

Abstract
Background
Binge-drinking is common in human adolescents. The adolescent brain is undergoing structural maturation and has a unique sensitivity to alcohol neurotoxicity. Therefore, adolescent binge ethanol may have long-term effects on the adult brain that alter brain structure and behaviors that are relevant to alcohol use disorders.
Methods
In order to determine if adolescent ethanol binge drinking alters the adult brain, male C57BL/6 mice were treated with either water or ethanol during adolescence (5g/kg/day i.g., post-natal days P28-37) and assessed during adulthood (P60-P88). An array of neurotransmitter-specific genes, behavioral tests (i.e. reversal learning, prepulse inhibition, and open field), and post-mortem brain structure using MRI and immunohistochemistry, were employed to assess persistent alterations in adult brain.
Results
At P38, 24 hours after adolescent ethanol (AE) binge, many neurotransmitter genes, particularly cholinergic and dopaminergic, were reduced by ethanol treatment. Interestingly, dopamine receptor type 4 mRNA was reduced and confirmed using immunohistochemistry. Normal control maturation (P38-P88) resulted in decreased neurotransmitter mRNA, e.g. an average decrease of 56%. Following adolescent ethanol treatment, adults showed greater gene expression reductions than controls, averaging 73%. Adult spatial learning assessed in the Morris water maze was not changed by adolescent ethanol treatment, but reversal learning experiments revealed deficits. Assessment of adult brain region volumes using MRI indicated that the olfactory bulb and basal forebrain were smaller in adults following adolescent ethanol. Immunohistochemical analyses found reduced basal forebrain area and fewer basal forebrain cholinergic neurons.
Conclusions
Adolescent binge ethanol treatment reduces adult neurotransmitter gene expression, particularly cholinergic genes, reduces basal forebrain and olfactory bulb volumes, and causes a reduction in the density of basal forebrain acetylcholine neurons. Loss of cholinergic neurons and forebrain structure could underlie adult reversal learning deficits following adolescent binge drinking.
Keywords: Adolescence, binge drinking, forebrain, reversal learning, ethanol
Introduction
Adolescence is best defined by characteristic behaviors including high social interaction and play behavior, high levels of risk-taking, high activity and exploration, impulsivity, and novelty and sensation seeking (Ernst et al., 2009; Spear, 2000). Adolescent behaviors are shared across many species, from humans (12 to 20–25 years of age) to mice (post-natal days 28 to 42) and others (Spear, 2000). Recent studies indicate that adolescent brain structural development coincides with consolidation of adult abilities (Blakemore and Choudhury, 2006), including intelligence (Shaw et al., 2006) and behavioral control of executive functions (Ernst et al., 2009). Adolescent brain maturation involves peaks in cholinergic, dopaminergic, and serotoninergic inputs to frontal cortex, as well as cortical width peaking during adolescence and then declining to stable adult levels (Giedd, 2004; Giedd et al., 2008; Gould et al., 1991; Kalsbeek et al., 1988; Kostovic, 1990; Rosenberg and Lewis, 1994; Spear, 2000). Developing systems are sensitive to ethanol neurotoxicity. Rat models of adolescent binge drinking find significant frontal neurodegeneration (Crews et al., 2000a) and loss of neurogenesis (Crews et al., 2006), suggesting that the adolescent brain is uniquely sensitive to ethanol neurotoxicity (Crews et al., 2007).
Current statistics indicate alcohol consumption during adolescence is common, with approximately 12% of 8th graders, 22% of 10th graders and 28% of 12th grade seniors reporting heavy episodic drinking (more than 5 drinks in a row) within the past 2 weeks of survey (Johnston et al., 2004; Masten et al., 2008). Heavy drinking increases among college students, 44% report binge drinking every 2 weeks and 19% report more than 3 binge-drinking episodes per week (O'Malley et al., 1998; Wechsler et al., 1995). Adolescents have low sedative responses to alcohol, enabling them to consume higher quantities of alcohol, leading to high blood alcohol levels (Silveri and Spear, 1998). Adolescents with alcohol use disorder average 14 drinks per occasion (Deas et al., 2000). The lifetime prevalence of past-year alcohol dependence peaks at 12% between the ages of 18 and 20, and declines across adulthood (Masten et al., 2008). The numerous changes in brain structure coupled with the high levels of alcohol consumption provide a strong rationale to conduct basic studies to understand the impact of underage drinking over the lifespan.
We utilized a mouse model of adolescent binge drinking to better understand adolescent brain development and the impact of binge drinking. C57BL/6 mice were evaluated both just after ethanol treatment towards the end of adolescence (P38) and weeks after treatment during young adulthood. Among subtypes of alcohol dependence, adolescent-youth limited adult alcohol dependence is the most common subtype (Jacob et al., 2005; Moss et al., 2008). We found that adolescent binge treatment altered adult gene expression, particularly cholinergic genes, reduced olfactory bulb and forebrain volume, decreased cholinergic neuron density and caused deficits in reversal, but not spatial learning.
Methods
Animal treatment
Male C57BL/6 mice were used for this study. Mice were ordered from Charles River Labs (Raleigh, NC), and were allowed to acclimate to the animal facilities for seven days in our animal facility prior to treatment. Adolescent mice were requested with the stipulation that all mice were the same weight, in order to reduce potential variability in brain size, and arrived on P21. At P28, animals were divided into control and ethanol treatment groups with five mice per cage. Prior to behavioral testing, 2-3 mice were housed per cage and given one week to acclimate prior to testing.
Mice were treated and tested as shown in Figure 1. Groups of mice were treated during adolescence (P28-37; Figure) and during adulthood (P88-97, Figure 1). Mice were given either water or ethanol (5g/kg, 25% ethanol w/v) intragastrically once per day for 10 days. Adolescent ethanol (AE) treatment resulted in an average blood alcohol level of 287.7 mg/dL ± 36.8 (mean±SEM) measured 1 hour after the last ethanol administration. This is similar to blood alcohol levels that we have observed previously in young adult mice (P56-65) treated with ethanol (5 g/kg/day i.g), where we observe blood alcohol levels of 316 mg/dL ± 11 (Qin et al., 2008). Binge ethanol treatment caused a delay in the adolescent growth spurt. In control mice, an increase in weight occurred from P28 to P32 (15.8 g to 17.5 g, 11%) followed by a plateau in weight from P32-P36 and another increase in weight from P36 to P49 (3.5 g, 19% increase) (Supplemental Figure 1). Mice that received the ethanol binge during adolescence were different from controls in that they did not experience a weight gain until P36, causing the average weight of ethanol treated mice to be significantly less than controls from P32 to P36 (Supplemental Figure 1; *p<0.05 ANOVA with Bonferroni post-tests). By P42, ethanol treated mice recovered and underwent a greater weight gain than controls from P36 to P49 (4.73 g, 31% increase) to arrive at weights by P49 that were not significantly different (Control: 21.7 ± 0.4, Ethanol: 20.23 ± 0.4, p>0.05).
Figure 1.
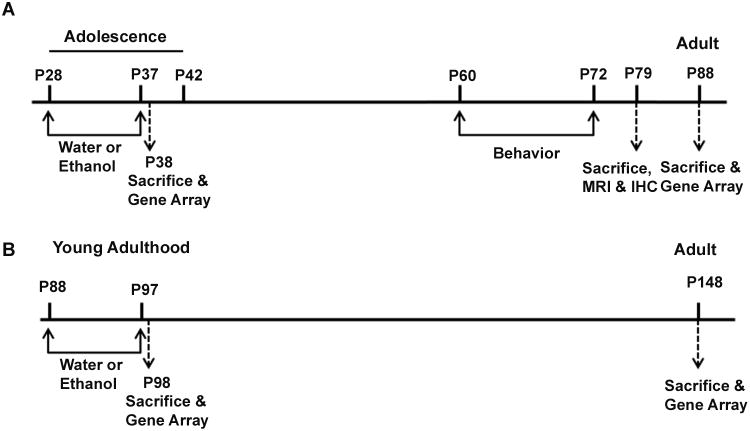
Experimental Design included adolescent treatment groups (A) and adult treatment groups (B) followed with assessments 40-50 days after treatment. (A) Adolescent mice (P28) were treated with either water or ethanol (5g/kg i.g.) once daily for ten days during adolescence (P28-37). One group underwent behavioral testing in adulthood (P60-72) and were sacrificed one week after the end of behavioral testing for postmortem brain magnetic resonance imaging (MRI) and immunohistochemistry (IHC) (N=10 Control, 8 Ethanol). A second group of adolescent mice was treated with either water or ethanol (5g/kg i.g.) once daily for ten days (P28-37)and assessed for neurotransmitter gene expression 24 hours after ethanol treatment (P38; N=6 Control, 6 Ethanol) as well as after a 50 day period of maturation to adulthood (P88; N=6 Control, 6 Ethanol). (B) Young adult (P88) treatment with either water or ethanol (5g/kg i.g.) once daily for ten days (P88-97). Mice were sacrificed either 24 hours (P98; N=6 Control, 6 Ethanol) or 50 days later (P148; N=5 Control, 6 Ethanol). The Neurotransmitter Receptor and Regulator Gene Superarray™ was used to measure gene expression 24 hours after ethanol treatment in both adolescent (P38) and young adult groups (P98). Similarly following a 50 day period of maturation the Neurotransmitter Receptor and Regulator Gene Superarray™ determined expression in young adult (P88) and mature adult (P148) groups.
