

Abstract
The goal of this study was to use X-ray crystallography to investigate the structural basis of resistance to human immunodeficiency virus type 1 (HIV-1) protease inhibitors. We overexpressed, purified, and crystallized a multidrug-resistant (MDR) HIV-1 protease enzyme derived from a patient failing on several protease inhibitor-containing regimens. This HIV-1 variant contained codon mutations at positions 10, 36, 46, 54, 63, 71, 82, 84, and 90 that confer drug resistance to protease inhibitors. The 1.8-angstrom (Å) crystal structure of this MDR patient isolate reveals an expanded active-site cavity. The active-site expansion includes position 82 and 84 mutations due to the alterations in the amino acid side chains from longer to shorter (e.g., V82A and I84V). The MDR isolate 769 protease “flaps” stay open wider, and the difference in the flap tip distances in the MDR 769 variant is 12 Å. The MDR 769 protease crystal complexes with lopinavir and DMP450 reveal completely different binding modes. The network of interactions between the ligands and the MDR 769 protease is completely different from that seen with the wild-type protease-ligand complexes. The water molecule-forming hydrogen bonds bridging between the two flaps and either the substrate or the peptide-based inhibitor are lacking in the MDR 769 clinical isolate. The S1, S1′, S3, and S3′ pockets show expansion and conformational change. Surface plasmon resonance measurements with the MDR 769 protease indicate higher koff rates, resulting in a change of binding affinity. Surface plasmon resonance measurements provide kon and koff data (Kd = koff/kon) to measure binding of the multidrug-resistant protease to various ligands. This MDR 769 protease represents a new antiviral target, presenting the possibility of designing novel inhibitors with activity against the open and expanded protease forms.
Treatment of human immunodeficiency virus type 1 (HIV-1)-infected persons with a combination of protease inhibitor and reverse transcriptase inhibitors has been highly effective, resulting in a reduction of plasma viral concentrations to below detectable levels (4). Antiretroviral treatment failure, typically defined as a significant rise from previously suppressed levels of circulating virus, is often associated with the emergence of virus strains resistant to antiviral drugs (12, 13). Many mutations in the protease and reverse transcriptase genes of HIV-1 have been associated with antiretroviral drug resistance (40). Protease inhibitor resistance mutations have been reported at codons 10, 20, 24, 30, 32, 33, 36, 46, 47, 48, 50, 53, 54, 63, 71, 73, 77, 82, 84, and 90 (5). Certain single mutations or combinations of point mutations confer cross-resistance to several protease inhibitors (40). Thus, new drugs are needed to treat patients harboring HIV-1 strains that are resistant to currently available protease inhibitors.
A major contributing factor to the design of effective antiviral compounds is the availability of structural information. Pioneering efforts have centered on inhibiting the viral protease that posttranslationally cleaves viral polyproteins (2, 9, 10, 15). Since these early investigations, several laboratories have studied the X-ray crystallographic structures of the proteases complexed with protease inhibitors. For understanding the structural basis of HIV-1 protease multidrug resistance, however, data regarding the crystal structures of uncomplexed protease enzymes and protease substrate and protease inhibitor multidrug-resistant (MDR) variants are needed.
There are data for four wild-type crystal structures of uncomplexed HIV-1 protease (determined at resolutions from 2.7 to 3.0 Å) deposited in the Protein Data Bank (Table 1). Currently, there are no data for the crystal structure of an uncomplexed MDR HIV-1 protease clinical isolate. The design of new compounds that inhibit MDR HIV-1 proteases will benefit from this work.
TABLE 1.
Crystal structures of uncomplexed HIV-1 protease isolates
Through modeling studies, our group has proposed the use of an active-site expansion model to explain HIV-1 protease inhibitor resistance. We suggested that HIV-1 MDR is associated with a decrease in the volume of amino acid side chains within the active-site cavity. The loss of van der Waals contacts and hydrogen bonds between the mutant HIV-1 proteases and inhibitors (due to the expansion of the active-site cavity of the enzyme) resulted in decreased binding affinity. In this study, we used X-ray crystallography analysis to evaluate changes in structure between a MDR protease and a wild-type protease.
MATERIALS AND METHODS
Characteristics of the HIV-1 protease clinical isolate.
The HIV-1 protease clinical isolate (MDR 769) was obtained from a patient who had received long-term antiretroviral therapy. The attending physician classified this patient as failing extensive antiretroviral therapy on the basis of persistent viremia, and the ensuing HIV-1 protease isolate was selected for study on the basis of clinical prevalence, high-level drug resistance (as measured by in vitro drug susceptibility assays), and cross-resistance to licensed inhibitors and experimental compounds currently in clinical development. The values of the in vitro drug susceptibility ratio (the ratio of the 50% inhibitory concentration of the MDR 769 variant to the 50% inhibitory concentration of the NL4-3 wild type) are as follows: 63-fold resistance for indinavir, 43-fold resistance for saquinavir, 47-fold resistance for nelfinavir, 14-fold resistance for amprenavir, 80-fold resistance for atazanavir, >100-fold resistance for DG-43, >100-fold resistance for palinavir, and >100-fold resistance for GS3333 (33). The licensed HIV-1 protease inhibitor lopinavir (48) was a generous gift from Abbott Laboratories, Chicago, Ill., and the water-soluble protease inhibitor DMP450 was kindly provided by Triangle Pharmaceuticals, Inc., Durham, N.C.
Expression, purification, and crystallization of an HIV-1 MDR 769 protease isolate.
Freshly transformed Escherichia coli strain BL21(DE3) (with the T7 expression vector containing the MDR 769 HIV-1 protease clinical isolate) was grown in liquid yeast tryptone ampicillin medium at 37°C in a shaking incubator, and the mid-log-phase bacterial culture was used to inoculate five 1-liter YT ampicillin medium flasks. This bacterial culture continued to grow for 18 to 20 h after inoculation (52).
