

Abstract
Glioblastoma multiforme (GBM) is a highly invasive brain tumour that is unvaryingly fatal in humans despite even aggressive therapeutic approaches such as surgical resection followed by chemotherapy and radiotherapy. Unconventional treatment options such as gene therapy provide an intriguing option for curbing glioma related deaths. To date, gene therapy has yielded encouraging results in preclinical animal models as well as promising safety profiles in phase I clinical trials, but has failed to demonstrate significant therapeutic efficacy in phase III clinical trials. The most widely studied antiglioma gene therapy strategies are suicide gene therapy, genetic immunotherapy and oncolytic virotherapy, and we have attributed the challenging transition of these modalities into the clinic to four major roadblocks: (1) anatomical features of the central nervous system, (2) the host immune system, (3) heterogeneity and invasiveness of GBM and (4) limitations in current GBM animal models. In this review, we discuss possible ways to jump these hurdles and develop new gene therapies that may be used alone or in synergy with other modalities to provide a powerful treatment option for patients with GBM.
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common and malignant primary brain tumour in adults.16 Today, the current standard of care consists of surgical resection followed by radiotherapy and chemotherapy.2 However, the effectiveness of surgical resection is often compromised due to the lack of a defined tumour margin and a tumour burden located at a close proximity to vital anatomical structures in the brain. Moreover, due to the limitations associated with current standard therapeutic options as well as the presence of a chemo-resistant and radio-resistant glioma stem cell (GSC) population, which play a major role in initiating clinical relapse,3 the median survival time for patients diagnosed with GBM is a meagre ~12–18 months with only ~3% of patients surviving longer than 5 years.4, 5 These statistics highlight the urgency of developing novel and effective therapeutic strategies against this devastating and uniformly fatal disease. As such, glioma has attracted a large amount of research attention as a target for gene therapy. ‘Gene therapy’ as related to brain tumours can be defined as the targeted transfer of genetic material into tumour cells for therapeutic purposes6 and has the ability to target invasive tumour cells that are resistant to conventional therapy and give rise to recurrent disease. Although gene therapy has shown promise in preclinical applications, it has not met clinical expectations due to various impediments related to the nature of the type of tumour and its location. The obstructions of gene therapy include: the anatomical barriers and physiological aspects of the brain that decrease transduction efficiency, tumour heterogeneity and invasiveness that challenge vector targeting and delivery,6, 7 as well as a lack of a satisfactory preclinical model to study glioma. Here, we review relevant gene therapy approaches for the treatment of glioma and discuss the pertinent shortcomings, modifications and future directions in the field.
Gene therapy strategies for glioma
In the last decade, efforts to develop more effective and innovative gene therapy to target GBM have led to the preclinical characterisation of many promising gene therapy approaches. Many of these methods demonstrate therapeutic efficacy against glioma xenografts in an animal model and have been tested in clinical trials. Retroviral and adenoviral vectors have been the most widely used vectors for delivery of antiglioma therapeutic genes.8 According to the Journal of Gene Medicine, replication-defective adenoviruses represent ~23% (n=424) and replication-deficient retroviruses ~20% (n=365) of all gene therapy clinical trials worldwide as of January 2012. In this section, we outline the most widely evaluated antiglioma gene therapy strategies which are discussed in figure 1.
Figure 1.
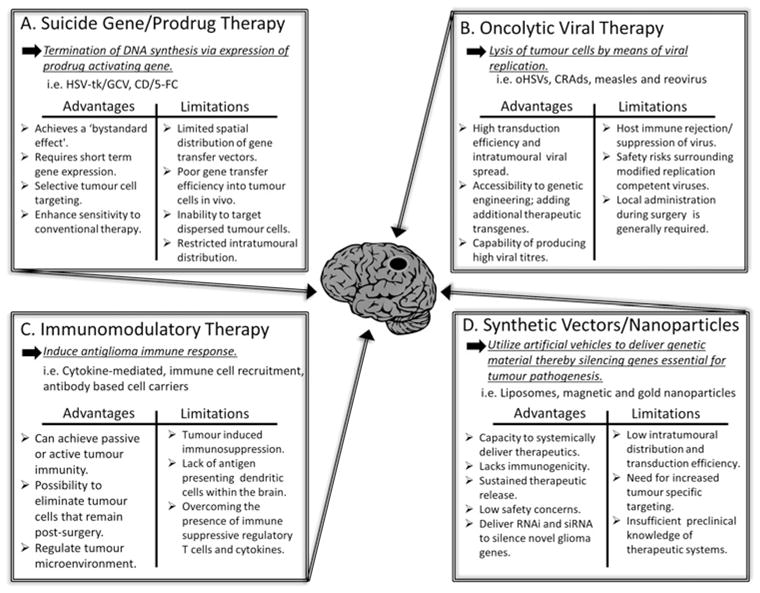
Highlights the advantages and limitations of the most commonly studied antiglioma gene therapies. (A) Suicide gene therapy inhibits cell division by blocking DNA replication. In this system, tumour cells are transfected by a gene that encodes for an enzyme that converts a systemically administered prodrug into an active drug toxic to glioma cells. (B) Oncolytic viral therapy takes advantage of viral infection and selective replication of virus in tumour cells through various genetic alterations of the virus genome thereby rendering the virus tumour specific and oncolytic. (C) Immunomodulatory gene therapy induces a host immune response to counteract the immune privileged central nervous system and immunosuppressive tumour microenvironment through various strategies. (D) Synthetic vectors such as nanoparticles are unique in their ability to be delivered systemically and cross the blood–brain barrier. This approach has been employed to deliver genetic material such as DNA plasmid, proteins, RNA interference (RNAi) and small interfering RNA (siRNA) that silence genes and provide the opportunity for the development of drugs against specific glioma targets.
Suicide gene therapy
The most commonly used gene therapy approach against GBM in the preclinical setting as well as in clinical trials is the enzyme-prodrug suicide gene therapy system. In this approach, viral vectors or cell carriers are genetically modified to express genes for an enzyme that converts an inactive prodrug, when administered systemically into toxic metabolites at the tumour sites, resulting in tumour cell killing. Such targeted cytotoxic gene delivery approaches are designed to achieve highly selective tumour cell destruction while sparing normal central nervous system (CNS) tissue from toxicity. A large number of enzyme-prodrug systems have been evaluated in 17 different clinical trials ranging from phase I to phase III in the USA and Europe. In all 17 trials, adenoviral, retroviral or non-viral vector based delivery methods were used and modest to no increase in median survival was demonstrated (figure 2).33, 34 Here, we briefly discuss some of the most commonly used suicide gene therapy systems against GBM.
Figure 2.

