

Abstract
Hypotrichosis simplex (HS) comprises a group of hereditary isolated alopecias that are characterized by a diffuse and progressive loss of hair starting in childhood and shows a wide phenotypic variability. We mapped an autosomal-dominant form of HS to chromosome 1q31.3-1q41 in a Spanish family. By direct sequencing, we identified the heterozygous mutation c.1A>G (p.Met1?) in SNRPE that results in loss of the start codon of the transcript. We identified the same mutation in a simplex HS case from the UK and an additional mutation (c.133G>A [p.Gly45Ser]) in a simplex HS case originating from Tunisia. SNRPE encodes a core protein of U snRNPs, the key factors of the pre-mRNA processing spliceosome. The missense mutation c.133G>A leads to a glycine to serine substitution and is predicted to disrupt the structure of SNRPE. Western blot analyses of HEK293T cells expressing SNRPE c.1A>G revealed an N-terminally truncated protein, and therefore the mutation might result in use of an alternative in-frame downstream start codon. Subcellular localization of mutant SNRPE by immunofluorescence analyses as well as incorporation of mutant SNRPE proteins into U snRNPs was found to be normal, suggesting that the function of U snRNPs in splicing, rather than their biogenesis, is affected. In this report we link a core component of the spliceosome to hair loss, thus adding another specific factor in the complexity of hair growth. Furthermore, our findings extend the range of human phenotypes that are linked to the splicing machinery.
Main Text
Hypotrichosis simplex (HS [MIM 146520, MIM 278150, MIM 146550, MIM 613981, and MIM 605389]) is a hereditary form of nonsyndromic, isolated alopecia that affects men and women equally. Both autosomal-dominant and autosomal-recessive patterns of inheritance have been observed in a number of families. Hair loss is diffuse and usually begins in early childhood, and then progresses until adulthood. The extent of scalp and body hair involvement can be very variable, within as well as between families, and ranges from partial alopecia to complete loss of scalp and body hair. The hair shaft characteristically shows no gross abnormality.
Mutations in six genes have been identified for isolated HS,1–6 and mutations in three of them—CDSN (MIM 602593),6 APCDD1 (MIM 607479),1 and RPL21 (MIM 603636)2—are responsible for autosomal-dominant forms. Mutations in these latter genes are the pathogenic cause for only a total of ten families with autosomal-dominant HS, and therefore a large number of families and simplex cases with hypotrichosis are so far unexplained.
To determine the molecular genetic basis of autosomal-dominant hypotrichosis simplex, we investigated a three-generation pedigree from Spain (Figure 1A). Most of the affected family members present with scanty or no eyebrows and additionally with a highly variable degree of alopecia since birth, ranging from slight thinning of scalp and axillary hair to complete loss of scalp and body hair (Figures 1D–1H and 1M and Figure S1 available online). Pubic hair remains mainly unaffected. Skin, nails, teeth, and sweating are normal in these individuals and no other abnormalities were detected. The clinical characteristics and pedigree structure of the family have been described in detail elsewhere.7
Figure 1.
