

Abstract
Triclosan has been previously shown to inhibit InhA, an essential enoyl acyl carrier protein reductase of mycolic acid biosynthesis, whose inhibition leads to the lysis of Mycobacterium tuberculosis. Using a structure-based drug design approach, a series of 5-substituted derivatives of triclosan was developed. Two groups of triclosan derivatives with alkyl and aryl substituents, respectively, were identified with dramatically enhanced potency against purified InhA. The most efficacious inhibitor displayed an IC50 value of 21 nM, which was 50-fold more potent than triclosan. X-ray crystal structures of InhA in complex with four triclosan derivatives revealed the structural basis for the inhibitory activity. Six selected triclosan derivatives were tested against isoniazid-sensitive and resistant strains of M. tuberculosis. Among those, the best inhibitor had an MIC value of 4.7 µg/mL (13 µM), which represents a tenfold improvement over the bacteriocidal activity of triclosan. A subset of these triclosan analogs was more potent than isoniazid against two isoniazid-resistant M. tuberculosis strains, demonstrating the significant potential for structure-based design in the development of next generation antitubercular drugs.
Keywords: antibiotics, drug resistance, fatty acids, triclosan, tuberculosis
Introduction
For nearly fifty years, isoniazid (INH) has been utilized as a front line drug to treat tuberculosis (TB).[1] INH is a key component of short course chemotherapy for TB cure, which entails six months of daily administration, and is also the sole component for TB prophylaxis, which includes nine months of daily administration. The emergence of strains resistant to INH has compromised TB control programs worldwide.[2, 3]
The Mycobacterium tuberculosis enoyl acyl carrier protein (ACP) reductase (InhA) has been demonstrated to be the target of INH and ethionamide (ETH),[4–7] validating it as an excellent antitubercular drug target. INH and ETH are prodrugs and their activities are dependent on activation in M. tuberculosis by KatG, a catalase/peroxidase enzyme, and EthA, a flavin monooxygenase, respectively.[8, 9] Upon activation, INH or ETH forms a covalent adduct with NAD cofactor, which inhibits InhA. As prodrugs, INH and ETH are highly specific and effective. However, mutations in activators katG and ethA have been linked to most of the clinical resistance in the diagnosed cases of drug-resistant TB.[9, 10] Compounds that do not require activation and directly target InhA represent a promising approach to circumvent this resistance mechanism. The first report of such small molecule InhA inhibitors came from our laboratories in 2003, featuring triclosan and two members of the Genzyme compound library.[11] Triclosan, for example, inhibits InhA without activation, although its use as an antitubercular may be limited by its sub-optimal bioavailability.[12] These small organic molecules, however, represent reasonable starting points for structure-based drug discovery efforts to afford effective InhA inhibitors. Aiding such an effort are several crystal structures, such as those of InhA:NADH,[13] InhA:NAD+:triclosan,[11] InhA:NAD+:Genz-10850,[11] InhA:NAD+:C16-substrate,[14] and InhA:INH-NAD[5] that were accessible at the outset of these investigations. During the course of this work, Sullivan et al. reported the X-ray structures of two triclosan analogs with 5-alkyl (n-pentyl and n-octyl) moieties, lacking the two B-ring chlorines.[6] This collection of structures provided a precisely defined active site of InhA and a thorough understanding of the ligand-enzyme interactions that render potent enzyme inhibition.
One promising route for the design of potent InhA inhibitors involves triclosan, a commercially available compound that has been reported to inhibit the enoyl acyl ACP reductases (ENR) from several species, including Plasmodium falciparum[15], Escherichia coli,[16] Bacillus subtilis,[17] Brassica napus,[18] and Pseudomonas aeruginosa.[19] The structures of ENR from Escherichia coli (FabI),[20] Brassica napus,[18] Plasmodium falciparum (PfENR),[21] and M. tuberculosis (InhA)[11] bound with triclosan have been characterized. In our laboratories, a series of 5-substituted triclosan derivatives was designed and synthesized in order to optimize the potency of triclosan in parallel against both purified PfENR[23] and InhA. This report discusses these efforts focused specifically on the SAR developed versus purified InhA and INH-sensitive and resistant M. tuberculosis.
Results and Discussion
Our efforts began with a survey of the complex structures of InhA bound with INH-NAD[5] and triclosan,[11] respectively. Two hydrophobic cavities that were capable of being filled were identified: the substrate-binding site and the pocket into which the isonicotinoyl group of INH-NAD protrudes. To date, all reported inhibitors of InhA occupied the hydrophobic cavity of the substrate-binding site, except the INH-NAD and ETH-NAD adducts. The isonicotinoyl moieties of the INH-NAD and ETH-NAD adducts were found in a hydrophobic pocket formed by movement of the side chain of Phe149. The pocket was underneath the fatty acyl substrate-binding site (as indicated in Figure 1) and, lined predominantly by hydrophobic groups from the side chains of Tyr158, Phe149, Met199, Trp222, Leu218, Met155, Met161, Gly192, and Pro193. This pocket may also serve as a portal to the external solvent at the left side of the active site (as indicated in Figure 1). The active forms of both INH and ETH occupied the same pocket and were extremely potent against InhA (Ki = 5 nM and 7 nM, respectively),[6, 7] validating this cavity as a suitable site to target with new inhibitors. Superimposition of the structures of InhA:INH-NAD and InhA:NAD+:triclosan indicated that the chlorine atom at the 5-position of the triclosan A-ring was about 2 Å away from the binding pocket of isonicotinoyl moiety of the INH-NAD adduct and was in van der Waals contact with Pro193, Met199, and Phe149. Based on this structural information, we hypothesized that it may be possible to replace the 5-chloro with various moieties to occupy this isonicotinoyl binding pocket and, thus, increase the in vitro activity against InhA. It should be noted that this strategy contrasts with that of Sullivan et al. to extend relatively long n-alkyl chains off what is essentially the triclosan 5-position (in molecules where the two B-ring chlorines have been excised) to mimic substrate analog trans-2-hexadecenoyl-(N-acetylcysteamine)-thioester, whose structure with InhA we reported in 1999.[14]
Figure 1.

