (17).
Figure 3.

Electron microscopy of mouse retinas. OS and retinal pigment epithelium (RPE) layers of retinas from 9-month-old mice of the indicated genotypes are shown. Note the shortening and disorganization of OS in the digenic retina. Also note the distortion of OS into whorl structures in the rds+/− retina. (×3,000.)
Reduced Retinal Photoresponse in rds-Mutant and Digenic Mice.
To probe the function of photoreceptors in vivo, we analyzed wild-type and mutant mice by electroretinography. ERG records the electrical response of the retina to a light flash on the corneal surface. The initial a-wave results from photoreceptor hyperpolarization. We computed the a-wave maximal amplitude (Rmp3; ref. 23) for each animal studied. A reduction in Rmp3 is associated with loss of functional discs due to abnormal development of OS or degeneration of photoreceptors (19). The average Rmp3 values obtained for mice of the indicated genotypes at approximately 6 months are shown in Fig. 4. Rmp3 was significantly reduced in L185P rds+/− monogenic and L185P rds+/−, rom1+/− (digenic) compared with wild-type mice. Rmp3 was further reduced in nontransgenic rds+/− mice. The amplification constants (S) was within normal limits for all mice tested.
Levels of Normal and Substituted rds and rom1 Proteins in Mice of Different Genotypes.
To determine the levels of rds and rom1, we analyzed retinal homogenates from transgenic and nontransgenic mice on several genetic backgrounds by quantitative immunoblotting. Mice were analyzed at 3 weeks of age, before the onset of significant photoreceptor death in any mutants. Immunoblots were performed in quintuplicate for rds and quadruplicate for rom1, using the rds and rom1 C-term Abs (15) and 125I-labeled protein A for detection. The results of a representative experiment are presented in Fig. 5a. Table 1 shows the levels of rds and rom1 in retinas from mice of each genotype expressed as a fraction of the level in wild-type retinas. The level of rds in rds+/− retinas was 26% that of wild-type retina, significantly less than the 50% predicted by levels of the endogenous mRNA in wild-type and rds+/− retinas (Fig. 1). The S231A substitution eliminates the single conserved site of N-glycosylation in rds, resulting in a protein of reduced molecular mass but normal function (18). Use of the S231A rds−/− background permitted immunoblot quantitation of other rds forms. Expression of S231A-substituted rds in transgenic mice completely rescued the rds+/− phenotype of disorganized OS (18). Accordingly, the level of endogenous (glycosylated) rds in rds+/− mice expressing the S231A transgene was close to the predicted value (45% vs. 50%). The level of total rds was significantly higher in L185P-transgenic rds+/− (62% of wild type) compared with nontransgenic rds+/− retinas, consistent with synthesis of additional L185P-substituted rds. Because OS were intact in these mice (Fig. 3), L185P-substituted rds must be transported to the OS where it must function normally. In contrast, expression of P216L-substituted rds in rds+/− retinas resulted in significantly reduced total rds (8% of wild type). OS in P216L-transgenic rds+/− retinas were considerably more dysplastic than nontransgenic rds+/− retinas, as shown (17). Finally, although rom1 was undetectable, rds was present at approximately normal levels in rds+/+, rom1−/− retinas (Fig. 5a), consistent with the relatively normal OS in mice of this genotype (8).
Figure 5.

Immunoblot analysis of rds and rom1 in mouse retinas. (a) Quantitative immunoblotting of homogenates from rds P216L-, L185P-, and S231A-transgenic retinas of the indicated genotypes at rds and rom1. Blots were incubated with antibodies against the rds C-term (rds) or rom1 C-term (rom1) antibodies. Note the reduced levels of rds in nontransgenic rds+/− and P216L rds+/− homogenates. Also note the lower mass of nonglycosylated, S231A-substituted rds. (b) Levels of rds in nontransgenic (nTG), L185P-, and P216L-transgenic rds−/− retinas. All mice were wild-type at rom1. Note the dramatically reduced levels of substituted rds in retinas from L185P- and P216L-transgenic mice. (c) Coimmunoprecipitation of normal, L185P-, P216L-, and S231A-substituted rds with rom1. The genotypes at rds are indicated, all mice were wild-type at rom1. The band of lower molecular mass in all lanes is S231A-substituted rds. Note the similar protein abundance profiles in the starting homogenates and rom1-immunoprecipitated samples.