An up-to-date overview of results obtained from glioma clinical trials that used virus. (A) Replication incompetent viruses or non-replicating viruses bearing suicide transgenes have been extensively studied and applied in clinical trials. Retro-mediated and adenoviral-mediated herpes simplex type 1 thymidine kinase (HSV-tk) gene therapies are the most commonly studied in clinical trials. Retrovirus: Prados et al,9 Rainov,10 Shand et al,11 Palu et al,12 Klatzmann et al,13 Izquierdo et al14 and Ram et al.15 Adenovirus: Trask et al,16 Sandmair et al,17 Smitt et al,18 Germano et al,19 Immonen et al20 and Lang et al.21 (B) Replication competent oncolytic virus such as conditionally replicating adenoviruses, herpes simplex virus (HSV) mutant vectors, Newcastle disease virus (NDV), and reovirus have all been tested in the clinical setting for treatment of glioma. HSV-1 (G207): Markert et al22 and Markert et al.23 HSV-1 (1716): Papanastassiou et al,24 Kesari et al25 and Rampling et al.26 NDV (MTH-68/H): Wagner et al,27 Csatary and Bakacs,28 and Csatary et al.29 NDV (NDV-HUJ): Freeman et al.30 Reovirus: Forsyth et al.31 AdV (ONYX-015): Chiocca et al.32
HSV-tk system
Herpes simplex type 1 thymidine kinase (HSV-tk) is the most extensively investigated suicide gene therapy system against GBM. HSV-tk converts the inactive prodrug ganciclovir (GCV) into a toxic metabolite called GCV-triphosphate.35 Induction of the ‘bystander effect’ is thought to be one advantage of this therapy,36 which can be observed when the toxic metabolite converted by HSV-tk is lethal to tumour cells at distant sites that were not originally transduced with the therapeutic gene. In a xenograft glioma model, significant therapeutic efficacy has been observed when only ~10% of total tumour cells in the disease burden are transduced with HSV-tk.17, 37 In the clinic, successful delivery of the HSV-tk system into the tumour cavity has been achieved by replication-defective retrovirus (RV), adenovirus (Adv), cell carrier and reovirus packing cells. One of the largest phase III randomised clinical trials was conducted by Rainov where retroviral packing cells were used to deliver HSV-tk in the tumour bed of patients with glioma. This study recruited 248 total patients with newly diagnosed and previously untreated GBM who were treated with standard chemotherapy and radiotherapy (n=124) or standard therapy in combination with adjuvant retrovirus-mediated HSV-tk/GCV gene therapy (n=124).10 Patients received a mean volume of 9.1 ml of retroviral producing cells into the margins of the tumour cavity at a concentration of 108 cells/ml during the craniotomy. Even though the clinical trial proved that adjuvant gene therapy was safe, patient median survival was 365 days versus 354 days and the 12-month survival rates were 50% versus 55% in the gene therapy and control groups, respectively. These data showed no significant therapeutic benefit between both groups.10 Sandmair et al reported a phase I clinical trial where 21 primary or recurrent GBM patients were injected with RV-mediated HSV-tk/GCV (n=7) or replication-defective adenovirus carrying HSV-tk/GCV (n=7) intraoperatively in the margins of the tumour cavity.17 In this clinical trial, the mean survival of the group that received Adv-mediated HSV-tk/GCV was significantly higher (15 months, p<0.012) as compared with the group that was administered RV-mediated HSV-tk/GCV injection (7.4 months), indicating that the adenoviral vector may be better suited for antiglioma gene delivery. The HSV-tk system has also been shown to enhance sensitivity to conventional chemotherapy and radiotherapy, which opens the possibility of combining such an approach with the standard of care for GBM patients.38, 39 Chiocca et al recently reported a phase IB clinical trial with 13 newly diagnosed GBM patients and observed that Adv-mediated HSV-tk/valacyclovir therapy in combination with conventional surgery and chemotherapy-radiotherapy can be clinically safe with no dose-limiting or significant added toxicity.40 The study also shed light on possible clinical efficacy in patients with an unmethylated O(6)-methylguanine-DNA methyltransferase (MGMT) promoter with one patient living up to 46.4 months. A phase II study is currently ongoing to further evaluate survival and MGMT independence trends.40 Furthermore, it has been observed that combining HSV-tk with pharmacological drugs can alter the pharmacokinetics of the administered prodrugs, and has also been shown to increase therapeutic efficacy when used in conjunction with conventional therapy. One study showed that scopadulciol enhanced prodrug activity through a HSV-tk specific mechanism and increased tumour cell killing through the bystander effect of acyclovir and GCV prodrugs.41
CD/5-FC system
The cytosine deaminase/5-fluorocytosine (CD/5-FC) gene therapy system has also been extensively investigated in the preclinical setting.42, 43 This system is also capable of inducing a strong bystander effect; significant therapeutic efficacy has been observed in a xenograft tumour model when only 2%–4% of tumour cells are transduced.44 A second generation non-lytic retroviral replicating vector (Toca 511) has demonstrated that stable delivery of CD resulted in long-term survival in two different immunocompetent brain tumour models.45 Toca 511 is currently under phase I–II clinical investigation in combination with 5-FC in patients with recurrent high-grade glioma (NCT01156584). The CD/5-FC system has also been reported to enhance conventional radiotherapy against glioma in an animal model,46 and a fusion gene of CD used in conjunction with HSV-tk has shown to provide an increased antiglioma effect when compared with each individual gene used alone.47, 48
Taken together, the antiglioma gene therapy approach using suicide genes is safe in treating patients with GBM, but has failed to achieve a consistent therapeutic benefit. These results can be attributed to limited spatial distribution of the viral vector, poor gene transfer efficiency into tumour cells and the inability to target disseminated tumour burden by the currently available gene transfer vectors. Moreover, with the exception of the Rainov trial,10 most of the early clinical trials treated a small number of patients, sometimes even without a control group. Therefore, it has been difficult to analyse whether these trials provided therapeutic efficacy in treated patients. Further optimisation of vectors used to deliver suicide gene therapy is essential for the improvement of clinical effectiveness. For the majority of antiglioma suicide gene therapy protocols, the short-term expression of therapeutic transgenes is sufficient to achieve tumour cell death. However, the restricted intratumoural distribution of the therapeutic payload still remains an issue for achieving optimal clinical efficacy. Greater viral vector stability as well as prolonged therapeutic transgene expression might result in more successful treatment of GBM. Thus, with use of adenovirus with superior glioma cell transduction capacity,17 and gutless adenovirus with reduced immunogenicity,49 conditionally replicating viral vectors might allow us to successfully translate antiglioma suicide gene therapy into the clinic because of their ability to amplify therapeutic transgenes via tumour-selective replication.
Oncolytic viral therapy
In order to address the issue surrounding the transduction efficiency of gene therapy vectors, researchers have engineered tumour-selective and conditionally replicating viral vectors referred to as oncolytic virus (OVs). OVs act by selective self-replication in tumour cells that leads to tumour cell lysis, as well as by amplifying therapeutic genes at tumour sites. It is evident from the current literature that tumour transduction efficiency is higher with replication competent viruses than with replication-deficient viruses, which highlights the potential of OVs as therapeutic gene delivery vehicles for anticancer gene therapy. Oncolytic herpes simplex virus (oHSV), conditionally replicating adenovirus (CRAd), reovirus, poliovirus, Newcastle disease virus and measles virus have all been evaluated or are currently being applied in antiglioma clinical trials (figure 2). Here, we describe some of the most commonly used antiglioma OV systems.
Oncolytic herpes simplex virus
oHSV was among the first OVs to be safely administered to patients with recurrent malignant glioma.50 Because HSV is a human pathogen with neurotropic properties, a critical issue in designing oHSVs is to provide tumour selectivity with an adequate safety profile. Since the first reported clinical trials using oHSV for the treatment of glioma in the late 1990s,51 at least eight different HSV-1 genes, including TK (UL23), ICP6 (UL39), γ34.5 and Us3, have been deleted/mutated to reduce neurovirulence and induce tumour selectivity.52 The most widely tested OV in clinical trials for antiglioma therapeutics is the oHSV vector G207, which is a genetically engineered HSV-1 vector that has a deleted γ34.5 gene at both alleles and a lacZ gene insertion that blocks the expression of the UL39 gene.53 Heretofore, three phase II and three phase I clinical trials have been conducted using the oHSV vector. Crusade Laboratories in Glasgow, Scotland, has begun a phase III clinical trial in Europe using HSV1716, an oHSV derived from the wild-type strain of ‘F’ containing attenuating mutation in both copies of the γ134.5 gene.53 In a recently reported phase IB clinical trial, six patients with resectable GBM received two injections of G207 during presurgery and postsurgery. Viral replication was observed but with limited evidence of antitumour activity.23 Results from early clinical trials have demonstrated high safety profiles of multiple oHSV vectors with no evidence of encephalitis but with limited therapeutic efficacy.54 Second generation oHSV vectors are currently under preclinical development where researchers have implemented various strategies to enhance oncolytic activity. Such strategies include those with a single copy of the γ34.5 gene reintroduced back into the vector that are genetically engineered to encode for therapeutic transgenes such as TNFα, vascular endothelial growth factor (VEGF) specific shRNA and the immunostimulatory gene interleukin (IL)-4.55–58 Others include surface retargeted HSVs that target glioma cells overexpressing human epidermal growth factor 259 and transcriptional targeting oHSVs that use tumour-selective promoters such as the HIF-responsive promoter.60 Development of new oHSVs provides optimism for the future.