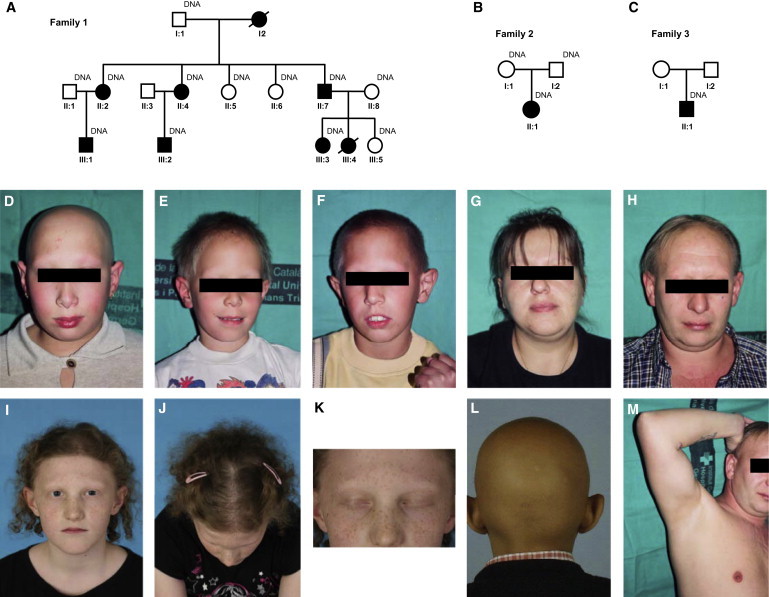
Pedigrees and Photographs of Affected Individuals from Three Different Hypotrichosis Simplex Families
(A) Pedigree of the Spanish hypotrichosis family. Affected family members are shown in black; circles and squares denote females and males, respectively.
(B) Pedigree of the British family.
(C) Pedigree of the Tunisian family.
(D) Index case (III:3) of the Spanish family. The picture was taken when she was 10 years old. She shows complete alopecia of the scalp and is devoid of eyebrows and eyelashes.
(E) Cousin of the index person at the age of 8 years. He has almost normal scalp hair, but sparse eyebrows and eyelashes.
(F) Cousin of the index person at the age of 10 years. His hair at the scalp is almost normal but he has no eyebrows and no eyelashes.
(G) Mother of the Spanish family. Her scalp hair is almost normal, but the eyebrows are sparse. Eyelashes are not visible.
(H and M) Father of the Spanish family. His scalp hair is sparse. Eyelashes and eyebrows are not present. Also, he does not have axillary hair.
(I–K) Affected female individual of the British family with HS at the age of 12 years. She has sparse scalp hair and eyelashes. Eyebrows are not present.
(L) Affected male individual of the Tunisian family at the age of 8 years. He shows complete alopecia of the scalp as well as absence of eyebrows and eyelashes.
We collected blood samples from 12 family members, 6 of whom are affected. Ethical approval was obtained from the ethics committee of the Medical Faculty of the University of Bonn and all participants provided written informed consent prior to blood sampling. The study was conducted in concordance with the Declaration of Helsinki Principles. DNA was extracted from peripheral blood leukocytes according to standard methods.
We performed a genome-wide linkage analysis approach by using 320 highly polymorphic microsatellite markers after excluding a number of candidate loci in this family (Table S1). Two-point LOD scores were calculated between each marker locus and hypotrichosis, under the assumption of autosomal-dominant inheritance with complete penetrance, a frequency of 10−5 for the disease allele, and equal allele frequencies for each marker, via the LINKAGE version 5.21 software.8 We found evidence for linkage to the marker D1S1660, with a maximum LOD score of 2.3 at recombination fraction (θ) 0 (Figure S2). By genotyping additional markers in the identified region, we narrowed the candidate region to 21 Mbp between markers D1S408 and D1S2141 (Figure S3). This region contained more than 100 annotated genes. Because the identified region did not contain any obvious gene that was functionally relevant for hair loss, we sequenced gene by gene. Therefore, PCR was performed in a total volume of 25 μl with 40 ng of genomic DNA with the REDTaq ReadyMix PCR Reaction Mix with MgCl2 (Sigma Aldrich, Saint Louis, MO). The PCR products were purified with the illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Freiburg, Germany) and directly sequenced on an ABI 3100 genetic analyzer (Applied Biosystems, Foster City, CA) with the BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems) and the DyeEx 2.0 Spin Kit for sample clean up (QIAGEN, Hilden, Germany).