L185P- and P216L-Substituted rds Are Dramatically Reduced in rds−/− Retinas.
To what extent does the function of L185P-substituted rds depend on the presence of normal rds? To address this question, we moved the L185P and P216L transgenes onto an rds−/− null genetic background. Immunoblot analysis showed dramatically reduced L185P- and P216L-substituted rds in rds−/− compared with wild-type retinas (Fig. 5b). These data indicate that in the absence of normal rds, the L185P- and P216L-substituted proteins are either unstable or not transported to OS. Consistent with these biochemical results, neither P216L- nor L185P-substituted rds rescued the phenotype of absent OS in rds−/− mice (data not shown). The low levels of both substituted proteins indicate that neither are accumulating within the endoplasmic reticulum (ER).
L185P- and P216L-Substituted rds Efficiently Coprecipitate with rom1.
Covalent homodimers of rds normally interact with rom1 homodimers to form a higher-order noncovalent complex in the disk rim (15, 24–27). To test the interaction of rom1 with L185P-substituted rds, we performed immunoprecipitation analysis with the rom1 C-term Ab on retinal homogenates from doubly transgenic mice that express S231A-substituted plus L185P- or P216L-substituted rds on a wild-type or rds−/− genetic background (Fig. 5c). The rom1 C-term Ab does not cross react with rds (15). Both L185P- and P216L-substituted rds coprecipitated with similar efficiency to normal rds. These data indicate that the L185P and P216L D2-loop substitutions do not disturb the interaction of rds with rom1.
Discussion
Digenic Inheritance of Retinal Degeneration in an Animal Model.
Digenic inheritance of RP was originally reported in patients doubly heterozygous for a mutation in RDS causing an L185P substitution plus an early frame-shift mutation in ROM1 (13, 14). To verify digenic inheritance of RP, we placed a transgene that expresses L185P-substituted rds at ≈50% the level of endogenous rds on an rds+/−, rom1+/− double heterozygous genetic background. The ERG results were in general agreement between the mouse and human models of digenic RP (14). In the mice, reduced ERG amplitudes were correlated with degeneration of photoreceptors by light microscopy. Together, these data validate digenic inheritance of RP in humans.
Photoreceptor Degeneration Is Correlated with Abnormal Development of OS.
We observed a good correlation between the extent of OS disorganization and the degree of photoreceptor degeneration. A progressive increase in the severity of both phenotypic parameters was observed in the series: (i) rom1+/−; (ii) L185P rds+/−; (iii) digenic; (iv) rds+/− (22); (v) P216L rds+/− (17); and (vi) rds−/− (9, 10). The mechanism of photoreceptor degeneration in rds mutants is unknown. Because no accumulation of substituted rds was observed in L185P- and P216L-transgenic rds−/− mice, ER stress can be ruled out. Oxygen toxicity is another possibility. According to this mechanism, reduced cation influx through cGMP-gated channels causes reduced activity of Na+/K+-ATPases and hence reduced uptake of O2 by mitochondria. The resulting increased pO2 causes oxidative damage to photoreceptors (28). The observed congruence between the extent of OS dysplasia and the rate of photoreceptor death here and in other studies (17, 22) supports this hypothesis. Mutations in the gene for rhodopsin are also responsible for a subset of autosomal dominant RP. The ultrastructural effects of several RP-associated mutations in the rhodopsin gene have been studied in transgenic mice, including the heterozygous-null, P347S, P23H, V20G, and P27L alleles (29–32). Shortening and disorganization of OS was observed in all cases. Thus, the principle of photoreceptor degeneration due to OS dysplasia may extend to mutations in multiple genes.
Covalent Homodimers of L185P- and P216L-Substituted rds Are Unstable.
