

Adenosine to inosine (A > I) RNA editing, which is catalyzed by the ADAR family of proteins, is one of the fundamental mechanisms by which transcriptomic diversity is generated. Indeed, a number of genome-wide analyses have shown that A > I editing is not limited to a few mRNAs, as originally thought, but occurs widely across the transcriptome, especially in the brain. Importantly, there is increasing evidence that A > I editing is essential for animal development and nervous system function. To more efficiently characterize the complete catalog of ADAR events in the mammalian transcriptome the authors developed a high-throughput protocol to identify A > I editing sites, which exploits the capacity of glyoxal to protect guanosine, but not inosine, from RNAse T1 treatment. Using this method they identified 665 editing sites in mouse brain RNA, including most known sites and suite of novel sites that include nonsynonymous changes to protein-coding genes and hyperediting of genes known to regulate p53.
Keywords: ADAR, noncoding RNA, ion channel, deep sequencing, genomics
Abstract
Adenosine to inosine (A > I) RNA editing, which is catalyzed by the ADAR family of proteins, is one of the fundamental mechanisms by which transcriptomic diversity is generated. Indeed, a number of genome-wide analyses have shown that A > I editing is not limited to a few mRNAs, as originally thought, but occurs widely across the transcriptome, especially in the brain. Importantly, there is increasing evidence that A > I editing is essential for animal development and nervous system function. To more efficiently characterize the complete catalog of ADAR events in the mammalian transcriptome we developed a high-throughput protocol to identify A > I editing sites, which exploits the capacity of glyoxal to protect guanosine, but not inosine, from RNAse T1 treatment, thus facilitating extraction of RNA fragments with inosine bases at their termini for high-throughput sequencing. Using this method we identified 665 editing sites in mouse brain RNA, including most known sites and suite of novel sites that include nonsynonymous changes to protein-coding genes, hyperediting of genes known to regulate p53, and alterations to non-protein-coding RNAs. This method is applicable to any biological system for the de novo discovery of A > I editing sites, and avoids the complicated informatic and practical issues associated with editing site identification using traditional RNA sequencing data. This approach has the potential to substantially increase our understanding of the extent and function of RNA editing, and thereby to shed light on the role of transcriptional plasticity in evolution, development, and cognition.
INTRODUCTION
Adenosine to inosine (A > I) RNA editing is a Metazoa-specific phenomenon (Jin et al. 2009) that is driven by the ADAR (adenosine deaminases acting on RNA) family of proteins (Bass 2002; Nishikura 2010), in which an adenosine is deaminated to generate an inosine. It is now becoming apparent that A > I editing is not a rare phenomenon but is instead the most common base nucleotide editing event in the mammalian transcriptome. Indeed, there is now substantial evidence that A > I editing tunes nervous system function by modifying the sequence of neuronal receptors in mammals, presumably to modulate the electrophysiological properties of the synapse (Sommer et al. 1991; Higuchi et al. 1993; Burns et al. 1997; Hoopengardner et al. 2003; Bhalla et al. 2004; Valente and Nishikura 2005; Ohlson et al. 2007; Daniel et al. 2010). A > I editing is also essential for normal embryological development (Higuchi et al. 2000; Wang et al. 2000; Walkley et al. 2012), and appears to affect stem cell differentiation decisions (Osenberg et al. 2010) and RNA localization (Prasanth et al. 2005; Chen et al. 2008), with editing enzymes themselves shuttled between the nucleus and cytoplasm (Strehblow et al. 2002; Fritz et al. 2009). Editing also modifies splicing pattern by creating new splice sites (Rueter et al. 1999), alters mRNA levels and translational availability by creating microRNA target sites (Borchert et al. 2009), and may modulate microRNA biogenesis through alteration of the pre-miRNA sequence (Luciano et al. 2004; Kawahara et al. 2007; de Hoon et al. 2010; Heale et al. 2010; Alon et al. 2012), although the latter may be due to mis-mapping (de Hoon et al. 2010).
ADARs exhibit strict tissue-specific and environment-dependent expression patterns (Paupard et al. 2000; Sansam et al. 2003). There are three orthologs: ADAR1 and ADAR2 occur in most animals, whereas ADAR3 is vertebrate- and brain-specific. ADAR1 is constitutively expressed in a wide range of tissues but can be induced by interferon (George and Samuel 1999). ADAR2 is preferentially expressed in neurons (Paupard et al. 2000; Jacobs et al. 2009) and its activity is dependent on IP6, which is complexed in the active site (Macbeth et al. 2005), suggesting a link to canonical cell signaling pathways.
Indeed, the existence of editing, as opposed to hard-wired genomic specification, indicates that it is a context-dependent process that allows environmental signals to alter the information in the transcriptome, with flow-on effects on the proteome and the regulome, although the full biological importance of editing has barely been explored. Recent analyses (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004; Macbeth et al. 2005; Schrider et al. 2012) have shown not only that A > I editing is far more widespread than had been anticipated from early cDNA cloning, but also that most RNA editing occurs in non-protein-coding sequences, suggesting that its effects are to alter RNA regulatory information, which in turn may modulate the epigenetic processes that underpin development, gene-environment interactions, and learning (Mattick et al. 2009; Mattick 2010). Moreover, the extent of RNA editing, especially in the brain, increases during mammalian and primate evolution (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004; Macbeth et al. 2005; Paz-Yaacov et al. 2010; Schrider et al. 2012), particularly in Alu sequences that now comprise >10% of the human genome, suggesting that expansion of the latter and editing-induced transcriptomic and epigenomic plasticity was central to the rise of human cognition (Mattick and Mehler 2008).
The characterization of editing events is a necessary step toward fully understanding the function and regulation of transcriptome, and the extent of its plasticity. Initial attempts to identify RNA editing events were based on in silico comparisons of EST databases with their cognate genomic sequences and querying for A > G mismatches (inosines are “read” as guanosines by both Sanger and high-throughput sequencing technologies, and by the translational machinery) (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). Although this approach was successful, it was limited, by definition, by the ESTs available, and it was also plagued by false positives—true editing sites are hard to differentiate from sequencing errors (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). Indeed, recent attempts to use high-throughput RNA-sequencing data sets to identify A > I editing events have shown that false positives, and the confounding influence of genomic polymorphisms, make it almost impossible to distinguish “noise” from “signal” in the absence of subsequent validation (Li et al. 2011; Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012; Schrider et al. 2012). Several other approaches, however, have been developed. For example, Ohlson and colleagues performed ADAR immunoprecipitations and identified the ADAR-associated RNAs by microarray. These results, however, were not only restricted to the transcripts with probes on the microarray, but also limited due to the fact that a transcript’s physical association with ADAR is not necessarily evidence of editing (Ohlson et al. 2005; Ohlson and Ohman 2007). More recently, Tseng et al. (2013) developed a method to detect RNA containing inosine by microarray and Sakurai and colleagues (Sakurai et al. 2010; Sakurai and Suzuki 2011) developed a protocol in which they used inosine cyanoethylation to block reverse transcription, which therefore allowed them to compare treated and untreated cDNAs to identify putative editing sites. While these protocols did not suffer the high false-positive rates seen in other experiments, like the Ohlson protocol they required preexisting knowledge of the transcripts known (or suspected) to harbor editing sites, although it could be adapted to high-throughput approaches.
We sought to generate a protocol capable of identifying A > I editing sites genome-wide that required no prior knowledge of the edited transcript, could be used with RNA from any source, and would have a low false-positive rate. To this end we co-opted aspects of a protocol previously published by Morse and Bass (Morse and Bass 1997; Morse 2004), which had shown that, in the presence of borate ions, glyoxal forms a stable adduct with guanosine but not with inosine, and that glyoxalated guanosines are resistant to RNase T1. This led us to speculate that a glyoxal modified RNA pool could be treated with RNase T1 to yield RNA fragments with inosine at their 3′ end, which could then be sequenced and bioinformatically queried to identify A > I editing sites genome-wide. Here we describe an initial set of proof of concept experiments to assess the enrichment of edited targets using this protocol, followed by its application to total RNA from mouse brain. The results indicate that this protocol is capable of not only robustly detecting known editing sites, but also identifying hundreds of novel editing sites throughout protein-coding and noncoding transcripts.
RESULTS
The iSeq protocol
Our inosine-specific sequencing protocol, which we have dubbed iSeq, is described in detail in the Supplemental Materials (see section titled Supplemental Protocol) and in outline form here (Fig. 1). Briefly, RNA (either poly(A)+ or total) is first biotinylated, treated with glyoxal, and bound to magnetic beads. Then, following the Morse and Bass protocol (Morse and Bass 1997; Morse 2004), the bead–RNA complex treated with glyoxal is successively treated with borate and RNAse T1. RNAse T1 normally cleaves after either guanosine or inosine. When the RNA is treated with glyoxal and boric acid, however, the guanosines form stable glyoxal conjugates which are resistant to RNAse T1 cleavage (Whitfeld and Witzel 1963; Morse and Bass 1997). Subsequent RNAse T1 treatment of the bead bound RNA then generates cleavage products with 3′ inosines (i.e., it liberates the 5′ end of the RNA to the point of the inosine base), which are then eluted and precipitated (inosine-containing RNA, I-RNA). RNA species still bound to the beads, which include both unedited RNAs and the 3′ ends of RNAs containing inosines, are also then eluted (bead-bound RNA, B-RNA). These two RNA pools can then be interrogated by any standard molecular biology approach, including PCR, Sanger sequencing, and high-throughput next-generation sequencing.