After excluding 60 genes by direct sequencing (Table S2), we identified a c.1A>G (p.Met1?) mutation in the start codon of SNRPE (also known as Sm-E; MIM 128260; RefSeq accession number NM_003094.2), which encodes a core protein of pre-mRNA processing U-rich small nuclear ribonucleoproteins (U snRNPs) (Figure 2A, left; primer sequences are given in Table S3). Because the mutation eliminates an NcoI restriction site, we performed an NcoI digestion with SNRPE PCR products of members of Family 1 and observed that the mutation segregated with the disorder (Figure S4). We then sequenced several cases of our hypotrichosis cohort and detected the same mutation in a simplex case from Great Britain (Figures 1I–1K). We could not identify the mutation in the healthy parents, indicating that the mutation occurred de novo in that individual (Figures 1B and S5). Paternity was confirmed by analyzing microsatellite markers that were consistent in the parents and their offspring (Figure S6). In another person originating from Tunisia, we identified a c.133G>A mutation (p.Gly45Ser) (Figure 2A, middle). Also in this family, the individual is a simplex case because the nonconsanguineous parents were reported as being unaffected; however, samples from the parents were not available (Figure 1C). We did not detect either of the two mutations in 880 control chromosomes of German origin and 598 control chromosomes of Spanish origin. The mutations were also not present in dbSNP and the 1000 Genomes database.
Figure 2.
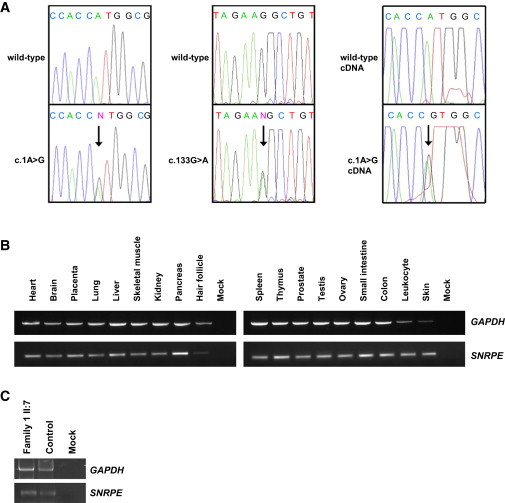
Sequence and Expression Analysis of SNRPE
(A) Sequence analysis of SNRPE in the Spanish family revealed a c.1A>G substitution that leads to the abolishment of the start codon (left). The same mutation was also identified in an affected individual from Great Britain. Sequence analysis in additional persons revealed a c.133G>A substitution in a boy from Tunisia (middle). Sequencing of cDNA samples of individual II:7 of the Spanish family and a control, indicating that the mutant RNA is present (right). Arrow indicates the mutation.
(B) Multiple tissue expression analysis of SNRPE revealed that the gene is expressed ubiquitously, including expression in hair follicles and skin.
(C) Expression analysis of SNRPE via immortalized lymphocytes of individual II:7 of the Spanish family and a healthy control. We isolated RNA from the respective cells and performed a reverse-transcriptase PCR. The obtained cDNA was used as template for a semiquantitative PCR with SNRPE- and GAPDH-specific primers. Negative control is shown on the right.
SNRPE consists of five coding exons with a 279 bp open reading frame encoding the 92 amino acid protein SNRPE,9 alternatively termed Sm-E, which constitutes a core component of U snRNPs. These RNA-protein particles are part of the spliceosome, which catalyzes the excision of introns from primary gene transcripts during the splicing process. SNRPE is one out of seven related Sm proteins (termed SNRPB [UniProtKB: P14678], SNRPD1 [UniProtKB: P62314], SNRPD2 [UniProtKB: P62316], SNRPD3 [UniProtKB: P62318], SNRPE, SNRPF [UniProtKB: P62306], and SNRPG [UniProtKB: P62308]) that assemble onto U snRNAs to from the toroidal Sm core domain of U snRNPs. This domain constitutes the structural framework of most spliceosomal U snRNPs (U6 and U6actac being two exceptions) and has been shown to be important for their biogenesis and function.