For neurotransmitter receptor gene array studies, mice were sacrificed by perfusion with saline either 24 hours (N=6 control, 6 ethanol) or 50 days (N=5 control, 5 ethanol) following AE treatment (Figure 1). Similarly, a group of young adult mice (P88) underwent a ten day treatment as adults (P88-97) and were assessed either 24 hours (N=6 control, 6 ethanol) or 50 days (N=5 control, 6 ethanol) after binge ethanol (Figure 1). A third group of mice that received adolescent binge ethanol treatment underwent behavioral testing in adulthood (P60-72) and were sacrificed one week after the end of behavioral testing for postmortem brain magnetic resonance imaging (MRI) and immunohistochemistry (IHC) (Figure 1, N=10 control, 8 ethanol). All protocols were approved by the University of North Carolina Institutional Animal Care and Use Committee and were ins accordance with the Congressional Animal Welfare Act.
Perfusion and brain tissue preparation for MRI and IHC
For MRI and IHC studies, mice were first anesthetized with pentobarbital (100mg/kg, i.p.) and then sacrificed by perfusion as described previously (Coleman et al., 2009; Crews et al., 2004). Briefly, mice were perfused with 0.1M phosphate buffered saline (PBS) followed by 4% PFA followed by post-fixation in 4% PFA at 4°C for 24 hours. The ears and skin were then removed from the head and the skull was re-immersed in 4% PFA for 24 hours. Mouse skulls with the brains intact were then submerged in a 1% PFA/PBS solution at 4°C until MRI. To ensure that time between imaging did not alter brain volume; four postmortem adult mice were scanned twice with each scan being three months apart. Importantly, there was no significant difference in total brain volume across this 3 month time period in these mice (t-test, p=0.62). Neither did we observe a correlation between brain volume and scan date (R= −0.0017, p=0.994).
Neurotransmitter receptor and regulator gene quantitative real-time PCR array
Mice were treated either during adolescence or adulthood for neurotransmitter receptor and regulator gene expression analysis (Figure1). Messenger RNA was extracted from whole brain using Trizol reagent, followed by purification with RNeasy column (Qiagen, Valencia, CA). The total RNA was converted into first stranded-cDNA using an RT first strand kit (SAB Bioscience Corporation, Frederick, MD). DNAase treatment was included in the kit to ensure the removal of genomic DNA. Quantitative real-time PCR was performed using a Bio-rad MyiQ Single-Color RT PCR Detection System. The Mouse Neurotransmitter Receptors and Regulators RT2 Profiler™ PCR Array, which assesses 84 genes (Supplemental Table 1) was used to perform the reaction (SABioscience Corporation; PAMM-060). The Sybr green DNA Real-Time PCR Mix was purchased from Applied Biosystems (Foster City, CA). The PCR mix was denatured at 95°C for 10 min before the first PCR cycle. The thermal cycle profile was programmed for denaturation for 15 s at 95°C and annealing for 60 s at 60°C. A total of 40 PCR cycles were performed. Data analysis was performed using the ΔCt method using the Data Analysis Template Excel file provided by SABioscience Corporation as published previously (Lee et al., 2008). Values for ΔCt were normalized to the average of five housekeeping genes: glucuronidase β, hypoxanthine guanine phosphoribosyl tranferase1, heat shock protein 90kDa α class b member 1, glyceraldehydes-3-phosphate dehydrogenase, and β actin. As recommended by SABioscience, a 2-Way ANOVA with Bonferroni post-tests (using average ΔCt values) was used to detect changes across the entire array as well as individual genes while accounting for multiple comparisons to reduce the probability of type II errors. The percent of expression relative to the adolescent control group (P38) is presented for each gene.
Figure 2.
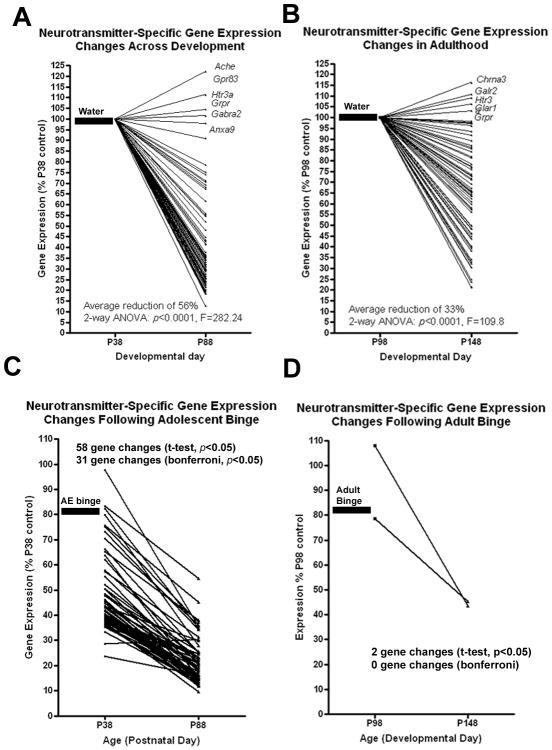
Additional details are within the methods. Adolescent ethanol (AE) binge treatment, but not adult ethanol binge, robustly alters the developmental trajectory of neurotransmitter-specific gene expression. Mice received water or ethanol (5g/kg, i.g.) once a day for ten days during either adolescence (P28-37) or adulthood (P88-97). Neurotransmitter-specific gene expression was assessed using a gene SuperArray either 24 hours (P38 or P88) or 50 days (P88 or P148) after the last alcohol treatment. Expression for each gene is compared to control expression levels 24 hours after treatment (P38 or P88). (A) Developmental changes in gene expression in young control mice. A developmental reduction in neurotransmitter receptor and regulator gene expression was observed across the entire array (Average of 56% reduction, 2-way ANOVA, ***p<0.001) with 36 genes reaching a p<0.05. Genes that showed either a potential increase in expression or no reduction at all are labeled. (B) Developmental changes in gene expression in adult control mice. A developmental reduction in neurotransmitter receptor and regulator gene expression was observed across all the genes tested (2-way ANOVA, ***p<0.0001) that was on average less than controls (33% average reduction) with 11 genes having a p<0.05. (C) Adolescent binge ethanol treatment potentiates the developmental reduction in gene expression, causing significant reductions in 59 genes (p<0.05, t-test of normalized ΔCt values vs P38 control) with 31 genes maintaining statistical significance following Bonferroni adjusted post-tests. (D) Adult binge ethanol resulted in only two genes showing reductions from P98 to P148 that reached a p<0.05, with zero changes following Bonferroni adjusted post-tests. Thick lines and corresponding labels illustrate which treatment was given prior to testing.
Adult behavioral testing: prepulse inhibition, elevated plus maze, locomotor behavior, and Morris water maze with reversal
Behavioral testing was performed in adulthood, beginning on P60 in the following order: P60-elevated plus maze, P61-locomotor behavior, P62-prepulse inhibition, P63-72-Morris Water Maze with reversal learning. All behavioral tests were performed in the UNC Neurodevelopmental Disorders Research Center Mouse Behavioral Phenotyping Core.
Elevated plus maze
The elevated plus maze was elevated 50 cm from the ground, and had two walled arms (20 cm in height) and two open arms. Animals with increased anxiety are predicted to spend less time in the open arms. Mice underwent one five-minute trial in the elevated plus maze. The observed measures were time on, and number of entries into, the open and closed arms.
Locomotion with center time in open field
Locomotor activity was used as a measure of overall activity and exploratory behavior in a novel environment. Reduced exploration was an index of anxiety-like behavior (Crawley, 1999). The open field chamber (40 cm × 40 cm × 30 cm, VersaMax system, AccuScan Instruments) was crossed by a grid of photobeams to detect motion. Mice were tested for one hour in the open field chamber. Measures of distance traveled, rearing movements, and time spent in the center were taken.
Prepulse inhibition
Prepulse inhibition of the acoustic startle response was used as a measure sensorimotor gating. Prepulse inhibition occured when a low pre-stimulus of varying intensity (20 ms duration; 74, 78, 82, 86, and 90 dB) was given 100ms prior to a louder startle stimulus (40 ms duration; 120 dB), diminishing the response to the louder stimulus. Mice were tested with a San Diego Instruments SR-Lab system as described previously (Moy et al., 2006; Paylor and Crawley, 1997). The peak startle amplitude during a 65-msec period beginning with the startle stimulus was measured. Each test session consisted of forty-two trials, following a five-minute habituation period.
