

Abstract
Although they lack a cell wall, mycoplasmas do possess a glycocalyx. The interactions between the glycocalyx, mycoplasmal surface proteins and host complement were explored using the murine pathogen Mycoplasma pulmonis as a model. It was previously shown that the length of the tandem repeat region of the surface lipoprotein Vsa is associated with susceptibility to complement-mediated killing. Cells producing a long Vsa containing about 40 repeats are resistant to complement, whereas strains that produce a short Vsa of five or fewer repeats are susceptible. We show here that the length of the Vsa protein modulates the affinity of the M. pulmonis EPS-I polysaccharide for the mycoplasma cell surface, with more EPS-I being associated with mycoplasmas producing a short Vsa protein. An examination of mutants that lack EPS-I revealed that planktonic mycoplasmas were highly susceptible to complement killing even when the Vsa protein was long, demonstrating that both EPS-I and Vsa length contribute to resistance. In contrast, the mycoplasmas were resistant to complement even in the absence of EPS-I when the cells were encased in a biofilm.
Introduction
Mycoplasmas are widely distributed throughout nature as parasites and pathogens that are dependent on their host for survival. These wall-less organisms cause a variety of chronic infections and generally are surface parasites, adhering to and colonizing the epithelial linings of the respiratory and urogenital tracts (Razin et al., 1998; Razin, 1999). Mycoplasma pulmonis is the causative agent of murine respiratory mycoplasmosis, and must have the capability to overcome the innate responses of host immunity such as the complement system (Lindsey et al., 1971; Taylor-Robinson et al., 1978). Typically, micro-organisms activate complement and become coated with complement components that can lead to phagocytosis or complement-mediated killing by formation of the membrane attack complex (MAC), a multimeric protein complex that forms a transmembrane channel and lyses the cells (Alitalo et al., 2001; Rother et al., 1997). One factor of M. pulmonis that has a known role in avoiding killing by complement is the Vsa (variable surface antigen) protein, a size- and phase-variable lipoprotein exposed at the cellular surface (Bhugra et al., 1995; Shen et al., 2000; Simmons et al., 1996). Phase variation results from isotype switching, which occurs when one of the silent vsa genes is combined with the vsa expression site by site-specific DNA inversion (Bhugra et al., 1995; Shen et al., 2000). Size variation results from slipped-strand mispairing, which occurs during DNA replication of the tandem repeat region of the vsa gene. The host immune response is thought to exert selection pressure for Vsa variants (Denison et al., 2005; Gumulak-Smith et al., 2001; Simmons & Dybvig, 2003; Simmons et al., 2004). The particular isotype that is expressed is not thought to be a factor in the avoidance of complement lysis. Only the length of the Vsa protein affects the ability of M. pulmonis to resist killing, with an inverse relationship between the length of the Vsa protein and susceptibility. Cells producing a long Vsa with 40–60 tandem repeats are resistant to complement, while cells producing a protein with five or fewer repeats are susceptible (Simmons & Dybvig, 2003; Simmons et al., 2004). Variability in the number of repeats in the Vsa protein has multi-faceted roles in immune avoidance, adherence and biofilm formation (Simmons & Dybvig, 2003; Simmons et al., 2004, 2007). Although mycoplasmas producing a long Vsa protein are protected from complement, the complement cascade is nevertheless activated. Bystander haemolysis assays demonstrate that the MAC is formed and complement component C3b binds to the mycoplasmal surface (Simmons et al., 2004).
The polysaccharide EPS-I of M. pulmonis is predicted to be a linear chain of alternating residues of glucose and galactose, with galactose being the terminal sugar (Daubenspeck et al., 2009). Mutants that do not produce EPS-I have a reduced ability to adhere to pulmonary epithelial cells but an enhanced ability to form a biofilm as compared with wild-type mycoplasmas producing a Vsa protein of equivalent length and isotype. Genetic complementation of the mutants results in full restoration of all observed phenotypes (Daubenspeck et al., 2009).
Although previous studies have not explored the role of mycoplasmal polysaccharides in protection from host defences, polysaccharides of numerous pathogenic bacteria can temper host responses by interfering with immune surveillance, phagocytosis, bacteriolysis, cellular adhesion, biofilm production and protection from complement (Alitalo et al., 2001; Comstock & Kasper, 2006; Keo et al., 2011; O’Riordan & Lee, 2004). We report here that mutants deficient in the production of EPS-I were killed by complement more readily than wild-type mycoplasmas that produced the same size and isotype of Vsa protein. We further demonstrate that the EPS-I mutants encased within a biofilm are protected from complement, affirming the role of mycoplasma biofilms in providing protection from innate immunity.
Methods
Mycoplasma strains.