The mRNA products of the L185P and P216L transgenes were present at ≈50% the level of the normal endogenous rds mRNA (ref. 17; Fig. 1). Thus, approximately equal levels of the mutant and normal rds mRNAs are present in L185P- and P216L-transgenic rds+/− retinas. If monomers of normal and substituted rds dimerized stochastically, and if each dimer form were equally stable, the distribution of normal–normal, normal–substituted, and substituted–substituted dimers should be 1:2:1. However, the capacity of L185P-substituted rds to rescue the rds phenotype was strongly dependent on the presence of normal rds. Also, the level of rds was close to wild type in L185P-transgenic rds+/− retinas, but undetectable in both L185P- and P216L-transgenic rds−/− retinas (Fig. 5b). These data suggest that covalent homodimers of L185P- and P216L-substituted rds do not form or are unstable. Consistent with these observations, it was shown (24) that expression of L185P-substituted rds in cultured cell membranes resulted in an impaired rds homointeraction, but normal interactions between rds and rom1. Given the absence of L185P–L185P homodimers, the maximum possible level of rds in L185P-transgenic rds+/− mice is 75% of wild type. The actual level observed was 62% (Table 1). Because OS are relatively intact in these mice (Fig. 3), we conclude that heterodimers of normal and L185P-substituted rds must also form functional complexes. Coimmunoprecipitation of L185P-substituted rds with a rom1 antibody (Fig. 5c) supports this conclusion. In contrast, the level of rds in P216L-transgenic rds+/− mice was only 8% of wild type. This suggests that with P216L, both normal–substituted and substituted–substituted dimers are unstable. A possible explanation for the profound OS disorganization and photoreceptor degeneration in P216L-transgenic mice (17), and the severity of RP in humans with a P216L mutation in RDS (4), is depletion of the normal rds protein due to degradation of normal–P216L heterodimers.
The L185P and P216L Mutations Cause a Deficiency of rds.
The abundance of rds is ≈2.5-fold that of rom1 (15, 27). This value, combined with the data in Table 1, permits us to calculate the total abundance of rds plus rom1 (rds + rom1) as a percent of wild type in mice of each genotype according to the equation:
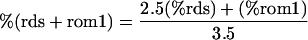 |
If we assume that rds and rom1 are functionally equivalent, we can compare rds + rom1 to the phenotype in several forms of rds-mediated retinal degeneration (Fig. 6). Mice were analyzed by quantitative immunoblotting at 3 weeks, before the onset of photoreceptor degeneration. The level of rds + rom1 in nontransgenic rom1+/− heterozygotes is 92% of wild type. We observed virtually no photoreceptor degeneration in these mice by histologic or ERG analysis. Consistently, no mutations in rom1 alone have been associated with any human disease (13). L185P rds+/− mice, with a level of rds + rom1 at 74%, exhibited mild photoreceptor degeneration. Humans of the corresponding genotype were asymptomatic, but showed slightly reduced ERG amplitudes and prolonged implicit times (14). Very mild photoreceptor degeneration was also observed in rom1−/− knockout mice (8), with an estimated level of rds + rom1 at 71% of wild type. We observed significant photoreceptor degeneration and OS dysplasia in digenic mice. The estimated level of rds + rom1 in these animals is 48%. Humans of the corresponding genotype have slowly progressive RP (14). Slightly more severe photoreceptor degeneration and OS dysplasia were seen in nontransgenic rds+/− mice, with a level of rds + rom1 at 38%. Slowly progressive RP in patients heterozygous for presumptive RDS null alleles have been described in several reports (33–38). Finally, rapid photoreceptor degeneration and profound OS dysplasia were observed in P216L-transgenic rds+/− mice (17). The level of rds + rom1 in these mice was 13%. This substitution causes dominant RP in humans (4). Together, these results suggest a critical threshold for the level of rds + rom1 in the range between 48% and 71% (average = 60%) of wild type. At levels below this approximate value, the extent of OS disorganization results in clinically significant photoreceptor degeneration. One prediction of this model is that transgenic overexpression of rom1 may rescue the rds+/− phenotype. In conclusion, these studies show that rds protein deficiency is a common etiologic factor in at least three forms of RDS-mediated retinal degeneration.
Figure 6.