FIGURE 1.
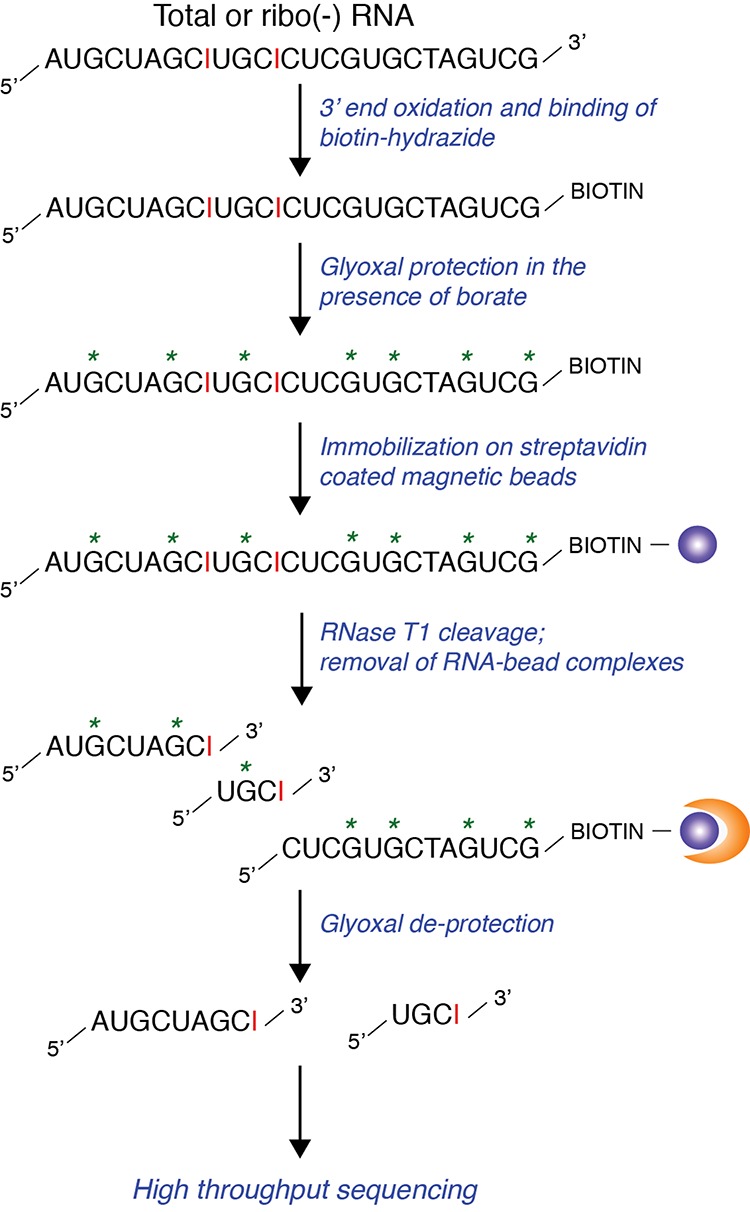
Overview of iSeq, a protocol for the isolation and sequencing of inosine containing RNA. Inosine bases are shown in red. The “*” represents guanosines protected by glyoxal during the RNase T1 cleavage process. The orange half-moon is representative of magnetic bead separation. Streptavidin beads are shown as purple circles.
Specific enrichment of known ADAR targets
To first assess the efficiency of inosilated RNA extraction using this method, the amount of known edited transcripts was assessed in the I-RNA and B-RNA pools extracted from mouse total brain RNA by quantitative PCR. The glutamate receptor Gria2, the serotonin receptor Htr2C, the potassium voltage-gated channel Kcna1, and the γ-aminobutyric acid receptor Gabra3 are reported as targets of the Adar proteins, and are all highly expressed in the mouse brain (Sommer et al. 1991; Higuchi et al. 1993; Burns et al. 1997; Hoopengardner et al. 2003; Bhalla et al. 2004; Ohlson et al. 2007; Daniel et al. 2010). Enrichment of these transcripts was compared with the relative enrichment of a set of negative controls, ß-Actin, Gapdh, Ppia, and Atp5e, which have no annotated editing sites in the DARNED database (Kiran and Baranov 2010), and Rplp0, which has been used as a negative control in previous A > I editing experiments (Ohlson et al. 2005; Ohlson and Ohman 2007). Gabra3, Htr2C, Gria2, and Kcna1 showed three- to sixfold enrichment in the I-RNA pool, whereas the levels of Atp5e, Ppia, Rplp0, and Gapdh were relatively depleted, demonstrating that the protocol facilitated an efficient enrichment of edited RNA species (Fig. 2A). Of the five negative controls, we only found one, ß-Actin, that showed enrichment in the I-RNA pool. However, this was less than twofold, which was more than a third lower than the most weakly enriched positive control (Gabra3) (Fig. 2A). Having established that the protocol successfully enriches for inosilated targets, deep sequencing was then performed to identify the full complement of A > I edited transcripts.
FIGURE 2.

Edited transcripts are selectively enriched by glyoxal protection and RNase T1 digestion. (A) The relative enrichment of Gria2, Htr2C, Kcna1, Gabra3, Rplp0, β-Actin, Gapdh, Ppia, and Atp5e in the I-RNA compared with B-RNA libraries derived from mouse brain is shown. The enrichment was assessed by qPCR in three replicates. Error bars represent the standard deviation of the triplicates. Note that the most weakly enriched known editing site, as assessed by this assay, Gabra3, is nonetheless still 3× above background and >1/3 more enriched than the most highly enriched negative control, β-actin. (B) The densities of nucleotides at the termini of the reads from the I-RNA library are shown; the cytosine density is in blue, the thymine density (i.e., uracil bases that are translated into thymines for high-throughput sequencing) is shown in red, the adenine density in green, and the guanine density in black. Note that we expect to detect inosines, which are read as guanosines by the sequencing machinery, at the 3′ end of the I-RNA reads—which is consistent with the peak in guanosine bases at the 3′ end shown here.
Deep-sequencing of I- and B-RNA pools
Deep sequencing was performed on 200 ng of I-RNA and 500 ng of B-RNA derived from mouse brain total RNA. The I-RNA was sequenced without fragmentation or size selection in order to ensure that the 3′ end of each species, which contained the inosine, would be sequenced. The B-RNA library was fragmented to remove the 3′ end biotin tag. The sequencing produced 34,623,034 and 83,049,769 pairs of 65-nt reads from the I-RNA and B-RNA libraries, respectively (Supplemental Table 1). The apparent discrepancy in the depth of the two libraries can be explained by the differences in their preparation. The B-RNA library preparation included fragmentation and size selection, and the library was therefore homogeneous and the sequencing optimal. In contrast, the I-RNA library did not include either fragmentation or size selection, producing a heterogeneous library that contained both long and short sequences, which reduced the sequencing efficiency. Both libraries, however, showed high levels of mapped tags (Supplemental Table 1), high quality metrics (data not shown), and low numbers of ambiguous bases. Importantly, analysis of the nucleotide content of the I-RNA reads showed a prominent 3′ end guanine bias, which was not observed at the 5′ end (Fig. 2B), consistent with inosine specific RNase T1 cleavage.
Identification of editing sites
To identify the exact position of the edited sites, the I-RNA and B-RNA libraries were mapped to the mouse genome using BWA (Li and Durbin 2009). We employed strict mapping parameters, allowing an edit distance of only 4% (three mismatches on a 65-nt read) and discarding all multimapping tags (i.e., all tags that did not map unambiguously to a single location). When examining the characteristics of the mapped reads, we noted that of all potential mismatch types (i.e., differences between the sequenced RNA and the reference genome), A > G was the most highly represented (Fig. 3), consistent with the iSeq protocol’s specific enrichment for RNA fragments containing a 3′ inosine. Using the BWA generated BAM file (Li et al. 2009a) we extracted the genomic location of the I-RNA 3′ ends and preferentially selected those with robust A > G matches as potential editing sites, yielding a total of 9151 loci (Supplemental Table 2). Each of these 9151 loci was then characterized according to their (i) genomic location, i.e., the coordinate of the last nucleotide of an I-RNA read, (ii) read coverage, defined as the number of reads from both the I-RNA and B-RNA libraries that mapped to the putatively edited location, (iii) frequency of cleavage, which was calculated as the number of I-RNA 3′ ends (i.e., number of cleavage sites) divided by the total number of reads covering the locus (i.e., coverage), and (iv) the frequency of editing, which we defined as the number of guanosines divided by the total number of nucleotides/reads at the locus (i.e., coverage).
FIGURE 3.