Conditionally replicating adenovirus
CRAds have also been extensively evaluated in both preclinical and clinical settings for antiglioma therapeutics, with ONYX-015 and Ad5-Delta24 being the most widely studied. These CRAds have been adapted to replicate and lyse tumour cells in different ways: ONYX-015 has a deletion in the E1B gene that permits its replication in tumours with a defective p53 pathway, while Ad5-Delta24 relies on a deletion in the retinoblastoma binding region of the EIA protein allowing the vector to replicate in GBM cells that have a defective retinoblastoma function.61 A phase I clinical trial conducted by Chiocca and colleagues show that ONYX-015 is safe to administer into the tumour bed cavity postsurgical resection.32 A phase I clinical trial is currently underway evaluating Ad5-Delta24 (NCT00805376). Our group is currently conducting a US Food and Drug Administration (FDA) guided preclinical study evaluating the CRAd-Survivin-pk7 vector, which contains a tumour specific survivin promoter that drives adenovirus E1A replication and an inserted pk7 fibre region that has a high affinity to heparin sulphate proteoglycans, which confers tumour-selective replication.62–64 One important advantage of CRAd viruses is they are naturally non-neurotropic and thus may possess an enhanced safety profile over the oHSV vector.
Oncolytic measles and reovirus vectors
Oncolytic measles virus and reovirus vectors are currently under preclinical evaluation for GBM virotherapy. Tumour specific reovirus replication is dependent on hyperactive RAS signalling and has shown efficacy against GBM in an orthotropic animal model.65 In a phase I clinical trial, reovirus was injected directly into the tumour of patients with glioma, and no participants showed any signs of clinical encephalitis.31 Strains of the attenuated measles virus derived from the Edmonston vaccine lineage (MV-Edm) are also under preclinical development and have yielded positive results.66 A phase I clinical trial for recurrent GBM patients using MV-CEA, a MV-Edm vector expressing the soluble peptide marker, carcinoembryonic antigen, is currently underway (NCT00390299).67 Although conditionally replicating viruses represent a major advantage over non-replicative viruses in terms of transduction efficiency, the host antivector immune response remains as the major obstacle for the translation of OVs into the clinic.
Immunomodulatory gene therapy
The objective of antiglioma immunomodulatory gene therapy is to induce or augment the T cell-mediated immune response against GBM. During tumourigenesis, glioma cells evolve to evade the host immune system. Moreover, the distinct immune privileged nature of the CNS also poses issues for generating effective antiglioma immune responses.68 Nevertheless, preclinical experimental evidence has demonstrated the feasibility of inducing immune responses against glioma cells as well as chemo-resistant and radio-resistant GSCs, which has laid the foundation for formulating antiglioma gene therapy based on immunomodulation. Such strategies include cytokine-mediated gene therapy, immune cell recruitment strategies and application of cell carriers expressing immunomodulatory genes.
Cytokine-mediated gene therapy
The rationale for cytokine gene therapy is that tumour-selective gene transfer and in situ expression of various immunostimulatory genes such as IL-2, -12, -4, interferon (IFN)-γ and IFN-β may induce potent immune responses restricted towards antigens specific to glioma cells, but not to normal brain tissue.69–74 Moreover, cytokine-mediated gene therapy compared with systemic administration of suicide gene therapy and OV gene therapy may allow us to achieve higher local concentrations, longer therapeutic gene persistence and reduce systemic toxicity. Type 1 interferon genes including IFN-γ, IFN-β and IFN-ω are primarily produced by specialised antigen presenting cells such as dendritic cells (DCs) postviral infection and have been shown to elicit robust antitumour effects.75 Among the IFN genes, the IFN-β gene has direct antiproliferative effects and has been the most extensively evaluated cytokine for anticancer gene therapeutics. A two stage phase I clinical trial in which the initial treatment of five patients with GBM comprised of tumour resection was followed by injection of cationic liposomes with the human IFN-β gene into the margin of the resection cavity reported minimal clinical toxicity with 50% reduction of tumour size in two patients.76 Another dose-escalating phase I clinical trial of stereotactic injection of an adenovirus vector expressing the IFN-β gene in 11 patients with GBM recently demonstrated safety as well as possible therapeutic effects due to an increased level of apoptosis in glioma cells.77
Immune cell recruitment strategies
In the preclinical setting, Castro and her colleagues have used the Ad-Fms-like thyrosine kinase 3 ligand to recruit antigen presenting cells such as DCs into the brain tumour mass. Their strategy used DC recruitment combined with suicide gene therapy by simultaneously administrating a second adenovirus vector with the TK gene. In this approach, dying tumour cells release endogenous tumour associated antigen as well as the high mobility group box 1 protein that acts as an agonist to toll-like receptor 2 leading to DC recruitment and an antitumour immune response.78, 79 This gene therapy approach has demonstrated tumour regression and long-term survival through its ability to induce an antiglioma immune response and immunological memory in several transplantable, orthotropic syngeneic models of GBM. In 2011, a phase I clinical trial was launched using this genetic immunotherapy approach.78
Cell carriers expressing immunomodulatory genes for antiglioma gene therapy
Stem cells or progenitor cells (SCs) have been evaluated extensively as therapeutic vehicles for antiglioma therapy due to their inherent tumour tropic properties. In the context of glioma, three types of SCs have been explored for their therapeutic use and are currently in preclinical development: neural, embryonic and mesenchymal. Embryonic stem cells have been modified to express and deliver mda-7/IL-24 and cause apoptosis in malignant glioma cells.80 Data also show similar apoptotic effects of embryonic stem cell-derived astrocyte-mediated delivery of TRAIL.81 Mesenchymal stem cells have been used to deliver a plethora of therapeutics to glioma including prodrugs, virus, cytokines and antibodies. One specific application is the genetic modification of human mesenchymal stem cells to express a single-chain antibody on their surface against the tumour specific antigen EGFRvIII. EGFRvIII was selected based on data showing that about ~20%–30% of human GBM express this genetic alteration.82 In an intracranial glioma xenograft model of U87-EGFRvIII, animals injected with human mesenchymal stem cells expressing the single-chain antibody against EGFRvIII showed a significant survival advantage when compared with mock animals.83
Synthetic vectors such as nanoparticles
Nanoparticles have been studied as a method to intravenously deliver vectors that can cross the blood–brain barrier. This gene therapy modality is based on coupling genetic material to nanoparticles or microparticles, and delivering genes to a targeted site by way of their size, charge, as well as high surface to volume ratio that provides a powerful force for diffusion.84, 85 Various genetic materials such as DNA plasmids, protein, RNA and siRNA have been conjugated onto or encapsulated inside nanoparticles to be delivered to tumour cells.85–87 Liposomes, due to their organic makeup, are the most widely investigated nanoparticles, and have been used to form artificial vesicles that encapsulate and deliver therapeutic agents such as RNA interference and small interfering RNA (siRNA). RNA interference has been used to silence specific messenger RNA and have led to the development of drugs against specific disease targets. Synthetic siRNAs have been shown to silence genes in vivo that are important for the pathogenesis of GBM.86 Therapeutics using siRNA represent a powerful tool for precise targeting of novel genes and have led to five different clinical trials that are currently ongoing.88
Challenges in developing effective antiglioma gene therapeutics
Each described strategy above has its own distinct advantages and disadvantages. Despite encouraging results in preclinical animal models and established safety profiles in phase I clinical trials, none of the gene therapies have demonstrated significant benefits in phase II and III clinical trials. The barriers limiting the efficient transition of gene therapy into the clinic include: anatomical barriers of the CNS that decrease the spatial distribution of the administered therapy, GBM heterogeneity and their invasiveness, cancer SCs, immunogenicity and limitations of established preclinical GBM models. In the following section, we discuss the various roadblocks of translation of antiglioma therapy from a preclinical setting to the clinic, and how the field of gene therapy has attempted to address them (table 1).