The MDR isolate 769 HIV-1 protease was purified and refolded from inclusion bodies, following previously published protocol (52). Briefly, after cell lysis the soluble protein fraction was removed from the inclusion bodies and discarded. The HIV protease inclusion bodies were washed with a series of buffer solutions followed by 20 min of centrifugation at 38,000 × g at 4°C between steps. After the final wash step, the protease inclusion bodies were solubilized using a buffer containing 6 M urea. After a final centrifugation, the solubilized inclusion bodies were applied to a Q-Sepharose ion exchange column preequilibrated with a buffer solution with 6 M urea. The flowthrough from the ion exchange column contained the purified HIV protease.
For refolding the purified MDR isolate 769 HIV-1 protease, the protein was diluted to a concentration of 0.1 to 0.2 mg/ml with a buffer solution containing 6 M urea followed by a series of dialysis buffer exchanges which replaced the urea buffer with 10 mM sodium acetate (pH 5.0)-1 mM dithiothreitol. The diluted protease was concentrated to 3 to 5 mg/ml.
A hanging drop vapor diffusion method (24) and a matrix screen (pH 5.5 to 7.5 versus 0.3 to 1.0 M sodium chloride) was used to obtain the protease crystals in approximately 3 days. The crystals were bipyramidal in morphology and formed from 2-μl drops at 22°C (52). After at least 14 days from the completion of the protease crystal formation, protease-inhibitor complexes were obtained by adding a small amount of solid inhibitor directly to the drop containing the protease crystals. Crystals were soaked with the inhibitor at saturating concentrations for at least 21 days prior to data collection.
X-ray diffraction data collection and structure solution.
The crystals of the uncomplexed MDR isolate 769 protease were placed in a cryoprotectant solution containing 30% (wt/vol) glucose and the artificial mother liquor and were frozen in liquid nitrogen for data collection at the Advanced Photon Source, Argonne National Laboratory (Argonne, Ill.). The data were collected (to a resolution of 1.86 Å), using a wavelength of 1.00 Å and a MAR CCD 165 detector, at station 5IDB at the DuPont-Northwestern-Dow Collaborative Access Team Synchrotron Research Center. HKL software (32) was used to process and scale the diffraction data.
The crystals of MDR isolate 769 protease-inhibitor complexes (DMP450 and lopinavir) were placed in the same cryoprotectant solution and were collected using a Rigaku FRD generator/HTC detector system. The data were collected (using a wavelength of 1.5418 Å) to a resolution of 2.6 Å for the MDR 769 protease-DMP450 complex and to a resolution of 2.8 Å for the MDR 769 protease-lopinavir complex. The data were processed and scaled using Crystal Clear version 1.3 software (36) from Rigaku/MSC (The Woodlands, Tex.).
The structures were determined (using the protein coordinates of the structure 1HXW [17] from the Protein Data Bank) by molecular replacement. Using CNS software (3) and MOLREP (49), a rotational search in the resolution range of 12 to 4 Å was performed using the default values in Lattman's θ+/θ− space (21). The translational search was then performed at 4-Å resolution, and a rigid body refinement was carried out at resolutions of 3.5, 3.0, 2.75, 2.5, 2.25, and 2.0 Å. The 2Fo-Fc electron density map (calculated using XTALVIEW) (25) revealed that the data for the final rigid body model of the protease, as well as that for the inhibitor in the protease active site, fit well into the electron density data. The positions of solvent molecules were determined and refined concurrently using ARP/wARP (mode solvent) software (35). The final refinement of the structure (for the uncomplexed high-resolution structure) was performed using program SHELXL software (44).
Binding studies by SPR.
For these initial studies performed using surface plasmon resonance (SPR), the protease inhibitor DMP450 was used for drug binding due to its high solubility in the SPR buffer. To prepare the protein samples for amide linkage to the carboxymethylated CM5 sensor chip (BIACORE), the MDR clinical variant (769 isolate) and the wild-type protease were diluted in 5 mM maleic acid (pH 6) to achieve a 10 μM concentration. Before the protein samples were applied to the CM5 chip, the carboxyl groups of the matrix were activated using N-ethyl-N′-(dimethylaminopropyl)-carbodiimide and N-hydroxysuccinimide for 7 min (23). The deactivation of the nonreactive N-hydroxysuccinimide esters was accomplished using 1 M ethanolamine for 7 min. The standard BIACORE-certified HEPES buffer saline (HBS) (0.01 M HEPES [pH 7.4], 0.15 M sodium chloride, 3 mM EDTA, 0.005% [vol/vol] polyoxyethylenesorbitan) was used for all sensorgram measurements at a flow rate of 30 μl/min. The DMP450 inhibitor (14, 19) was prepared at concentrations of 1, 10, 100, and 1,000 nM in standard BIACORE HBS buffer from a 100 μM stock solution.
Protein Data Bank accession numbers.
The accession numbers for the crystal structures discussed in the paper are as follows: 1RPI for the MDR isolate 769 uncomplexed structure, 1RQ9 for MDR 769 complexed with DMP450, and 1RV7 for MDR 769 complexed with lopinavir.
RESULTS
Structure solution of the uncomplexed HIV-1 MDR 769 protease isolate and of its DMP450 and lopinavir complexes.
The key results for the structure of the MDR 769-DMP450 and MDR 769-lopinavir complexes are summarized in Table 2 and Table 3.
TABLE 2.