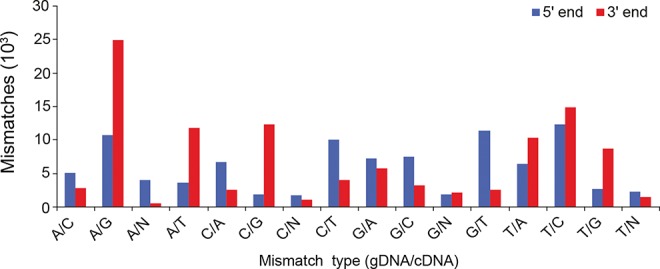
The iSeq protocol enriches for A > G mismatch at the 3′ end of the reads from the I-RNA library. The graph displays the distribution of I-RNA mismatches when mapped to the mouse genome. 5′ end mismatches are shown in red, while 3′ end mismatches are shown in blue. Note the preponderance of A > G mismatches at the 3′ end, consistent with sites of inosine editing.
Despite seeming specificity of this protocol, we were mindful of the fact that the A > G mismatches that defined this initial set of putative editing sites could be the result of experimental or bioinformatic artifacts. For example, the glyoxal protection step is not 100% efficient, and therefore we would expect some 3′ guanosine RNAse T1 products in the I-RNA library. The vast majority of these reads would be filtered out because they would not contain a 3′ A > G mismatch, but some may be included if they mapped to an A > G SNP. Likewise, if the 3′ extremity of a read spans a splice junction, and there is mis-mapping to the adjacent intron instead of the next exon, it can lead to the false discovery of an A > G mismatch (Kleinman and Majewski 2012; Lin et al. 2012). Finally, inaccurate A > G mismatches may also be detected due to sequencing error (Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012; Schrider et al. 2012).
To reduce these biases, four levels of selection filters were applied to the potentially edited loci (Fig. 4): one coverage-specific, two related to the quality of the sequencing at each site, and the last related to the annotation of the locus in the genome. First, the frequency of cleavage was compared with the frequency of random cleavage as a function of the locus coverage (Fig. 5). The I-RNA library contained 384,050 loci that mapped perfectly to the genome and whose 3′ end was a G, meaning that these cleavage sites were generated from the cleavage of guanosines that were not protected by glyoxal. These reads were used to estimate the random cleavage background to be expected when measuring the cleavage frequency of loci presenting with an A > G mismatch. The cleavage frequency of these perfectly mapped loci was estimated as a function of their coverage (from one read to more than 6 million reads). The 95th percentile of the distribution of the cleavage frequencies was used as threshold to select the editing sites with a confidence of P-value <0.05. This selection step facilitated the removal of 5006 potentially edited loci (Supplemental Table 2) that did not have a cleavage frequency significantly higher than random cleavage frequency.
FIGURE 4.
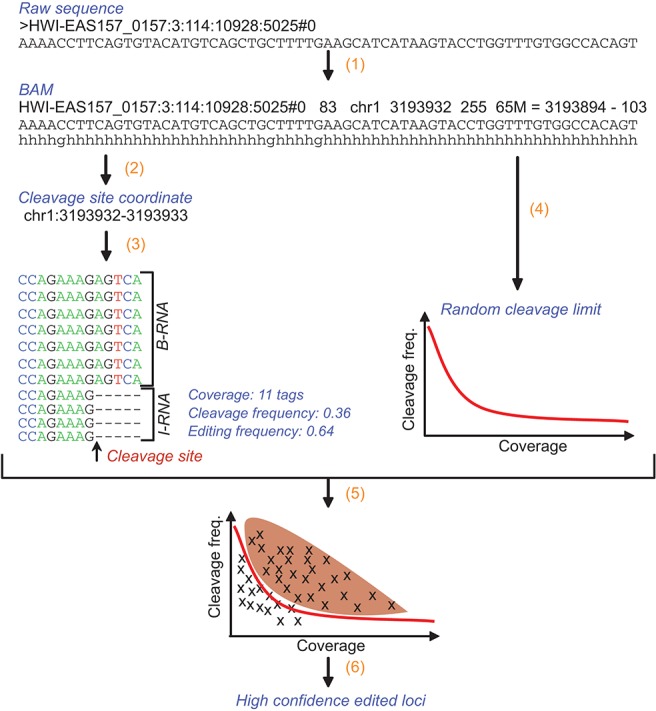
Outline of the algorithm used to detect editing sites and editing frequencies. First the I-RNA and B-RNA deep-sequencing data are mapped to the mouse genome using BWA (1); the coordinates of the tags from the I-RNA library containing an A > G mismatch at their 3′ end are determined—they correspond to the potential editing sites (2). For each position, the coverage by both I-RNA and B-RNA libraries is used to calculate the cleavage frequency and editing frequency (3). At the same time, the tags from the I-RNA library mapping to the genome with no mismatch and having a G in the 3′ end are used to calculate the frequency limit of random cleavage as a function of the coverage (4). Following this, the potential editing sites presenting a cleavage frequency higher than the limit of random cleavage were selected (5). Among the remaining editing sites, the ones (i) presenting an editing frequency higher than the 95th percentile of other mismatches' frequencies from 10 nt upstream of to 10 nt downstream from the locus, (ii) presenting a number of A > G mismatches significantly superior to the number of A > C and A > T mismatches, and (iii) not overlapping with known SNP or splice junction (±5 nt) were considered as high-confidence editing sites (6).
FIGURE 5.

Frequency of cleavage as a function of the coverage for each high-confidence editing site. Each blue cross indicates a high-confidence editing site. The red line indicates the limit of random cleavage at a P-value of 0.05.
Second, the frequency of editing (i.e., A > G mismatches) was compared with the frequency of all the other mismatches found immediately adjacent to the putative editing site. For each locus, the frequency of all mismatches was measured from 10 nt before the locus to 10 nt after the locus. The putative editing loci presenting an editing frequency higher than the 95th percentile of the distribution of the frequencies of all mismatches were selected. This selection step removed an additional 64 potentially edited loci (Supplemental Table 2).
Third, the frequency of editing (i.e., A > G mismatch) was compared with the frequency of the other possible mismatches (i.e., A > C and A > T mismatches) at the putative editing locus itself. The loci presenting an editing frequency significantly higher (P-value <0.05) than the sum of the frequencies of the two other mismatches were selected. This selection step removed 3383 potentially edited loci (Supplemental Table 2). The second and third selection steps were particularly potent at removing loci with a high coverage that had generated putative editing sites because of sequencing errors.
Finally, all the potentially edited loci were compared against all known mouse SNPs present in the SNPdb (Sherry et al. 2001), and all annotated splice junctions in the UCSC KnownGene data set (Hsu et al. 2006). Only two putative editing sites were likely SNP artifacts and were removed at this step, although we noted that a further nine had been removed in the previous three filtering steps. Likewise, we removed a further 31 sites within ±5 nt of splice sites, but found that a further 255 splice-site proximal sites were removed in the previous filtering steps. Overall, we found this set of filtering criteria to be both robust and conservative, and it allowed us to parse our 9151 putative sites down to a set of 665 high-confidence editing events (listed in Supplemental Table 3).
The potency of the discriminative power of the iSeq protocol in combination with this set of filtering steps is illustrated by the editing sites detected in the 3′ UTR of Ebna1bp2, a gene thought to be required for the processing of the 27S pre-rRNA. Five reads from the I-RNA library located in the Ebna1bp2 3′ UTR show A > G mismatches at their 3′ ends (Supplemental Fig. S1A), consistent with two discrete editing sites—“Site 1” covered by three reads, and “Site 2” covered by two reads (Supplemental Fig. S1B). To determine the coverage and editing frequency, all the reads in both I-RNA and B-RNA libraries were aligned and the total coverage and number of mismatches calculated (Supplemental Fig. S1C). As per the filtering criteria, the site-specific mismatch threshold was calculated by summing the frequency T and C mismatches at the cleavage site (0.0 for both Sites 1 and 2), and the surrounding mismatch threshold was estimated by calculating the frequency of all mismatch types within 10 nt of the putative editing sites (Supplemental Fig. S2A–C). Both sites showed an editing frequency higher than both site-specific and surrounding mismatch limits (editing frequency of Site 1 was 90.9% A > G vs. 0% for all others, and the editing frequency at Site 2 was 37.5% vs. 0% for all others). However Sites 1 and 2 differed dramatically in their cleavage frequency: Site 1 exceeded the background cleavage limit (27.3% vs. a random cleavage limit of 18.2% for a coverage of 11 tags) while Site 2 did not (25.0% vs. a random cleavage limit of 25.0% for a coverage of eight tags). Thus, the filtering algorithm designated Site 1 as a high-confidence editing site, while Site 2 was rejected (Supplemental Table 4). Sanger sequencing of Ebna1bp2 genomic DNA (gDNA) and cDNA confirmed the presence of Site 1 in Ebna1bp2 transcript (i.e., an A > G discrepancy between the cDNA and gDNA was observed), and failed to detect any editing at Site 2 (Supplemental Fig. S2D).