Unless stated otherwise, M. pulmonis was cultured in mycoplasma broth (MB) and assayed for colonies on mycoplasma agar (MA) (Dybvig et al., 2000; Simmons & Dybvig, 2003). The strains of M. pulmonis used in this study are summarized in Table 1. CTp12 is a wild-type strain (Simmons & Dybvig, 2003). All other strains are derived from strain CT and contain transposon Tn4001T at a genomic location described previously (Dybvig et al., 1995, 2000; French et al., 2008). Mutants lacking the EPS-I polysaccharide, CTG1291 and CTG1701, contain transposon disruptions in the MYPU_7420 and MYPU_7410 genes, respectively. The complemented mutant CTG1701-C contains the operon consisting of MYPU_7410 and 7420 cloned into CTG1701, restoring EPS-I production. CTG38 contains the transposon at an innocuous genomic location.
Table 1. Summary of strains used in this study.
| Strain | Vsa type | Vsa repeat length | Complement susceptibility | Produces EPS-I | Reference |
| CTG-R5 | G | R5 (short) | + | + | Daubenspeck et al. (2009); Simmons et al. (2004) |
| CTG38 | G | R40 (long) | − | + | Daubenspeck et al. (2009) |
| CTG1291 | G | R40 (long) | + | − | Daubenspeck et al. (2009) |
| CTG1701 | G | R40 (long) | + | − | Daubenspeck et al. (2009) |
| CTG1701-C | G | R40 (long) | − | + | Daubenspeck et al. (2009) |
| CTG-R40 | G | R40 (long) | − | + | Simmons et al. (2004) |
| CTp12 | A | R40 (long) | − | + | Simmons & Dybvig (2003) |
Complement killing of planktonic cells.
Mycoplasmas were cultured in 3 ml MB until stationary phase was reached. Cells were harvested by centrifugation, washed three times with 1 ml PBS, and suspended in 1 ml freezing medium (MB 80 %, glycerol 20 %). Cells were sonicated at 50 % power with a 50 % duty cycle on a Branson Sonifier 450 for 30 s to gently disrupt cell aggregates. Aliquots were frozen at −80 °C. One frozen aliquot of each strain was thawed and assayed for c.f.u. to determine the titre of the stocks of mycoplasma. Lyophilized guinea pig serum (GPS; Colorado Serum Company) was reconstituted with deionized water, aliquoted, and stored at −80 °C. Half of the aliquots were heat-inactivated (HIA) at 56 °C for 30 min prior to freezing. The reaction solution consisted of GPS or HIA-GPS diluted to 20 % with 50 µl normal saline (0.9 % NaCl) plus 104 c.f.u. of the mycoplasma test strain in 50 µl MB. Divalent cations were added to a final concentration of 5 mM Mg2+ and 1 mM Ca2+ in a total volume of 100 µl. After incubation in a water bath at 37 °C for 30 min, the reaction mixtures were placed on ice and assayed for c.f.u. Three samples were assayed for each strain in each experiment. The percentage c.f.u. recovered for each replicate tube was represented by the fraction, multiplied by 100, of the c.f.u. recovered after treatment with GPS relative to the c.f.u. recovered after treatment with HIA-GPS (Simmons & Dybvig, 2003, 2007; Simmons et al., 2004; Taylor-Robinson et al., 1978).
Complement killing within a biofilm.
M. pulmonis strains CTG-R5 and CTG1291 were cultivated in nine-well tissue culture plates. The lots and concentrations of the GPS, the HIA-GPS and the cations were the same as used for killing of planktonic mycoplasmas as described above, except that the volume of the reaction solution was increased to 200 µl in order to cover the bottom of the wells. After incubation at 37 °C for 30 min, the wells were gently washed three times with PBS and scraped. The cells were transferred to microcentrifuge tubes and placed on ice. Tubes were sonicated under the conditions described above to break up aggregates and assayed for c.f.u. (Simmons & Dybvig, 2007). The percentage of mycoplasmas surviving treatment with complement was represented by the fraction, multiplied by 100, of the c.f.u. recovered after treatment with GPS relative to the c.f.u. recovered after treatment with HIA-GPS.
Complement deposition analysis.