X-ray diffraction data for MDR isolate 769 and its complexes
| Condition or parameter | Value
|
||
|---|---|---|---|
| Uncomplexed isolate 769 | Complex
|
||
| 769-DMP450 | 769-LPVb | ||
| Experimental conditions | |||
| X-ray source | APS (DND-CAT) | Rigaku FRD | Rigaku FRD |
| Wavelength (Å) | 1.00 | 1.5418 | 1.5418 |
| Sample temp (K) | 100 | 100 | 100 |
| Crystal analysis parameters | |||
| Resolution range (Å) | 35.00-1.86 | 23.36-2.60 | 33.97-2.70 |
| Unit cell (Å) | 45.07, 45.07, 105.52 | 45.10, 45.10, 102.96 | 44.98, 44.98, 103.68 |
| Space group | P41 | P41 | P41 |
| Mosaicity (degrees) | 0.4 | 0.8 | 1.5 |
| Percent solvent | 43 | 43 | 46 |
| Data processing parameters | |||
| No. of reflections used | 74,727 | 48,192 | 16,880 |
| No. of unique reflections | 17,571 | 6,422 | 5,316 |
| Redundancy | 4.25 | 7.50 | 3.18 |
| Ι/σ(I) | 21.6 (4.1) | 3.2 (2.1) | 7.1 (3.2) |
| Completeness (%) | 99.3 (99.4) | 100 (100) | 99.6 (100) |
| Rsyma (%) | 4.2 (25.9) | 17.0 (32.9) | 10.8 (29.7) |
Rsym = Σ|Ι − <Ι>|/ΣΙ.
LPV, lopinavir.
TABLE 3.
Refinement statistics for MDR isolate 769 structures and its complexes
| Parameter, factor, deviation, or category | Value
|
||
|---|---|---|---|
| Uncomplexed isolate 769 | Complex
|
||
| 769-DMP450 | 769-LPVf | ||
| Refinement parameters | |||
| Resolution range (Å) | 20.00-1.86 | 20.00-2.60 | 20.00-2.70 |
| No. of protein atoms | 1,552a | 1,558 | 1,558 |
| No. of inhibitor atoms | 0 | 40 | 46 |
| No. of water molecules | 235 | 41 | 36 |
| No. of glucose molecules | 1b | 0 | 0 |
| R factors | |||
| Rcrystc | 0.217 | 0.254 | 0.214 |
| Rfreed | 0.296 | 0.335 | 0.316 |
| σ Cutoff | 0.0 | 2.0 | 2.0 |
| Average atomic temp factors (Å2) | |||
| Protein | 28.84 | 40.16 | 40.25 |
| Side chains | 31.79 | 39.96 | 40.28 |
| Main chains | 26.17 | 40.34 | 40.23 |
| Inhibitor | NAe | 13.74 | 21.47 |
| Solvent | 38.80 | 39.48 | 39.70 |
| Root mean square deviations from ideal geometry | |||
| Bonds (Å) | 0.006 | 0.010 | 0.009 |
| Angles (°) | 1.9 | 1.5 | 1.6 |
| Ramachandran plot category | |||
| Favorable (%) | 92.9 | 83.3 | 85.3 |
| Additional (%) | 7.1 | 16.0 | 12.8 |
| Generous (%) | 0.0 | 0.6 | 1.9 |
| Forbidden (%) | 0.0 | 0.0 | 0.0 |
The refinement statistics include residues in alternate conformations. The true number of protein atoms is 1,508.
The occupancy of glucose is 0.58.
Rcryst = Σ||Fobs| − |Fcalc||/Σ|Fobs|
Rfree = Σ||Fobs| − |Fcalc||/Σ|Fobs|, where Fobs values are test set amplitudes (851 reflections) not used in refinement.
NA, not applicable.
LPV, lopinavir.
Data processing and diffraction analysis indicated an Rsym = 4.2% (where Rsym = 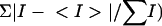 and a tetragonal P41 space group with unit cell parameters of a = 45.07 Å, b = 45.07 Å, c = 105.52 Å, and α = β = γ = 90°, with one HIV-1 protease dimer in the crystallographic asymmetric unit. Cα represents a central carbon atom for each amino acid residue in the polypeptide chain. The crystal structure of the MDR isolate 769 variant was determined by molecular replacement using the wild-type HIV-1 protease-ritonavir complex crystal structure 1HXW (17) as an initial model; the structure solution statistics are summarized in Table 2. The crystallographic refinement converged after 35 cycles to values of Rcryst = 21.7% and Rfree = 29.6% (where Rcryst =
and a tetragonal P41 space group with unit cell parameters of a = 45.07 Å, b = 45.07 Å, c = 105.52 Å, and α = β = γ = 90°, with one HIV-1 protease dimer in the crystallographic asymmetric unit. Cα represents a central carbon atom for each amino acid residue in the polypeptide chain. The crystal structure of the MDR isolate 769 variant was determined by molecular replacement using the wild-type HIV-1 protease-ritonavir complex crystal structure 1HXW (17) as an initial model; the structure solution statistics are summarized in Table 2. The crystallographic refinement converged after 35 cycles to values of Rcryst = 21.7% and Rfree = 29.6% (where Rcryst = 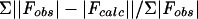 and Rfree =
and Rfree = 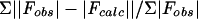 ) (Fobs values are test set amplitudes [851 reflections] not used in refinement) in the resolution range of 20.00 to 1.86 Å, with no σ cutoff (Table 3). The examination of the 2F0-to-Fc electron density map revealed a glucose molecule bound to the protease. This glucose molecule was present in the cryoprotectant and was included in the last 10 cycles of the refinement calculations.
) (Fobs values are test set amplitudes [851 reflections] not used in refinement) in the resolution range of 20.00 to 1.86 Å, with no σ cutoff (Table 3). The examination of the 2F0-to-Fc electron density map revealed a glucose molecule bound to the protease. This glucose molecule was present in the cryoprotectant and was included in the last 10 cycles of the refinement calculations.
Structural comparison of the HIV-1 protease MDR isolate 769 structure with the wild-type 3PHV crystal structure.
The conformational changes of the Cα superposition of the uncomplexed crystal structures of the wild-type and the MDR 769 isolates and the distribution of resistance mutations from the wild-type protease and the MDR 769 clinical isolate protease are illustrated in Fig. 1. Note that the MDR 769 dimer is wider than the wild-type dimer structure. The “flaps” of MDR 769 are also further apart than in the wild type. For example, the distance between the Cα atoms of residues I50 and I150 located in the flaps of the dimer wild type is 4.07Å whereas in the MDR 769 the distance is 12.51Å. Although the RMS Cα deviation is only 1.18 Å when individual monomers are overlapped, this becomes 1.86 Å when the dimers are compared.