Table 1.
Potential strategies to overcome current limitations of glioma gene therapy
| Challenges to overcome | Prospective solutions | Novelty | Representative study |
|---|---|---|---|
| 1. Heterogeneity and invasive properties of GBM | Stem cell (SC) carriers | Exploit intrinsic tumour tropic properties of SCs to reach distant tumour foci; target radio-resistant and chemo-resistant GSCs. | 89, 90 |
| Adjuvant viral therapy | Combine conventional and gene therapy approaches that provide therapeutic synergy; intervene against multiple tumour cell types; cytotoxic to GSCs. | 91 | |
| Nanoparticles | Offer precise interference and silencing of novel genes. | 87 | |
| Next generation OV vectors | Express new novel transgenes such as TNFα, VEGF specific shRNA and IL-4; retargeted vectors that increase glioma cell and GSC targeting. | 59 | |
| 2. Anatomical and physiological features of central nervous system and GBM | SC carriers | Increase spatial distribution of gene therapeutics. | 83 |
| Nanoparticles | Ability to cross BBB permits systemic administration. | 92 | |
| Convection-enhanced delivery | Achieve high virus/vector concentrations over large volumes of targeted tissue; enhanced levels of transduction. | 93 | |
| Next generation OV vectors | Increased vector penetration and transduction efficiency. | 62 | |
| 3. Host immune system | Genetic immunotherapy and vaccination | Modulate tumour microenvironment to stimulate host immune response against tumour cells; achieve higher local and long-term concentration of therapeutic genes. | 78, 79 |
| SC carriers | Mask gene therapy vectors from host immune system clearance. | 94 | |
| 4. Inadequacy of preclinical models | Advanced imaging protocols | Non-invasive real time imaging technology; provide a new tool to study and optimise current gene therapy strategies. | 95 |
| Superior animal models | Mimic human glioblastoma properties such as the tumour microenvironment, heterogeneity, growth pattern, histopathology and antitumour immune response; more representative of human brain and tumour size, for better assessment of pharmacokinetic properties and delivery strategies. | 96 |
BBB, blood–brain barrier; GBM, glioblastoma multiforme; GSC, glioma stem cell; IL-4, interleukin 4; OV, oncolytic virus; TNFα, tumour necrosis factor α; VEGF, vascular endothelial growth factor; shRNA, short hairpin RNA.
Limited spatial distribution of the therapeutic payload
One of major hurdles for achieving clinically relevant therapeutic efficacy by antiglioma gene therapeutic approaches is the limited tissue penetration and spatial distribution of the therapeutic payload in GBM tissue. To achieve clinically relevant therapeutic efficacy, any given anticancer therapy must effectively access the tumour site and destroy as many tumour cells as possible without affecting the surrounding healthy tissue. Physiologically, the CNS is protected by a unique anatomical barrier, the blood–brain barrier, which has been considered the major impediment to any systemic treatment of CNS diseases including glioma.97 Thus, most antiglioma gene therapeutic approaches are applied during craniotomy directly in the tumour bed or into the margins of the tumour cavity itself. Despite direct delivery, the transduction efficiency of glioma cells with the currently available viral and non-viral vectors remains poor. One reason contributing to the poor transduction efficiency is because only a small percentage of primary GBM cells express the cognate receptor for the viral vector that allow them to enter into the target tumour cells efficiently. For example, Ad5-based gene therapy for malignant glioma is limited due to the poor expression of the adenovirus entry receptor CAR on primary GBM.98 To overcome this problem, researchers have developed retargeted gene therapy strategies, which use receptors that are only expressed in glioma cells but not in normal neural tissue. Our laboratory has been using a CRAd with a fibre modification containing an inserted polylysine (pk7) motif that binds with a high affinity to heparin sulphate proteoglycans which has shown to confer glioma-selective internalisation. Another major limitation of gene therapy vectors is poor tissue penetration of the therapeutic virus after injection into glioma tissue. A clinical study demonstrated that the distribution of the viral vector was limited to an average range of 5 mm from the needle track.21 Researchers have been exploring a new delivery method known as convection-enhanced delivery (CED),99 which relies on continuous infusion of drugs and virus via intracranial catheters, enabling convective distribution of high virus/drug concentrations over large volumes of the targeted tissue.100 CED has been applied in a glioma clinical trial to administer large molecules, including immunotoxins,101 as well as to achieve enhanced transduction efficiency of the viral vector in a glioma xenograft model.102 These studies have shown that CED has the potential to improve the therapeutic efficacy of antiglioma gene therapy, but the success of this approach remains to be resolved. Furthermore, in the majority of antiglioma gene therapy clinical trials viral vectors were administered into the resection cavity or remaining tumour bed by a single injection, through a catheter or by multiple injections of a rather small volume of vector suspensions.18, 20, 21 Such injection protocols can be technically demanding, requiring precise estimation of the correct depth of the injection with respect to the extent of parenchyma-invading tumour cells. The accuracy and targeting capacity of therapeutic payload delivery protocols can be significantly improved if such injections are carried out with the help of robotic technology and guided by advanced imaging systems.
The host immune system and targeting the heterogenic and invasive properties of GBM
In theory, OVs should provide a solution for poor gene transfer efficiency as progeny released from the initial infected tumour cells should laterally spread to the tumour burden and amplify OV killing effects. However, results from early clinical trials using antiglioma OVs showed limited success due to the inability of currently available OVs to target disseminated glioma burdens as well as the host immune response interfering with viral vectors. The use of SCs has recently received a great deal of attention as possible cell carriers for targeted antiglioma therapy. In the last decade, many in vitro and in vivo studies demonstrated that SCs have unique inherent properties to migrate throughout the brain, target and home to metastatic invasive solid tumours, including gliomas.89, 103, 104 Aboody and colleagues have used prodrug systems to modify HB1.F3 neural SC (NSC) lines and were able to show a 70%–80% decrease in tumour volume of mice bearing orthotopic gliomas or intracranial melanoma.90 Based on the encouraging preclinical results, the FDA recently approved Aboody and colleagues to conduct the first clinical study of genetically modified neural SCs (HB1. F3-CD) for patients with recurrent high-grade glioma. This clinical trial began recruiting with the goal of enrolling 12–20 patients. Similarly, our lab has extensively investigated the possibility of using the inherent tumour tropic properties of NSCs to deliver glioma restricted oncolytic adenovirus selectively to disseminated tumour burdens. Our recent data indicates that distant delivery of NSCs loaded with oncolytic adenovirus significantly prolonged survival of animals in several orthotopic murine models of human glioblastoma when compared with mice treated with virus alone.63, 64 We proved that the increased survival was due to amplified therapeutic virus at distant tumour sites in the presence of NSCs. Also, we have reported that a bone marrow mesenchymal SC carrier was able to protect the oncolytic viral therapeutic payload from the host immune system in a cotton rat model.105 There is also an abundance of preclinical data that suggest that in vivo transplanted NSCs can act as an immunosuppressant.106 It has been shown that NSCs lack the expression of major histocompatibility complex class II and express low levels of the co-stimulatory molecules CD80 and CD86 which provide them with protection from immune-mediated killing.94 NSCs have also been shown to express immunosuppressive cytokines such as IL-10 in the context of OV infection/loading.64 In the future, it will be crucial to gain a better understanding of the molecular mechanism underlying the tumour tropic properties of NSCs in order to increase their migratory capacity and improve the efficacy of this gene therapy system. Recent advancements in molecular imaging protocols using PET and/or MRI are providing us with the capacity to study SC migration in a non-invasive longitudinal manner, and may allow us to precisely delineate the mechanism of tumour tropism. Our lab has used ferumoxides-protamine sulphate labelled NSCs to visually track the migration of NSCs towards human glioma in an orthotropic mouse model. Information gathered from this technique may provide us with the insight to increase the migration of NSCs towards glioma in the future.