Morris water maze spatial and reversal learning in adults
The Morris water maze task was used to test spatial learning and memory as well as the ability to reverse a learned response as described previously (Moy et al., 2007). The water maze (122 cm in diameter) was filled with 45 cm deep water at 24-26° C. The room had numerous visual cues. Water in the maze was tinted white with nontoxic poster paint so the escape platform was not visible. The experimenter was blinded as to the treatment group of the mice. The measures taken were swimming distance, swimming velocity, and latency to find the platform by an automated tracking system (Noldus Ethovision). In each test, mice that displayed significant floating behavior (average swimming velocity of <10cm/s) were removed from the analysis. Criterion for acquisition of learning was set at a 15-second average latency of all the mice to find the platform.
Visual Cue
Mice were first tested for their ability to find the 12 cm diameter visible platform having a tall cylinder as the visual cue. This was used to identify whether any mice had visual or motor deficits that could prohibit them from learning the hidden platform tasks. Mice were given four trials per day until criterion was achieved. Each mouse was placed in the pool at one of four possible randomly ordered locations, and then given 60 seconds to find the cued visible platform.
Initial Learning Acquisition
Mice were evaluated in the hidden platform task using the same procedure as described above. The platform was submerged under the water in Quadrant 1. C57BL/6 mice have been found to acquire learning by day six of testing (Moy et al., 2007). Mice were given four trials per day to learn the position of the hidden platform. Once the 15 second criterion was reached for the entire group, mice were given a one-minute probe trial in the pool without the platform in place. During the probe trial, the time spent in each quadrant of the pool was measured.
Reversal Learning
Mice were then tested for their ability to ‘unlearn’ the previous location of the platform and to learn its new location. The location of the hidden platform was moved to the opposite quadrant of the water maze (Quadrant 3), and mice were tested for acquisition of the new location. After reaching the 15-second group-wide criterion, mice underwent a second probe trial to assess quadrant preference after reversal. During the reversal probe trial, normal C57/BL6 mice were shown to spend more time in the most recent quadrant (Q3), than in the initial learning quadrant (Q1) (Moy et al., 2007). Thus, mice with perseverative reversal learning deficits were expected to spend more time in the former quadrant (Q1) than unimpaired mice.
Postmortem MRI and Image Segmentation
Following behavioral testing and sacrifice, whole mouse skulls with brain intact were scanned at the UNC Biomedical Research Imaging Center (BRIC) on a 9.4 Tesla Bruker BioSpec spectrometer (Bruker Biospin Inc., Billerica, MA). High-resolution MRI images (0.12mm × 0.12mm × 0.12mm) were acquired using the following diffusion weighted 3D RARE sequence: TR=0.7s, TEeff =23.662ms, Rare Factor = 3, RARE echo spacing = 11.3067ms, diffusion gradient time δ =6ms, diffusion gradient separation Δ = 12.422, b value – 1000s/mm2, matrix size = 200×125×80, FOV=24.0mm × 15mm × 9.6mm. Total scan time was 15 hours per animal. Each head was scanned individually.
Mean diffusivity images, which were computed from the reconstructed diffusion tensor data, were aligned and brain structures were segmented using an automatic segmentation protocol developed at the UNC Neuro Image Research and Analysis Laboratory (NIRAL) (Lee et al., 2009). Following automatic segmentation, blinded investigators manually post-segmented selected regions of interest identified from the automatic segmentation, as well as measuring region volume using ITK-SNAP™ software version 1.8. Brain region boundaries were determined using the Brookhaven National Library 3-D MRI Digital Atlas Database of the adult C57BL/6 mouse (www.bnl.gov/medical/RCIBI/mouse).
Immunohistochemistry and Image Analysis
Immunohistochemical images were captured using an Olympus BX51TRF microscope and ProgRes® CapturePro 2.5 digital camera system (Jenoptik, Germany). The Bioquant Nova Advanced Image Analysis System™ was used to assess histological area and neuron density (immuno-positive neurons/mm2) using a cell counting technique described previously (Coleman et al., 2009; Crews et al., 2004). We have shown previously that this method results in nearly identical percent changes between treatment and control groups as other unbiased stereological techniques (Crews et al., 2004). In order to confirm MRI volume findings, area measurements of the basal forebrain were taken using Bioquant on sections labeled for parvalbumin. Parvalbumin interneurons filled the nuclei of the basal forebrain, making landmarks and boundaries more easily identifiable. Up to six sections in three different regions, defined by distance from bregma, were measured. Anatomical landmarks were identified using the mouse atlas (Franklin and Paxinos, 2001) in the same regions as those analyzed by MRI. The landmarks for each bregma and the corresponding nuclei were as follows: +1.34 to +1.1-concentric horns of the forceps minor of the corpus callosum to the genu of the corpus callosum including the medial septum, vertical limb of diagonal band, and ventral pallidum; +0.86 to +0.14-narrowing of the corpus callosum and appearance of the lateral septum to the decussation of the anterior commissure which included the lateral septal regions, medial septum, ventral pallidum, substantia innominata, horizontal diagonal band, magnocellular preoptic, and lateral preoptic areas; +0.02 to −0.22-separation of the anterior commissure decussation to the disappearance of the anterior commissure posterior, using the posterior wings of the anterior commissure and the internal capsule as landmarks in conjunction with parvalbumin staining to triangulate the basal forebrain nuclei which included the ventral pallidum, substantia innominata, horizontal diagonal band, bed nucleus of striatum, magnocellular preoptic nucleus, anterior amygdaloid area dorsal, and the interstitial nucleus of the posterior limb of the anterior commissure.
To determine the density of choline acetyltranferase (ChAT) immuno-reactive (IR) neurons, a technique using Bioquant imaging software was used by a blinded investigator with three to six sections per mouse (Crews et al., 2004). The region of the basal forebrain that had the greatest volumetric reduction as measured by MRI, (bregma +0.14 to −0.22) was identified on the sections using the adult mouse atlas (Franklin and Paxinos, 2001). This has been designated previously in rodents as the Ch4 region (Mesulam et al., 1983). Sections from three bregma locations were analyzed (+0.14,−0.10, and −0.22). These bregma locations were identified reliably using major landmarks in the mouse atlas (+0.14-the decussation of the anterior commissure, −0.10 shape of the posterior region of the anterior commissure and the triangular configuration of the fornices and dorsal third ventricle, −0.22 reduced size of the posterior region of the anterior commissure and the third ventricle). ChAT+IR neurons per immunohistochemical area of the region including the horizontal diagonal band, substantia innominata, and ventral pallidum was determined. The number of ChAT+IR neurons was counted manually using bioquant, and the area of the circumscribed region was reported in mm2 by Bioquant. The density of ChAT+IR neurons/mm2 was averaged between both hemispheres at each of the three distances from bregma and then averaged across the entire region investigated (+0.14 to −0.22).
Statistical Analysis
Gene array statistical analysis
In order to analyze the Superarray data, the company recommended using a 2-way ANOVA with average ΔCt values followed by Bonferroni post-tests to identify statistically significant changes of individual genes while accounting for the multiple comparisons. To gain additional insight, simple t-tests of normalized ΔCt values were also performed on each gene using the company's spreadsheet, in order to identify candidate genes that may not reach statistical significance following the stricter Bonferroni method. The t-test analyses may result in type II errors resulting in false positives due to the multiple comparisons, but these statistics are presented in order to more completely identify candidate genes. Values in groups that received treatment during adolescence (i.e. P38 ethanol group, P88 control group, and P88 ethanol group) were compared to the P38 control group (Figure 1). Similarly, groups that underwent adult treatment (i.e. P98 ethanol group, P148 control, and P148 ethanol) were compared to the P98 control group.
Structural MRI Image statistics
For the automatic segmentation volume data, simple t-tests were performed to identify candidate regions for further manual segmentation (p<0.05). Following manual correction, t-tests performed and corrected corrected for multiple comparisons using the Bonferroni method using the following formula: p<0.05/4 comparisons. Therefore a p<0.0125 was required to be considered statistically significant.
Behavioral and Immunohistochemistry statistics
A Student's t-test or two-Way ANOVA were used for most behavioral and immunohistochemical comparisons (p<0.05 for significance). For prepulse inhibition and locomotor activity, a repeated measures 2-way ANOVA was used.
Results
Decreased Neurotransmitter-Specific Gene Expression During Adolescent Brain Maturation and Binge Ethanol Treatment
Synaptic connections increase during brain development, reaching a developmental peak associated with high levels of plasticity that mature to lower stable adults levels. To investigate broad changes in neurotransmitter gene expression during adolescent brain maturation to young adulthood (P38 - P88), we used an RT-PCR Superarray™ of 84 neurotransmitter-specific mRNAs. Eleven genes were removed from the analysis due to insufficient yield (Table S1). Interestingly, there was a significant main effect of age on the expression of neurotransmitter-specific genes in controls across the entire array from P38 to P88(Figure 2A; average reduction of 56%, p<0.0001, F=282.4). The expression of thirty-five mRNAs was lower in P88 young adults as compared to P38 adolescents (Table 1; t-test, *p<0.05, **p<0.01, § p<0.05 Bonferroni post-test). Only six of the 73 neurotransmitter-specific mRNA did not decline between P38 and P88 (See Figure 2A). Adolescent brain maturation showed large decreases in peptide, γ-amino-butyric acid (GABA), cholinergic and dopaminergic gene expression (Fig. 3,Fig. 4 and Table 1). Examples include peptides such as NPY and somatostatin receptors, which decreased 70-80% during brain maturation across receptor subtypes (Table 1). These peptides are often found in inhibitory GABAergic interneurons. Between P38 and P88, GABAA receptor subunit mRNA and the GABA synthetic enzyme, glutamate decarboxylase, decreased 70-80%. Also, acetylcholine receptor mRNA, both muscarinic and nicotinic, and the acetylcholine synthetic enzyme ChAT decreased approximately 60-80% during adolescent brain maturation (Table 1), although acetylcholinesterase (Ache), the acetylcholine inactivating enzyme, maintained a constant level of expression during adolescent brain development (Figure 2A). A reduction in neurotransmitter mRNA expression between adolescence and young adulthood is consistent with maturation of neurocircuitry.