FIG. 1.

Structural comparison of HIV-1 wild-type protease and the MDR isolate 769 clinical variant. Cα superposition of the wild-type HIV-1 protease (green) and the MDR 769 HIV-1 protease (red). The green and red side chains represent the catalytic residue at position 25. The blue spheres represent changes in MDR 769 from the wild type.
The van der Waals surface diagrams of the wild-type monomer (Fig. 2A) and MDR isolate 769 monomer (Fig. 2B) are shown to point out the MDR 769 protease structural changes from the structure of the wild type. As can be seen in the figure, residues 50 and 81 are 3.23 Å further apart in the MDR 769 clinical isolate than in the wild-type protease. Moreover, the L10I mutation causes the residues near position 7 to move further away from the catalytic residue at position 25.
FIG. 2.

Close-up view of MDR isolate 769 and wild-type HIV-1 protease active sites. (A) The ribbon diagram of the wild-type protease (green) and MDR 769 protease (red) indicates the side chain difference between the active-site region by the V82A and I84V mutations in the wild type and the MDR 769 protease variant. The volume in the active site has increased not only as a result of dimer flap movements but also as a result of side chain shrinkage in the individual monomers. (B) Stereo diagram of the 2Fo-Fc electron density map for the residue range 81 through 85 in the MDR 769 protease. This electron density map is representative throughout the molecule.
As indicated for Fig. 2A, there is a significant decrease in the size of the amino acid side chains of both resistance mutations (V82A and I84V) in the MDR isolate 769 compared to the results seen with the wild type. To examine the three-dimensional characteristics of the residues, a stereographic view of residues 81 to 85, along with corresponding electron density data, is shown in Fig. 2B. The V82A and I84V mutations are associated with multidrug resistance to licensed protease inhibitors (5).
As determined on the basis of the crystal structures of the MDR isolate 769 protease variant reported here and of wild-type HIV-1 protease (1), the changes in the binding mode of the inhibitor (DMP450) are illustrated in Fig. 3. The electron density data for the DMP450 inhibitor bound to the MDR 769 HIV-1 protease isolate are shown in Fig. 4. The DMP450 inhibitor binds in a completely different mode relative to the binding of the same inhibitor to the wild-type protease.
FIG. 3.

HIV-1 protease structural comparison of MDR isolate 769 and the wild type complexed with DMP450. Cα traces of the wild-type HIV-1 protease and the DMP450 molecule are in green, and the MDR 769 and its cognate DMP450 are shown in red.
FIG. 4.

DMP450 electron density map. A 2Fo-Fc electron density map for the inhibitor DMP450 bound to MDR isolate 769 at the protease active site is shown. Colors for the ball-and-stick model are as follows: carbon is shown in gray, nitrogen is shown in black, and oxygen is shown in red.
The structural change in the lopinavir binding mode is shown in Fig. 5, and the conformations are based on the crystal structures of the wild-type isolate (48) and the MDR 769 protease isolate reported here. Data regarding the lopinavir electron density within the active-site cavity are shown in Fig. 6. Data regarding the molecular recognition of the lopinavir inhibitor by the MDR 769 HIV-1 protease are shown in Fig. 7, illustrating the network of noncovalent interactions between the ligand and the protein. The analysis of these interactions is based on HBPLUS/LIGPLOT software developed by Janet Thornton's group (53). The network of inhibitor interactions is completely different when compared to that of the wild-type protease. The mutations at positions 82 and 84 and possibly at other positions led to the expansion of pockets S1 and S1′. Lopinavir forms stronger interactions with the S1 pocket alone; residues 82 and 84 from the S1′ pocket no longer interact with the inhibitor due to active-site expansion. The flaps of the protease that form critical interactions with the ligand in the wild type are no longer capable of forming these strong interactions in the MDR protease.
FIG. 5.

HIV-1 protease structural comparison of MDR isolate 769 and the wild type complexed with lopinavir. Cα traces of the wild-type HIV-1 protease and the lopinavir molecule are shown in green. The MDR 769 protease variant and its respective lopinavir molecule are shown in red.
FIG. 6.

Lopinavir electron density map. A 2Fo-Fc electron density map for the inhibitor lopinavir bound to the MDR isolate 769 protease is shown. Colors for the ball-and-stick model are as follows: carbon is shown in gray, nitrogen is shown in black, and oxygen is shown in red.
FIG. 7.

Molecular recognition of lopinavir by the MDR isolate 769 protease variant. The van der Waals contacts of the protease with lopinavir are represented by the purple residue side chains, while the gold residue side chain represents the hydrogen bond to the inhibitor. The lopinavir molecule in the protease active site is shown in blue.
Binding studies of the HIV-1 proteases.
SPR was used to examine the interaction of the DMP450 inhibitor with wild-type (Fig. 8A) and MDR 769 (Fig. 8B) protease variants. In these reactions, the HIV-1 proteases were covalently coupled to a (CM5) sensor chip surface and the binding of DMP450 in solution was examined. The inhibitor was injected (at various concentrations) for 30 s, representing the association phase of the binding reaction. In Fig. 8A, the increase in response units is indicated by the four different DMP450 concentrations, confirming the binding of the inhibitor to the wild-type protease. In contrast, no appreciable binding was observed for the MDR variant except at the highest DMP450 concentration (1,000 nM). After the inhibitor injection, dissociation of the complexes was examined for several minutes. Note a more robust binding (at all concentrations tested) of the wild-type protein compared to the results seen with the mutant variant. Our analysis suggests that binding of DMP450 to the wild-type protease is a high-affinity interaction (Fig. 8A), but significantly weaker binding was observed for the MDR isolate 769 (Fig. 8B).
FIG. 8.