Serum from DBA2/NCr mice (Frederick Cancer Research) was diluted 10-fold in normal saline and supplemented with Mg2+ and Ca2+ to final concentrations of 5 and 1 mM, respectively. Samples of 20 µl were withdrawn from cultures of CTp12 that were grown for 3 days, placed onto glass microscope slides and dried. One hundred microlitres of the mouse serum, or HIA-mouse serum as a control, was placed onto the glass microscope slides and incubated at 37 °C for 30 min. The mycoplasmas were fixed by incubating with 100 µl of 10 % neutral buffered formalin for 30 min at room temperature. To detect the deposition of complement, the slides were incubated with goat anti-murine C3 (Immunology Consultant Laboratory) and Alexa Fluor 594-conjugated donkey anti-goat (Invitrogen) as primary and secondary antibodies, respectively. The slides were incubated with Hoechst 33342 at 10 µg ml−1 to detect the DNA of the mycoplasmas. The slides were washed three times with PBS in between each incubation. The slides were examined at a magnification of ×640 with a Leica HC epifluorescence microscope fitted with the Chroma 86012v2 filter set. The DAPI and Texas red filter sets were used for image acquisition. In some cases, the image was acquired in multiple focal planes, and the Depth From Focus function on the ImageJ application (v1.45, National Institutes of Health) was used to make a composite image showing an extended depth of field.
Vsa length determination.
Total protein from each strain of mycoplasma was separated by SDS-PAGE, transferred to a nitrocellulose membrane (Millipore), and exposed to the monoclonal antibody 7.1-2, which recognizes the conserved portion of all Vsa proteins, as previously described (Bhugra et al., 1995; Denison et al., 2005). Bands that reacted with the antibody were visualized and photographed after exposure to 5-bromo-4-chloro-3-indolyl phosphate–nitro blue tetrazolium chloride (Sigma).
GC/MS.
For analysis of EPS-I polysaccharide by GC, mycoplasmas were grown under conditions designed to minimize the presence of glycomoieties from the horse serum that is a component of MB. These glycomoieties can adsorb to the surface of the mycoplasma, resulting in noise on the gas chromatogram. Tissue culture flasks (75 cm2) containing 20 ml MA were overlaid with 20 ml serum-free medium (SFM) that was prepared similarly to that described by Yus et al. (2009). Unless stated otherwise, the SFM reagents were obtained from Sigma. Mixed in 1 l of water were 6 g glucose, 13.5 g Dulbecco’s modified Eagle’s medium (Mediatech), 6 ml IsoVitaleX (Becton Dickinson), 1.2 ml 20 % degraded herring sperm DNA, ampicillin (final concentration 50 µg ml−1), 2.5 g Select Peptone 140 (Life Technologies), 0.2 mg α-lipoic acid, 20 mg uracil, 20 mg spermine, 0.5 g glycerol and 6.8 g of a 136 g amino acid mixture. The amino acid mixture consisted of 7.1 g l-alanine, 12.5 g l-arginine, 10.6 g l-asparagine, 5.4 g glycine, 11.8 g l-histidine, 8.4 g l-isoleucine, 8.4 g l-leucine, 9.3 g l-lysine, 23.2 g l-methionine, 2.0 g l-phenylalanine, 9.2 g l-proline, 1.3 g l-serine, 7.6 g l-threonine, 2.0 g l-tryptophan, 0.4 g l-tyrosine and 16.8 g l-valine. SFM was adjusted to pH 7.8 and sterilized by filtration.
After incubation to the late exponential growth phase, the mycoplasmas in the SFM overlay were harvested by centrifugation and washed three times with 1 ml buffer (100 mM Tris/HCl, 10 mM MgCl2, pH 7.8). The washes were saved and pooled. The cells were sonicated at full power at 90 % duty cycle on a Branson Sonifier 450 for 30 s to lyse the cells. Samples consisting of cell lysate or washes were henceforth treated identically. The samples were heated to 80 °C for 15 min to denature proteins and digested overnight at 37 °C with 25 µg DNase I and 25 µg Rnase A followed by digestion overnight again at 37 °C with 25 µg proteinase K. The samples were dialysed with 5000 vols water (Millipore) in 2000 molecular-mass cut-off dialysis cassettes (Pierce). The samples were dried, treated with acidic methanol (1 : 10 acetyl chloride to methanol) for 16 h and dried again. The resulting material was dissolved in 150 µl methanol, transferred to Polyspring inserts, dried, and sealed under argon. The samples were derivatized with 50 µl of the reagent HMDS+TMCS+pyridine, 3 : 1 : 9 (Sylon HTP kit) (Sigma). Samples were analysed by GC/MS with an Agilent Technologies 6890N Network GC System and a 5973 Network Mass Selective Detector with MSD Productivity Chemstation Software Rev. D.00.00. The relative amount of EPS-I in each sample was determined by calculating the area under the peaks of the glucose and galactose methyl glycosides (Daubenspeck et al., 2009).
Staining of mycoplasma cultures with India ink.