Cross-section through the surface of the InhA active site of superimposed structures of InhA in complex with the INH-NAD adduct and triclosan. The carbon atoms of the INH-NAD adduct and triclosan are colored in gold and white, respectively. Other atoms are colored according to the atom type as following: oxygen atoms are red, nitrogen atoms are blue, chloride atoms are cyan, and phosphorus atoms are purple. This figure was made using the program Spock.[27]
A series of triclosan derivatives with modifications at the 5-position of triclosan was evaluated for their inhibition of purified InhA. These small molecules were prepared during the course of a concurrent program to investigate 5-substituted triclosan analogs as PfENR inhibitors.[23] Inhibitors with hydrophobic substituents, such as alkyl groups (compounds 2 and 7) were much more potent than those with hydrophilic substituents (compounds 3, 4 and 5) (Table 2). This result is consistent with our proposal that the 5-substituent of triclosan projects into a hydrophobic cavity of InhA. It is interesting to note the lack of activity of phenyl 6 which may be explained by a potential steric clash with Phe149 (as clarified below).
Table 2.
In vitro activities of select triclosan derivatives against M. tuberculosis InhA.[a]
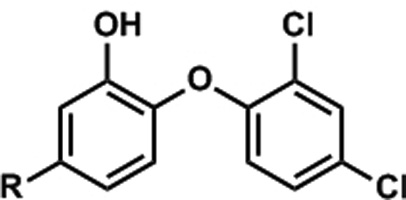 | ||
|---|---|---|
| Cmpd | R | InhA IC50 (nM) |
| triclosan | Cl | 1100 ± 180 |
| 2 | Me | 800 ± 99 |
| 3 | 2H-tetrazol-5-yl | >10000 |
| 4 | COOH | >10000 |
| 5 | C(O)NH2 | >10000 |
| 6 | Ph | >10000 |
| 7 | CH2(C6H11) | 110 ± 31 |
Values reported as the mean ± standard error for at least three independent measurements.
Following the initial modest activity of compounds 2 and 7, a series of 5-alkyl triclosan derivatives was examined for their inhibitory activity against InhA. Their potency versus InhA appeared to increase with the chain length of the 5-alkyl groups (Table 3). Highest inhibitory potencies were observed for compounds 10 and 12 with four-carbon chains. Sullivan observed the same trend with 5-alkyl substituted triclosan derivatives (des-chloro analogs) where the two chlorines of the B-ring were excised.[22] It was found that the in vitro activity of those inhibitors was optimal at a carbon chain length of eight. Triclosan inhibitors show better in vitro activity than their des-chloro counterparts, with 5-substituents of the same carbon chain length, from Sullivan’s report (IC50 = 120 ± 19 nM versus 2000 ± 700 nM for derivatives with an ethyl substituent and IC50 = 55 ± 20 nM versus 80 ± 15 nM for derivatives with an n-butyl substituent), suggesting that the two chlorine atoms on the B-ring contribute to the binding of the inhibitor to the enzyme. Molecular modeling suggests that this may be due to favorable van der Waals interactions of the 4'-Cl with Phe97 and Met103. These results imply that the hypothesis of Sullivan[22] that the B-ring chlorines do not contribute to efficacy may not be correct.
Table 3.
In vitro activities of select triclosan derivatives against M. tuberculosis InhA.[a]
 | |||
|---|---|---|---|
| Cmpd | R1 | R2 | InhA IC50 (nM) |
| 8 | CH2CH3 | Cl | 120 ± 19 |
| 9 | (CH2)2CH3 | Cl | 91 ± 15 |
| 10 | (CH2)3CH3 | Cl | 55 ± 20 |
| 11 | CH2CH(CH3)2 | Cl | 96 ± 46 |
| 12 | (CH2)2CH(CH3)2 | Cl | 63 ± 9 |
| 13 | CH2CH(CH3)CH2CH3 | Cl | 130 ± 56 |
| 14 | 2-pyridyl | CN | >10000 |
| 15 | 3-pyridyl | Cl | >10000 |
| 16 | 4-pyridyl | CN | >10000 |
| 17 | CH2(2-pyridyl) | Cl | 29 ± 11 |
| 18 | CH2(3-pyridyl) | Cl | 42 ± 10 |
| 19 | CH2(4-pyridyl) | CN | 75 ± 16 |
| 20 | o-CH3-Ph | Cl | 1300 ± 77 |
| 21 | o-CH3-Ph | CN | >10000 |
| 22 | m-CH3-Ph | Cl | 870 ± 110 |
| 23 | p-F-Ph | Cl | >10000 |
| 24 | CH2Ph | Cl | 51 ± 6 |
| 25 | (CH2)2Ph | Cl | 21 ± 8 |
| 26 | (CH2)3Ph | Cl | 50 ± 14 |
Values reported as the mean ± standard error for at least three independent measurements.