SPR measurements of DMP450 binding to the wild-type (A) and MDR 769 (B) HIV-1 proteases. The proteases were coupled to a CM5 sensor chip surface, and binding of DMP450 at concentrations of 1 nM (lowest curve), 10 nM, 100 nM, and 1,000 nM (highest curve) was examined. Nonspecific binding to a control (uncoupled) sensor surface was determined for each concentration and subtracted from the data presented. The x axis represents time in seconds, and the y axis represents response units. The start and end times of DMP450 injection are indicated.
DISCUSSION
The HIV-1 protease clinical isolate (MDR 769) was overexpressed, purified, and crystallized, and the crystallographic structure solved to 1.86-Å resolution. The MDR 769 crystal structure is the first uncomplexed MDR HIV-1 protease clinical isolate to be characterized, and the sequence contains 10 mutations (distributed throughout the entire molecule) relative to the wild-type sequence. The MDR protease represents a novel drug target for antiretroviral therapy.
Note that the active-site expansion data reported for the MDR isolate 769 are consistent with both the substrate envelope hypothesis (37, 38, 55; M. Prabu-Jeyabalan, N. M. King, E. Nalivaika, W. R. P. Scott, and C. A. Schiffer, abstract from the abstract from the XIth International HIV Drug Resistance Workshop 2002, Antivir. Ther. 7:S36, 2002) and thermodynamic measurements of ligand binding (18). Regarding the substrate envelope hypothesis, Schiffer and coworkers have determined a series of crystal structures of HIV protease-substrate complexes. The authors propose a hypothesis according to which inhibitors that fit within the substrate envelope of HIV-1 protease might be more effective and less susceptible to drug resistance mutations. Microcalorimetric measurements by Freire et al. indicate that drug-resistant mutants lower the affinity of the licensed inhibitors by 2 or 3 orders of magnitude (22, 30, 50, 51). From the thermodynamic standpoint, the combined effects of the drug-resistant mutations 82 and 84 (in combination with other mutations) involve significant alterations in the enthalpy and entropy changes for inhibitor binding (28; A. Velazquez-Campoy, V. Sonia, and E. Freire, Abstr. 17th Annu. Meet. Groups Studying Struct. AIDS-Related Systems. Their Application Targeted Drug Design, p. 7, 2003).
The MDR isolate 769 crystal structure reveals several major differences relative to the wild-type structure. First, the V82A and I84V mutations lead to an expansion of the active-site cavity. The loss of a sigma carbon-carbon bond results in an approximate change of 1.5 Å in each of amino acid residues 82, 182, 84, and 184. Since these four residues are in opposite corners of the active sites, there is an approximately 3.0-Å expansion inside the active-site cavity. These are frequently occurring drug resistance mutations, involving a codon change from a longer to a shorter amino acid side chain. The V82A mutation confers resistance to five (indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir) of the six licensed inhibitors, and the I84V mutation confers resistance to six licensed protease inhibitors (including amprenavir). A second difference is the distance increase between residues 50 and 81, as illustrated by the side view of the thicker MDR 769 variant (Fig. 9). The widened gap between residues 50 and 81 is in concert with the 82 and 84 mutations and further contributes to active-site expansion of the protease. A third structural difference regards the open flaps of the MDR 769 protease. The 8.44 Å difference between the wild-type structure and the variant structure contributes to a significant widening of the already open flaps of the uncomplexed HIV-1 protease.
FIG. 9.

A comparison of electrostatic potential surface diagrams of the wild-type HIV-1 protease monomer (A) and the MDR isolate 769 HIV-1 protease monomer (B). Red represents van der Waals surface regions that are negatively charged, while blue represents van der Waals surface regions that are positively charged. In panel B there is a marked increase in distance between amino acid residues 50 and 81.
Our X-ray crystallographic analysis of the two inhibitor complexes of the MDR HIV-1 protease indicates structural changes in several areas. Neither lopinavir nor DMP450 binds to MDR isolate 769 HIV-1 protease in the same way as these two protease inhibitors bind to the wild-type HIV-1 protease (19, 48) (Fig. 3 and 5). There is active-site expansion in the S1 and S1′ pockets due to mutations from longer to shorter amino acid side chains at residues 82 and 84. There are conformational changes in the S2 and S2′ pockets, and there is active-site expansion in the S3 and S3′ pockets. Finally, the water molecule that forms (bridging hydrogen bonds between the flap amino acid residues I50 and I150 and the ligand) is lost in the MDR HIV-1 protease.
The active-site expansion is associated with the in vitro susceptibility data, indicating an up to 100-fold increase in resistance to licensed inhibitors and experimental compounds (33). For example, the resistance to indinavir of the MDR isolate 769 is increased 63-fold and the sequence contains the indinavir resistance mutations L10I, M46L, I54V, A71V, V82A, I84V, and L90 M. The enlargements at the 82 and 84 regions, 50 and 81 regions, and 50 and 150 regions all contribute to the expansion of the active-site cavity. While other MDR isolates have a phenotype similar to that of the MDR isolate 769 (33), the active-site expansion for these proteases remains to be examined.
Compensatory mutations from smaller to larger amino acid side chains in the substrate cleavage sites have been reported (8). For example, the HIV-1 protease I84V mutant contains the compensatory mutation of leucine to phenylalanine at the P1′ position of the p1/p6 cleavage site. Dauber et al. reported an alanine-to-valine change at position P2 of the NC/p1 cleavage site in drug-resistant patient isolates (6). The authors propose that these mutations might represent a mechanism by which severely compromised, drug-resistant viral strains can increase the fitness levels of these HIV strains. While the protease active site is expanding, the substrate cleavage sites mutate to fill the expanded active-site cavity. These observations are consistent with the active-site expansion model.
To correlate binding of inhibitors to the altered HIV-1 protease, we measured the binding of a water-soluble inhibitor, DMP450, to the MDR HIV-1 protease. SPR (BIACORE) measurements indicate that DMP450 does not bind as tightly to MDR 769 variant as the wild-type protein.