Mycoplasmas were grown in broth in 15 ml polypropylene tubes for 3 days. India ink (Speedball Art Products) was diluted twofold in PBS and centrifuged at 12 000 g for 1 min to remove clumps of carbon black. Seven-microlitre samples of the mycoplasmas were mixed with an equal volume of India ink on glass microscope slides, covered with 22 mm×22 mm glass coverslips, and immediately imaged with a Leica HC microscope equipped with optics for differential interference contrast microscopy. In some experiments, the mycoplasmas were washed by centrifugation three times in PBS before placing on the slides and applying the India ink. In other experiments, the mycoplasma cultures were incubated with Hoechst 33342 at a final concentration of 20 µg ml−1.
Statistical analysis.
Statistical analysis was performed with the JMP version 8 software package (SAS Institute). Analysis of variance (ANOVA) was used with the Student–Newman–Keuls posthoc test to compare the level of killing of each strain with those of the other strains. Student’s t test was used in analysing the complement killing data from mycoplasmas that were allowed to grow in biofilms. All data reported as statistically significant have a P value of less than 0.01.
Results
EPS-I is required for planktonic cells to resist complement-mediated killing
Strain CTG-R5 produces a short VsaG protein with five tandem repeats. The remaining strains used in this study all produce a long Vsa protein with approximately 40 tandem repeats of isotypes as listed in Table 1. The percentage survival of mycoplasmas incubated with complement was calculated from the ratio of c.f.u. recovered from cells incubated with GPS to those incubated with HIA-GPS and is reported as percentage survival along with the standard error of the mean (Fig. 1). In concordance with previous studies, wild-type mycoplasmas producing a long Vsa, such as strain CTG38, were resistant to complement killing (79 % survival), while those producing a short Vsa protein, such as CTG-R5, were readily killed (6 % survival). The survival rates for mutants that lacked EPS-I, strains CTG1291 and CTG1701, were 24 and 30 %, respectively. EPS-I production in the CTG1701 mutant was restored by complementation, resulting in strain CTG1701-C (Daubenspeck et al., 2009). CTG1701-C survived exposure to complement at the same level as the wild-type organism: 85 % survival. The percentage survival of CTG-R5 was statistically significantly lower than that of all other strains, while the percentage survival of CTG38 and CTG1701-C was significantly higher than all other strains. The two mutants CTG1291 and CTG1701 exhibited significantly lower percentage survival than CTG38 and CTG1701-C, but not from one another. These data demonstrate that EPS-I, in addition to a long Vsa protein, is involved in the protection of the mycoplasma from complement.
Fig. 1.
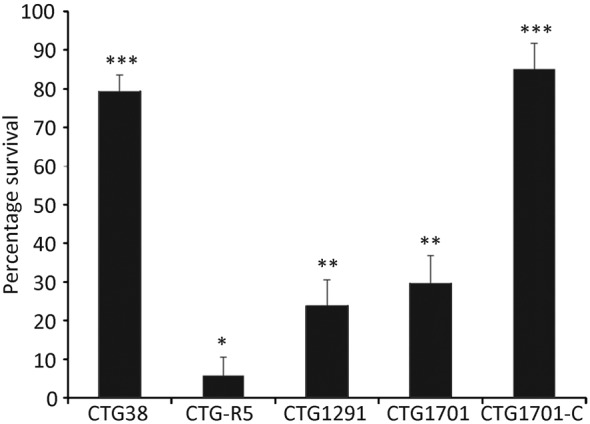
Susceptibility of EPS-I to complement-mediated killing. Cells (104 c.f.u.) of the wild-type strains CTG38 and CTG-R5, the CTG1291 and CTG1701 mutants that lack EPS-I, and the complemented mutant CTG1701-C were assayed for complement susceptibility. Data represent multiple experiments and the total number of samples assayed for each strain was 27 for CTG1701-C and 36 for all other strains. Means with the same number of asterisks were not significantly different from each other, but there were significant differences between means with a different number of asterisks. Error bars, sem.
EPS-I is not required for protection from complement for cells encased in a biofilm
The above experiments were performed with planktonic cells. Although planktonic cells that produce a short Vsa protein are readily killed by complement, these cells are resistant to complement when encased within the tower region of a biofilm (Simmons & Dybvig, 2007). The wild-type mycoplasma is proficient at biofilm formation only when the Vsa protein is short (Simmons et al., 2007), but mutants lacking EPS-I can form a biofilm even when the Vsa protein is long (Daubenspeck et al., 2009). When biofilms of the EPS-I mutant CTG1291, which produces a long VsaG protein, and of CTG-R5, which produces a short VsaG protein, were incubated with GPS, both strains resisted killing about equally. Recovered from the biofilms were 76 % of the c.f.u. of strain CTG1291 (sem = 0.089, n = 18, reproduced in six experiments) and 70 % of the c.f.u. of strain CTG-R5 (sem = 0.075, n = 15, reproduced in five experiments), compared with control biofilms (containing 107 c.f.u.) treated with HIA-GPS. We have previously shown that CTG-R5 cells encased within a biofilm and protected from complement are readily killed when the cells are dispersed (Simmons & Dybvig, 2007).