The alkyl substituents in previous studies were all unbranched. In the present report, compounds 11, 12, and 13, with β- or γ-branched methyl substituents, failed to improve upon the potency of their straight-chain counterparts (compounds 9 and 10) of the same chain length. Similarly, cyclohexylmethyl analog 7 exhibited an IC50 = 110 nM. The 2.8 Å crystal structure of InhA in complex with compound 7 (Figure 2) was solved to provide a structural basis for the activity difference amongst the alkyl substituents. The crystal belonged to I4122, a space group for InhA that has not been reported previously. It is worth noting that the substrate binding loop (residues 195–205) was ordered in the structure of InhA bound with compound 7, while it has been disordered in the structures of InhA bound with substrate, triclosan, and all other triclosan derivatives solved to date. The structure was readily superimposed on those of InhA in complex with 5-pentyl-2-phenoxyphenol,[22] which intriguingly features a somewhat strained C4-C5 portion of its pendant alkyl chain, and the C16 substrate analog[14] (overlapping the position of carbons 4 through 7 of the U-shaped acyl chain). The 5-cyclohexylmethyl group formed predominantly hydrophobic interactions with the side chains of Phe149, Ile215, Leu218, Met155, Tyr158, and Met199. Based on this structure, we hypothesized that 5-substituents with a longer alkyl chain would create more extensive hydrophobic interactions in this pocket. Therefore, it was not surprising that the potency of the 5-alkyl triclosan analogs increased with the chain length. However, it is not so obvious from a structural point of view why our limited subset of methyl-branched inhibitors do not exhibit greater potency than the unbranched inhibitors of the same chain length. A possible explanation could be steric clashes with one or more of the side chains of Phe149, Tyr158, and Met199 with the branched methyl groups. Compared to the structure of InhA bound with Sullivan’s 5-pentylphenol, the side chain of Met199 on the substrate-binding loop flipped approximately 100° to form a hydrophobic interaction with the cyclohexyl group at a distance of 3.6 Å. In addition, the B-ring of compound 7 also rotated about 30° from its position in 5-pentyl-2-phenoxyphenol to allow the 4'-chloride to form a weak hydrogen bonding interaction with the amide N-H of Met98 at a distance of 3.2 Å.
Figure 2.
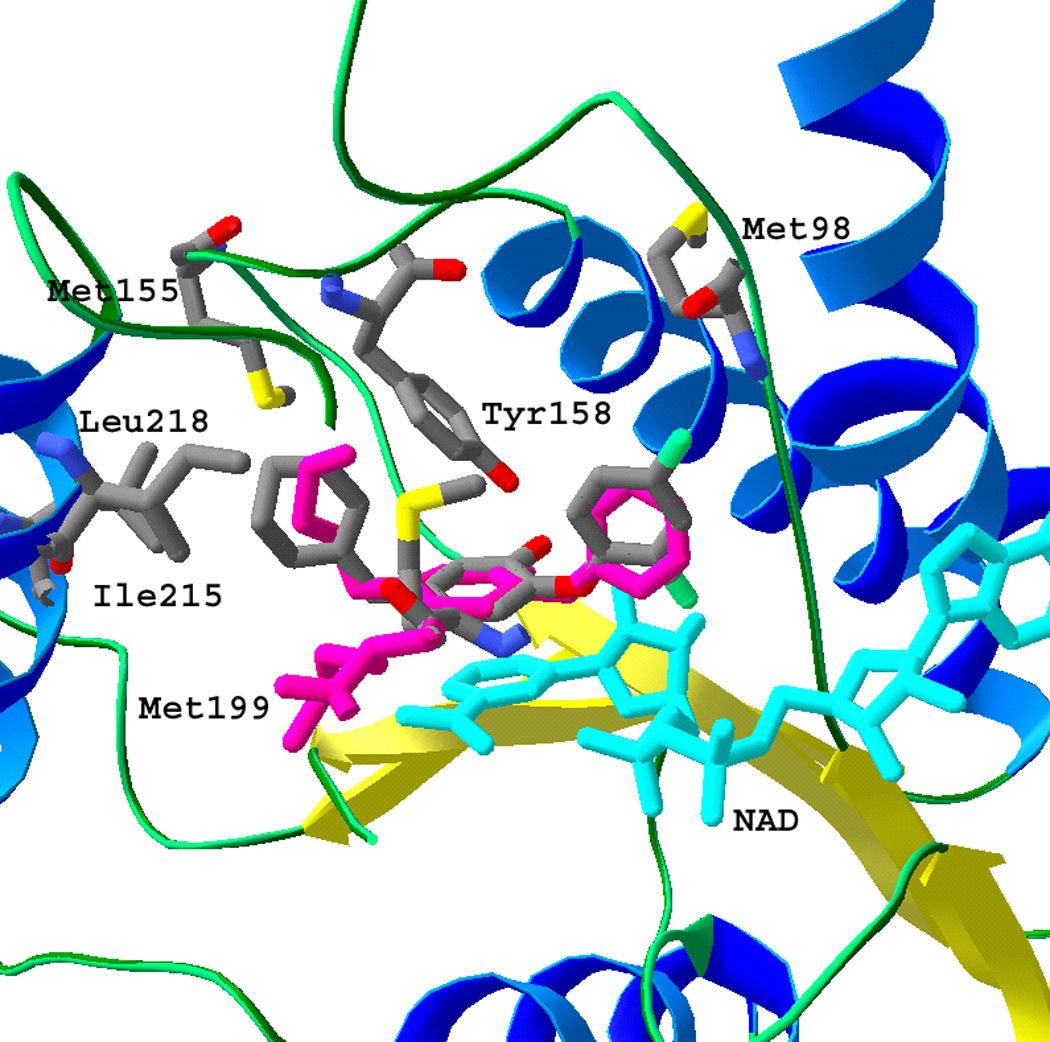
The superposition of crystal structures of InhA (ribbon and tube with key residues in stick format) in complex with 5-pentyl-2-phenoxyphenol[22] (stick drawing, colored in purple) and compound 7 (stick drawing, carbon atoms colored in gray) in the presence of NAD+ (stick drawing, colored in cyan). The B-ring of the 5-pentyl triclosan analog was in a different orientation from 7, because of the lack of two chlorides. The cyclohexylmethyl group of 7 was in a similar position to the pentyl group of 5-pentyl triclosan analog. This figure, in addition to Figures 3 and 4was made using the program SwissPDB viewer.[28]
Analogs of phenyl 6 were examined to improve upon its surprisingly poor enzyme inhibition (Table 3). Pyridyl derivatives 14 – 16 and simple phenyl analogs 20 – 23 were also quite unsatisfactory InhA inhibitors. Although the crystal structures for InhA bound with 5-phenyl or 5-pyridyl triclosan analogs have not been obtained to date, the inhibitors were modeled[24] into the active site based on the structure of InhA:triclosan. A directly attached aryl group at the 5-position appeared to be too close to the side chain of Phe149 (shortest distance was about 1.4 Å between the aryl substituent and the phenyl group of Phe149), leading to steric clashes. The hydrophobic pocket of interest was separated from the 5-position by a distance of 2.5 Å.