To analyze the expression of SNRPE and genes encoding other Sm proteins, we used the Human MTC Panel I and II (BD Biosciences, Franklin Lakes, NJ) and also RNA isolated from additional human tissues. After reverse-transcriptase PCR, we performed semiquantitative PCRs by using gene-specific primers (Table S3). Expression analysis in different human tissues and cells revealed that SNRPE is expressed ubiquitously including skin and hair follicle cells (Figure 2B). Consistent with a role in splicing, not only SNRPE but also all other genes encoding Sm proteins are expressed ubiquitously in the human body, including skin, lymphocytes, and hair follicle cells of the scalp and the eyebrow (Figure S7). To determine whether the mutant allele is present in the RNA of affected individuals and not degraded, we isolated RNA from immortalized lymphocytes of an affected individual from Family 1 and a control. After cDNA synthesis and PCR with gene-specific primers, we sequenced the obtained products and detected both the wild-type and the mutant allele (Figure 2A, right, and 2C).
Via immunohistochemistry, we investigated in which cells of the skin SNRPE is present. In sections of human scalp skin we detected SNRPE immunoreactivity in the nuclei of all cells of the hair follicle, the epidermis, and the dermis (Figure S8), which could be completely blocked by adding the immunogenic peptide to the antibody solution (data not shown). An identical pattern of SNRPE immunoreactivity was observed in dorsal skin of 9-day-old C57BL/6J mice (Figure S9). The nuclear localization of the SNRPE immunoreactivity would be expected for a protein involved in splicing.
The mutations identified in our affected individuals with HS are predicted to have a major impact on gene expression or structure of SNRPE. The p.Gly45Ser substitution leads to a change of a highly conserved glycine (Figures S10 and S11). This amino acid lies within the so-called Sm motif 1 that, together with an adjacent Sm motif 2, allows formation of the Sm fold characteristic of the family of Sm and LSm proteins (Figure S11).10 Structure-based examination of the p.Gly45Ser substitution predicted a clash with Leu58 in the Sm fold. Furthermore, the substituted serine is supposed to disrupt the hydrophobic core of the protein (Figure S12).
In contrast, the c.1A>G mutation alters the start codon of SNRPE. The use of alternative initiator codons has been suggested, but these non-ATG triplets are generally less efficient and show a lower activity than the regular ATG start codon.11 On the other hand, there is the possibility that a downstream ATG might be used for the initiation of translation. The growing number of start codon mutations that have been described in the literature for various genes and diseases points to their pathogenicity in humans.12,13
Next, we assessed whether the pathogenic mutants are expressed in vivo. For this purpose, the mutant and wild-type genes were cloned into the eukaryotic expression vector pcDNA3.1/V5-His (Invitrogen, Paisley, UK) and transiently transfected in HEK293T cells (European Collection of Cell Cultures [ECACC]). Wild-type and mutant (c.1A>G and c.133G>A) constructs allowed the translation of an 11 kDa protein, which corresponds to the full-length SNRPE (Figure 3C), indicating that the initial ATG as well as GTG can be used as a start codon during translation. Interestingly, c.1A>G also allowed the production of a weaker truncated version of 9 kDa, which most probably arises from the initiation of translation at the second in-frame ATG codon at position 14 (Figure 3C). The second in-frame ATG is not embedded in a Kozak consensus sequence, but the purine in position −3, which is functionally the most important position in the Kozak consensus sequence, as well as the G in position +4, which is also highly conserved, are still present and might facilitate initiation of translation.14 Compared to the full-length protein, however, the truncated form is less efficiently translated (Figure 3E) and is mostly insoluble (compare protein abundance in pellet versus soluble fraction [Figure S13] upon cell extract preparation).
Figure 3.