Table 1.
The effect of adolescent binge ethanol on neurotransmitter specific gene expression.
| Symbol | Description | P38 Control | P38 AE binge | P88 Control | P88 AE binge |
|---|---|---|---|---|---|
| Peptide receptors | |||||
| Brs3 | Bombesin-like receptor 3 | 100 | 66* | 45* | 25** |
| Cckar | Cholecystokinin A receptor | 100 | 64 | 43* | 15**†§ |
| Cckbr | Cholecystokinin B receptor | 100 | 55 | 45* | 18**†§ |
| Galr1 | Galanin receptor 1 | 100 | 50 | 28 | 15*§ |
| Grpr | Gastrin releasing peptide receptor | 100 | 83 | 102 | 55*† |
| Gpr103 | G protein-coupled receptor 103 | 100 | 39 | 26* | 17* |
| Npffr1 | Neuropeptide FF receptor 1 | 100 | 36 | 20* | 12**§ |
| Npffr2 | Neuropeptide FF receptor 2 | 100 | 37 | 13 § | 10**§ |
| Nmur1 | Neuromedin U receptor 1 | 100 | 36 | 36* | 23* |
| Nmur2 | Neuromedin U receptor 2 | 100 | 24* | 18**§ | 16**§ |
| Npy1r | Neuropeptide Y receptor Y1 | 100 | 44 | 32 | 30* |
| Npy2r | Neuropeptide Y receptor Y2 | 100 | 35 | 19* | 16**§ |
| Npy5r | Neuropeptide Y receptor Y5 | 100 | 40 | 21* | 21** |
| Ntsr1 | Neurotensin receptor 1 | 100 | 48 | 25 | 19*§ |
| Prokr1 | Prokineticin receptor 1 | 100 | 45 | 20* | 20** |
| Prokr2 | Prokineticin receptor 2 | 100 | 40 | 19*§ | 12**§ |
| Sstr2 | Somatostatin receptor 2 | 100 | 48 | 36* | 26* |
| Sstr4 | Somatostatin receptor 4 | 100 | 33 | 19** | 14**§ |
| Tacr1 | Tachykinin receptor 1 | 100 | 41 | 22* | 24* |
| Tacr3 | Tachykinin receptor 3 | 100 | 40 | 23 | 15*§ |
| Cholinergic receptors | |||||
| Chat | Choline acetyltransferase | 100 | 45 | 36* | 15**§ |
| Chrm1 | Cholinergic receptor, muscarinic 1 | 100 | 38 | 29* | 16**§ |
| Chrm2 | Cholinergic receptor, muscarinic 2 | 100 | 35 | 29* | 17**§ |
| Chrm3 | Cholinergic receptor, muscarinic 3 | 100 | 42 | 23* | 15**§ |
| Chrm4 | Cholinergic receptor, muscarinic 4 | 100 | 40 | 30 | 17*§ |
| Chrm5 | Cholinergic receptor, muscarinic 5 | 100 | 46 | 24 | 12*§ |
| Chrna3 | Cholinergic receptor, nicotinic, α3 | 100 | 76 | 75 | 45** |
| Chrna4 | Cholinergic receptor, nicotinic, α4 | 100 | 70 | 68 | 36* |
| Chrna5 | Cholinergic receptor, nicotinic, α5 | 100 | 58 | 48* | 25** |
| Chrna6 | Cholinergic receptor, nicotinic, α6 | 100 | 52 | 42* | 22* |
| Chrna7 | Cholinergic receptor, nicotinic, α7 | 100 | 44 | 30* | 14*§ |
| Chrnb1 | Cholinergic receptor, nicotinic, β1 | 100 | 43 | 35 | 15*§ |
| Chrnb2 | Cholinergic receptor, nicotinic, β2 | 100 | 37 | 31* | 16**§ |
| Chrnb3 | Cholinergic receptor, nicotinic, β3 | 100 | 39 | 24* | 15**§ |
| Chrnb4 | Cholinergic receptor, nicotinic, β4 | 100 | 39 | 36 | 19**§ |
| Monamine Receptors | |||||
| Comt | Catechol-O-methyltransferase | 100 | 98 | 71 | 36** |
| Drd1a | Dopamine receptor D1A | 100 | 82 | 69 | 35* |
| Drd2 | Dopamine receptor 2 | 100 | 75 | 71 | 37* |
| Drd3 | Dopamine receptor 3 | 100 | 62 | 44* | 20* |
| Drd4 | Dopamine receptor 4 | 100 | 29* | 45 | 30* |
| Drd5 | Dopamine receptor 5 | 100 | 43 | 30* | 15**§ |
| GABA receptors | |||||
| Gabra1 | GABA-A receptor, α1 | 100 | 38 | 42 | 25* |
| Gabra2 | GABA-A receptor, α2 | 100 | 41 | 22* | 13**§ |
| Gabra3 | GABA-A receptor, α3 | 100 | 40 | 26* | 15**§ |
| Gabra4 | GABA-A receptor, α4 | 100 | 43 | 26 | 16**§ |
| Gabra5 | GABA-A receptor, α5 | 100 | 50 | 32 | 23* |
| Gabrb3 | GABA-A receptor, β3 | 100 | 73 | 62* | 34* |
| Gabrd | GABA-A receptor, δ | 100 | 57 | 62 | 31* |
| Gabrg1 | GABA-A receptor, γ1 | 100 | 48 | 43* | 21** |
| Gabrg2 | GABA-A receptor, γ2 | 100 | 43 | 34* | 23* |
| Gabrq | GABA-A receptor, θ | 100 | 36 | 26* | 13** |
| Gabrr1 | GABA-A receptor, ρ1 | 100 | 39 | 23* | 16*§ |
| Gabrr2 | GABA-A receptor, ρ2 | 100 | 40 | 23* | 21* |
| Gad1 | Glutamic acid decarboxylase 1 | 100 | 42 | 25 | 16**§ |
| Glycine Receptors | |||||
| Glra1 | Glycine receptor, α1 subunit | 100 | 80 | 55 | 28**† |
| Glra3 | Glycine receptor, α3 subunit | 100 | 45 | 33** | 18**§ |
| Glra4 | Glycine receptor, α4 subunit | 100 | 39 | 27* | 14*§ |
| Glrb | Glycine receptor, β subunit | 100 | 39 | 35* | 23*§ |
Adolescent alcohol binge robustly alters neurotransmitter gene expression across development. Mice received either water or ethanol (5g/kg) once a day for 10 days during adolescence (P28-37). The expression of 84 neurotransmitter receptor genes was assessed in whole brain either 24 hours after treatment (P38; N=6 Control, 6 Ethanol) or in adulthood (P88; N=5 Control, 5 Ethanol) using an RT2 Profiler Neurotransmitter Receptor and Regulator Superarray™. Gene expression levels are given as the percent of P38 control. Genes that changed in at least one treatment group are presented. Data analysis was performed using the ΔCt method using the Data Analysis Template Excel file provided by SABioscience Corporation as published previously (Lee et al., 2008). Values for ΔCt were normalized to the average of five housekeeping genes: glucuronidase β, hypoxanthine guanine phosphoribosyl transferase 1, heat shock protein 90kDaα class b member 1, glyceraldehydes-3-phosphate dehydrogenase, and β actin. A 2-way ANOVA with Bonferroni post-tests to account for multiple comparisons was used to determine statistical significance of individual genes (§ p<0.05). Student t-tests were also performed in order to gain insight, however t-test analysis may produce type II errors. (t-test versus P38 control:
p<0.05;
p<0.01;
t-test versus P88 control:
p<0.05)
Figure 3.