Therefore, to reorient and better position the hydrophobic aryl group into the target pocket, a linker varying from 1 to 3 carbons was designed between the aryl substituent and the 5-position carbon on the A-ring (Table 3). Consistent with molecular modeling, an evaluation of 5-position substituents of the type (CH2)nAr demonstrated that carbon linkers significantly increased the inhibitory potency versus purified enzyme. Among them, compound 25 had the highest potency of IC50 = 21 nM, representing a 50-fold increase compared to triclosan. It is the most potent triclosan derivative against purified InhA we have studied to date. Compounds 24 and 26, with similar activities against InhA (IC50 equal to 51 nM and 50 nM, respectively), were also active inhibitors. The crystal structures of InhA bound with 24 and 25 showed that the (CH2)nPh group extended into the pocket and formed hydrophobic interactions with residues Leu218, Ile215, Phe149, Met199, and Pro193 (Figure 3). The triclosan backbone atoms of 24 and 25 were in nearly identical positions, but their 5-substituents were clearly different. The phenyl group of 24 was positioned in the center of the binding pocket, while the phenyl group of 25 protruded about 2 Å deeper into, and was closer to the end of, the binding pocket due to the longer carbon linker. Thus, 25 additionally engaged Trp222 in a hydrophobic interaction. The side chains of Leu218 and Ile215 rotated 30° and shifted 1.5 Å, respectively, to accommodate the phenyl ring of 25, which also flipped ca. 70° from the position of the phenyl group of 24. All of these conformational changes suggest that there were more hydrophobic interactions between the active site residues and the 5-substituent of 25 than that of 24, which may explain why 25 was twice as potent than 24 against InhA.
Figure 3.

The superposition of crystal structures of InhA (ribbon and tube with key residues in stick format) in complex with triclosan (stick drawing, carbon atoms colored in gray), 24 (stick drawing, colored in purple), and 25 (stick drawing, colored in gold) in the presence of NAD+ (stick drawing, colored in cyan). The 5-phenyl groups of 24 and 25 are shown making van der Waals contacts with the side chains of Tyr158, Pro193, Leu218, Ile215, and Phe149. The movements of Ile215 and Leu218 in response to the triclosan 5-substituent are depicted by showing this residue in stick format with the coloring of the respective ligand. It should be noted that Met-199 was not depicted for the sake of clarity.
To further increase the potency of the 5-(CH2)nPh series through potential mimicry of the INH-NAD isonicotinoyl moiety, derivatives with 5-CH2(n-pyridyl) (n = 2, 3, and 4) substituents (compounds 17 – 19) were examined for in vitro activity. In the crystal structure of InhA:INH-NAD,[5] the pyridyl group of the INH-NAD adduct formed a hydrogen bonding interaction with a buried water molecule. Without a carbon linker, the 5-pyridyl triclosan derivatives 14 – 16 showed very low activity versus InhA (IC50 > 10 µM), presumably due to a steric clash with Phe149. In contrast, with a linker of one carbon, pyridyl analogs 17 – 19 were all potent inhibitors (IC50 < 80 nM), and their IC50 values were in the order 17 < 18 < 19. Compared to benzyl derivative 24, the activity of 17 increased ca. twofold while the activity of 18 increased only slightly, and 19 was slightly less potent. The structure of InhA:NAD+:17 demonstrated that the pyridyl ring of 17 extended into the substrate-binding site to form hydrophobic interactions with the side chains of Tyr158 and Phe149 (Figure 4). These interactions are similar to those of the phenyl group in InhA:NAD+:24. However, the nitrogen atom on the 2-position of the pyridyl ring also formed a hydrogen-bonding interaction with the side chain carboxylate of Glu219 through a water molecule. Modeling studies indicated that similar interactions could also exist for 18, but were not likely for 19, based on the distance between the pyridyl nitrogen atom and the side chain oxygen of Glu219. The nitrogen atom of 19 potentially pointed to the hydrophobic side chains of residues Met155 and Leu218, which may not be favored energetically. The superimposition of InhA:NAD+:17 and InhA:NAD+:triclosan,[11] demonstrated movement of both Glu219 and Leu218 to accommodate the 5-pyridylmethyl moiety.