Another inherent characteristic of GBM is its heterogeneous makeup that exists due to the diverse genetic and epigenetic changes that accumulate in the pathogenesis of the different tissue subtypes found in GBM. This tumour property makes it exceptionally difficult to select one appropriate therapeutic approach against all tissue types in GBM.107 The use of drugs in combination with viral vectors has been applied to target multiple tumour cell types or tumour pathways to achieve a synergistic outcome. Bevacizumab (BEV) or Avastin, an anti-angiogenic monoclonal antibody against VEGF, has been approved by the FDA for the treatment of GBM but has yielded no survival benefits in humans. Results of a study conducted by Zhang et al have shown that a local injection of G47Δ-mAngio, an HSV-derived OV expressing angiostatin, in conjunction with systemic administration of BEV increases virus spread throughout the brain, tumour killing and angiostatin inhibition of VEGF expression. Furthermore, this therapy synergises BEVs inhibitory activity of invasion markers such as matrix metal-loproteinases-2 (MMP-2), MMP-9 and collagen. This adjunct therapy has led to increased survival in an intracranial mouse model of human glioma (U87) through increasing antiangiogenesis and reducing the invasiveness of GBM.108 Researchers are also using multiple viral vectors to target GBM heterogeneity and achieve therapeutic viral synergy. A current example of such is combining the vesicular stomatitis virus (VSV) with vaccinia virus (VV). VSVand VV were shown to enhance viral replication and infiltration throughout tumour cells of one another. Boeuf et al observed a 10- to 10 000-fold increase in VSV titres following co-infection of tumour samples with VV in 33 out of 44 tumour samples.109
Other approaches that target GBM heterogeneity focus on targeting cells that make it uniquely invasive and resistant to conventional cancer therapies, when compared with other human cancers. Research has attributed GBM’s resistance to treatment and high rate of recurrence to a small subpopulation of cells called GSCs. GSCs have unique phenotypic properties which include relative quiescents as well as an ability to differentiate, self-renew, and resist chemotherapy and radiotherapy.110 Since a majority of investigated gene therapies focus on targeting properties retained in the main tumour bulk (ie, rapidly dividing cells) and not specific GSC properties, GSCs survive therapy and give rise to new tumour formation and re-initiate the disease. By using SC specific promoters such as, Cox-2, hTERT and mdr, Bauerschmitz et al were able to show a reduction in breast cancer SC population after the treatment with Ad5/3-mdr-Δ24.111 Research on brain specific cancer SCs has shown that tumour-selective oncolytic adenovirus Delta-24-RGD replicates and induces cell death in GSCs. A phase I clinical trial for patients with malignant gliomas is currently underway.112 oHSV has also been used to target GSCs. G47Δ has been tested in combination with a low-dose etoposide and showed increased tumour cell apoptosis and increased survival of mice with etoposide-insensitive intracranial human GSC-derived tumours.113 G47Δ has also been shown to cooperate with temozolomide in killing GSCs through viral manipulation of DNA damage response pathways in preclinical models.114 A modified oHSV, MG18L, containing a Us3 deletion and an inactivating LacZ insertion in UL39, replicates in GSCs and has antitumour activity in GBM cells in vivo. Furthermore, when MG18L was used in combination with phosphoinositide-3-kinase/Akt inhibitors, increased GSC and glioma apoptosis were observed and survival of GBM-bearing mice was prolonged when compared with treatment with either single therapeutic agent alone.115 Other groups have used OV vectors carrying an exogenous Endo-Angio fusion gene (VAE) to infect and lyse GSCs and have shown the significance of this modality in vitro.116 Moreover, GSCs have been shown to overexpress ATP-binding cassette (ABC) transporters, especially ABCG2 that can pump out active prodrugs and resist suicide gene therapy.117 Combining an ABCG2 blocker, such as gefitinib, with other suicide gene therapy vectors may provide an opportunity to further target GSCs and improve the therapeutic efficacy of antiglioma suicide gene therapy systems.117
Preclinical animal model
The therapeutic efficacy of most antiglioma gene therapeutic approaches is commonly evaluated in immunocompromised animal models using xenogeneic cells for tumour implantation with only a short interval of time between engraftment and treatment. The circumstances in human GBM do not closely mimic those in animal models as tumour initiation is usually sporadic and clinical symptoms can be observed months to years after initial establishment, resulting in increased heterogeneity. Thus, for the successful investigation of gene therapies, it is essential to build good animal models that are both reliable and representative of human glioma. To date, many models exist including: implantation of rodent glioma cells into immunocompetent rodents, implantation of human GBM into immunocompromised nude mice and endogenous brain tumour animal models.118 These models have been used due to their high level of reproducibility and characteristics that accurately recapitulate the tumour microenvironment, heterogeneity, growth pattern, histopathology and antitumour immune response represented in human GBM.33 Although these models are widely used and have generated vast amounts of data to lead to the development of novel gene therapies, the failure of the studied therapies transition into the clinic can be partially attributed to a need for a superior glioma model. As one of the possible options, the spontaneous GBM model in the brachycephalic canine has been reported.119 Canine GBM is highly invasive and mimics human GBM characteristics such as necrosis with pseudopalizading, neovascularisation and endothelial proliferation.5 Stoica et al have reported that GSCs are present in dog GBM and have a high capacity for self-renewal, proliferation and differentiation similar to human GBM.96 The most important aspect of the canine model is its comparable brain size to the human brain. This characteristic is essential for a good preclinical model in order to precisely assess such pharmacokinetic properties as toxicity, dosage, side effects, as well as measure delivery strategies. For example, NSCs have been shown to deliver gene therapies to targeted tumour sites beyond the primary tumour in small animal models. But can NSCs withstand the test of distance and deliver to metastatic sites far away from the site of injection in a human brain? The failures of gene therapy can be undoubtedly linked to the inaccessibility to animal models that recapitulate human GBM and therefore answer prudent questions about an antiglioma gene therapy before its translation into clinical trials. It is essential to collaborate with veterinarian institutions that receive glioma bearing canines and cancer gene therapy laboratories with a need for this model, in order to bring antiglioma gene therapy closer to achieving clinical relevance.