Comparison of the association of EPS-I with cells of mycoplasmas producing a long or short Vsa protein
The amount of EPS-I that remained adhered to the mycoplasmas after washing and the amount in the washes was assessed by GC/MS (Fig. 2). Washed cells of the long Vsa-producing strain CTG38 retained 41 % of the EPS-I, while the washes contained 59 %. Washed cells of the short Vsa-producing strain CTG-R5 retained 58 % of the EPS-I, while the washes contained 42 %. Thus, cells producing a short Vsa protein are sensitive to complement even though the majority of the polysaccharide remains associated with the cells after washing.
Fig. 2.
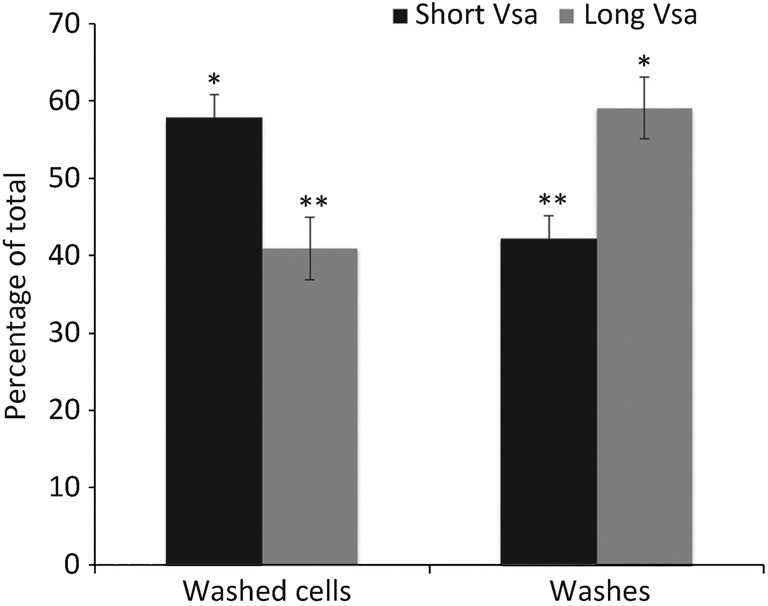
Distribution of EPS-I for the long VsaG-producing strain CTG38 and the short VsaG-producing strain CTG-R5 (n = 2), showing the relative amount of EPS-I associated with washed cells and the washes. The values are represented as a percentage of the total polysaccharide (washed cells plus the washes). Means with the same number of asterisks were not significantly different from each other, but there were significant differences between means with a different number of asterisks. Error bars, sem.
An extracellular matrix on M. pulmonis
Mycoplasmas producing a long Vsa protein form aggregates referred to as microcolonies when grown in broth (Simmons et al., 2007). Individual cells are present in the cultures as well. Negative staining of the cultures with India ink revealed a halo around the individual cells of strain CTG-R40 (Fig. 3c), a feature typical of capsular material. An exclusion zone (halo) was also observed around microcolonies of strains CTp12 (Fig. 3a, d and e) and CTG-R40 (Fig. 3f), suggesting the presence of an extracellular matrix. When the microcolonies were stained with Hoechst 33342, which binds to the DNA of mycoplasma cells, the cellular mass could be observed within the exclusion zone (Fig. 3a, b). Microcinemaphotography of CTp12 revealed that the material corresponding to the exclusion zone was shed (Supplementary Movie File S1, also see Fig. 3d, e). The exclusion zones often extended as strands of negative-staining material between microcolonies and other groups of mycoplasma cells, as shown for the wild-type strain CTG-R40 (Fig. 3g, h). When the microcolonies of CTG-R40 were washed in PBS, the cell masses dispersed and the exclusion zones were no longer prominent (Fig. 3i). The EPS-I mutants lacked this exclusion zone (Fig. 3j, k). These data are consistent with the GC data of Fig. 2, suggesting that the negatively stained material is composed of EPS-I and is loosely associated with the cells.
Fig. 3.

Staining of mycoplasmas with India ink. Black arrows indicate negative-staining material, while white arrows indicate the mycoplasmas in the microcolonies. Exclusion zones (haloes, see black arrows) are observed surrounding the microcolonies of strains CTp12 (a, d and e), CTG-R40 (f) and individual mycoplasma cells of strain CTG-R40 (c). (a, b) The same microcolony stained with India ink and Hoechst, respectively. The Hoechst binds to the DNA of the mycoplasmas and indicates the location of the cellular mass relative to the negative-staining material. (g, h) Strain CTG-R40 stained with India ink, showing negatively staining material running in strands between a microcolony and other cells. (i) Microcolonies of strain CTG-R40 that lack an exclusion zone after washing in PBS. The negative-staining material is not observed extending between the microcolonies of mutants CTG1291 (j) and CTG1702 (k), nor is it observed forming a halo around the microcolonies. Scale bars: (a, b, d, e, g, h, j and k) 50 µm; (f, i) 25 µm; (c) 10 µm.