Figure 4.

The superposition of crystal structures of InhA (ribbon and tube with key residues in stick format) in complex with triclosan (stick drawing, all atoms colored in purple) and 17 (stick drawing, carbon atoms in gray) in the presence of NAD+ (stick drawing, colored in cyan). The nitrogen atom on the pyridyl ring formed a hydrogen bonding interaction with the side chain of Glu219 through a water molecule. The side chain of Glu219 rotated 90° from its original position (colored in purple) to form this hydrogen bonding interaction. It should be noted that Phe-149 was not depicted for the sake of clarity.
A comparison of the X-ray crystal structures of bound 7, 17, 24, and 25 with that of INH-NAD (PDB code 1ZID) is instructive. At the outset, our goal was to reach from the 5-position of triclosan into the isonicotinoyl-binding pocket. The Phe149 side chains from the four triclosan analog structures overlapped well with each other, but not that of 1ZID. In 1ZID, the isonicotinoyl group of INH was stabilized by the rotation of Phe149. The various triclosan 5-substituents, however, did not occupy the same volume as the isonicotinoyl group, orienting similarly to the C16 chain of the structurally characterized substrate analog[14] and the appended alkyl substituent in Sullivan’s structures,[22] and thus did not perturb Phe149. In the 1ZID structure, the movement of Phe149 evidently displaced Tyr158. While this motion was tolerated in 1ZID, in the triclosan structures, the phenol moiety is positioned through an interaction with the hydroxyl of Tyr158. Loss of the triclosan phenol-Tyr158 interaction would most likely result in complete abrogation of inhibitor binding as des--phenol triclosan analogs have displayed little if any activity versus InhA. Thus, while the triclosan analogs can attempt to place substituents proximal to the INH binding pocket, they cannot occupy the exact volume of the isonicotinoyl moiety, as this would ultimately require the movement of Phe149 and Tyr158, thus losing a key hydrogen-bonding interaction with Tyr158.
The activities of three 5-alkyl (7, 10, and 11) and three 5-aryl triclosan derivatives (24, 25, and 26) against six different M. tuberculosis strains were assayed (Table 4). Among these strains, H37Rv is the M. tuberculosis wild-type, sensitive to isoniazid. The InhA(S94A) strain, carrying the inhA(S94A) allele, has been demonstrated to confer resistance to isoniazid through weakening of co-factor binding.[6] The InhA(C-15T) strain is an M. tuberculosis spontaneous mutant carrying the inhA expression region mutation (C-15T).[6] It has been shown that the C-15T inhA promoter mutation mediates enhanced transcription of inhA, resulting in INH and ETH resistance. The ΔkatG mutant features the complete deletion of the katG gene and, thus, obviates the potential for INH activation. Most significantly, strains 12081 and 5071 are multi-drug resistant clinical isolates from Mexico that are INH resistant without known mutations in katG, inhA, ndh, and ahpC. 12081 (SCG V) was, in our hands, resistant to INH, streptomycin, and rifampicin, while 5071 (SCG 3b) was resistant to both INH and streptomycin.
Table 4.
Activities of triclosan derivatives against wild-type and mutant M. tuberculosis strains.
| Cmpd | MIC H37Rv (µg/ml) (µM) |
MIC H37Rv inhA (S94A) (µg/ml) (µM) |
MIC H37Rv PmabAinhA (C-15T) (µg/ml) (µM) |
MIC H37Rv ΔkatG (µg/ml) (µM) |
MIC clinical isolate 12081 (µg/ml) (µM) |
MIC clinical isolate 5071 (µg/ml) (µM) |
|---|---|---|---|---|---|---|
| INH | 0.060 (0.44) | 0.50 (3.6) | 0.80 (5.8) |
>200 (>1400) | 1.0 (7.3) |
8.0 (56) |
| triclosan | 40 (140) | |||||
| 7 | 9.4 (27) |
37 (110) | 37 (110) |
19 (55) |
37 (110) |
4.7 (13) |
| 10 | 9.4 (30) |
37 (120) | 37 (120) |
19 (60) |
37 (120) |
4.7 (15) |
| 11 | 19 (60) |
37 (120) | 37 (120) |
37 (120) |
75 (240) |
19 (60) |
| 24 | 9.4 (27) |
75 (220) | 37 (110) |
19 (55) |
75 (220) |
9.4 (27) |
| 25 | 19 (52) |
75 (210) | 75 (210) |
37 (110) |
75 (210) |
19 (52) |
| 26 | 4.7 (13) |
19 (50) |
19 (50) |
9.4 (26) |
37 (100) |
4.7 (13) |
The selected six compounds with high potency against InhA in vitro (IC50 ≤ 110 nM) all demonstrated good antitubercular activity against the wild-type H37Rv strain, exhibiting improvements over the whole-cell efficacy of triclosan. Clearly, the 5-chloro group of triclosan is not optimal and the use of larger and more hydrophobic moieties is advantageous both in terms of InhA inhibition and whole-cell potency. The most active compound, 26 (MIC = 13 µM), was superior to that of the second line drug ethionamide (MIC = 18 µM),[6] while being greater than ten times as potent as triclosan. The activities of these compounds against purified InhA and cultured tuberculosis strains generally correlated well. For example, compound 10, which had twofold higher activity against InhA than 11, also exhibited twofold better antitubercular activity. This supports a hypothesis that the unbranched 5-alkyl substituent has an advantage over its branched counterpart. In addition, compound 26, which displayed the highest antitubercular activity in the series, also had the second highest potency against InhA. Exceptions may be noted such as with 25, which showed the highest potency among the (CH2)nPh series and yet the worst antitubercular activity. As may be expected, a number of factors other than in vitro efficacy versus purified InhA, including pharmacokinetic parameters, contribute to the whole-cell inhibition of mycobacterial growth.