These MDR HIV-1 protease crystal structures will be useful in HIV-AIDS research in a number of ways. For example, the structure could be used for increasingly powerful docking experiments to identify novel protease inhibitors. DOCK software (11, 26, 27, 31, 45, 46) is designed to find favorable orientations of a ligand in a receptor and has already been used successfully for the design of protease inhibitors with activity against the wild-type HIV-1 protease (7, 39). The crystal structure of MDR HIV-1 protease will be useful as a template for homology modeling experiments to predict the structures of numerous other MDR HIV-1 variants deposited in the Stanford HIV RT and Protease Sequence Database (16, 42, 43), in the Los Alamos HIV Drug Resistance Database (34), and in other HIV sequence databases.
Another possible application of the MDR HIV-1 protease crystal structure is the examination of flap movement by molecular dynamics simulation. Scott and Schiffer reported curling of the flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance (41). The authors reported that the flaps of HIV-1 protease opened completely during a 10-ns solvated molecular dynamics simulation starting from the unliganded crystal structure. The molecular dynamic simulation experiment with the MDR isolate 769 HIV-1 crystal structure would be very interesting, due in particular to the fact that the 48-G-G-I-G-G-52 sequences in the two monomers of the MDR 769 variant are much further apart compared to the distance in the wild-type flap tips. Hoffman et al. (14a) reported covariation among positions in HIV-1 protease sequences for 536 protease inhibitor-treated persons. Two different statistical tests indicated linkage between 25 pairs of sites, eight of which involved position 10. Crystal structures such as that of the MDR 769 protease may provide elements for experimental validation of the observation that numerous pairs could interaction within a local environment.
In summary, the MDR isolate 769 structure is the first crystal structure of an uncomplexed HIV-1 MDR protease to be described and is the highest-resolution crystal structure of an uncomplexed HIV-1 protease. The structure reveals an expanded active-site cavity representing an altered receptor. The crystal structures of the MDR 769 protease-inhibitor complexes reveal unexpected binding modes which provide a structural basis for protease inhibitor resistance. Therefore, these crystal structures identify a target for a new class of protease inhibitors designed specifically to inhibit the MDR form of the enzyme.
Acknowledgments
This work was supported by a National Institutes of Health grant (GM62990) to L.C.K. and a National Foundation for Cancer Research grant to T.C.M. The Michigan Life Science Corridor provided funding to enhance the structural biology facility at Wayne State University. Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E. I. DuPont de Nemours & Co., The Dow Chemical Company, the U.S. National Science Foundation (through grant DMR-9304725), and the State of Illinois (through the Department of Commerce and the Board of Higher Education grant IBHE HECA NWU 96). The use of the APS was supported by the Office of Energy Research, Basic Energy Sciences, U.S. Department of Energy, under contract W-31-102-Eng-38. We also acknowledge the Center for Molecular and Cellular Toxicology with Human Applications in Michigan (funded by NIEHS grant 1 P30 ESO 6639).
REFERENCES
- 1.Ala, P. J., R. J. DeLoskey, E. E. Huston, P. K. Jadhav, P. Y. Lam, C. J. Eyermann, C. N. Hodge, M. C. Schadt, F. A. Lewandowski, P. C. Weber, D. D. McCabe, J. L. Duke, and C. H. Chang. 1998. Molecular recognition of cyclic urea HIV-1 protease inhibitors. J. Biol. Chem. 273:12325-12331. [DOI] [PubMed] [Google Scholar]
- 2.Blundell, T. L., R. Lapatto, A. F. Wilderspin, A. M. Hemmings, P. M. Hobart, D. E. Danley, and P. J. Whittle. 1990. The 3-D structure of HIV-1 proteinase and the design of antiviral agents for the treatment of AIDS. Trends Biochem. Sci. 15:425-430. [DOI] [PubMed] [Google Scholar]
- 3.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905-921. [DOI] [PubMed] [Google Scholar]
- 4.Carpenter, C. C., D. A. Cooper, M. A. Fischl, J. M. Gatell, B. G. Gazzard, S. M. Hammer, M. S. Hirsch, D. M. Jacobsen, D. A. Katzenstein, J. S. Montaner, D. D. Richman, M. S. Saag, M. Schechter, R. T. Schooley, M. A. Thompson, S. Vella, P. G. Yeni, and P. A. Volberding. 2000. Antiretroviral therapy in adults: updated recommendations of the International AIDS Society-USA Panel. JAMA 283:381-390. [DOI] [PubMed] [Google Scholar]
- 5.D'Aquila, R., J. Schapiro, F. Brun-Vezinet, B. Clotet, B. Conway, L. Demeter, C. Loveday, R. M. Grant, V. A. Johnson, D. Kuritzkes, R. W. Shafer, and D. D. Richman. 2002. Drug resistance mutations in HIV-1. Top. HIV Med. 10:21-25. [PubMed] [Google Scholar]
- 6.Dauber, D. S., R. Ziermann, N. Parkin, D. J. Maly, S. Mahrus, J. L. Harris, J. A. Ellman, C. Petropoulos, and C. S. Craik. 2002. Altered substrate specificity of drug-resistant human immunodeficiency virus type 1 protease. J. Virol. 76:1359-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DesJarlais, R. L., G. L. Seibel, I. D. Kuntz, P. S. Furth, J. C. Alvarez, P. R. Ortiz de Montellano, D. L. DeCamp, L. M. Babe, and C. S. Craik. 1990. Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc. Natl. Acad. Sci. USA 87:6644-6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyon, L., G. Croteau, D. Thibeault, F. Poulin, L. Pilote, and D. Lamarre. 1996. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 70:3763-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erickson, J., D. J. Neidhart, J. VanDrie, D. J. Kempf, X. C. Wang, D. W. Norbeck, J. J. Plattner, J. W. Rittenhouse, M. Turon, N. Wideburg, and et al. 1990. Design, activity, and 2.8 Å crystal structure of a C2 symmetric inhibitor complexed to HIV-1 protease. Science 249:527-533. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald, P. M., B. M. McKeever, J. F. VanMiddlesworth, J. P. Springer, J. C. Heimbach, C. T. Leu, W. K. Herber, R. A. Dixon, and P. L. Darke. 1990. Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0-Å resolution. J. Biol. Chem. 265:14209-14219. [PubMed] [Google Scholar]