Complement deposition
Immunofluorescence microscopy of strain CTp12 incubated with mouse serum showed that complement component C3 deposited on globular material that often did not co-localize with the mycoplasma cells (Fig. 4a, b). This material even appeared to exclude areas where the mycoplasma could deposit on the microscope slide (Fig. 4d, g). In cases where this material did co-localize with the cellular masses, only small amounts of C3 were bound to the mycoplasmas (Fig. 4e, h). C3 deposition was not detected in cultures of cells that were incubated with HIA-mouse serum, demonstrating that the antibody was specific for C3 (Fig. 4c, f and i).
Fig. 4.
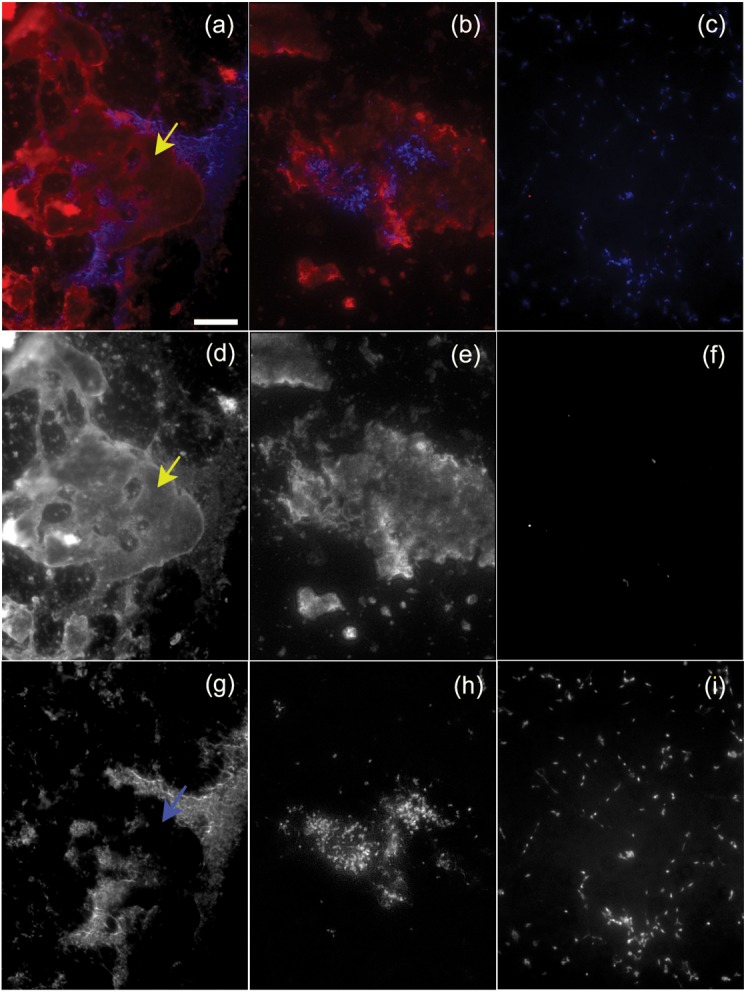
Complement deposition on strain CTp12. (a, b and c) Localization of C3 (shown in red) relative to the mycoplasma cells stained with Hoechst (shown in blue). (a, b) Strain CTp12 incubated with mouse serum; (c) CTp12 incubated with HIA-mouse serum. (d, e and f) Separate immunofluorescent signal for C3 localization; (g, h and i) separate fluorescent signal for DNA localization. The yellow arrows in (a, d) show an area where material binds to C3 and is devoid of cellular material, as shown by the blue arrow in (g). The area represented by the mycoplasma cells (g, h) is smaller than the area where C3 is deposited (d, e). (b, e and h) Composite images showing the extended depth of field. The mycoplasma cells incubated with HIA-mouse serum did not bind C3 (c, f, i). Scale bar, 25 µm for all panels.