It is interesting to compare the H37Rv data for 5-n-butyltriclosan 10 and the corresponding des-chloro analog of Sullivan and co-workers. While 10 (IC50 = 55 nM) was slightly more active than its des-chloro relative versus InhA (IC50 = 80 nM),[22] the des-chloro compound (MIC = 10.8 µM) was more efficacious against H37Rv than 10 (MIC = 30 µM). It should be noted that the most potent triclosan analog reported to date is Sullivan’s n-octyl analog (IC50 = 5.0 nM vs. InhA; MIC = 6.5 µM vs. H37Rv).[22]
Triclosan has previously been reported to display promising activity against both INH sensitive and resistant M. tuberculosis strains.[11, 22] We next examined five strains that had not previously been tested against the triclosan family. Compared to their efficacy versus the wild-type strain, five of the six analogs examined showed 4 – 8 fold lower activity against the InhA(S94A) and InhA(C-15T) strains. This was slightly lower, yet still comparable in magnitude, to the 8- and 13-fold losses in potency for INH versus these two strains, respectively. The InhA(C-15T) strain has a higher expression of InhA, which leads to resistance to compounds that target the protein. The resistance to triclosan by InhA(S94A) strains of either M. tuberculosis or other bacteria has not been previously reported to the best of our knowledge. A G93V FabI (residue conserved in InhA) mutation has previously been found to confer E. coli resistance to triclosan.[25] The inhA(S94A) mutation has been demonstrated to decrease the efficacy of INH by weakening the binding of INH-NAD.[6] Comparatively, the binding of triclosan and its analogs to InhA highly depend on the interaction between the diaryl ether scaffold and NAD+. Quemard and co-workers reported that the S94A mutation weakened the binding of NADH by about 500-fold.[26] It is proposed that the same mutation may decrease the stability of the InhA:NAD+:triclosan-derivative complex and, thereby, abrogate inhibition. The resistance of triclosan derivatives by the InhA(S94A) and InhA(C-15T) strains is, thus, supportive of our proposal that the target of these triclosan relatives is indeed InhA.
The activity of the triclosan analog subset versus the ΔkatG mutant further substantiates the potential advantage of these InhA inhibitors over INH. While INH was more than 3000-times less active against the ΔkatG mutant, the triclosan analogs merely doubled their respective MICs. This is consistent with our genetic, enzymatic, and X-ray crystallographic data that demonstrate InhA inhibition by the triclosan family without the need for activation. It, therefore, follows that one would expect a lack of cross-resistance with many INH-resistant M. tuberculosis strains, where the katG mutations represent the dominant allele conferring resistance. Sullivan and colleagues have also demonstrated their phenol diaryl ethers to not suffer significant losses in potency versus clinical isolates with varying degrees of INH resistance.[22]
The two clinical strains in Table 5 were much more susceptible to the triclosan analogs than INH. Strain 12081 afforded 17-fold resistance to INH, which was reduced to a 4–8 factor resistance versus the triclosan subset. Against clinical isolate 5071, the triclosan subset maintained or improved (in two cases by two-fold) their respective wild-type potencies. These results should be contrasted to the greater than two orders of magnitude resistance conferred against INH. Thus, triclosan analogs may, in general, offer a high degree of activity against INH resistant M. tuberculosis.
Conclusion
The efficacy of these 5-substituted triclosan analogs has been shown to be tunable through a structure-driven optimization of the 5-position. Novel analogs afforded gains of 50-fold in InhA inhibition and tenfold in bacteriocidal activity. Most significantly, these triclosan derivatives demonstrated efficacy versus INH-resistant laboratory and clinical strains of M. tuberculosis. Further studies of triclosan and other small molecule inhibitors of InhA hold significant promise for the delivery of novel antituberculars that are effective against drug-resistant M. tuberculosis. Current efforts are underway to examine the physiochemical properties of our most promising analogs. Replacements for the phenol will be sought to avoid potential liabilities due to rapid Phase II metabolism,[12] preceding clearance. In addition, other modifications to the A- and/or B-rings of the diaryl ether scaffold can be envisioned that would potentially increase the resulting molecule’s stability to oxidative metabolism, while not sacrificing efficacy.