- 11.Gschwend, D. A., and I. D. Kuntz. 1996. Orientational sampling and rigid-body minimization in molecular docking revisited: on-the-fly optimization and degeneracy removal. J. Comput.-Aided Mol. Des. 10:123-132. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch, M. S., F. Brun-Vezinet, R. T. D'Aquila, S. M. Hammer, V. A. Johnson, D. R. Kuritzkes, C. Loveday, J. W. Mellors, B. Clotet, B. Conway, L. M. Demeter, S. Vella, D. M. Jacobsen, and D. D. Richman. 2000. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society-USA panel. JAMA 283:2417-2426. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch, M. S., B. Conway, R. T. D'Aquila, V. A. Johnson, F. Brun-Vezinet, B. Clotet, L. M. Demeter, S. M. Hammer, D. M. Jacobsen, D. R. Kuritzkes, C. Loveday, J. W. Mellors, S. Vella, and D. D. Richman. 1998. Antiretroviral drug resistance testing in adults with HIV infection: implications for clinical management. International AIDS Society-USA panel. JAMA 279:1984-1991. [DOI] [PubMed] [Google Scholar]
- 14.Hodge, C. N., P. E. Aldrich, L. T. Bacheler, C. H. Chang, C. J. Eyermann, S. Garber, M. Grubb, D. A. Jackson, P. K. Jadhav, B. Korant, P. Y. Lam, M. B. Maurin, J. L. Meek, M. J. Otto, M. M. Rayner, C. Reid, T. R. Sharpe, L. Shum, D. L. Winslow, and S. Erickson-Viitanen. 1996. Improved cyclic urea inhibitors of the HIV-1 protease: synthesis, potency, resistance profile, human pharmacokinetics and X-ray crystal structure of DMP 450. Chem. Biol. 3:301-314. [DOI] [PubMed] [Google Scholar]
- 14a.Hoffman, N., C. A. Schiffer, and R. Swanstrom. Virology, in press.
- 15.Jaskolski, M., A. G. Tomasselli, T. K. Sawyer, D. G. Staples, R. L. Heinrikson, J. Schneider, S. B. Kent, and A. Wlodawer. 1991. Structure at 2.5 Å resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry 30:1600-1609. [DOI] [PubMed] [Google Scholar]
- 16.Kantor, R., R. Machekano, M. J. Gonzales, K. Dupnik, J. M. Schapiro, and R. W. Shafer. 2001. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database: an expanded data model integrating natural language text and sequence analysis programs. Nucleic Acids Res. 29:296-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kempf, D. J., K. C. Marsh, J. F. Denissen, E. McDonald, S. Vasavanonda, C. A. Flentge, B. E. Green, L. Fino, C. H. Park, X. P. Kong, et al. 1995. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. USA 92:2484-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurt, N., W. R. Scott, C. A. Schiffer, and T. Haliloglu. 2003. Cooperative fluctuations of unliganded and substrate-bound HIV-1 protease: a structure-based analysis on a variety of conformations from crystallography and molecular dynamics simulations. Proteins 51:409-422. [DOI] [PubMed] [Google Scholar]
- 19.Lam, P. Y., Y. Ru, P. K. Jadhav, P. E. Aldrich, G. V. DeLucca, C. J. Eyermann, C. H. Chang, G. Emmett, E. R. Holler, W. F. Daneker, L. Li, P. N. Confalone, R. J. McHugh, Q. Han, R. Li, J. A. Markwalder, S. P. Seitz, T. R. Sharpe, L. T. Bacheler, M. M. Rayner, R. M. Klabe, L. Shum, D. L. Winslow, D. M. Kornhauser, C. N. Hodge, et al. 1996. Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2′ structure-activity relationship, and molecular recognition of cyclic ureas. J. Med. Chem. 39:3514-3525. [DOI] [PubMed] [Google Scholar]
- 20.Lapatto, R., T. Blundell, A. Hemmings, J. Overington, A. Wilderspin, S. Wood, J. R. Merson, P. J. Whittle, D. E. Danley, K. F. Geoghegan, et al. 1989. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature 342:299-302. [DOI] [PubMed] [Google Scholar]
- 21.Lattman, E. E., and W. E. Love. 1970. A rotational search procedure for detecting a known molecule in a crystal. Acta Crystallogr. Sect. B Struct. Sci. 26:1854-1857. [Google Scholar]
- 22.Leavitt, S., and E. Freire. 2001. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 11:560-566. [DOI] [PubMed] [Google Scholar]
- 23.Markgren, P. O., M. Hamalainen, and U. H. Danielson. 1998. Screening of compounds interacting with HIV-1 proteinase using optical biosensor technology. Anal. Biochem. 265:340-350. [DOI] [PubMed] [Google Scholar]
- 24.McPherson, A., Jr. 1976. The growth and preliminary investigation of protein and nucleic acid crystals for X-ray diffraction analysis. Methods Biochem. Anal. 23:249-345. [DOI] [PubMed] [Google Scholar]
- 25.McRee, D. E. 1999. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125:156-165. [DOI] [PubMed] [Google Scholar]
- 26.Meng, E. C., D. A. Gschwend, J. M. Blaney, and I. D. Kuntz. 1993. Orientational sampling and rigid-body minimization in molecular docking. Proteins 17:266-278. [DOI] [PubMed] [Google Scholar]
- 27.Meng, E. C., B. K. Shoichet, and I. D. Kuntz. 1992. Automated docking with grid-based energy evaluation. J. Comp. Chem. 13:505-524. [Google Scholar]
- 28.Muzammil, S., P. Ross, and E. Freire. 2003. A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry 42:631-638. [DOI] [PubMed] [Google Scholar]
- 29.Navia, M. A., P. M. Fitzgerald, B. M. McKeever, C. T. Leu, J. C. Heimbach, W. K. Herber, I. S. Sigal, P. L. Darke, and J. P. Springer. 1989. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 337:615-620. [DOI] [PubMed] [Google Scholar]
- 30.Ohtaka, H., A. Velazquez-Campoy, D. Xie, and E. Freire. 2002. Overcoming drug resistance in HIV-1 chemotherapy: the binding thermodynamics of amprenavir and TMC-126 to wild-type and drug-resistant mutants of the HIV-1 protease. Protein Sci. 11:1908-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshiro, C. M., I. D. Kuntz, and J. S. Dixon. 1995. Flexible ligand docking using a genetic algorithm. J. Comput.-Aided Mol Des. 9:113-130. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 33.Palmer, S., R. W. Shafer, and T. C. Merigan. 1999. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 13:661-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh, U., C. Calef, B. Larder, R. F. Schinazi, and J. W. Mellors. 2001. HIV sequence compendium, p. 191-277. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory Press, Los Alamos, N.Mex.