CONCLUSIONS AND FUTURE DIRECTION
Although antiglioma gene therapies have demonstrated promising efficacy in preclinical glioma models with favourable safety profiles in phase I clinical trials, they have ultimately failed to provide significant benefits in both phase II and III clinical trials. Since gene therapy has demonstrated great promise in the preclinical setting, we must accept the initial discouraging outcomes of clinical trials with a grain of salt. A majority of antiglioma phase I clinical trials have been conducted on patients with advanced stage cancer, and this may contribute to their low success rate. In order to adequately judge efficacy, clinical trials need to be conducted on patients with earlier stages of cancer. Furthermore, many phase I clinical trials are designed to determine the safety profile of a treatment modality and not clinical efficacy. Others have suggested that the failure of phase III clinical trials can be attributed to the lack of ‘preclinical robustness,’ a term coined to describe the need for more stringent experimental protocols that address whether a therapy will be well translated into the clinical setting.120 As the field of gene therapy moves forward, it is vital that we modify current gene therapy approaches and adopt new ways to overcome the formidable obstacles GBM has presented. A growing level of attention has been given to therapeutic synergies. Antiglioma gene therapies such as OVs and genetically modified SCs have the potential to cooperate with standard modes of treatment.121 An optimal combination therapy would include a well-designed strategy that uses multiple therapies to target heterogeneous GBM.91 Multiple treatment modalities will have the power to target different parts of the tumour such as the tumour bulk or GSCs, which address the importance of strategically targeting tumour heterogeneity. The synergistic advantages between multiple therapies need to be further evaluated to attain optimal results. Given the highly variable and evolving nature of GBM, advancements in non-invasive imaging protocols and cancer genomics will allow neuro-oncologists to acquire information such as the molecular, cellular, genetic and epigenetic makeup of a specific tumour. This information will provide the clinician with the powerful tools to continually provide personalised gene therapy treatment protocols that can be adjusted based on specific and real-time information gathered on an individual basis. Although the road ahead is challenging, if we can overcome the obstacles and ameliorate current anti-glioma gene therapies, one day it may be possible that gene therapy can be used as the standard of care for GBM patients.
Acknowledgments
Funding Research by the authors was supported by the NCI (R01CA122930, R01CA138587), the National Institute of Neurological Disorders and Stroke (U01NS069997) and the American Cancer Society (RSG-07-276-01-MGO).
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.
Contributors AT wrote and organised the manuscript. AA summarised the data on current gene therapy trials. K-SM provided an overview on current challenges. MSL read, reviewed, funded and approved the final review for submission.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. New Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Roila F. Malignant glioma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):126–8. doi: 10.1093/annonc/mdp151. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Grossman SA, Ye X, Piantadosi S, et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–9. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Candolfi M, Kroeger KM, Muhammad AK, et al. Gene therapy for brain cancer: combination therapies provide enhanced efficacy and safety. Curr Gene Ther. 2009;9:409–21. doi: 10.2174/156652309789753301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bansal K, Engelhard HH. Gene therapy for brain tumors. Curr Oncol Rep. 2000;2:463–72. doi: 10.1007/s11912-000-0067-z. [DOI] [PubMed] [Google Scholar]
- 7.Roth JA, Cristiano RJ. Gene therapy for cancer: what have we done and where are we going? J Natl Cancer Inst. 1997;89:21–39. doi: 10.1093/jnci/89.1.21. [DOI] [PubMed] [Google Scholar]
- 8.Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–98. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 9.Prados MD, McDermott M, Chang SM, et al. Treatment of progressive or recurrent glioblastoma multiforme in adults with herpes simplex virus thymidine kinase gene vector-producer cells followed by intravenous ganciclovir administration: a phase I/II multi-institutional trial. J Neurooncol. 2003;65:269–78. doi: 10.1023/b:neon.0000003588.18644.9c. [DOI] [PubMed] [Google Scholar]
- 10.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389–401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 11.Shand N, Weber F, Mariani L, et al. A phase 1–2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Hum Gene Ther. 1999;10:2325–35. doi: 10.1089/10430349950016979. [DOI] [PubMed] [Google Scholar]
- 12.Palu G, Cavaggioni A, Calvi P, et al. Gene therapy of glioblastoma multiforme via combined expression of suicide and cytokine genes: a pilot study in humans. Gene Ther. 1999;6:330–7. doi: 10.1038/sj.gt.3300805. [DOI] [PubMed] [Google Scholar]
- 13.Klatzmann D, Valery CA, Bensimon G, et al. A phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma. Study group on gene therapy for glioblastoma. Hum Gene Ther. 1998;9:2595–604. doi: 10.1089/hum.1998.9.17-2595. [DOI] [PubMed] [Google Scholar]
- 14.Izquierdo M, Martin V, de Felipe P, et al. Human malignant brain tumor response to herpes simplex thymidine kinase (HSVtk)/ganciclovir gene therapy. Gene Ther. 1996;3:491–5. [PubMed] [Google Scholar]
- 15.Ram Z, Culver KW, Oshiro EM, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354–61. doi: 10.1038/nm1297-1354. [DOI] [PubMed] [Google Scholar]
- 16.Trask TW, Trask RP, Aguilar-Cordova E, et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195–203. doi: 10.1006/mthe.2000.0030. [DOI] [PubMed] [Google Scholar]
- 17.Sandmair AM, Loimas S, Puranen P, et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197–205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- 18.Smitt PS, Driesse M, Wolbers J, et al. Treatment of relapsed malignant glioma with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by ganciclovir. Mol Ther. 2003;7:851–8. doi: 10.1016/s1525-0016(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 19.Germano IM, Fable J, Gultekin SH, et al. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncology. 2003;65:279–89. doi: 10.1023/b:neon.0000003657.95085.56. [DOI] [PubMed] [Google Scholar]
- 20.Immonen A, Vapalahti M, Tyynela K, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967–72. doi: 10.1016/j.ymthe.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Lang FF, Bruner JM, Fuller GN, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–18. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- 22.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 23.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34. 5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 25.Kesari S, Randazzo BP, Valyi-Nagy T, et al. Therapy of experimental human brain tumors using a neuroattenuated herpes simplex virus mutant. Lab Inves. 1995;73:636–48. [PubMed] [Google Scholar]
- 26.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34. 5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 27.Wagner S, Csatary CM, Gosztonyi G, et al. Combined treatment of pediatric high-grade glioma with the oncolytic viral strain MTH-68/H and orl valpoic acid. APMIS. 2006;114:731–43. doi: 10.1111/j.1600-0463.2006.apm_516.x. [DOI] [PubMed] [Google Scholar]
- 28.Csatary LK, Bakacs T. Use of Newcastle disease virus vaccine (MTH-68/H) in a patient with high-grade glioblastoma. JAMA. 1999;281:1588–9. doi: 10.1001/jama.281.17.1588-a. [DOI] [PubMed] [Google Scholar]
- 29.Csatary LK, Gosztonyi G, Szeberenyi J, et al. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- 30.Freeman AI, Zakay-Rones Z, Gomori JM, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221–8. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Forsyth P, Roldan G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16:627–32. doi: 10.1038/sj.mt.6300403. [DOI] [PubMed] [Google Scholar]
- 32.Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958–66. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 33.Kroeger KM, Muhammad AK, Baker GJ, et al. Gene therapy and virotherapy: novel therapeutic approaches for brain tumors. Discov Med. 2010;10:293–304. [PMC free article] [PubMed] [Google Scholar]