Experimental Section
Triclosan analog synthesis
The small molecules tested versus InhA and the mycobacterial strains were synthesized as described previously in the literature.[23]
Cloning, expression, and purification for Mycobacterium tuberculosis inhA
M. tuberculosis inhA was cloned into E. coli BL21 (DE3) as described previously.[13] The transformed E. coli were cultured in Terrific Broth media with 50 µg/mL carbenicillin at 37 °C until an OD600 of 0.8 was observed. inhA expression was induced for 20 h at 16 °C through addition of 1 mM isopropyl β-D-thiogalactopyranoside. The resulting cells harvested through centrifugation were re-suspended in 50 mM PIPES (pH 6.8) and 1 mM phenylmethylsulfonyl fluoride and lysed via French press. Exposure to DnaseI was followed by removal of insoluble material through centrifugation. The supernatant was subjected to a HiTrap Blue Sepharose column (AP Biotech), pre-equilibrated with the same buffer, using a fast protein liquid chromatography system and eluted through a NaCl gradient (0 – 2 M). Elution with 0.9 M NaCl afforded fractions containing InhA that were subsequently subjected to an octyl-sepharose column (AP Biotech), pre-equilibrated with 1 M NaCl, and eluted through a NaCl gradient (1 – 0 M). Pooling of InhA fractions and gel filtration through a Superdex 200 column were carried out to separate monomeric protein from aggregated material. SDS-PAGE and Coomassie Blue staining were consistent with homogeneous InhA in the yield of 40 mg/L from E. coli culture.
InhA enzyme assay
Experiments were carried out utilizing a Cary100 Bio spectrophotometer at 25 °C, through monitoring the oxidation of NADH to NAD+ at 340 nm. Reactions were initiated via addition of dodecenoyl-CoA (50 µM) substrate to mixtures containing InhA (5 nM), NADH (100 µM), and inhibitor (1 – 10000 nM). The IC50 was determined from a dose-response plot of enzyme fractional activity versus the concentration of inhibitor.
Antimycobacterial assays
The relevant M. tuberculosis strains were obtained from laboratory stocks and grown in Middlebrook 7H9 medium (Difco), supplemented with 10% (v/v) OADC enrichment (Difco), 0.2% (v/v) glycerol, and 0.05% (v/v) tyloxapol to an OD600 of ca. 1.0. The cultures were diluted 4 logs and 0.1 mL of the resulting dilutions were inoculated into 2 mL of Middlebrook 7H9 media with varying concentrations of inhibitor. The cultures were incubated with shaking at 37 °C for four weeks. The MIC was defined as the concentration of inhibitor that prevented visible mycobacterial growth.
Crystallization of InhA with selected inhibitors
The hanging drop vapor diffusion method was utilized. Typically, InhA was incubated with an inhibitor and NAD+ in the molar ratio of 1:2:100 for two hours. Co-crystallization was then attempted in hanging droplets consisting of 2 µL of 10 mg/mL protein solution and 2 µL of buffer (20% PEG 3350, 6% DMSO, 0.1 M N-(2-acetamido)iminodiacetic acid pH 6.8, 0.08 M NH4OAc) at 16 °C in Linbro plates against 0.5 mL of the same buffer. Protein crystals were observed after about four days.
Data collection and processing
Data were collected at 121 K utilizing cryo-protection solution with reservoir solution with an added 30% ethylene glycol. A crystal of InhA:17 diffracted X-rays to 1.98 Å at beam line 23ID at the Advanced Photo Source (APS), Argonne National Laboratory. Crystals of InhA:24, InhA:25 and InhA:7 diffracted X-rays to 2.30 Å, 2.80 Å and 1.98 Å, respectively, using a Rigaku Raxis detector coupled to an X-ray generator with a copper rotating anode (CuKα, λ = 1.54 Å). Diffraction data was obtained from a single crystal with 0.5° degree oscillation widths for a range of 180°. The data were integrated and reduced using HKL2000 (Table 1).[29]
Table 1.
Data collection and refinement statistics for reported X-ray structures.[a]
| InhA:7 | InhA:17 | InhA:24 | InhA:25 | |
|---|---|---|---|---|
| Maximum resolution (Å) | 1.97 | 1.98 | 2.30 | 2.80 |
| Space group | I4(1)22 | C2 | C2 | I4(1)22 |
| a (Å) | 90.0 | 125.6 | 125.6 | 90.0 |
| b (Å) | 90.0 | 92.3 | 92.4 | 90.0 |
| c (Å) | 183.9 | 103.0 | 102.4 | 183.1 |
| α (°) | 90.0 | 90.0 | 90.0 | 90.0 |
| β (°) | 90.0 | 106.4 | 106.5 | 90.0 |
| γ (°) | 90.0 | 90.0 | 90.0 | 90.0 |
| Unique reflections[b] | 27204 (2964) | 71784 (6260) | 43113 (3799) | 9708 (1063) |
| Rsym (%) | 6.0 (74.3) | 9.9 (70.5) | 10.3 (88.3) | 12.8 (82.3) |
| Completeness (%) | 99.8 (100) | 87.3 (65.5) | 99.1 (98.5) | 99.8 (100) |
| Redundancy | 10.2 (10.0) | 3.7 (2.1) | 5.3 (4.9) | 8.5 (8.6) |
| I/σ | 42.5 (4.3) | 15.7 (1.3) | 28.6 (3.3) | 22.3 (3.7) |
| Resolution range (Å) | 19.92-1.97 | 19.96-1.98 | 19.85 - 2.30 | 19.85-2.8 |
| # reflections | 25755 | 68885 | 46976 | 9157 |
| # atoms / subunit | ||||
| Protein | 1994 | 7826 | 7739 | 1994 |
| Cofactor (NAD) | 52 | 176 | 176 | 52 |
| Ligand | 22 | 88 | 88 | 23 |
| Solvent | 168 | 703 | 253 | 11 |
| Rcryst (%) | 20.7 | 20.0 | 20 | 22.1 |
| Rfree (%) | 25.2 | 25.3 | 26 | 26.8 |
| Average B-factors (Å2) | 35.0 | 1.98 | 40.2 | 54.8 |
PDB accession code are AAAA (7), BBBB (17), CCCC (24), and DDDD (25).