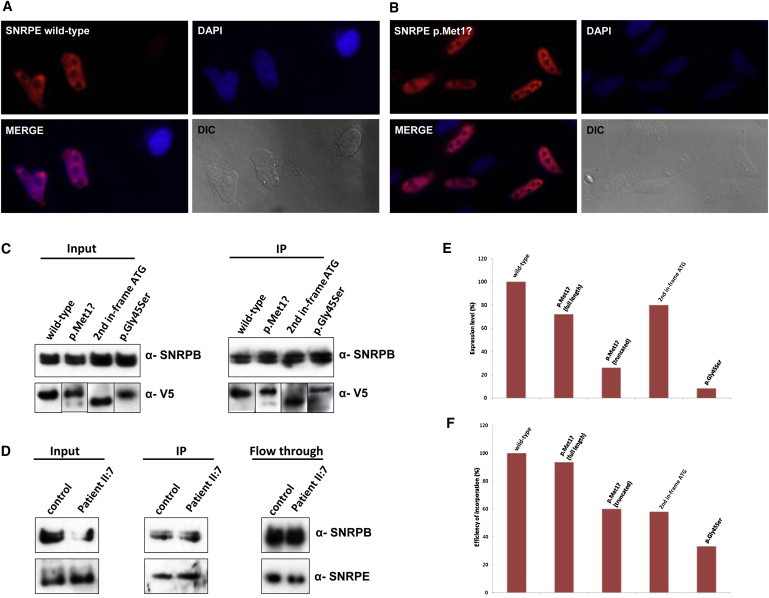
Protein Analyses of SNRPE Wild-Type and Mutant Constructs
(A and B) Subcellular localization of SNRPE wild-type (A) or p.Met1? (B) by immunofluorescence analysis. SNRPE shows a speckled pattern within the nucleus of COS7 cells. In addition, the protein is enriched in discrete spots, which most probably represent Cajal bodies. No significant difference in the localization of the wild-type and mutant SNRPE protein was observed. The nuclei are stained with DAPI; DIC, differential interference microscopy.
(C) Immunoprecipitation of snRNPs with m3G-cap antibody (H20) from transfected HEK293T cells followed by western blot analysis with V5 antibody. Western blotting with SNRPB antibody was used as a control for purification of the snRNPs. We observed that in the case of p.Met1?, both full-length and truncated forms of the protein are translated, albeit at lower levels than the wild-type, and incorporated into the Sm core. We conclude that the initial GTG can be used as a start codon during translation. Integration of the truncated form of the protein (second in-frame ATG) proves that this alteration does not affect the early incorporation of the protein into the Sm core. Interestingly, the substitution p.Gly45Ser is also incorporated into the Sm Core, although with a lower efficiency.
(D) Immunoprecipitation of snRNPs with m3G-cap antibody (H20) from lymphocytes of a control and individual II:7 of the Spanish family (p.Met1?) followed by western blot analysis with SNRPE antibody. Western blotting with SNRPB antibody was used as a control for purification of the snRNPs. Interestingly, in cells of affected individuals, only full-length protein is produced and incorporated into the Sm core with almost equal efficiency to the wild-type. Becauses of identical sizes, one cannot distinguish between wild-type and full-length mutant protein in the cell line of the affected person.
(E) Normalized protein levels of SNRPE wild-type and mutant proteins in HEK293T cells. Quantification has been performed with the results from three separate experiments. In each lane, the protein level has been normalized according to the signal of SNRPB in the same lane and been shown as a percentage of the wild-type protein. Quantifications have been performed by the ImageJ program.
(F) Normalized efficiency of incorporation of wild-type and mutant SNRPE proteins into the Sm core in HEK293T cells. The efficiency of incorporation in each lane has been normalized according to the signal of SNRPB in the same lane and also the protein level in the same experiment. Quantification has been performed with the results from three separate experiments by the ImageJ program.