Discussion
Resistance to the lytic effects of the complement cascade system of the innate immune response provides the opportunity for bacteria to heighten chances of survival and bolster virulence (Rother et al., 1997). Polysaccharides serve bacteria in a number of capacities across a large spectrum of genera, and notably are often associated with avoidance of innate immunity (Alitalo et al., 2001; Comstock & Kasper, 2006; Keo et al., 2011; O’Riordan & Lee, 2004; Rother et al., 1997; Simmons et al., 2004; Simmons & Dybvig, 2007). Prior studies have demonstrated that the number of tandem repeats of the Vsa lipoprotein determines the level of susceptibility of M. pulmonis to killing by complement (Simmons & Dybvig, 2003; Simmons et al., 2004). The current study identifies the EPS-I polysaccharide as an additional contributing factor. Mutants that did not produce EPS-I were more susceptible to killing than were wild-type mycoplasmas or complemented mutants that produced the same isotype and length of Vsa protein. EPS-I alone is not responsible for protection because mycoplasmas producing a short Vsa protein have an abundance of EPS-I but are nevertheless killed. It should be noted that we have not been able to generate an EPS-I mutant that produces a short Vsa protein and that null mutants lacking any Vsa protein have also never been obtained (French et al., 2008). Perhaps the Vsa proteins and EPS-I shield the mycoplasmas not only from complement but also from toxic factors present in the culture medium.
Wild-type M. pulmonis cells grow as a biofilm only when the Vsa protein is short, but the EPS-I mutants form a biofilm even though they produce a long Vsa protein (Daubenspeck et al., 2009). The cells within the densely packed towers of the biofilm are resistant to complement but cells in honeycombed regions are killed (Simmons & Dybvig, 2007). It is the structure of the biofilm that imparts resistance. When dispersed from the biofilm, nearly all the cells are readily killed by complement. We find here that the biofilm formed by the EPS-I mutants is similarly resistant to complement, while planktonic mycoplasmas are killed effectively. The factors that contribute to the matrix of the biofilm that render the cells resistant to complement are unknown, but it appears that the EPS-I polysaccharide is not one of them.
Why are both a long Vsa protein with many tandem repeats and the EPS-I polysaccharide required for resistance of planktonic cells to complement? A possible explanation is that the tandem repeats of the Vsa protein have a role in tethering EPS-I to the mycoplasma cell surface. This explanation is false. If anything, cells producing a short Vsa protein with few repeats retained more EPS-I upon washing than did cells producing a long Vsa protein. Perhaps the mycoplasma produces a lectin that is anchored to the membrane and binds EPS-I. When the Vsa protein is short, the lectin may be exposed and available for binding. On these cells, EPS-I may be considered capsular. When the Vsa protein is long, the lectin may be buried, and EPS-I may be thought of as forming a slime layer that is loosely associated with the cell surface. However, not all of the polysaccharide is washed away from cells producing a long Vsa protein. Because the washed cells are less sensitive to complement than are the EPS-I mutants, the polysaccharide that remains after washing contributes to the resistance phenotype.
It has been proposed that the Vsa proteins act in the capacity of a shield that mediates surface interactions through steric hindrance (Simmons et al., 2004). The convergence of evidence suggests that EPS-I also contributes to the shielding. The mutants lacking EPS-I do not produce the material that excludes India ink from the cellular masses, suggesting that EPS-I contributes to this abundant material. Much of the complement component C3 is deposited on this material and shed away from the cells. Such shedding may render much of the complement unavailable to lyse or opsonize the mycoplasma cells.
Acknowledgements
We thank David S. Jordan for technical assistance. This work was supported by NIH grants AI63909, AI64848 and T32 AI007041.
Abbreviations:
- GPS
guinea pig serum
- HIA
heat-inactivated
- SFM
serum-free medium
Footnotes
A supplementary movie file, showing microcinematography of India ink-stained microcolonies of strain CTG-R40, is available with the online version of this paper.