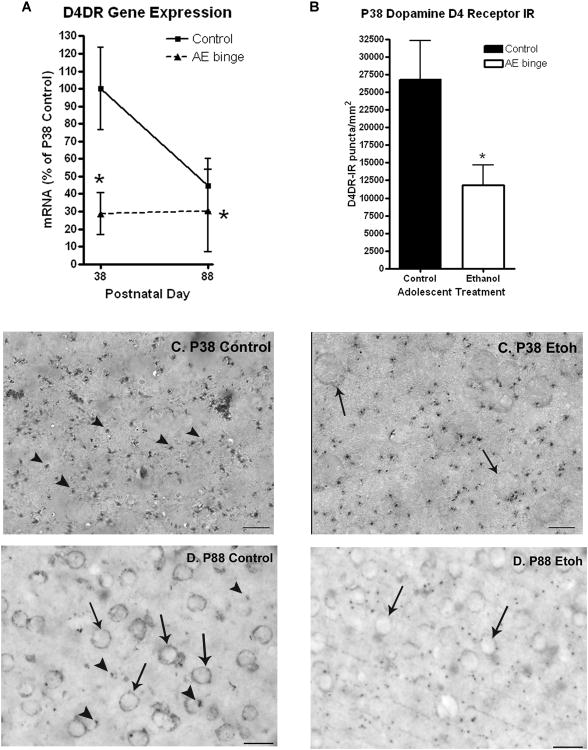
Effects of adolescent binge ethanol treatment ondopamine D4 receptor (D4DR) gene and protein expression. Adolescent mice received either water or binge ethanol (5g/kg once daily, i.g.) for ten days (P28-37). Mice were sacrificed 24 hours (P38) or 50 days (P88) after the last treatment for RT-PCR and immunohistochemistry. (A) The adolescent ethanol binge treatment caused a 71% (*p<0.05 vs P38 controls) reduction in D4DR gene expression in whole brain 24 hours after treatment at P38 that remained reduced at P88. Control mice underwent a 55% developmental reduction in D4DR gene expression (P38 to P88). (B) Quantification of punctate D4DR immunoreactivity (D4DR+IR) in the orbitofrontal cortex in adolescent mice (P38). Adolescent mice that received binge ethanol had 44% less D4DR+IR in the orbitofrontal cortex than controls (Controls: 26800 ± 5500 D4DR+IR puncta/mm2; Ethanol: 11790 ± 2900 D4DR+IR puncta/mm2, *p<0.05, t-test; N=3 Control, N=4 Ethanol). Although gene expression is whole brain and D4DR+IR protein levels are orbital frontal cortex, both show significant decreases following adolescent ethanol binge treatment. (C and D) Images of orbitofrontal cortex from adolescent (C: P38) and adult (D: P88) mice. C: Images of punctate D4DR+IR staining in P38 control and P38 binge ethanol (Etoh) treated mice (magnification 120×). D4DR+IR puncta are identified by the arrow heads (Δ). Adult D4DR+IR are both punctate and somatic. Somatic staining is identified by arrows. D: Images of orbitofrontal cortex from adult (P88) mice. Note D4DR+IR staining in P88 adult is both punctate and somatic. Quantification of P88 D4DR+IR was 6800 ± 4500 D4DR+IR puncta/mm2 for controls and 4790 ± 2900 D4DR+IR puncta/mm2 for ethanol treated animals (N=3 Control, N=4 Ethanol). (t-test; vs P38 controls, Scale bar: 20 micron).
Figure 4.
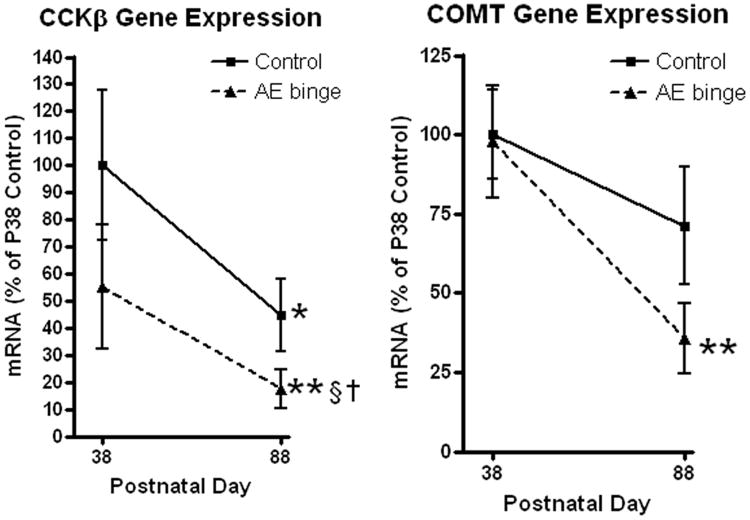
Adolescent alcohol binge differentially alters developmental trajectory of neurotransmitter receptor and regulator gene expression. RT-PCR was performed using an RT2 Profiler PCR Array for neurotransmitter receptor and regulator genes. mRNA levels are presented as percent of P38 control to demonstrate the effects across development Patterns of adolescent ethanol binge (AE binge) treated animal developmental trajectory for Cholecystokininβ (CCKβ) and Catechol-O-methyltransferase (COMT) are shown. (Left) Binge ethanol treatment reduces P38 CCKβ expression by 64% of P38 control. Both control and ethanol groups show a similar developmental decline during the fifty days after the adolescent ethanol binge. In controls, CCKβ receptor gene expression decreases 55% across development from P38 (100%) to P88 (45% of P38 control, *p<0.05). In ethanol binge animals CCKβ mRNA levels were reduced 82% (**p<0.01 vs P38 control). However, the developmental decline from P38 AE binge (55% of P38 control) to P88 AE binge (18% of P38 control) is comparable to the control developmental decline. Adult P88 AE binge animals express 39% of P88 control CCKβ mRNA expression (†p<0.05, vs P88 control) similar to the 45% P38 AE binge animals suggesting the developmental decline continued a similar course following the initial AE binge insult. (Right) Catechol-O-methyltransferase (COMT) shows a different pattern. Binge ethanol treatment does not change P38 COMT gene expression. The 50 day developmental decline in COMT brain gene expression in controls was not statistically significant (29% reduction, p=0.24). However, AE binge treated animals showed a 64% developmental reduction in COMT gene expression (p<0.01 vs P38 controls). AE binge treatment appears to alter the developmental trajectory of COMT gene expression.
To compare maturation changes in gene expression across adolescent to young adulthood (P38-P88) to a similar period, (i.e., 50 days across young adulthood), we also investigated neurotransmitter gene changes between P98 to P148. There was a significant main effect of age on gene expression between P98 and P148 (Figure 2B; average reduction of 33%, 2-Way ANOVA: p<0.0001, F=109.8). Using t-tests, 11 genes showed continuing developmental reductions, mostly peptide receptors, muscarinic cholinergic receptors and GABAA subunits (Table 2, Figure 2B; t-test *p<0.05), with four genes reaching statistical significance following Bonferroni post-tests: GABAR α4, GAD1, Galantin receptor 1, Prokineticin receptor 2, and Neuropeptide FF receptor 2 (Table 2). Linear plots of gene expression show different patterns of gene expression across all the genes in the array during the two different developmental time periods (Figure 2A and 2B). Following adolescent control treatment, adolescent maturation (P38-P88) shows a dark band of many genes with reductions greater than 60% (Figure 2A; overall average reduction of 56%), whereas young adult maturation (P98-P148) shows less robust declines (Figure 2B; overall average reduction of 33%).
Table 2.
The effect of adult binge ethanol on neurotransmitter receptor and regulator gene expression.
| Symbol | Description | P88 Control | P98 (Adult Ethanol) | P148 Control | P148 (Adult Ethanol) |
|---|---|---|---|---|---|
| Peptide receptors | |||||
| Cckar | Cholecystokinin α receptor | 100 | 108 | 66 | 44* |
| Galr1 | Galanin receptor 1 | 100 | 45 | 33* | 43 |
| Galr2 | Galanin receptor 2 | 100 | 79 | 111 | 45* |
| Npffr2 | Neuropeptide FF receptor 2 | 100 | 70 | 21*§ | 36 |
| Npy2r | Neuropeptide Y receptor Y2 | 100 | 94 | 33* | 65 |
| Npy5r | Neuropeptide Y receptor Y5 | 100 | 89 | 39* | 80 |
| Prokr2 | Prokineticin receptor 2 | 100 | 87 | 24*§ | 47 |
| Tacr3 | Tachykinin receptor 3 | 100 | 54 | 31* | 51 |
| Cholinergic receptors | |||||
| Chrm3 | Cholinergic receptor, muscarinic 3 | 100 | 88 | 33* | 57 |
| Chrm4 | Cholinergic receptor, muscarinic 4 | 100 | 83 | 38* | 79 |
| Chrm5 | Cholinergic receptor, muscarinic 5 | 100 | 81 | 31* | 58 |
| GABA receptors | |||||
| Gabra4 | GABA-A receptor, α4 | 100 | 71 | 25*§ | 60 |
| Gabrr2 | GABA-A receptor, ρ2 | 100 | 111 | 65 | 87 |
| Gad1 | Glutamic acid decarboxylase 1 | 100 | 70 | 21*§ | 51 |
Adult alcohol binge slightly alters neurotransmitter gene expression across development. Mice received either water or ethanol (5g/kg) once a day for 10 days during adulthood (P88-97). The expression of 84 neurotransmitter receptor genes was assessed in whole brain either 24 hours after treatment (P98; N=6 Control, 6 Ethanol) or in adulthood (P148; N=5 Control, 6 Ethanol) using an RT2 Profiler Neurotransmitter Receptor and Regulator Superarray™. Gene expression levels are given as the percent of P88 control. Genes that changed in at least one treatment group are presented. A 2-way ANOVA with Bonferroni post-tests to account for multiple comparisons was used to determine statistical significance of individual genes (§ p<0.05). Student t-tests were also performed in order to gain insight into other genes that might be changing, however t-test analysis may produce type II errors.(t-test versus P88 control:
p<0.05).