- 35.Perrakis, A., T. K. Sixma, K. S. Wilson, and V. S. Lamzin. 1997. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple refined dummy atomic models. Acta Crystallogr. Sect. D Biol. Crystallogr. 53:448-455. [DOI] [PubMed] [Google Scholar]
- 36.Pflugrath, J. W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. Sect. D Biol. Crystallogr. 55:1718-1725. [DOI] [PubMed] [Google Scholar]
- 37.Prabu-Jeyabalan, M., E. Nalivaika, and C. A. Schiffer. 2002. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure (Cambridge) 10:369-381. [DOI] [PubMed] [Google Scholar]
- 38.Prabu-Jeyabalan, M., E. A. Nalivaika, N. M. King, and C. A. Schiffer. 2003. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J. Virol. 77:1306-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rutenber, E., E. B. Fauman, R. J. Keenan, S. Fong, P. S. Furth, P. R. Ortiz de Montellano, E. Meng, I. D. Kuntz, D. L. DeCamp, R. Salto, et al. 1993. Structure of a non-peptide inhibitor complexed with HIV-1 protease. Developing a cycle of structure-based drug design. J. Biol. Chem. 268:15343-15346. [PubMed] [Google Scholar]
- 40.Schinazi, R. F., B. A. Larder, and J. W. Mellors. 2000. Mutations in retroviral genes associated with drug resistance: 2000-2001 update. Int. Antivir. News 8:61-91. [Google Scholar]
- 41.Scott, W. R., and C. A. Schiffer. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Struct. Fold Des. 8:1259-1265. [DOI] [PubMed] [Google Scholar]
- 42.Shafer, R. W., D. R. Jung, B. J. Betts, Y. Xi, and M. J. Gonzales. 2000. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 28:346-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shafer, R. W., D. Stevenson, and B. Chan. 1999. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database. Nucleic Acids Res. 27:348-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheldrick, G. M., and T. R. Schneider. 1997. SHELXTL: high resolution refinement. Methods Enzymol. 277:319-343. [PubMed] [Google Scholar]
- 45.Shoichet, B. K., D. L. Bodian, and I. D. Kuntz. 1992. Molecular docking using shape descriptors. J. Comp. Chem. 13:380-397. [Google Scholar]
- 46.Shoichet, B. K., and I. D. Kuntz. 1991. Protein docking and complementarity. J. Mol. Biol. 221:327-346. [DOI] [PubMed] [Google Scholar]
- 47.Spinelli, S., Q. Z. Liu, P. M. Alzari, P. H. Hirel, and R. J. Poljak. 1991. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie 73:1391-1396. [DOI] [PubMed] [Google Scholar]
- 48.Stoll, V., W. Qin, K. D. Stewart, C. Jakob, C. Park, K. Walter, R. L. Simmer, R. Helfrich, D. Bussiere, J. Kao, D. Kempf, H. L. Sham, and D. W. Norbeck. 2002. X-ray crystallographic structure of ABT-378 (lopinavir) bound to HIV-1 protease. Bioorg. Med. Chem. 10:2803-2806. [DOI] [PubMed] [Google Scholar]
- 49.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 30:1022-1025. [Google Scholar]
- 50.Velazquez-Campoy, A., Y. Kiso, and E. Freire. 2001. The binding energetics of first- and second-generation HIV-1 protease inhibitors: implications for drug design. Arch. Biochem. Biophys. 390:169-175. [DOI] [PubMed] [Google Scholar]
- 51.Velazquez-Campoy, A., M. J. Todd, and E. Freire. 2000. HIV-1 protease inhibitors: enthalpic versus entropic optimization of the binding affinity. Biochemistry 39:2201-2207. [DOI] [PubMed] [Google Scholar]
- 52.Vickrey, J. F., B. C. Logsdon, G. Proteasa, S. Palmer, M. A. Winters, T. C. Merigan, and L. C. Kovari. 2003. HIV-1 protease variants from 100-fold drug resistant clinical isolates: expression, purification and crystallization. Protein Expr. Purif. 28:165-172. [DOI] [PubMed] [Google Scholar]
- 53.Wallace, A. C., R. A. Laskowski, and J. M. Thornton. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127-134. [DOI] [PubMed] [Google Scholar]
- 54.Wlodawer, A., M. Miller, M. Jaskolski, B. K. Sathyanarayana, E. Baldwin, I. T. Weber, L. M. Selk, L. Clawson, J. Schneider, and S. B. Kent. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616-621. [DOI] [PubMed] [Google Scholar]
- 55.Wu, T. D., C. A. Schiffer, M. J. Gonzales, J. Taylor, R. Kantor, S. Chou, D. Israelski, A. R. Zolopa, W. J. Fessel, and R. W. Shafer. 2003. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J. Virol. 77:4836-4847. [DOI] [PMC free article] [PubMed] [Google Scholar]