In parentheses is the value of the highest resolution shell.
Structure determination and model refinement
Initial phases of the InhA:inhibitor complexes were obtained through molecular replacement utilizing the apo-InhA structure (PDB code 1ENY) and refined with CCP4[30] (Table 1). Electron density maps were calculated and additional density was found, consistent with the respective inhibitor. The inhibitor was fit into the additional density and the whole model was rebuilt utilizing XtalView.[24] In the final refinement cycles, water molecules were added into peaks above 3-σ of the Fo – Fc electron density maps, such that the water molecules were within hydrogen-bonding distance from the appropriate protein atoms. Final statistics are in Table 1.
Acknowledgements
We would like to thank John Kim (AECOM) and Dr. Tsungda Hsu (AECOM) for preparing the M. tuberculosis ΔkatG strain and Professor David Alland (UMDNJ) for donation of the 5071 and 12081 clinical isolates.
This work has been supported by funding from the Medicines for Malaria Venture, the National Institutes of Health (PO1A1068135), and the Robert A. Welch Foundation (A-0015).
References
- 1.Vilchèze C, Jacobs WR., Jr Ann. Rev. Microbiol. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- 2.CDC. MMWR Morb. Mortal. Wkly. Rep. 2006;55:301–305. [PubMed] [Google Scholar]
- 3.Crofton J, Chaulet P, Maher D, Grosset J, Harris W, Norman H, Iseman M, Watt B. Guidelines for the management of Multidrug-resistant Tuberculosis (World Health Organization, Geneva) 1997
- 4.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 5.Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 6.Vilchèze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR., Jr Nat. Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 7.Wang F, Langley R, Gulten G, Dover LG, Besra GS, Jacobs WR, Jr, Sacchettini JC. J Exp Med. 2007;204:73–78. doi: 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE., 3rd Proc. Natl. Acad. Sci. U. S. A. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. Antimicrob. Agents Chemother. 2003;47:3799–3805. doi: 10.1128/AAC.47.12.3799-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hazbón MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, García-García L, León CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de León A, Alland D. Antimicrob. Agents Chemother. 2006;50:2640–2649. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr, Sacchettini JC, Fidock DA. J Biol Chem. 2003;278:20851–20859. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- 12.Wang L-Q, Falany CN, James MO. Drug Metab. Dispos. 2004;32:1162–1169. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- 13.Dessen A, Quemard A, Blanchard JS, Jacobs WR, Jr, Sacchettini JC. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 14.Rozwarski DA, Vilchèze C, Sugantino M, Bittman R, Sacchettini JC. J Biol Chem. 1999;274:15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 15.Surolia N, Surolia A. Nature Medicine. 2001;7:167–173. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 16.Heath RJ, Yu Y-T, Shapiro MA, Olson E, Rock CO. J. Biol. Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 17.Heath RJ, Su N, Murphy CK, Rock CO. J. Biol. Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 18.Roujeinikova A, Levy CW, Rowsell S, Sedelnikova S, Baker PJ, Minshull CA, Mistry A, Colls JG, Camble R, Stuitje AR, Slabas AR, Rafferty JB, Pauptit RA, Viner R, Rice DW. J. Mol. Biol. 1999;294:527–535. doi: 10.1006/jmbi.1999.3240. [DOI] [PubMed] [Google Scholar]
- 19.Hoang TT, Schweizer HP. J. Bacteriol. 1999;181:5489–5497. doi: 10.1128/jb.181.17.5489-5497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy CW, Roujeinikova A, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Nature. 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 21.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, Fidock DA, Sacchettini JC. J Biol Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. ACS Chem Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 23.Freundlich JS, Wang F, Tsai H-C, Kuo M, Shieh H-M, Anderson JW, Nkrumah LJ, Valderramos J-C, Yu M, Kumar TRS, Valderramos SG, Jacobs WR, Jr, Schiehser GA, Jacobus DP, Fidock DA, Sacchettini JC. J. Biol. Chem. 2007;282:25436–25444. doi: 10.1074/jbc.M701813200. [DOI] [PubMed] [Google Scholar]
- 24.McRee DE. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 25.McMurry LM, Oethinger M, Levy SB. Nature. 1998;394:531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 26.Quemard A, Sacchettini JC, Dessen A, Vilchèze C, Bittman R, Jacobs WR, Jr, Blanchard JS. Biochemistry. 1995;34:8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 27.Christopher JA. Program Manual. College Station, TX: The Center for Macromolecular Design, Texas A&M University; 1998. SPOCK: The Structural Properties Observation and Calculation Kit. [Google Scholar]
- 28.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. Number 4. [Google Scholar]