Neurotransmitter-specific gene expression allows for a global assessment of the impact of ethanol on the brain. The impact of the ethanol binge drinking procedure was assessed in both adolescent and young adult groups one day after treatment and 50 days later. There was a significant main effect of AE binge treatment (P28-37, Figure 1A) on neurotransmitter-specific gene expression 24 hours after treatment on P38 (Figure 2C; average reduction of 57%, p<0.0001, F=161.2). However, there was not a significant main effect of binge ethanol treatment during adulthood (P88-97) on neurotransmitter-specific gene expression 24 hours after treatment (Figure 2D; p = 0.39, F=0.74). AE binge enhanced the developmental downregulation of neurotransmitter-specific genes. A significant main effect of AE treatment was observed (p<0.0001, F=650.9), with a larger percent reduction than controls (73% reduction vs. 57% in controls). Of the 73 neurotransmitter-specific genes measured in the array, 58 were persistently reduced by binge ethanol treatment at P88 (Table 1, *p<0.05, **p<0.01 t-test, P38 control vs P88 ethanol), and 31 after Bonferroni correction for multiple comparisons (Table 1, §p<0.05 Bonferroni post-test). Regarding the 15 cholinergic genes, expression in young adult controls (P88) averaged 37% (range 23-75%) of control adolescent (P38) levels with no single gene significantly decreased (Table 1). AE binge treatment significantly altered cholinergic genes. Cholinergic gene expression in young adults (P88) that received AE binge averaged 20% (range 14-45%) of control P38 levels, with 11 of the 15 cholinergic genes showing significant reductions (Table 1, §p<0.05 Bonferroni post-test). Adults (P148) 50 days following an earlier adulthood ethanol binge (P88-P97, Figure 1) also showed signs of an enhanced reduction in neurotransmitter-specific gene expression (Table 2; p<0.0001, F=40.18). However, the reduction was much less than that found in the adolescent binge treated group (average reduction of 37% vs 73% reduction following AE binge). Also, only two individual genes were suppressed persistently at P148 following young adult binge ethanol treatment (P88-P97): CCKα and Galantin receptor 2 (Table 2; 56% and 55% reduction respectively, *p<0.05 t-test). The statistical significance of these two reductions was lost after Bonferroni correction for multiple comparisons (Table 2). These findings indicate that adolescent binge ethanol treatment causes more pronounced and persistent decreases in brain neurotransmitter-specific gene expression than young adult binge ethanol treatment.
Expression levels of most neurotransmitter-specific genes declined during late adolescence to early adulthood (P38-P88), with AE binge treatment enhancing this developmental downregulation. Interestingly, AE treatment altered the developmental trajectories of different genes in different patterns, suggesting unique responses of each gene (Fig. 3(r), Fig. 4). Dopamine receptor 4 (D4DR) mRNA was decreased about 71% on P38 following adolescent binge treatment (Fig. 3, Table 1, *p<0.05, t-test of normalized ΔCt values). Interestingly, D4DR underwent a 70% developmental decline between P38 and P88, whereas binge ethanol treated animals showed no additional developmental reduction, i.e. expression on P38 and P88 following binge ethanol treatment were both 70% below P38 control levels (Table 1, Fig. 3). Since dopamine circuitry continues to develop during adolescence in frontal cortex we used immunohistochemistry (IHC) to assess D4DR expression. The orbitofrontal cortex (OFC) of adolescent rats showed strong punctate D4DR+IR that was 44% less in the OFC of P38 binge ethanol treated mice compared to P38 adolescent control mice (Figure 3, *p<0.05). Since the developmental decrease in D4DR mRNA mimicked the AE induced decrease and no developmental change occurred in ethanol treated animals, D4DR gene expression was similar in P88 controls and P88 ethanol treated animals (Figure 3A). Both punctate and somatic D4DR+IR staining were observed in adults, suggesting developmental regulation of D4DR subcellular localization (Figure 3). There was no effect of adult ethanol binge treatment (P88-P97) on D4DR gene expression either immediately (P98) or after 50 days (P148). Thus, D4DR development is disrupted by adolescent binge ethanol treatment, although expression levels in adults appear normal.
Other patterns of AE-induced neurotransmitter-specific gene responses involve persistent changes in adulthood, but varied adolescent brain responses (Fig. 4, Table 1). Although AE decreased expression in general, gene specific reductions in expression levels were often not statistically significant at P38. Following ethanol treatment, most genes followed a decreasing developmental trajectory similar to that found in controls (Fig. 4 and Table 1). Gastrin releasing peptide receptor, Grpr, was unique among the transmitter genes studied, in that its expression did not change during adolescence/early adulthood (P38-P88) or during adulthood (P98-P148) (Figure 2). Neither was Grpr expression altered 24 hours following adolescent binge ethanol, or at all (24h or 50 days) following adult binge ethanol treatment (Tables 1 and 2). However, in adults (P88) 50 days after AE binge treatment, Grpr expression was 55% of P88 controls (†p<0.05, t-test, P88 control vs P88 ethanol group), suggesting a unique adolescent-specific ethanol effect. Other mRNAs that were not markedly altered on P38 just after AE binge treatment, but showed statistically significant reductions 50 days after AE treatment (P88) were cholecystokinin α and β (CCKα and CCKβ), and the glycine receptor α1 subunit (†p<0.05, t-test of normalized ΔCt values, P88 control vs P88 ethanol group). The pattern of developmental expression for these genes is illustrated by the peptide receptor CCKβ (Figure 4). AE binge ethanol caused a 45% decrease in CCKβR mRNA 24 hours after the end of the binge. Control animals underwent a 55% reduction in CCKβR mRNA from P38 to P88 (*p<0.05) while, ethanol treated animals underwent an additional 37% reduction in gene expression from P38 to P88, resulting in an overall 82% significant reduction from P38 control levels (§ p<0.05 Bonferroni post-test vs P38 control, †p<0.05 t-test vs P88 control). Catechol-O-methyltransferase (COMT) showed a particularly interesting pattern of altered developmental trajectory (Figure 4). Adolescent binge ethanol did not cause any change COMT gene expression at P38. Although there was a non-significant developmental reduction in controls (29% reduction, p=0.24), adolescent binge treatment resulted in a 64% significant decrease in COMT mRNA expression in P88 adults (Table 1, Fig 4 **p<0.01 t-test). Similar developmental effects were observed with two other genes associated with the dopaminergic system, dopamine receptors D1a and D2. Thus, although alcohol showed no immediate effect on COMT, dopamine D1a or D2 expression, the developmental trajectories were altered resulting in persistent reduced expression in adults. Therefore, adolescent binge ethanol treatment alters neurotransmitter-specific gene developmental trajectories, contributing to persistent changes in adult neurotransmitter gene expression.
Young Adult Behavior Following Adolescent Binge Ethanol Treatment: Reversal Learning Deficit
To investigate global behaviors in adults following AE binge treatment (P28-37), we assessed young adult mice (P63-P72; Figure 1) for locomotor open field activity, Morris Water Maze acquisition and reversal learning, as well as prepulse inhibition and anxiety-like behavior in the elevated plus maze. In the water maze, mice from both groups easily learned with a visual cue on the platform (Figure S2A, <10 seconds). Similarly, in the hidden platform task, both groups learned in parallel over four days of testing, as indicated by reduced time to find the hidden platform (Figure S2B). Results from a probe trial also supported equivalent learning ability in both groups, since both spent significantly more of their search time in the correct quadrant, and less than 20% in the incorrect quadrants, with no difference between the treatment groups (Figure S2C). Both groups of mice received 3 days of reversal learning training with the platform in the opposite quadrant, with both groups showing a modest reversal (Figure S2D). The reversal learning probe trial indicated significant differences between young adult control and young adults following adolescent binge ethanol treatment (Figure 5).
Figure 5.