To further determine the subcellular localization of wild-type and mutant SNRPE proteins, we performed immunofluorescence analysis with transiently transfected COS7 cells. As expected for a core splice factor, SNRPE was found in a speckled pattern within the nucleus of cells. In addition, the protein localized to discrete spots, which most probably represent Cajal bodies. Notably, we found no significant differences in the localization of the mutant proteins in comparison to the wild-type protein (Figures 3A and 3B).
Next, we investigated whether mutant SNRPE incorporates into U snRNPs and is hence part of spliceosomal complexes. For this, we expressed V5-tagged wild-type and mutant SNRPE in HEK293T cells. Extracts from these cells were then subjected to immunoprecipitation with an antibody directed against the m3G-cap of U snRNPs. Note that the truncated version of p.Met1? and the p.Gly45Ser mutant were less abundant in these extracts (Figure 3E). As shown in Figure 3C, these antibodies coprecipitated wild-type and mutant SNRPE, indicating that the proteins have incorporated into U snRNPs. In the case of p.Met1?, only a minor reduction in the efficiency of U snRNP incorporation could be observed. However, in the case of p.Gly45Ser, the reduction in the incorporation efficiency is more substantial (Figure 3F).
Next, we investigated the presence of SNRPE in immortalized lymphocytes of individuals harboring the p.Met1? substitution. By using a monoclonal antibody, we were able to detect only full-length SNRPE in these cells, which is incorporated into the Sm core with almost equal efficiency to the wild-type protein of a healthy control individual (Figure 3D). Because these cells contain the mutant as well as the wild-type allele, it is currently unclear whether they express the protein from the mutant locus. However, the truncated version of SNRPE, which is translated upon overexpression (see Figures 3C and S13), was not detectable in these cells. This may indicate that the protein is unstable in the cell line of the affected person.
SNRPE is part of spliceosomal U snRNPs. Therefore, it was interesting to test whether the SNRPE mutants would interfere with splicing in vivo. To test this possibility, we performed splicing analyses by using artificial minigenes as well as lymphocyte cell lines of affected individuals. When analyzing these model substrates, no major splicing abnormalities were observed (see Figures S14 and S15). However, at present, we cannot exclude the possibility that SNRPE mutations cause subtle changes in the splicing pattern of the target organ, i.e., the hair follicle of affected persons.
By using a genetic linkage approach, we could associate mutations in SNRPE with autosomal-dominant hypotrichosis simplex. SNRPE encodes a core splicing factor and therefore is required in all nucleated cells of the body for pre-mRNA maturation. The two mutations identified here appear to affect the solubility of SNRPE and therefore may be pathogenic because they reduce the soluble fraction that can be incorporated into U snRNPs. Interestingly, however, for both mutants we observed a small fraction of soluble protein, which can efficiently assemble into U snRNPs. Hence, it is possible that the mutant SNRPE proteins become part of the spliceosome but interfere at an as-yet-to-be-identified step with the splicing process. Of note, Weiss et al. reported a dominant Snrpe mutation in a mouse strain that was identified in a mutagenesis screen for hypogonadism. However, these mice show no obvious skin or hair phenotype, which might be due to the position of the mutation or due to different physiological processes in mouse and human.15
In this report we identified a spliceosomal small nuclear ribonucleoprotein that is involved in a specific hair phenotype. Interestingly, until now, mutations in a few genes encoding core splicing factors were known to cause human pathogenic phenotypes. Among them are SNRNP200 (RP33, MIM 610359),16 PIM1-associated protein (RP9 or PAP1, MIM 607331),17 PRPF31 (RP11, MIM 606419),18 PRPC8 (RP13, MIM 600059),19 and HPRP3 (RP18, MIM 601414)20 that have been linked to autosomal-dominant retinitis pigmentosa (RP, MIM 268000). Interestingly, it has been shown that mutations in the RP-associated splice factors do not lead to a general defect in splicing. Rather, tissue-specific alterations in the expression of a relatively small number of transcripts have been found, most of them being implicated in photoreceptor maintenance or development. Thus, these data provide insight into how mutations in general, and splice factors in particular, cause a tissue-specific disease.21 Along the same lines, we may speculate that mutations in SNRPE also lead to subtle defects in the splicing machinery in a way that compromises hair growth/development and thus leads to the mild and specific phenotype. Analyzing the splicing pattern in hair follicle cells of affected individuals in comparison to unaffected individuals will provide further insight into the etiology of HS. Alternatively, the reduced protein levels of some mutant forms of SNRPE, as shown in in vitro protein analyses, may also contribute to the etiology of the disease.