Editing site annotation
To characterize the 665 high-confidence editing sites, they were first intersected with transcripts from the UCSC knownGene database (Hsu et al. 2006), the Refgene database (Pruitt et al. 2005), the Ensembl genes database (Hubbard et al. 2002), the GenBank database for mouse and other species (Benson et al. 2004, 2011), and RepeatMasker. Six-hundred forty-two loci were located in transcripts described in at least one of these databases (Supplemental Table 3). The majority of the sites (540 loci) were also located, as expected based on prior research (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004; Carmi et al. 2011; Danecek et al. 2012) in repeats including LINE1s and the B1 and B2 SINE elements. The analysis of the 5′ and 3′ neighbors of the editing sites showed an enrichment in U > A > C > G in 5′, which is concordant with previous reports (Eggington et al. 2011). For the 3′, the enrichment was not as distinct with the following preference A ≈ G ≈ C > U, which is not concordant with previous reports but is due to the difference in site preference between Adar1 and Adar2 and the nondistinction between the sites edited by the two enzymes (Lehmann and Bass 2000; Eggington et al. 2011).
A GO-term enrichment analysis was performed on the genes containing the high-confidence editing sites, using all the genes with I-RNA and B-RNA read coverage as the background. This analysis revealed that the genes with high-confidence editing sites in mouse brain are associated with ion transporter activity, synaptic transmission, and are enriched in the synapse (Supplemental Table 5). This is consistent with the localization of editing enzymes to the neurons in the brain (Jacobs et al. 2009), and the strong impact of editing on neuronal receptors and ions channels (for review, see Jepson and Reenan 2008).
Comparison with previous reports of editing sites
The experiments described here were restricted to RNA isolated from mouse brain, which rendered comparisons between our data and previous A > I editing studies, which were almost exclusively performed in human, difficult. For instance, the DARNED A > I editing database lists 42,042 editing sites in the human genome (Kiran and Baranov 2010), of which only 1794 have orthologous positions in the mouse genome. Indeed, only two of the DARNED database entries overlapped with our high-confidence sites. For the few studies that included mouse transcripts, Kim and colleagues (Kim et al. 2004) reported 90 edited genes, of which 13 were found in our data set; Osenberg and colleagues predicted editing in 98 mouse genes (Osenberg et al. 2009), two of which were confirmed by our study; Neeman and colleagues predicted 833 editing sites (Neeman et al. 2006), six of which are present in our data set; and, finally, Danecek and colleagues reported 7389 editing sites (Danecek et al. 2012), 92 of which are present in our data set (see Supplemental Table 3 for complete details). Overall, our set of high-confidence editing sites in mouse brain includes 99 that were previously reported, and 566 novel editing sites. The low overlap between previous reports of editing sites in mouse and our data may indicate that the number of editing sites is more extended than found so far.
Validation of editing sites by Sanger sequencing
Among the 566 new editing sites, 20 sites were randomly selected for validation by Sanger sequencing (Fig. 6; Supplemental Fig. S3): ID_5463 (Fig. 6A) is located in a B1 repeat in the 3′ UTR of Ebna1bp2, ID_6291–94 (Fig. 6B) are located in the large first intron of an alternative transcript of Kcnip4, ID_984–86 (Fig. 6C) are located in the 3′ UTR of Rpa1, ID_4361 (Fig. 6D) is located in a B1 repeat in the first intron of Zc3h6, ID_3216 and ID_3217 (Supplemental Fig. S3A) are located in the exonic region of Ak138184, ID_7659 (Supplemental Fig. S3B) is located in the first intron of Csmd1, ID_209 (Supplemental Fig. S3C) is located in the second intron of Grik2, ID_2089 (Supplemental Fig. S3D) is located in the large intron of Hs6st3, ID_6208 (Supplemental Fig. S3E) is located in a L1 repeat in the 5′ UTR of AK036806, ID_3921–23 (Supplemental Fig. S3F) are located in the 3′ UTR of NM_029909, ID_1317 (Supplemental Fig. S3G) is located in the small nucleolar RNA SNORA28, and ID_524 (Supplemental Fig. S3H) is located in the 3′ UTR of Tbc1d16. To verify that these sites were edited, the genomic DNA and RNA associated with each locus were sequenced. Genetic material was isolated from a mouse brain not used in the original study. Eighteen out of 20 loci (90%) presented clear A > G polymorphisms in the cDNA sequence that were not observed in the gDNA sequence (Fig. 6A–D; Supplemental Fig. S3A–H), thus confirming editing at these loci. The two remaining sites did not show significant polymorphism in the cDNA (Supplemental Fig. S3B,G). This is the highest validation rate reported for a de novo editing site identification protocol.
FIGURE 6.
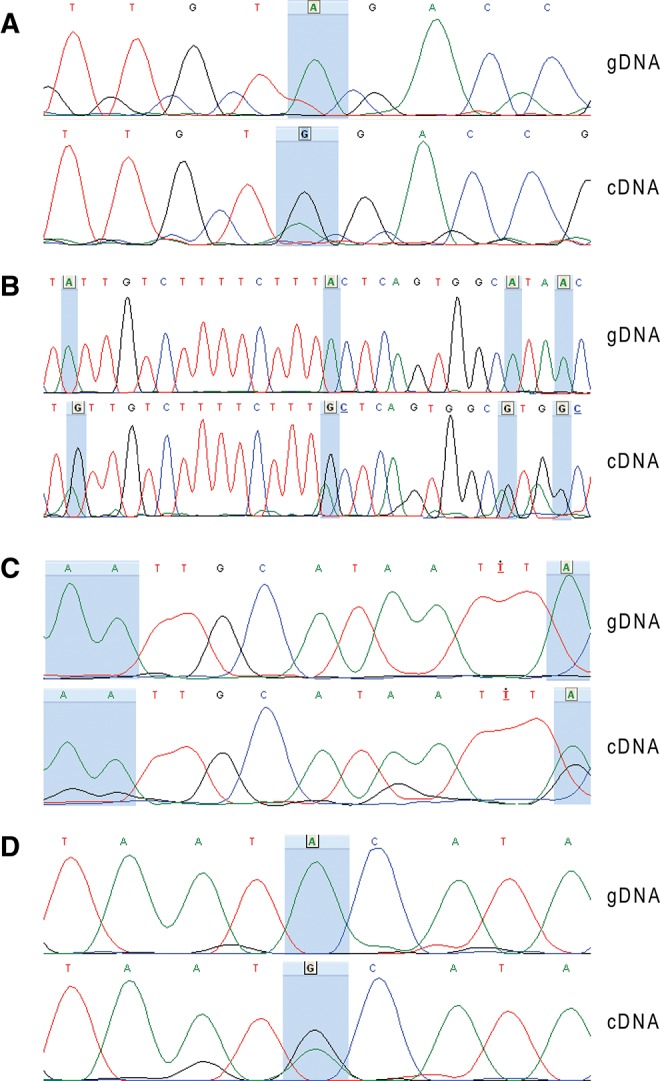
Chromatograms of the sequences of the genome and transcript at nine high-confidence editing loci. The loci are located in five different transcripts: (A) the site ID_5463 is in Ebna1bp2, (B) the sites ID_6291–94 (from right to left) are in Kcnip4_bis, (C) the sites ID_984–86 (from right to left) are in Rpa1, (D) and the site ID_4361 is in Zc3h6. For each candidate, the chromatogram of the genomic sequence (gDNA) is aligned to the chromatogram of the transcript’s sequence (cDNA). The level of adenine is represented in green, the level of guanine is in black, the level of thymine in red, and the level of cytosine in blue. The blue highlights indicate the high-confidence editing sites characterized by iSeq.
Editing sites in CDS
We identified a total of eight editing sites in protein-coding regions. Six have been previously described: the site I/V in Kcna1 (Bhalla et al. 2004), the site A (I/V) in Htr2C (Burns et al. 1997; Fitzgerald et al. 1999; Niswender et al. 1999), the site Y/C in Grik2 (Kohler et al. 1993), the site K/E in Cyfip2 (Levanon et al. 2005), the site I/M in Gabra3 (Ohlson et al. 2007; Daniel et al. 2010), and the site E/G in Cadps (Li et al. 2009b). The editing frequencies at these sites were nearly identical to what has been previously reported. For example, we found that Kcna1 site I/V was 50% edited, compared with a report of 47% editing rate in mouse brain (Hoopengardner et al. 2003; Bhalla et al. 2004). Likewise, we found GABRA3 site I/M editing at 94%, compared with a prior report of 100% of editing in adult mouse (Ohlson et al. 2007). 5Ht2C site A was 73% edited in our data and was previously reported at ∼80% (Burns et al. 1997). Lastly, Grik2 site Y/C and Cyfip2 site K/E were measured at 80% and 86% respectively, consistent with prior reports of 80% and 90% editing (Table 1; Kohler et al. 1993; Riedmann et al. 2008). Novel editing sites were found in the coding regions of the histones genes Hist2h2ab and Hist2h2ac (Supplemental Fig. S4; Supplemental Table 6). The editing site in Hist2h2ac generates a nonsynonymous change—an asparagine is altered to become a serine (Table 1). Further work is needed to investigate the possible ramifications of this alternation, but is worthwhile noting that (i) the asparagine is conserved across the metazoan spectrum at least back to boney fish and that (ii) serines within histones are frequently modified, perhaps suggesting that editing of this histone CDS could facilitate further downstream epigenetic modifications and remodeling.