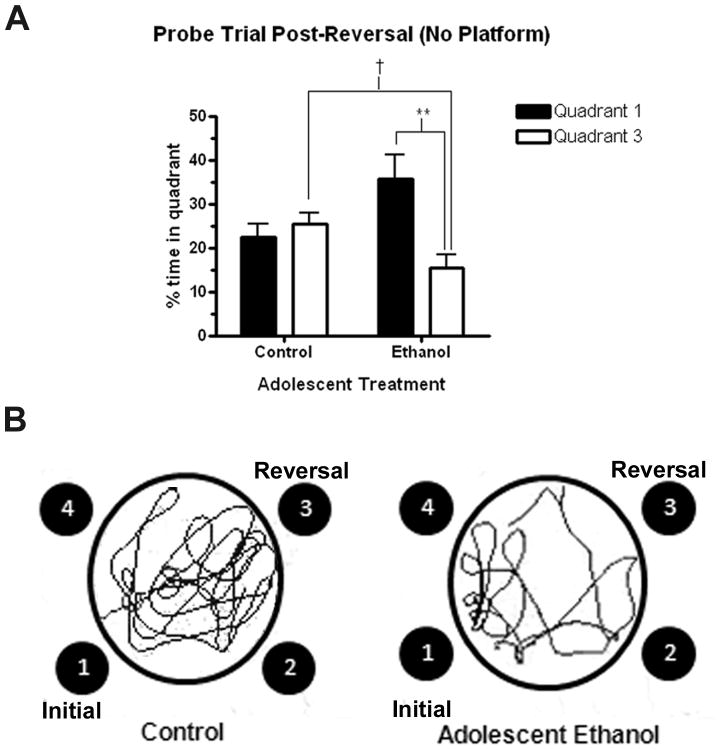
Mice that received ethanol binge during adolescence had a deficit in reversal learning as adults. Following acquisition of reversal learning, mice were tested in a 60 second probe trial in the absence of the hidden platform. (A) Control mice spent a nearly equal amount time in the initial learning quadrant (Quadrant 1, 22.4%±3.02) and the reversal learning quadrant (Quadrant 3, 25.5%±2.5). Adult mice were treated with ethanol during adolescence spent more than twice as much time in the quadrant where the platform was during initial learning (Quadrant 1, 35.6%±5.5) than where the platform was during reversal learning (Quadrant 3, 15.5%±7.7) (**p < 0.01, t-test). Also, control mice spent significantly more time in the reversal quadrant (Quadrant 3, 25.5%±2.5) than the mice that received alcohol during adolescence (15.5%±7.7) († p<0.05, t test). Data is presented as mean ± SEM. N = 7 Control, 7 Ethanol. (B) Representative tracings of the reversal probe trial from one animal in each treatment group, depicting the increased time spent by control mice in the reversal learning quadrant (Quadrant 3) compared to adolescent ethanol treated mice, which spent more time in the initial learning quadrant (Quadrant 1).
Following reversal training, the control mice no longer demonstrated a strong preference for the original platform location in quadrant 1, spending roughly an equal amount of time also searching quadrant 3, where the platform had been re-located during reversal. In contrast, the ethanol-treated mice significantly spent more than twice as much time in quadrant 1 (35.6%) than in the quadrant with the new platform location (15.5%), demonstrating a perseverative response pattern (Figure 5A; **p<0.01). Further, the ethanol-treated group spent significantly less time than the control mice searching quadrant 3, where the platform had been re-located during reversal († p<0.05, compared to controls). These findings demonstrate adolescent binge ethanol treatment of C57BL/6 mice does not cause young adult spatial learning deficits, but disrupts reversal learning.
We tested a spectrum of other behaviors to determine if adolescent binge ethanol caused other differences in adult mice. Behaviors associated with sensorimotor gating, locomotor activity, and anxiety showed no significant differences between groups. Prepulse inhibition is a measure of sensorimotor gating. Adult mice that received alcohol during adolescence showed nonsignificant reductions in prepulse inhibition across all the prepulse decibels tested (Figure S3, 2-way ANOVA: p=0.11, F=2.931). Reductions at two of the prepulse decibels measured (78 and 90dB) reached statistical significance using a t-test (*p<0.05). Locomotor activity in the open field was the same in both groups across a one-hour period (Figure S3B, 2-way ANOVA: p =0.27, F=1.30). In the elevated plus maze, a measure of anxiety-like behavior, both groups spent similar amounts of time in the open arms (Figure S3C, control: 35.9 sec ± 8.3, ethanol 39.4 sec ± 4.5, p=0.73, t test) and the closed arms (control: 203.6 ± 16.1, ethanol 210.7 ± 7.1, p=0.72, t test). Center time in the open field, another measure of anxiety-like behavior and activity in a novel environment, was also unchanged between groups (Figure S3D, 2-way ANOVA: p=0.94, F=0.005). Thus, C57BL/6 mice are behaviorally resilient and adolescent binge drinking procedures do not alter global locomotor, spatial learning, sensorimotor gating or anxiety assessments in young adults. Only the reversal learning probe trial indicated persistent young adult dysfunction following adolescent binge drinking.
Adolescent Binge Ethanol Reduces MRI Adult Basal Forebrain and Olfactory Bulb Brain Regional Volume
To investigate adult brain structure following adolescent binge ethanol treatment, we performed MRI on postmortem adult mouse brains. Twenty brain regions were initially segmented in an automatic fashion to identify candidate regions for further manual segmentation (Fig. 6, Fig. 7, Fig. 8, Fig. 9). Although not statistically different, mean total brain volume and neocortex volume were 1-2% lower in AE treatment groups, being 441mm3 vs 435 mm3 and 152 mm3 vs 148 mm3 in young adult control and adolescent alcohol treated animals (both P79) respectively. Further segmentation of the frontal cortex (from the genu of the corpus callosum forward) found no differences in volume (not shown). Some brain regions such as the globus pallidus and internal capsule tended to show increased mean volumes in adults following adolescent binge alcohol treatment that were not statistically significant. Four brain regions in adolescent binge treated young adults showed >4% decreases in mean volume, basal forebrain−4.4% (p<0.02), olfactory bulb −6.5% (p<0.046), anterior commissure −5.1% (p<0.04) and hippocampus −4.5% (p=0.11) (Table 3). These findings suggest that adolescent binge alcohol treatment has brain region specific effects.
Figure 6.
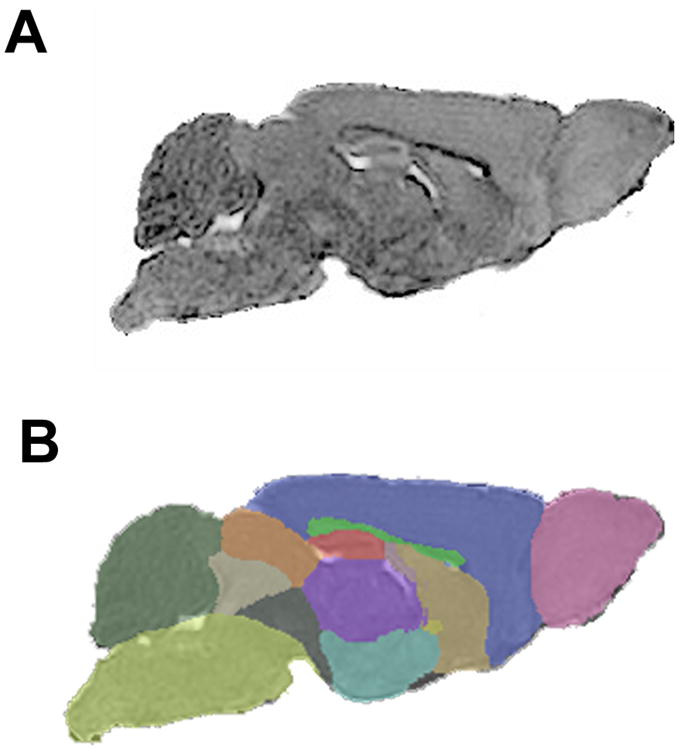
Representative automatic segmentation of structural MRI. (A) Representative structural MRI of a control mouse. Mean diffusivity images were computed from reconstructed diffusion tensor data. (B) Brain regions were segmented in an automated fashion in order to identify candidate regions for further manual segmentation. Key: pink-olfactory bulb, blue-neocortex, brown-basal forebrain, yellow-anterior commissure, bright green-corpus callosum, red-hippocampus, purple-thalamus, turquoise-hypothalamus, dark gray-rest of midbrain, light gray- central gray, orange-superior colliculi, dark green-cerebellum, yellow green-brain stem.
Figure 7.
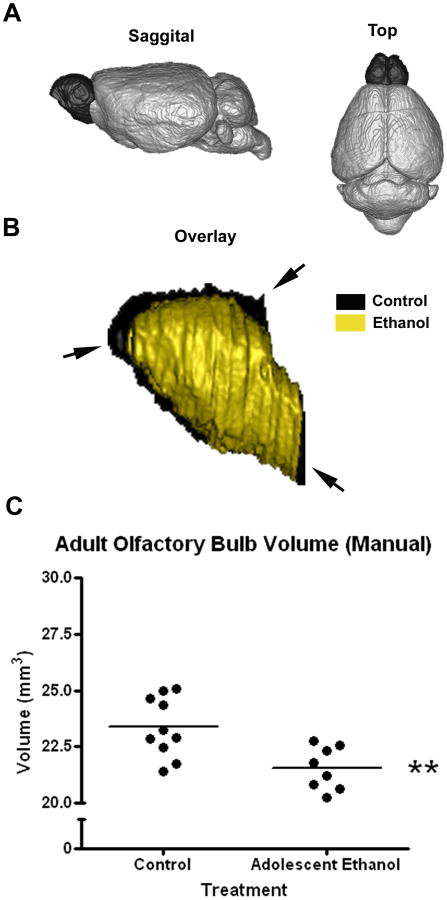
Adolescent ethanol binge causes a reduction in the volume of the olfactory bulb in adults. Adolescent mice received either water or ethanol (5g/kg) once a day for ten days (P28-37). Postmortem MRI was performed on adults (P79). Following automatic segmentation, manual segmentation of the olfactory was performed by blinded investigators. (A) 3D representation of the brain showing large relative size of the olfactory bulb in the rodent brain (darkened). (B) Overlay of 3D renderings of representative control and ethanol treated adult olfactory bulbs. Arrowheads highlight volume differences between control (black) and ethanol treated (white) groups (C) Adult olfactory bulb volume quantification following manual correction. Adult mice that received binge ethanol treatment during adolescence showed a 7.8% reduction in the olfactory bulb volume (Control: 23.37 ± 0.42 mm3; Ethanol: 21.54 ± 0.33 mm3; **p<0.005; N=10 Control, 8 Ethanol).