Although the vast majority of SNRPE is part of splicing U snRNPs, the protein is also an essential part of the U7 snRNP implicated in 3′ end processing of replication-dependent histone mRNAs. Thus, it is a possibility that malfunction of U7 snRNP also contributes to the HS phenotype. Clearly, future investigation of the transcriptome of affected persons will give a deeper insight into the underlying mechanisms.
In summary, mutations in SNRPE add to the growing list of snRNP genes known to be responsible for different human diseases. Our results contribute to a better understanding of the biology of the hair follicle and identify that snRNPs play an important role in hair development.
Acknowledgments
We would like to thank the families for participating in this study. The technical support by J. Pforr, G. Eversloh, and M. Michels is gratefully acknowledged. The H20 antibody was a generous gift of R. Lührmann. Constructs for analysis of differential splicing of various genes are a kind gift of Y. Fu and K. Ohno. E.P. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. M.M.N. was supported by the Alfried Krupp von Bohlen und Halbach-Stiftung. The work presented here was supported by a DFG grant to U.F. (FI 573/8-1) and an Emmy Noether research grant of the German Research Foundation (DFG) to R.C.B. (BE 2346/3). R.C.B. is a recipient of a Heisenberg Professorship of the DFG (BE 2346/4-1).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
ClustalW2 - Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
USCS Genome Bioinformatics, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Shimomura Y., Agalliu D., Vonica A., Luria V., Wajid M., Baumer A., Belli S., Petukhova L., Schinzel A., Brivanlou A.H. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature. 2010;464:1043–1047. doi: 10.1038/nature08875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou C., Zang D., Jin Y., Wu H., Liu Z., Du J., Zhang J. Mutation in ribosomal protein L21 underlies hereditary hypotrichosis simplex. Hum. Mutat. 2011;32:710–714. doi: 10.1002/humu.21503. [DOI] [PubMed] [Google Scholar]
- 3.Pasternack S.M., von Kügelgen I., Al Aboud K., Lee Y.A., Rüschendorf F., Voss K., Hillmer A.M., Molderings G.J., Franz T., Ramirez A. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 4.Kljuic A., Bazzi H., Sundberg J.P., Martinez-Mir A., O’Shaughnessy R., Mahoney M.G., Levy M., Montagutelli X., Ahmad W., Aita V.M. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 2003;113:249–260. doi: 10.1016/s0092-8674(03)00273-3. [DOI] [PubMed] [Google Scholar]
- 5.Kazantseva A., Goltsov A., Zinchenko R., Grigorenko A.P., Abrukova A.V., Moliaka Y.K., Kirillov A.G., Guo Z., Lyle S., Ginter E.K., Rogaev E.I. Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science. 2006;314:982–985. doi: 10.1126/science.1133276. [DOI] [PubMed] [Google Scholar]
- 6.Levy-Nissenbaum E., Betz R.C., Frydman M., Simon M., Lahat H., Bakhan T., Goldman B., Bygum A., Pierick M., Hillmer A.M. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat. Genet. 2003;34:151–153. doi: 10.1038/ng1163. [DOI] [PubMed] [Google Scholar]
- 7.Just M., Ribera M., Fuente M.J., Bielsa I., Ferrándiz C. Hereditary hypotrichosis simplex. Dermatology (Basel) 1998;196:339–342. doi: 10.1159/000017909. [DOI] [PubMed] [Google Scholar]