Abundance of rds plus rom1 in mouse models of human RDS-mediated retinal degenerations. The levels of rds plus rom1 as a fraction of wild-type [%(rds + rom1)] in mice of the indicated genotypes were calculated from the data in Table 1, assuming a 2.5:1 abundance ratio of rds to rom1 (15, 27). Minimal photoreceptor degeneration was seen in monogenic (rom1+/− and L185P-transgenic rds+/−) mice. Humans of the corresponding genotypes were asymptomatic (14). Slow photoreceptor degeneration was seen in L185P-transgenic rds+/−, rom1+/− (digenic) mice. Humans of the corresponding genotype had slowly progressive digenic RP (14). Slightly faster retinal degeneration was seen in rds+/− mice and in humans of the corresponding genotype (33–38). Finally, severe photoreceptor degeneration was seen in P216L-transgenic rds+/− mice. Humans heterozygous for an RDS P216L mutation have dominant RP (4). Thus, the level of rds + rom1 required to prevent disease is ≈60% of wild type. When the combined abundance of these proteins falls below this value (indicated by the dashed line), photoreceptor degeneration becomes clinically recognizable as RP.
Acknowledgments
We gratefully acknowledge Walid Moghrabi for generating rds and rom1 C-term Abs, and Marsha Lloyd, Mark Pennesi, and Roxana Radu for their outstanding technical assistance. This work was supported by grants from the National Eye Institute (EY08043, EY00444, and EY00331), the Foundations Fighting Blindness of United States and Canada, and the Canadian Genetic Disease Network. R.M. is an International Research Scholar of the Howard Hughes Medical Institute. D.B. is the Dolly Green Professor of Ophthalmology at the University of California, Los Angeles, and a Research to Prevent Blindness Senior Scientific Investigator. G.H.T. is the Charles Kenneth Feldman Professor of Ophthalmology at the University of California, Los Angeles.
Abbreviations
- ERG
electroretinogram
- L185P
leucine 185 to proline substitution in rds
- ONL
outer nuclear layer
- OS
outer segment
- rds
rds/peripherin protein
- Rmp3
a-wave maximal response by ERG
- RP
retinitis pigmentosa
- rds C-term Ab and rom1 C-term Ab
antisera against residues 296–346 from the carboxy terminus of rds and residues 296–351 from the carboxy terminus of rom1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Heckenlively J R. In: Retinitis Pigmentosa. Heckenlively J R, editor. Philadelphia: Lippincott; 1988. pp. 221–252. [Google Scholar]
- 2.Keen T J, Inglehearn C F. Hum Mutat. 1996;8:297–303. doi: 10.1002/(SICI)1098-1004(1996)8:4<297::AID-HUMU1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B. Acta Anatomica. 1998;162:75–84. doi: 10.1159/000046471. [DOI] [PubMed] [Google Scholar]
- 4.Kajiwara K, Hahn L B, Mukai S, Travis G H, Berson E L, Dryja T P. Nature (London) 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 5.Connell G, Bascom R, Molday L, Reid D, McInnes R R, Molday R S. Proc Natl Acad Sci USA. 1991;88:723–726. doi: 10.1073/pnas.88.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travis G H, Sutcliffe J G, Bok D. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- 7.Bascom R A, Manara S, Collins L, Molday R S, Kalnins V I, McInnes R R. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- 8.Clarke G, Goldberg A F X, Vidgen D, Collins L, Ploder L, Schwarz L, Molday L L, Rossant J, Szel A, Molday R S, Birch D G, McInnes R R. Nat Genet. 2000;25:67–73. doi: 10.1038/75621. [DOI] [PubMed] [Google Scholar]
- 9.van Nie R, Ivanyi D, Demant P. Tissue Antigens. 1978;12:106–108. doi: 10.1111/j.1399-0039.1978.tb01305.x. [DOI] [PubMed] [Google Scholar]
- 10.Sanyal S, Jansen H G. Neurosci Lett. 1981;21:23–26. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- 11.Travis G H, Brennan M B, Danielson P E, Kozak C A, Sutcliffe J G. Nature (London) 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- 12.Bascom R A, Liu L, Heckenlively J R, Stone E M, McInnes R R. Hum Mol Genet. 1995;4:1895–1902. doi: 10.1093/hmg/4.10.1895. [DOI] [PubMed] [Google Scholar]