- 34.Iwami K, Natsume A, Wakabayashi T. Gene therapy for high-grade glioma. Neurol Med Chir (Tokyo) 2010;50:727–36. doi: 10.2176/nmc.50.727. [DOI] [PubMed] [Google Scholar]
- 35.Gomez-Manzano C, Jiang H, Alonso M, et al. Gene therapy. Handb Clin Neurol. 2012;104:331–8. doi: 10.1016/B978-0-444-52138-5.00021-9. [DOI] [PubMed] [Google Scholar]
- 36.Matuskova M, Hlubinova K, Pastorakova A, et al. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010;290:58–67. doi: 10.1016/j.canlet.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Chen CY, Chang YN, Ryan P, et al. Effect of herpes simplex virus thymidine kinase expression levels on ganciclovir-mediated cytotoxicity and the “bystander effect”. Hum Gene Ther. 1995;6:1467–76. doi: 10.1089/hum.1995.6.11-1467. [DOI] [PubMed] [Google Scholar]
- 38.Chiocca EA, Broaddus WC, Gillies GT, et al. Neurosurgical delivery of chemotherapeutics, targeted toxins, genetic and viral therapies in neuro-oncology. J Neurooncol. 2004;69:101–17. doi: 10.1023/b:neon.0000041874.02554.b3. [DOI] [PubMed] [Google Scholar]
- 39.Rainov NG, Fels C, Droege JW, et al. Temozolomide enhances herpes simplex virus thymidine kinase/ganciclovir therapy of malignant glioma. Cancer Gene Ther. 2001;8:662–8. doi: 10.1038/sj.cgt.7700355. [DOI] [PubMed] [Google Scholar]
- 40.Chiocca EA, Aguilar LK, Bell SD, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29:3611–19. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayashi K, Lee JB, Maitani Y, et al. The role of a HSV thymidine kinase stimulating substance, scopadulciol, in improving the efficacy of cancer gene therapy. J Gene Med. 2006;8:1056–67. doi: 10.1002/jgm.931. [DOI] [PubMed] [Google Scholar]
- 42.Miller CR, Williams CR, Buchsbaum DJ, et al. Intratumoral 5-fluorouracil produced by cytosine deaminase/5-fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res. 2002;62:773–80. [PubMed] [Google Scholar]
- 43.Lengler J, Omann M, Duvier D, et al. Cytochrome P450 reductase dependent inhibition of cytochrome P450 2B1 activity: implications for gene directed enzyme prodrug therapy. Biochem Pharmacol. 2006;72:893–901. doi: 10.1016/j.bcp.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Trinh QT, Austin EA, Murray DM, et al. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55:4808–12. [PubMed] [Google Scholar]
- 45.Ostertag D, Amundson KK, Lopez Espinoza F, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012;14:145–59. doi: 10.1093/neuonc/nor199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kambara H, Tamiya T, Ono Y, et al. Combined radiation and gene therapy for brain tumors with adenovirus-mediated transfer of cytosine deaminase and uracil phosphoribosyltransferase genes. Cancer Gene Ther. 2002;9:840–5. doi: 10.1038/sj.cgt.7700506. [DOI] [PubMed] [Google Scholar]
- 47.Aghi M, Kramm CM, Chou TC, et al. Synergistic anticancer effects of ganciclovir/thymidine kinase and 5-fluorocytosine/cytosine deaminase gene therapies. J Natl Cancer Inst. 1998;90:370–80. doi: 10.1093/jnci/90.5.370. [DOI] [PubMed] [Google Scholar]
- 48.Chang JW, Lee H, Kim E, et al. Combined antitumor effects of an adenoviral cytosine deaminase/thymidine kinase fusion gene in rat C6 glioma. Neurosurgery. 2000;47:931–8. doi: 10.1097/00006123-200010000-00026. [DOI] [PubMed] [Google Scholar]
- 49.Thomas CE, Schiedner G, Kochanek S, et al. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc Natl Acad Sci United States America. 2000;97:7482–7. doi: 10.1073/pnas.120474397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aghi M, Martuza RL. Oncolytic viral therapies - the clinical experience. Oncogene. 2005;24:7802–16. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- 51.Aghi M, Rabkin S. Viral vectors as therapeutic agents for glioblastoma. Curr Opinion Mol Ther. 2005;7:419–30. [PubMed] [Google Scholar]
- 52.Kanai R, Zaupa C, Sgubin D, et al. Effect of gamma 34. 5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. 2012;86:4420–31. doi: 10.1128/JVI.00017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cassady KA, Parker JN. Herpesvirus vectors for therapy of brain tumors. Open Virol J. 2010;4:103–8. doi: 10.2174/1874357901004010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Todo T, Martuza RL, Rabkin SD, et al. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA. 2001;98:6396–401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aboitiz F, Montiel J. Co-option of signaling mechanisms from neural induction to telencephalic patterning. Rev Neurosci. 2007;18:311–42. doi: 10.1515/revneuro.2007.18.3-4.311. [DOI] [PubMed] [Google Scholar]
- 56.Danks MK, Yoon KJ, Bush RA, et al. Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma. Cancer Res. 2007;67:22–5. doi: 10.1158/0008-5472.CAN-06-3607. [DOI] [PubMed] [Google Scholar]
- 57.Terada K, Wakimoto H, Tyminski E, et al. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006;13:705–14. doi: 10.1038/sj.gt.3302717. [DOI] [PubMed] [Google Scholar]
- 58.Han ZQ, Assenberg M, Liu BL, et al. Development of a second-generation oncolytic herpes simplex virus expressing TNFalpha for cancer therapy. J Gene Med. 2007;9:99–106. doi: 10.1002/jgm.999. [DOI] [PubMed] [Google Scholar]
- 59.Gambini E, Reisoli E, Appolloni I, et al. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol Ther. 2012;20:994–1001. doi: 10.1038/mt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Longo SL, Griffith C, Glass A, et al. Development of an oncolytic herpes simplex virus using a tumor-specific HIF-responsive promoter. Cancer Gene Ther. 2011;18:123–34. doi: 10.1038/cgt.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dey M, Ulasov IV, Tyler MA, et al. Cancer stem cells: the final frontier for glioma virotherapy. Stem Cell Rev. 2011;7:119–29. doi: 10.1007/s12015-010-9132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulasov IV, Zhu ZB, Tyler MA, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther. 2007;18:589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- 63.Ahmed AU, Tyler MA, Thaci B, et al. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm. 2011;8:1559–72. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmed AU, Thaci B, Alexiades NG, et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011;19:1714–26. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coffey MC, Strong JE, Forsyth PA, et al. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–4. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 66.Msaouel P, Opyrchal M, Galanis E. Translational research in oncolytic measles virotherapy: early discoveries and future steps. Future Microbiol. 2011;6:125–8. doi: 10.2217/fmb.10.170. [DOI] [PubMed] [Google Scholar]
- 67.Myers R, Harvey M, Kaufmann TJ, et al. Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas. Hum Gene Ther. 2008;19:690–8. doi: 10.1089/hum.2008.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lowenstein PR. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 2002;23:23–30. doi: 10.1016/s1471-4906(01)02063-4. [DOI] [PubMed] [Google Scholar]
- 69.Borden EC, Lindner D, Dreicer R, et al. Second-generation interferons for cancer: clinical targets. Semin Cancer Biol. 2000;10:125–44. doi: 10.1006/scbi.2000.0315. [DOI] [PubMed] [Google Scholar]
- 70.Freeman SM, Abboud CN, Whartenby KA, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–83. [PubMed] [Google Scholar]
- 71.Markert JM, Cody JJ, Parker JN, et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing Il-12. J Virol. 2012;86:5304–13. doi: 10.1128/JVI.06998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ryu CH, Park SH, Park SA, et al. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011;22:733–43. doi: 10.1089/hum.2010.187. [DOI] [PubMed] [Google Scholar]
- 73.Candolfi M, Xiong W, Yagiz K, et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc Natl Acad Sci USA. 2010;107:20021–6. doi: 10.1073/pnas.1008261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Denbo JW, Williams RF, Orr WS, et al. Continuous local delivery of interferon-beta stabilizes tumor vasculature in an orthotopic glioblastoma xenograft resection model. Surgery. 2011;150:497–504. doi: 10.1016/j.surg.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang BX, Rahbar R, Fish EN. Interferon: current status and future prospects in cancer therapy. J Interferon Cytokine Res. 2011;31:545–52. doi: 10.1089/jir.2010.0158. [DOI] [PubMed] [Google Scholar]
- 76.Wakabayashi T, Natsume A, Hashizume Y, et al. A phase I clinical trial of interferon-beta gene therapy for high-grade glioma: novel findings from gene expression profiling and autopsy. J Gene Med. 2008;10:329–39. doi: 10.1002/jgm.1160. [DOI] [PubMed] [Google Scholar]