TABLE 1.
High-confidence editing sites located in coding exons of protein coding genes
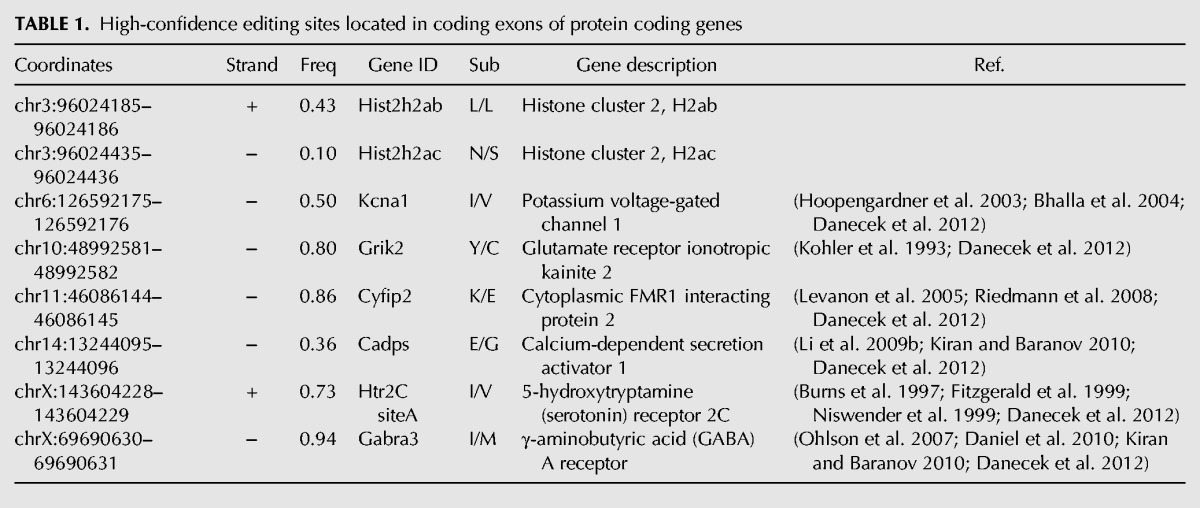
We failed to identify the Gria2 R/G (Maas et al. 1996; Melcher et al. 1996; Yang et al. 1997), Gria2 Q/R (Sommer et al. 1991; Higuchi et al. 1993), or Blcap (Levanon et al. 2005) editing sites, despite the fact that they have been reported to be highly edited. Manual curation of these sites revealed that our data do in fact detect them, but that they were excluded by the filtering algorithm due to low coverage. For example, for Blcap site we detected only eight reads from the I-RNA library and one cleavage site. This further indicates that our protocol is conservative and robust, and that the number of editing sites we have detected in protein-coding regions and overall is likely a lower bound.
Editing in repeats
The vast majority, 81%, of the high-confidence editing sites are located in genomic regions annotated as repetitive elements. Since transcripts derived from repeats are usually difficult to sequence and bioinformatically analyze, we reexamined our BWA analysis to allow us to test if multimapping between repeats was affecting our results and to examine if we could discriminate between repeat elements of the same family. First, the reads from the I-RNA library were remapped with an edit distance of 0.08 (i.e., twice the edit distance allowed for the initial analysis, or five mismatches for a read of 65 nt), next the data were filtered to select only uniquely mapping reads, which were then queried for the presence of the previously identified editing sites and whose alignment metrics were compared with the results of the initial strict I-RNA read mapping. This analysis returned 658 (99%) of the high-confidence set, suggesting that multimapping tags were not deleteriously affecting our analysis strategy and that editing site association with repeat classes can be robustly measured using the iSeq and associated informatics protocols. Of the 534 sites annotated in RepeatMasker, the vast majority were located in B1, B2, and L1 elements (Supplemental Fig. S5). Seventy-two sites located in repeats were described in previous reports (Supplemental Table 3).
Hyperediting
Genes containing two repeats from the same family, with one in the sense and the other in the antisense orientation, can form double-stranded structures that are targeted by Adar and hyperedited (Morse et al. 2002; Kawahara and Nishikura 2006; Carmi et al. 2011). Hyperediting can lead to transcript sequestration in paraspeckles, which can be later released by cleavage under stress conditions (Lunyak et al. 2007). Osenberg et al. (2009) predicted hyperediting of 107 mouse genes, of which 16 show evidence of editing by iSeq (Supplemental Table 7) including high-confidence sites in Mark3 and Tapbp. To investigate hyperediting in mouse brain we systematically screened for genes containing at least four high-confidence editing sites. The transcript containing the highest number of high-confidence editing sites was Kcnip4 (ENSMUST00000087395), with 27 editing sites spread over its first intron. Since the intron is large (>1 Mb), it was unclear if the editing sites were located on the intron of the pre-mRNA or on transcripts derived from the same region. This was particularly true for the editing sites ID_6208, ID_6211, ID_6212, and ID_6214, which overlapped transcript AK036806 (Supplemental Figs. S6, S7A) and the editing sites ID_6223–25, ID_6227, and ID_6230 that overlapped the putatively noncoding transcript AK148828 (Supplemental Figs. S6, S8A). Read coverage analysis revealed, however, that I-RNA and B-RNA read density across AK036806 and AK148828 was not significantly higher than the surrounding region, but was instead relatively homogenous throughout the intron (Supplemental Fig. S6), suggesting that the editing sites detected were derived from the Kcnip4 pre-mRNA.
This conclusion was further supported by investigation of RNA secondary structures. Indeed, we found that the minimum free energy of folding regions corresponding to both transcripts was substantially lowered when the adjacent Kcnip4 intronic region was included. For example, RNAfold estimated the minimum free energy of AK036806 at −1027.94 kcal/mol by itself, and −2100.06 kcal/mol when including the adjacent L1; and when the values were normalized as a function of length, the transcript alone had a relative minimum free energy of −0.27 kcal/mol/nt and the transcript with the additional L1 in the 3′ end had a relative minimum free energy of −0.36 kcal/mol/nt. For AK148828, RNAfold estimated the minimum free energy of the transcript at −519.82 kcal/mol by itself (relative minimum free energy: −0.27 kcal/mol/nt) and at −1484.57 kcal/mol with an adjacent L1 element at the 5′ end (relative minimum free energy: −0.39 kcal/mol/nt). Moreover, the addition of the regions adjacent to AK036806 and AK148828 generated the ideal secondary structure for hyperediting by promoting the formation of long double-stranded structures that were not observed when folding the transcripts alone (Supplemental Figs. S7B,C, S8B,C). Interestingly, all the editing sites found on AK036806 and AK148828 were located in double-stranded structures when folded with the adjacent L1 repeats, but were scattered in poorly structured regions when folded on their own (Supplemental Figs. S7C and S8C, respectively). These results suggest that the first intron of the Kcnip4 pre-mRNA is heavily edited en masse in mouse brain.
We also identified two further cases of extensive hyperediting. Fgf14 encodes two splice isoforms, Fgf14a and Fgf14b, which differentially regulate voltage-gated sodium channels (Laezza et al. 2009). Fgf14b’s first intron is >500 kb and contains 294 LINE elements and 115 SINE elements. Fifteen high-confidence editing sites were identified in this large intron, including 10 in LINE1 elements and three in B1 elements (Supplemental Fig. S9). Like Kcnip4 above, read coverage was continuous throughout the intron, with only one site ambiguous as to its origin (site ID_2117 overlaps with AK016500). This strongly suggests that these editing events were localized to the Fgf14b pre-mRNA, and not other aberrant or cryptic transcripts derived from the locus.
Usp29, a deubiquitinating enzyme that is expressed in response to stress and stabilizes p53 and facilitates apoptosis (Liu et al. 2011), showed four high-confidence editing sites in its 3′ UTR. Secondary structure prediction indicated that the sites were located in a double-stranded structure formed by a B2 and adjacent B3 element (Supplemental Fig. S10). Intriguingly, we found that an antagonist of Usp29, Gnl3l, which prevents ubiquitylation of MDM2 (Meng et al. 2011) and in return promotes the ubiquitylation of p53 (Moll and Petrenko 2003), also contains four high-confidence editing sites in its 3′ UTR. Like Usp29, they are located in double-stranded structure formed by two B1 elements of opposite orientation, with the sense oriented B1 containing one editing site and the antisense B1 three editing sites. Secondary structure prediction indicated that, although ∼1 kb separates the solitary editing site from the cluster of three, after folding, all are immediately adjacent (Supplemental Fig. S11). It is possible that hyperediting may play a role in the regulation and expression not only of these genes, but also the p53 pathway.