- 8.Lathrop G.M., Lalouel J.M., Julier C., Ott J. Strategies for multilocus linkage analysis in humans. Proc. Natl. Acad. Sci. USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neiswanger K., Stanford D.R., Sparkes R.S., Nishimura D., Mohandas T., Klisak I., Heinzmann C., Wieben E.D. Assignment of the gene for the small nuclear ribonucleoprotein E (SNRPE) to human chromosome 1q25-q43. Genomics. 1990;7:503–508. doi: 10.1016/0888-7543(90)90192-w. [DOI] [PubMed] [Google Scholar]
- 10.Hermann H., Fabrizio P., Raker V.A., Foulaki K., Hornig H., Brahms H., Lührmann R. snRNP Sm proteins share two evolutionarily conserved sequence motifs which are involved in Sm protein-protein interactions. EMBO J. 1995;14:2076–2088. doi: 10.1002/j.1460-2075.1995.tb07199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peabody D.S. Translation initiation at non-AUG triplets in mammalian cells. J. Biol. Chem. 1989;264:5031–5035. [PubMed] [Google Scholar]
- 12.Patten J.L., Johns D.R., Valle D., Eil C., Gruppuso P.A., Steele G., Smallwood P.M., Levine M.A. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N. Engl. J. Med. 1990;322:1412–1419. doi: 10.1056/NEJM199005173222002. [DOI] [PubMed] [Google Scholar]
- 13.Wang C.P., Chen T.C., Chang Y.L., Ko J.Y., Yang T.L., Lo F.Y., Hu Y.L., Chen P.L., Wu C.C., Lou P.J. Common genetic mutations in the start codon of the SDH subunit D gene among Chinese families with familial head and neck paragangliomas. Oral Oncol. 2012;48:125–129. doi: 10.1016/j.oraloncology.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 14.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss J., Hurley L.A., Harris R.M., Finlayson C., Tong M., Fisher L.A., Moran J.L., Beier D.R., Mason C., Jameson J.L. ENU mutagenesis in mice identifies candidate genes for hypogonadism. Mamm. Genome. 2012;23:346–355. doi: 10.1007/s00335-011-9388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C., Bellur D.L., Lu S., Zhao F., Grassi M.A., Bowne S.J., Sullivan L.S., Daiger S.P., Chen L.J., Pang C.P. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am. J. Hum. Genet. 2009;85:617–627. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keen T.J., Hims M.M., McKie A.B., Moore A.T., Doran R.M., Mackey D.A., Mansfield D.C., Mueller R.F., Bhattacharya S.S., Bird A.C. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur. J. Hum. Genet. 2002;10:245–249. doi: 10.1038/sj.ejhg.5200797. [DOI] [PubMed] [Google Scholar]
- 18.Vithana E.N., Abu-Safieh L., Pelosini L., Winchester E., Hornan D., Bird A.C., Hunt D.M., Bustin S.A., Bhattacharya S.S. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest. Ophthalmol. Vis. Sci. 2003;44:4204–4209. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]
- 19.McKie A.B., McHale J.C., Keen T.J., Tarttelin E.E., Goliath R., van Lith-Verhoeven J.J., Greenberg J., Ramesar R.S., Hoyng C.B., Cremers F.P. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum. Mol. Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 20.Chakarova C.F., Hims M.M., Bolz H., Abu-Safieh L., Patel R.J., Papaioannou M.G., Inglehearn C.F., Keen T.J., Willis C., Moore A.T. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 21.Linder B., Dill H., Hirmer A., Brocher J., Lee G.P., Mathavan S., Bolz H.J., Winkler C., Laggerbauer B., Fischer U. Systemic splicing factor deficiency causes tissue-specific defects: a zebrafish model for retinitis pigmentosa. Hum. Mol. Genet. 2011;20:368–377. doi: 10.1093/hmg/ddq473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.