- 77.Chiocca EA, Smith KM, McKinney B, et al. A phase I trial of Ad. hIFN-beta gene therapy for glioma. Mol Ther. 2008;16:618–26. doi: 10.1038/sj.mt.6300396. [DOI] [PubMed] [Google Scholar]
- 78.Curtin JF, Liu N, Candolfi M, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PloS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Candolfi M, Yagiz K, Foulad D, et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res. 2009;15:4401–14. doi: 10.1158/1078-0432.CCR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Germano IM, Emdad L, Qadeer ZA, et al. Embryonic stem cell (ESC)-mediated transgene delivery induces growth suppression, apoptosis and radiosensitization, and overcomes temozolomide resistance in malignant gliomas. Cancer Gene Ther. 2010;17:664–74. doi: 10.1038/cgt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Uzzaman M, Keller G, Germano IM. Enhanced proapoptotic effects of tumor necrosis factor-related apoptosis-inducing ligand on temozolomide-resistant glioma cells. J Neurosurg. 2007;106:646–51. doi: 10.3171/jns.2007.106.4.646. [DOI] [PubMed] [Google Scholar]
- 82.Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748–54. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 83.Balyasnikova IV, Ferguson SD, Sengupta S, et al. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. Plos One. 2010;5:e9750. doi: 10.1371/journal.pone.0009750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dobson J. Gene therapy progress and prospects: magnetic nanoparticle-based gene delivery. Gene Ther. 2006;13:283–7. doi: 10.1038/sj.gt.3302720. [DOI] [PubMed] [Google Scholar]
- 85.Jin S, Ye K. Nanoparticle-mediated drug delivery and gene therapy. Biotechnol Prog. 2007;23:32–41. doi: 10.1021/bp060348j. [DOI] [PubMed] [Google Scholar]
- 86.Germano IM, Binello E. Gene therapy as an adjuvant treatment for malignant gliomas: from bench to bedside. J Neurooncol. 2009;93:79–87. doi: 10.1007/s11060-009-9869-5. [DOI] [PubMed] [Google Scholar]
- 87.Jin J, Bae KH, Yang H, et al. In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjugate Chem. 2011;22:2568–72. doi: 10.1021/bc200406n. [DOI] [PubMed] [Google Scholar]
- 88.de Fougerolles AR. Delivery vehicles for small interfering RNA in vivo. Hum Gene Ther. 2008;19:125–32. doi: 10.1089/hum.2008.928. [DOI] [PubMed] [Google Scholar]
- 89.Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA. 2000;97:12846–51. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aboody KS, Bush RA, Garcia E, et al. Development of a tumor-selective approach to treat metastatic cancer. Plos One. 2006;1:e23. doi: 10.1371/journal.pone.0000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karapanagiotou E, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18:2080–9. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu W, Sun Q, Wan J, et al. Cationic albumin-conjugated pegylated nanoparticles allow gene delivery into brain tumors via intravenous administration. Cancer Res. 2006;66:11878–87. doi: 10.1158/0008-5472.CAN-06-2354. [DOI] [PubMed] [Google Scholar]
- 93.White E, Bienemann A, Megraw L, et al. Evaluation and optimization of the administration of a selectively replicating herpes simplex viral vector to the brain by convection-enhanced delivery. Cancer Gene Ther. 2011;18:358–69. doi: 10.1038/cgt.2011.2. [DOI] [PubMed] [Google Scholar]
- 94.Pluchino S, Zanotti L, Rossi B, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266–71. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 95.Jacobs A, Voges J, Reszka R, et al. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet. 2001;358:727–9. doi: 10.1016/s0140-6736(01)05904-9. [DOI] [PubMed] [Google Scholar]
- 96.Stoica G, Lungu G, Martini-Stoica H, et al. Identification of cancer stem cells in dog glioblastoma. Vet Pathol. 2009;46:391–406. doi: 10.1354/vp.08-VP-0218-S-FL. [DOI] [PubMed] [Google Scholar]
- 97.Neuwelt EA. Reversible osmotic blood-brain barrier disruption in humans: implications for the chemotherapy of malignant brain tumors. Neurosurgery. 1980;7:204. doi: 10.1097/00006123-198008000-00018. [DOI] [PubMed] [Google Scholar]
- 98.Miller CR, Buchsbaum DJ, Reynolds PN, et al. Differential susceptibility of primary and established human glioma cells to adenovirus infection: targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Res. 1998;58:5738–48. [PubMed] [Google Scholar]
- 99.Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91:2076–80. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mardor Y, Rahav O, Zauberman Y, et al. Convection-enhanced drug delivery: increased efficacy and magnetic resonance image monitoring. Cancer Res. 2005;65:6858–63. doi: 10.1158/0008-5472.CAN-05-0161. [DOI] [PubMed] [Google Scholar]
- 101.Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med. 1997;3:1362–8. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- 102.Sanftner LM, Sommer JM, Suzuki BM, et al. AAV2-mediated gene delivery to monkey putamen: evaluation of an infusion device and delivery parameters. Exp Neurol. 2005;194:476–83. doi: 10.1016/j.expneurol.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benedetti S, Pirola B, Pollo B, et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med. 2000;6:447–50. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- 104.Dvorak HF, Brown LF, Detmar M, et al. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–39. [PMC free article] [PubMed] [Google Scholar]
- 105.Ahmed AU, Rolle CE, Tyler MA, et al. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus Suppress the anti-adenoviral immune response in the cotton rat model. Mol Ther. 2010;18:1846–56. doi: 10.1038/mt.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Einstein O, Ben-Hur T. The changing face of neural stem cell therapy in neurologic diseases. Arch Neurol. 2008;65:452–6. doi: 10.1001/archneur.65.4.452. [DOI] [PubMed] [Google Scholar]
- 107.Van Meir EG, Hadjipanayis CG, Norden AD, et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–93. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang W, Fulci G, Buhrman JS, et al. Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther. 2012;20:37–45. doi: 10.1038/mt.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Le Boeuf F, Diallo JS, McCart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18:888–95. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Ann Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 111.Bauerschmitz GJ, Ranki T, Kangasniemi L, et al. Tissue-specific promoters active in CD44+CD24−/low breast cancer cells. Cancer Res. 2008;68:5533–9. doi: 10.1158/0008-5472.CAN-07-5288. [DOI] [PubMed] [Google Scholar]
- 112.Alonso MM, Jiang H, Gomez-Manzano C, et al. Targeting brain tumor stem cells with oncolytic adenoviruses. Methods Mol Biol. 2012;797:111–25. doi: 10.1007/978-1-61779-340-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cheema TA, Kanai R, Kim GW, et al. Enhanced antitumor efficacy of low-dose etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res. 2011;17:7383–93. doi: 10.1158/1078-0432.CCR-11-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104:42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kanai R, Wakimoto H, Martuza RL, et al. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17:3686–96. doi: 10.1158/1078-0432.CCR-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhu G, Su W, Jin G, et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59–69. doi: 10.1016/j.brainres.2011.03.050. [DOI] [PubMed] [Google Scholar]
- 117.Hu WW, Liu WG. Side populations of glioblastoma cells are less sensitive to HSV-TK/GCV suicide gene therapy system than the non-side population. In Vitro Cell Dev Biol Anim. 2010;46:497–501. doi: 10.1007/s11626-010-9274-6. [DOI] [PubMed] [Google Scholar]
- 118.Hu X, Holland EC. Applications of mouse glioma models in preclinical trials. Mutat Res. 2005;576:54–65. doi: 10.1016/j.mrfmmm.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 119.Candolfi M, Curtin JF, Nichols WS, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–48. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lowenstein PR, Castro MG. Uncertainty in the translation of preclinical experiments to clinical trials. Why do most phase III clinical trials fail? Curr Gene Ther. 2009;9:368–74. doi: 10.2174/156652309789753392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nandi S, Ulasov IV, Tyler MA, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68:5778–84. doi: 10.1158/0008-5472.CAN-07-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]