References
- Alitalo A., Meri T., Rämö L., Jokiranta T. S., Heikkilä T., Seppälä I. J. T., Oksi J., Viljanen M., Meri S. (2001). Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect Immun 69, 3685–3691. 10.1128/IAI.69.6.3685-3691.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhugra B., Voelker L. L., Zou N., Yu H., Dybvig K. (1995). Mechanism of antigenic variation in Mycoplasma pulmonis: interwoven, site-specific DNA inversions. Mol Microbiol 18, 703–714. 10.1111/j.1365-2958.1995.mmi_18040703.x [DOI] [PubMed] [Google Scholar]
- Comstock L. E., Kasper D. L. (2006). Bacterial glycans: key mediators of diverse host immune responses. Cell 126, 847–850. 10.1016/j.cell.2006.08.021 [DOI] [PubMed] [Google Scholar]
- Daubenspeck J. M., Bolland J. R., Luo W., Simmons W. L., Dybvig K. (2009). Identification of exopolysaccharide-deficient mutants of Mycoplasma pulmonis. Mol Microbiol 72, 1235–1245. 10.1111/j.1365-2958.2009.06720.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison A. M., Clapper B., Dybvig K. (2005). Avoidance of the host immune system through phase variation in Mycoplasma pulmonis. Infect Immun 73, 2033–2039. 10.1128/IAI.73.4.2033-2039.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybvig K., Gasparich G. E., King K. W. (1995). Artificial transformation of mollicutes via polyethylene glycol- and electroporation-mediated methods. In Molecular and Diagnostic Procedures in Mycoplasmology, pp. 179–184. Edited by Razin S., Tully J. G. Orlando, FL: Academic Press; 10.1016/B978-012583805-4/50018-2 [DOI] [Google Scholar]
- Dybvig K., French C. T., Voelker L. L. (2000). Construction and use of derivatives of transposon Tn4001 that function in Mycoplasma pulmonis and Mycoplasma arthritidis. J Bacteriol 182, 4343–4347. 10.1128/JB.182.15.4343-4347.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. T., Lao P., Loraine A. E., Matthews B. T., Yu H., Dybvig K. (2008). Large-scale transposon mutagenesis of Mycoplasma pulmonis. Mol Microbiol 69, 67–76. 10.1111/j.1365-2958.2008.06262.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumulak-Smith J., Teachman A., Tu A.-H. T., Simecka J. W., Lindsey J. R., Dybvig K. (2001). Variations in the surface proteins and restriction enzyme systems of Mycoplasma pulmonis in the respiratory tract of infected rats. Mol Microbiol 40, 1037–1044. 10.1046/j.1365-2958.2001.02464.x [DOI] [PubMed] [Google Scholar]
- Keo T., Collins J., Kunwar P., Blaser M. J., Iovine N. M. (2011). Campylobacter capsule and lipooligosaccharide confer resistance to serum and cationic antimicrobials. Virulence 2, 30–40. 10.4161/viru.2.1.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey J. R., Baker H. J., Overcash R. G., Cassell G. H., Hunt C. E. (1971). Murine chronic respiratory disease. Significance as a research complication and experimental production with Mycoplasma pulmonis. Am J Pathol 64, 675–708. [PMC free article] [PubMed] [Google Scholar]
- O’Riordan K., Lee J. C. (2004). Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev 17, 218–234. 10.1128/CMR.17.1.218-234.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin S. (1999). Adherence of pathogenic mycoplasmas to host cells. Biosci Rep 19, 367–372. 10.1023/A:1020204020545 [DOI] [PubMed] [Google Scholar]
- Razin S., Yogev D., Naot Y. (1998). Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev 62, 1094–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother K., Till G. O., Hansch G. M. (1997). The Complement System. New York: Springer. [Google Scholar]
- Shen X., Gumulak J., Yu H., French C. T., Zou N., Dybvig K. (2000). Gene rearrangements in the vsa locus of Mycoplasma pulmonis. J Bacteriol 182, 2900–2908. 10.1128/JB.182.10.2900-2908.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons W. L., Dybvig K. (2003). The Vsa proteins modulate susceptibility of Mycoplasma pulmonis to complement killing, hemadsorption, and adherence to polystyrene. Infect Immun 71, 5733–5738. 10.1128/IAI.71.10.5733-5738.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons W. L., Dybvig K. (2007). Biofilms protect Mycoplasma pulmonis cells from lytic effects of complement and gramicidin. Infect Immun 75, 3696–3699. 10.1128/IAI.00440-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons W. L., Zuhua C., Glass J. I., Simecka J. W., Cassell G. H., Watson H. L. (1996). Sequence analysis of the chromosomal region around and within the V-1-encoding gene of Mycoplasma pulmonis: evidence for DNA inversion as a mechanism for V-1 variation. Infect Immun 64, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons W. L., Denison A. M., Dybvig K. (2004). Resistance of Mycoplasma pulmonis to complement lysis is dependent on the number of Vsa tandem repeats: shield hypothesis. Infect Immun 72, 6846–6851. 10.1128/IAI.72.12.6846-6851.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons W. L., Bolland J. R., Daubenspeck J. M., Dybvig K. (2007). A stochastic mechanism for biofilm formation by Mycoplasma pulmonis. J Bacteriol 189, 1905–1913. 10.1128/JB.01512-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Robinson D., Schorlemmer H. U., Furr P. M., Allison A. C. (1978). Macrophage secretion and the complement cleavage product C3a in the pathogenesis of infections by mycoplasmas and L-forms of bacteria and in immunity to these organisms. Clin Exp Immunol 33, 486–494. [PMC free article] [PubMed] [Google Scholar]
- Yus E., Maier T., Michalodimitrakis K., van Noort V., Yamada T., Chen W. H., Wodke J. A., Güell M., Martínez S. & other authors (2009). Impact of genome reduction on bacterial metabolism and its regulation. Science 326, 1263–1268. 10.1126/science.1177263 [DOI] [PubMed] [Google Scholar]