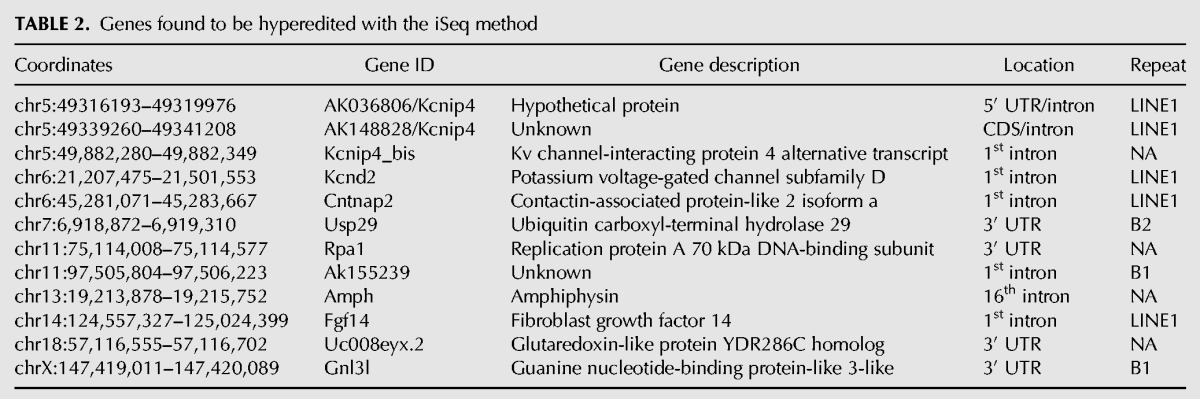
Overall, we identified a total of seven additional genes that were robustly hyperedited (Table 2): Kcnd2, Cntnap2, Rpa1, Ak155239, Amph, Kcnip4_bis, and uc008eyx.2. All sites of hyperediting correlated with predicted double-stranded regions (Table 2 and structures in Supplemental Figs. S12–14), even when no repeats were present (Supplemental Fig. S12A–D). Additionally, we observed consistent localization of editing sites within the secondary structure, similar to the conformational arrangement of Gnl3l described above. For example, Kcnip4_bis contains two regions with a single and four editing events, respectively, that are 1 kb apart in linear genomic space but <30 nt in the predicted secondary structure (Supplemental Fig. S12A). Likewise, Kcnd2 contains editing sites separated by 3.5 kb of genomic sequence, but that are <150 nt from each other in the predicted secondary structure (Supplemental Fig. S13A); and similar editing site proximity alterations were observed for Amph (Supplemental Fig. S12B), Rpa1 (Supplemental Fig. S12D), and Ak155239 (Supplemental Fig. S14A).
TABLE 2.
Genes found to be hyperedited with the iSeq method
Editing in intergenic regions
Twenty-three editing sites were located in regions that were not annotated as containing expressed transcripts in any of the following databases: the UCSC KnownGene database (Hsu et al. 2006), Refgene (Pruitt et al. 2005), Ensembl (Hubbard et al. 2002), or GenBank (Benson et al. 2004, 2011) (Supplemental Table 3). To test if these editing sites were derived from weakly expressed transcripts that had not yet been included in any publicly available database, we interrogated a large cohort of publicly available RNAseq data sets. De novo transcriptomes were generated using Tophat and Cufflinks (Trapnell et al. 2009) from a data set spanning 19 mouse tissues (GSE29278) (Shen et al. 2012), data derived from stranded whole brain data set (SRX003743) (Parkhomchuk et al. 2009), and a paired-end unstranded whole brain data set (ERS028664) (Keane et al. 2011). We were able to ascertain the transcripts associated with a further four editing sites, for which no evidence of transcription was available elsewhere. The 19 remaining sites, however, were not able to be identified with any known transcriptional unit. This suggests that there is likely to be weakly expressed poly(A)+ transcripts, or cryptic non-poly(A)+ transcripts that have not been completely polled, that are specifically edited in mouse brain. Additional experimental work, however, is required to fully characterize this small, but interesting, subset of high-confidence editing sites.
DISCUSSION
Here we have described a novel high-throughput experimental approach to identify sites of A > I editing. RNAs are immobilized on magnetic beads, treated with glyoxal and RNAse T1 to specifically cleave at inosine bases, and RNA fragments with inosines at their 3′ terminus are then analyzed by next-generation sequencing. We have partnered these laboratory steps with a bioinformatic algorithm that selects sites showing enrichment in cleavage frequency and A > G mismatches with a low background of other nucleotide permutations. We successfully deployed this system to identify 665 high-confidence editing sites—520 were present with the transcriptional bounds of known genes, and seven of these were located in coding regions and generated nonsynonymous mutations.
One of the limitations of this protocol was the depth of the sequencing. Since our algorithm applies two levels of selection which rely highly on the coverage of each locus, many potential editing sites were discarded because of low coverage (e.g., 35% of the potential editing sites were covered by less than three reads). The poor coverage was explained partially by the use of total RNA (as opposed to poly(A)+ RNA), and thus 58.3% of the tags in the I-RNA library came from rRNA (71.9% in the B-RNA library). Although this facilitated the identification of novel editing sites in rRNA (ID_3350, ID_3352, and ID_3476 map to the 45S pre-ribosomal RNA), it considerably reduced the coverage of other loci which has almost certainly resulted in an underestimate of the number of “true” editing sites detected. Despite the high number of reads that mapped to rRNA, the vast majority of editing sites were nonribosomal (83%), illustrating the robustness and efficiency of this protocol. Moreover, this experiment was conducted on a tissue of high complexity (brain), which we suspect has a high number of editing events in transcripts that are expressed in a minority of cells, again suggesting that the number of editing sites we have identified here is likely a lower bound. To increase the discovery rate, efforts should be made to increase the depth of the I-RNA sequencing and to reduce the complexity of the tissue or the transcriptome investigated by dissecting specific region of tissues and/or removing the rRNA from the total RNA, particularly if working with human brain RNA, which contains 20 times more inosine than mouse brain RNA (Eisenberg et al. 2005).
To conclude, iSeq represents a powerful tool to enrich for edited RNA, identify A > I editing sites, and quantify the level of editing de novo transcriptome-wide fashion. Its application on RNA from knockout mutants for the different ADAR in cell lines or whole organisms (Riedmann et al. 2008) would allow a clear identification of the enzyme targeting each site. In addition, the initial input of iSeq is purified RNA, which means that, given minor adjustments of the protocol for the length of the input, any kind of pre-purification step can be applied to the sample to interrogate specific family of RNA such as small RNA, poly-adenylated RNA, or rRNA. We submit that this protocol will have significant value in enriching editing sites, given the likely complexity and cell specificity of the transcriptome (Mercer et al. 2012) and the editome, and thereby assist in uncovering this new dimension of molecular genetic plasticity.
MATERIALS AND METHODS
Preparation of the I-RNA and B-RNA
A single mouse brain was harvested from an eight-week-old male C57Bl/6 mouse and total RNA was extracted using TRIzol (Invitrogen #15596-026) according to manufacturer’s instructions. Total RNA was then treated as follows (a detailed protocol is available in Supplemental Protocol): Briefly, 30 μg total RNA was biotinylated, coated with glyoxal and boric acid, and bound to streptavidin-coated magnetic beads. The RNA–magnetic bead complexes were then treated with RNAse T1 to cleave the inosinylated RNA (I-RNA) off the beads, which was collected. The RNA bound to the beads (B-RNA) was eluted using formamide. The glyoxal was removed by heating from both I-RNA and B-RNA. In order to perform 3′ end sequencing on the I-RNA, it was treated with TAP to remove any 5′ cap-like modifications and with PNK to repair the extremities.
PCR
To assess the efficiency of the enrichment in inosilated RNA, the quantity of the following transcripts was queried by PCR in B-RNA and I-RNA: Gria2, Htr2C, Kcna1, Gabra3, Rplp0, β-actin, Gapdh, Ppia, and Atp5E. The primers (Supplemental Table 8) were designed upstream of the editing sites for Gria2, Htr2C, Kcna1, and Gabra3; 30 ng of I-RNA and 30 ng of B-RNA were reverse-transcribed with the Superscript III kit (Invitrogen #18080-051) according to manufacturer’s instructions using random hexamers. Real time PCR was then performed in triplicate using SYBR Green PCR Master Mix (Applied Biosystems #4309155), 0.5 µL of cDNA and 0.25 μM primers in 10 μL on the ViiA7 Real-Time PCR System (Applied Biosystems). The enrichment of a given target in I-RNA compared with B-RNA was calculated according to the following formula:
The experiments were performed in triplicate on RNA from the same mouse brain.
Library preparation and deep sequencing
The library preparation and deep sequencing were performed by GeneWorks (Adelaide, Australia; http://www.geneworks.com.au). The I-RNA library was prepared with 200 ng I-RNA using the Illumina TruSeq Small RNA kit (Illumina #RS-200-0012) and size selection performed using AMPure beads (Beckman Coulter #A63880) to select for inserts >100 nt. The I-RNA library was sequenced on two lanes, generating paired-end 65-nt reads. The B-RNA library was prepared with 500 ng B-RNA using the Illumina TruSeq RNA kit (Illumina #FC-122-1001) and sequenced on two lanes to also generate paired-end 65-nt reads. All sequencing was done on an Illumina Genome Analyzer IIx.