- 13.Dryja T P, Hahn L B, Kajiwara K, Berson E L. Invest Ophthalmol Visual Sci. 1997;38:1972–1982. [PubMed] [Google Scholar]
- 14.Kajiwara K, Berson E L, Dryja T P. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 15.Kedzierski W, Weng J, Travis G H. J Biol Chem. 1999;274:29181–29187. doi: 10.1074/jbc.274.41.29181. [DOI] [PubMed] [Google Scholar]
- 16.Travis G H, Groshan K R, Lloyd M, Bok D. Neuron. 1992;9:113–119. doi: 10.1016/0896-6273(92)90226-4. [DOI] [PubMed] [Google Scholar]
- 17.Kedzierski W, Lloyd M, Birch D G, Bok D, Travis G H. Invest Ophthalmol Visual Sci. 1997;38:498–509. [PubMed] [Google Scholar]
- 18.Kedzierski W, Bok D, Travis G H. J Neurochem. 1999;72:430–438. doi: 10.1046/j.1471-4159.1999.0720430.x. [DOI] [PubMed] [Google Scholar]
- 19.Hood D C, Birch D G. Invest Ophthalmol Visual Sci. 1994;35:2948–2961. [PubMed] [Google Scholar]
- 20.Lamb T D, Pugh E N., Jr J Physiol. 1992;449:719–758. doi: 10.1113/jphysiol.1992.sp019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Norton J C, Allen A C, Burns J B, Hasel K W, Burns J L, Sutcliffe J G, Travis G H. Genomics. 1995;28:212–219. doi: 10.1006/geno.1995.1133. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins R K, Jansen H G, Sanyal S. Exp Eye Res. 1985;41:701–720. doi: 10.1016/0014-4835(85)90179-4. [DOI] [PubMed] [Google Scholar]
- 23.Hood D C, Birch D G. Visual Neurosci. 1993;10:857–871. doi: 10.1017/s0952523800006076. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg A F, Molday R S. Proc Natl Acad Sci USA. 1996;93:13726–13730. doi: 10.1073/pnas.93.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moritz O L, Molday R S. Invest Ophthalmol Visual Sci. 1996;37:352–362. [PubMed] [Google Scholar]
- 26.Goldberg A F X, Loewen J R, Molday R S. Biochemistry. 1998;37:680–685. doi: 10.1021/bi972036i. [DOI] [PubMed] [Google Scholar]
- 27.Loewen C J R, Molday R S. J Biol Chem. 2000;275:5370–5378. doi: 10.1074/jbc.275.8.5370. [DOI] [PubMed] [Google Scholar]
- 28.Travis G. Am J Hum Genet. 1998;62:503–508. doi: 10.1086/301772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphries M M, Rancourt D, Farrar G J, Kenna P, Hazel M, Bush R A, Sieving P A, Sheils D M, McNally N, Creighton P, et al. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 30.Naash M I, Hollyfield J G, al-Ubaidi M R, Baehr W. Proc Natl Acad Sci USA. 1993;90:5499–5503. doi: 10.1073/pnas.90.12.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li T, Snyder W, Olsson J, Dryja T. Proc Natl Acad Sci USA. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X R, Wu T H, Stowe S, Matsushita A, Arikawa K, Naash M I, Williams D S. J Cell Sci. 1997;110:2589–2597. doi: 10.1242/jcs.110.20.2589. [DOI] [PubMed] [Google Scholar]
- 33.Kajiwara K, Sandberg M A, Berson E L, Dryja T P. Nat Genet. 1993;3:208–212. doi: 10.1038/ng0393-208. [DOI] [PubMed] [Google Scholar]
- 34.Meins M, Gruning G, Blankenagel A, Krastel H, Reck B, Fuchs S, Schwinger E, Gal A. Hum Mol Genet. 1993;2:2181–2182. doi: 10.1093/hmg/2.12.2181. [DOI] [PubMed] [Google Scholar]
- 35.Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, et al. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 36.Lam B L, Vandenburgh K, Sheffield V C, Stone E M. Am J Ophthalmol. 1995;119:65–71. doi: 10.1016/s0002-9394(14)73815-2. [DOI] [PubMed] [Google Scholar]
- 37.Apfelstedt-Sylla E, Theischen M, Ruther K, Wedemann H, Gal A, Zrenner E. Br J Ophthalmol. 1995;79:28–34. doi: 10.1136/bjo.79.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobson S G, Cideciyan A V, Kemp C M, Sheffield V C, Stone E M. Invest Ophthalmol Visual Sci. 1996;37:1662–1674. [PubMed] [Google Scholar]