Deep-sequencing data analysis
Mapping of the raw data
The nucleotide density of the raw sequences for each library was measured using FastQC, developed by Babraham Bioinformatics (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/).
Both I-RNA and B-RNA libraries were mapped as paired-end libraries to the mouse genome assembly NCBI37/mm9 (ftp://ftp.ensembl.org/pub/release-64/fasta/mus_musculus/dna/) using BWA for short queries (Li and Durbin 2009) allowing an edit distance of 4%. Uniquely mapping pairs of reads were then selected using the samtools package (Li et al. 2009a) to remove all reads that were not aligned (Flag 256). All the pairs of reads containing 65-nt long sequences (CIGAR = 65 M) were then selected, and for each pair of reads from the I-RNA library, the 3′ end nucleotide and its position were extracted using a custom AWK script. Each nucleotide was compared with its genomic counterpart, which was deduced using the bed coordinate of the 3′ end of each pair of reads from the I-RNA library using fastaFromBed from the BEDTools package (Quinlan and Hall 2010). All the loci presenting an A > G mismatch (cDNA/gDNA) were identified as potential editing sites.
Determination of the high-confidence editing sites
The cleavage frequency was estimated by counting the number of read pairs from the I-RNA library whose 3′ end matched the potential editing site coordinate, and calculating the ratio (cleavage reads/total reads) using reads from both the I-RNA and B-RNA data sets.
To estimate the level of the nonspecific cleavage, the 3′ ends of all reads from the I-RNA library that did not present an A > G mismatch at their 3′ end were determined. For each position, the frequency of cleavage and the coverage by both I-RNA and B-RNA libraries were estimated. The 95th percentile of the unspecific cleavage frequency was calculated as a function of the coverage as follows: The frequencies of cleavage were sorted according to their values, and the lower value of the top 5% of the frequencies represented the 95th percentile of the distribution of the nonspecific cleavage as a function of the coverage.
To estimate the level of background due to sequencing error, the nucleotide composition of the surrounding area (10 nt upstream and downstream) of each site was determined by first extracting all the reads mapping to the site in both the B-RNA and I-RNA libraries using samtools view (Li et al. 2009a), then all these reads were aligned to each other, and the nucleotide composition of each position 10 nt upstream of and downstream from the site was estimated using a custom AWK script. For each site the 95th percentile of the distribution of mismatch frequency was determined as follows: All the mismatch frequencies of the area surrounding the editing site were sorted according to their values, and the lower limit of the top 5% represented the 95th percentile of the distribution of the mismatch frequencies surrounding each site. The G frequency at the site was defined as the “editing frequency.”
The high-confidence editing sites were then selected as follows: The cleavage frequency needed to be higher than the 95th percentile of the distribution of the nonspecific cleavage events; the editing frequency needed to be higher than the 95th percentile of the distribution of the mismatch frequencies surrounding each site; and the editing frequency needed to be significantly higher than the sum of the frequencies of the two other possible mismatches (A > T and A > C) at the site. For this last criterion a one-tailed two proportion z-test was performed as to measure the significance of the difference. The Z-score was then determined with the following formula, with N being the number of tags covering the locus:
 |
The P-values were determined using a Z-score table.
To remove the sites for which A > G polymorphisms were confounding, the locations of the editing sites were intersected with the locations of all reported mouse SNPs (Sherry et al. 2001) using intersectBed from the BEDTools package (Quinlan and Hall 2010). Sites located <5 nt from a splice junction with the read spanning this splice junction were also removed. The location of each splice junction and the next 5 nt was determined using AWK based on the BED12 coordinate sets of all genes reported in the UCSC gene database (Hsu et al. 2006). intersectBed from the BEDTools package (Quinlan and Hall 2010) was used to determine which editing sites were located in these regions.
All the sites passing these criteria were called high-confidence loci.
Annotation of the editing sites
The bed coordinates of the UCSC gene database (Hsu et al. 2006), the RefSeq database (Pruitt et al. 2005), the Ensembl database (Hubbard et al. 2002) and the GenBank database for mouse and other species (Benson et al. 2004, 2011), and RepeatMasker (Smit et al. 1996–2010) entries were intersected successively with the coordinates of the high-confidence loci using intersectBed (Quinlan and Hall 2010). Each locus was then annotated as follows: “Database”: UCSC, RefSeq, Ensembl, Genbank, GenbankOther, and, if none, N/A; “Database Gene ID” and, if none, N/A; “Location in the transcript”: exonic if located in UTR or other exon, otherwise intronic, N/A if not available; “Repeat”: when applicable to the name of the repeat and when its genomic orientation was notified.
All loci were also compared with the stranded transcriptomes generated by Shen et al. (2012) for 19 tissues from C57BL/6 mice and primary cell types (GSE29278), the stranded transcriptome (SRX003743) generated with whole brain RNA from C57BL/6 mouse by Parkhomchuk et al. (2009), and the paired-end unstranded transcriptome generated on mouse brain RNA from C57BL/6 mouse (ERS028664) (Keane et al. 2011). For each library, the alignment file (.bam) was generated with Tophat (Trapnell et al. 2009) or downloaded directly from the authors if available. Cufflinks (Trapnell et al. 2010) was used to generate a de novo transcriptome. The UCSC Table browser (Karolchik et al. 2004) was then used to intersect the editing sites with the Cufflinks assembled transcripts. The annotation of each high-confidence locus is available in Supplemental Table 3.
The 5′ and 3′ neighbors were determined by extracting the sequences surrounding each editing site, and the nucleotide preference was determined using WebLogo (Crooks et al. 2004).
Ontology analysis
The genes containing high-confidence editing sites assessed for GO-term enrichment, using all genes with I-RNA or B-RNA coverage as the background set. The analysis was performed using GOrilla (Eden et al. 2009). The P-value threshold was set at 10−5.
Characterization of hyperediting
High-confidence loci were crossed with the hyperediting predictions from Osenberg et al. (2009) using intersectBed (Quinlan and Hall 2010). A systematic investigation of hyperedited genes was performed as follows: A gene was considered “hyperedited” if one of its components (UTR, intron, and exon) contained four or more editing sites. For each candidate, the secondary structure of the region containing the edited loci and the minimum free energy of the structure were determined using RNAfold (Hofacker and Stadler 2006) and the secondary structure was annotated using VARNA (Darty et al. 2009). For Kcnip4, to compare the stability of two structures of transcripts of various lengths, the minimum free energy per nucleotide (Hughes and McElwaine 2006) was estimated by calculating the ratio [minimum free energy given by RNAfold/length of the transcript being folded].
Confirmation of A > I editing by Sanger sequencing
RNA and genomic DNA were extracted from a unique mouse brain (eight-week-old male C57Bl/6) using Trizol (Invitrogen #15596-026). The RNA was DNAse-treated twice with Turbo DNAse (Ambion #AM2238), cleaned with RNeasy MinElute Cleanup Kit (Qiagen #74204), and converted to cDNA using SuperScript III First-Strand Synthesis System (Invitrogen # 18080-051) with random hexamers.
Primers were designed around the editing sites located in Ak138184, Csmd1, Ebnabp2, Grik2, Hs6st3, Ak036806, Kcnip4_bis, NM_029909, Rpa1, snoRA28, Tbc1d16, and Zc3h6 (Supplemental Table 8) and PCR were performed with the Phusion High-fidelity PCR kit (NEB #E0553S) according to the manufacturer instruction on the mouse brain cDNA and gDNA with the following program: 98°C for 30 sec (98°C for 10 sec, 59°C for 15 sec, 72°C for 30 sec) × 35 and 72°C for 10 min. The PCR products were then run in an agarose gel 1.5%/TBE 1× in TBE 1× and stained in Ethidium bromide 0.5 µg/mL of TBE 1× for 10 min. Under UV light, the bands corresponding to the PCR product were cut from the gel, and the PCR products were extracted from the gel’s bands using the QIAquick Gel Extraction Kit (Qiagen #28706) according to manufacturer instructions. The purified PCR amplicons were then sequenced by Sanger sequencing by GATC (http://www.gatc-biotech.com) using the sequencing probe indicated in Supplemental Table 8.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Australian Research Council (project grant DP0988851 and Federation Fellowship grant FF0561986) and the Australian National Health & Medical Research Council (Australia Fellowship 631668; J.S.M.). R.J.T. is supported by an Australian Research Council Discovery Early Career Researcher Award. We thank GeneWorks for generating the deep-sequencing libraries and optimizing the GAIIx system to work with our samples.
REFERENCES
- Alon S, Mor E, Vigneault F, Church GM, Locatelli F, Galeano F, Gallo A, Shomron N, Eisenberg E 2012. Systematic identification of edited microRNAs in the human brain. Genome Res 22: 1533–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadis A, Rich A, Maas S 2004. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2: e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL 2004. GenBank: Update. Nucleic Acids Res 32: D23–D26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW 2011. GenBank. Nucleic Acids Res 40: D48–D53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJ, Holmgren M, Reenan R 2004. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol 11: 950–956 [DOI] [PubMed] [Google Scholar]
- Blow M, Futreal PA, Wooster R, Stratton MR 2004. A survey of RNA editing in human brain. Genome Res 14: 2379–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, Davidson BL 2009. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet 18: 4801–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387: 303–308 [DOI] [PubMed] [Google Scholar]
- Carmi S, Borukhov I, Levanon EY 2011. Identification of widespread ultra-edited human RNAs. PLoS Genet 7: e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL, DeCerbo JN, Carmichael GG 2008. Alu element-mediated gene silencing. EMBO J 27: 1694–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE 2004. WebLogo: A sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Nellaker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ 2012. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 13: r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C, Wahlstedt H, Ohlson J, Bjork P, Ohman M 2010. Adenosine-to-inosine RNA editing affects trafficking of the γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem 286: 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y 2009. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25: 1974–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H, Kawano M, Kishima M, Lassmann T, Faulkner GJ, Mattick JS, et al. 2010. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res 20: 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z 2009. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY 2005. Is abundant A-to-I RNA editing primate-specific? Trends Genet 21: 77–81 [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Iyer G, Conklin DS, Krause CM, Marshall A, Patterson JP, Tran DP, Jonak GJ, Hartig PR 1999. Messenger RNA editing of the human serotonin 5-HT2C receptor. Neuropsychopharmacology 21: 82S– 90S [DOI] [PubMed] [Google Scholar]
- Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch MF 2009. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol 29: 1487–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE 1999. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci 96: 4621–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale BS, Eulalio A, Schulte L, Vogel J, O’Connell MA 2010. Analysis of A to I editing of miRNA in macrophages exposed to Salmonella. RNA Biol 7: 621–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH 1993. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81 [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Stadler PF 2006. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 22: 1172–1176 [DOI] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R 2003. Nervous system targets of RNA editing identified by comparative genomics. Science 301: 832–836 [DOI] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC Known Genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. 2002. The Ensembl genome database project. Nucleic Acids Res 30: 38–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TA, McElwaine JN 2006. Mathematical and biological modelling of RNA secondary structure and its effects on gene expression. Comput Math Methods Med 7: 37–43 [DOI] [PubMed] [Google Scholar]
- Jacobs MM, Fogg RL, Emeson RB, Stanwood GD 2009. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev Neurosci 31: 223–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Reenan RA 2008. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta 1779: 459–470 [DOI] [PubMed] [Google Scholar]
- Jin Y, Zhang W, Li Q 2009. Origins and evolution of ADAR-mediated RNA editing. IUBMB Life 61: 572–578 [DOI] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Nishikura K 2006. Extensive adenosine-to-inosine editing detected in Alu repeats of antisense RNAs reveals scarcity of sense-antisense duplex formation. FEBS Lett 580: 2301–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K 2007. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A 2004. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res 14: 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran A, Baranov PV 2010. DARNED: A DAtabase of RNa EDiting in humans. Bioinformatics 26: 1772–1776 [DOI] [PubMed] [Google Scholar]
- Kleinman CL, Majewski J 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302; author reply 1302 [DOI] [PubMed] [Google Scholar]
- Kohler M, Burnashev N, Sakmann B, Seeburg PH 1993. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: Diversity by RNA editing. Neuron 10: 491–500 [DOI] [PubMed] [Google Scholar]
- Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM 2009. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci 42: 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39: 12875–12884 [DOI] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E 2005. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res 33: 1162–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009a. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM 2009b. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324: 1210–1213 [DOI] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG 2011. Widespread RNA and DNA sequence differences in the human transcriptome. Science 333: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302; author reply 1302 [DOI] [PubMed] [Google Scholar]
- Liu J, Chung HJ, Vogt M, Jin Y, Malide D, He L, Dundr M, Levens D 2011. JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO J 30: 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano DJ, Mirsky H, Vendetti NJ, Maas S 2004. RNA editing of a miRNA precursor. RNA 10: 1174–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, Prefontaine GG, Nunez E, Cramer T, Ju BG, Ohgi KA, Hutt K, Roy R, Garcia-Diaz A, Zhu X, et al. 2007. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science 317: 248–251 [DOI] [PubMed] [Google Scholar]
- Maas S, Melcher T, Herb A, Seeburg PH, Keller W, Krause S, Higuchi M, O’Connell MA 1996. Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J Biol Chem 271: 12221–12226 [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL 2005. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309: 1534–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS 2010. RNA as the substrate for epigenome-environment interactions: RNA guidance of epigenetic processes and the expansion of RNA editing in animals underpins development, phenotypic plasticity, learning, and cognition. Bioessays 32: 548–552 [DOI] [PubMed] [Google Scholar]
- Mattick JS, Mehler MF 2008. RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci 31: 227–233 [DOI] [PubMed] [Google Scholar]
- Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF 2009. RNA regulation of epigenetic processes. Bioessays 31: 51–59 [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M 1996. A mammalian RNA editing enzyme. Nature 379: 460–464 [DOI] [PubMed] [Google Scholar]
- Meng L, Hsu JK, Tsai RY 2011. GNL3 L depletion destabilizes MDM2 and induces p53-dependent G2/M arrest. Oncogene 30: 1716–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, Mattick JS, Rinn JL 2012. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol 30: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, Petrenko O 2003. The MDM2-p53 interaction. Mol Cancer Res 1: 1001–1008 [PubMed] [Google Scholar]
- Morse DP 2004. Identification of substrates for adenosine deaminases that act on RNA. Methods Mol Biol 265: 199–218 [DOI] [PubMed] [Google Scholar]
- Morse DP, Bass BL 1997. Detection of inosine in messenger RNA by inosine-specific cleavage. Biochemistry 36: 8429–8434 [DOI] [PubMed] [Google Scholar]
- Morse DP, Aruscavage PJ, Bass BL 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci 99: 7906–7911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeman Y, Levanon EY, Jantsch MF, Eisenberg E 2006. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 12: 1802–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E 1999. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem 274: 9472–9478 [DOI] [PubMed] [Google Scholar]
- Ohlson J, Ohman M 2007. A method for finding sites of selective adenosine deamination. Methods Enzymol 424: 289–300 [DOI] [PubMed] [Google Scholar]
- Ohlson J, Enstero M, Sjoberg BM, Ohman M 2005. A method to find tissue-specific novel sites of selective adenosine deamination. Nucleic Acids Res 33: e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlson J, Pedersen JS, Haussler D, Ohman M 2007. Editing modifies the GABAA receptor subunit α3. RNA 13: 698–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Dominissini D, Rechavi G, Eisenberg E 2009. Widespread cleavage of A-to-I hyperediting substrates. RNA 15: 1632–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Paz Yaacov N, Safran M, Moshkovitz S, Shtrichman R, Sherf O, Jacob-Hirsch J, Keshet G, Amariglio N, Itskovitz-Eldor J, et al. 2010. Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS One 5: e11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupard MC, O’Connell MA, Gerber AP, Zukin RS 2000. Patterns of developmental expression of the RNA editing enzyme rADAR2. Neuroscience 95: 869–879 [DOI] [PubMed] [Google Scholar]
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G 2010. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci 107: 12174–12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Gilad Y, Pritchard JK 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302; author reply 1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR 2005. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33: D501–D504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14: 1110–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80 [DOI] [PubMed] [Google Scholar]
- Sakurai M, Suzuki T 2011. Biochemical identification of A-to-I RNA editing sites by the inosine chemical erasing (ICE) method. Methods Mol Biol 718: 89–99 [DOI] [PubMed] [Google Scholar]
- Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T 2010. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol 6: 733–740 [DOI] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB 2003. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci 100: 14018–14023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Gout JF, Hahn MW 2012. Very few RNA and DNA sequence differences in the human transcriptome. PLoS One 6: e25842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K 2001. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29: 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P 1996–2010. RepeatMasker Open-3.0 http://www.repeatmasker.org
- Sommer B, Kohler M, Sprengel R, Seeburg PH 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11–19 [DOI] [PubMed] [Google Scholar]
- Strehblow A, Hallegger M, Jantsch MF 2002. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell 13: 3822–3835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C, Chang H, Stocker J, Wang H, Lu C, Wu C, Yang J, Cho C, Huang H 2013. A method to identify RNA A-to-I editing targets using I-specific cleavage and exon array analysis. Mol Cell Probes 27: 38–45 [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K 2005. ADAR gene family and A-to-I RNA editing: Diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol 79: 299–338 [DOI] [PubMed] [Google Scholar]
- Walkley CR, Liddicoat B, Hartner JC 2012. Role of ADARs in mouse development. Curr Top Microbiol Immunol 353: 197–220 [DOI] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768 [DOI] [PubMed] [Google Scholar]
- Whitfeld PR, Witzel H 1963. On the mechanism of action of Takadiastase ribonuclease T1. Biochim Biophys Acta 72: 338–341 [PubMed] [Google Scholar]
- Yang JH, Sklar P, Axel R, Maniatis T 1997. Purification and characterization of a human RNA adenosine deaminase for glutamate receptor B pre-mRNA editing. Proc Natl Acad Sci 94: 4354–4359 [DOI] [PMC free article] [PubMed] [Google Scholar]