Figure 8.
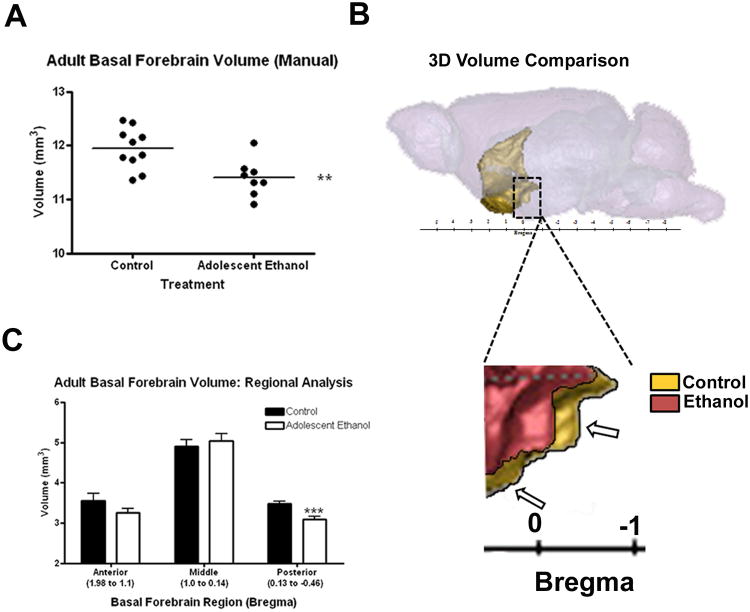
Adolescent ethanol binge causes a reduction in the volume of the basal forebrain/medial septum brain region in adults. Adolescent mice received either water or ethanol (5g/kg) once a day for ten days (P28-37). Postmortem MRI was performed on adults (P79), the basal forebrain region was manually segmented by blinded investigators, and its volume was measured. (A) 3D schematic of the location of the basal forebrain/medial septum region from the top, sagittal, and front/coronal perspectives. (B) Basal forebrain volumes from manual segmentation. Adult mice that previously received ethanol showed a 4.6% reduction in the volume of the basal forebrain/medial septum region (Control: 11.95 ± 0.12 mm3; Ethanol: 11.40 ± 0.12 mm3, **p<0.01; N=10 Control, 8 Ethanol). (C) A regional analysis of the basal forebrain volume shows that the majority of the volume reduction (11% reduced from controls) was to the posterior segment of the basal forebrain (bregma + 0.13 to −0.46), ***p<0.001. (D) sagittal 3D rendering of two representative cases showing volume reduction in adult mice that received adolescent binge ethanol.
Figure 9.
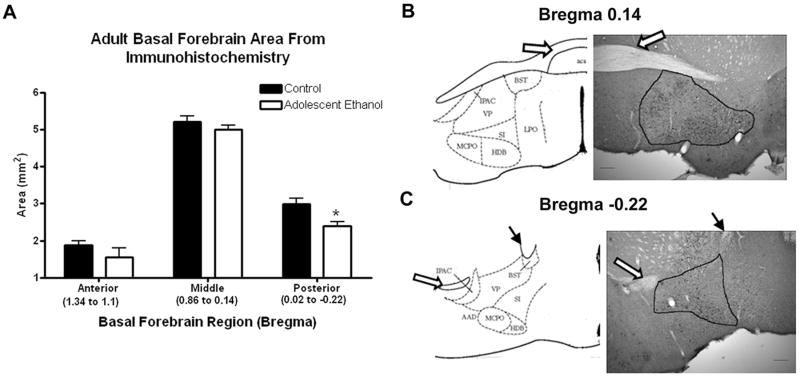
Basal forebrain nuclei area reduction in adults following adolescent ethanol treatment. In order to confirm MRI volume reductions, the area of the basal forebrain region nuclei was measured microscopically on sections immunolabeled for parvalbumin using bioquant software, in three different regions defined by their distances from bregma (1.34 to 1.1 mm, 0.86 to 0.14 mm, and 0.02 to −0.22 mm). Area measures were taken on up to three different immunolabeled sections per mouse, N=4-7 mice for each region. (A) The basal forebrain area was significantly reduced in the posterior bregma of the basal forebrain: 20% difference, 0.02 to −0.22 mm, *p<0.05; N=5 Control and 5 Ethanol mice. Basal forebrain areas measured at bregma 0.14mm (B) and −0.22mm (C) are shown from representative sections. The anterior commissure (open arrow) and internal capsule (closed arrow) were used as landmarks and are labeled on the atlas and image to help with orientation. Key: VP-ventral pallidum, SI-substantia innominata, HDB-horizontal diagonal band, MCPO-magnocellular preoptic nucleus, LPO-lateral preoptic area, IPAC-interstitial nucleus of the posterior limb of the anterior commissure, BST-bed nucleus of stria terminalis, AAD-anterior amygdaloid area dorsal, aca-anterior commissure anterior.
Table 3.
Adult brain MRI volumes using automatic segmentation following adolescent binge ethanol.
| Brain Region | Control (mm3) | Ethanol (mm3) | % Change | p value |
|---|---|---|---|---|
| Hippocampus | 24.6 | 23.5 | −4.5 | 0.11 |
| Corpus Callosum & External Capsule | 13.2 | 13.2 | 0.0 | 0.95 |
| Caudate & Putamen | 22.3 | 22.1 | −0.9 | 0.69 |
| Anterior Commissure | 1.2 | 1.1 | −5.1* | 0.04 |
| Globus Pallidus | 2.4 | 2.6 | 6.6 | 0.39 |
| Internal capsule | 1.9 | 2.1 | 9.9 | 0.45 |
| Thalamus | 24.6 | 24.5 | −0.4 | 0.91 |
| Cerebellum | 53.0 | 54.1 | 2.1 | 0.42 |
| Superior Colliculi | 8.30 | 8.26 | −0.5 | 0.90 |
| Ventricle | 1.2 | 1.2 | 0.0 | 0.96 |
| Hypothalamus | 10.2 | 10.0 | −1.1 | 0.84 |
| Inferior colliculi | 5.1 | 5.2 | 2.8 | 0.52 |
| Central Gray | 4.0 | 3.9 | −2.0 | 0.63 |
| Neocortex | 151.5 | 147.5 | −2.6 | 0.23 |
| Amygdala | 12.6 | 12.7 | 0.4 | 0.91 |
| Olfactory Bulb | 24.6 | 23.0 | −6.5* | 0.046 |
| Brain Stem | 55.4 | 55.8 | 0.7 | 0.85 |
| Rest of Midbrain | 11.13 | 11.15 | 0.2 | 0.94 |
| Basal Forebrain & Septum | 11.9 | 11.4 | −4.4* | 0.02 |
| Fimbria | 1.8 | 1.9 | 4.9 | 0.55 |
| Total Volume | 440.9 | 435.2 | −1.3 | 0.37 |
The effect of adolescent binge ethanol on adult regional brain volume using Magnetic Resonance Imaging (MRI). Postmortem brains were scanned using a 9.4T MRI scanner. Brain regions were segmented on the images using an automatic segmentation protocol. Volumes of the individual structures (mm3) in control and ethanol groups, percent change from control, and the p value (t-test) are shown. The anterior commissure, olfactory bulb, hippocampus and basal forebrain & septum (bold) showed changes that were investigated further with manual segmentation (*p<0.05). Since this was an initial screening, a statistical correction for multiple comparisons was not performed, so that potential regions of interest would not be discarded prior to a more thorough manual segmentation. Therefore displayed p values may contain type II errors, resulting in false positives.
Basal forebrain, olfactory bulb, anterior commissure, and hippocampus were manually segmented by blinded investigators to refine segmentation volumes. Manual segmentation reduced variation and mean differences between groups. For example, control hippocampal automated segmentation volumes ranged from 22 to 27.5 mm3 (coefficient of variation = 0.66) compared to manual hippocampal volumes that ranged from 24.5 to 28 mm3 (coefficient of variation = 0.45), likely due to the difficulty in resolving the most anterior and ventral regions of the hippocampus. Both automated and blinded manual segmentation indicated AE treated animals have slightly smaller, but not significantly different hippocampal volumes (Figure S4A). Though automatic segmentation reported significantly smaller anterior commissure in mice that underwent adolescent binge, no significant difference was found when segmented manually (p=0.31), indicating that AE treated animals may have slightly smaller, but not significantly different anterior commissure volumes (FigureS4B). However, manual segmentation of the olfactory bulb (Figure 7) and the basal forebrain/septum (Figure 8) confirmed significant reductions in volume in adult mice that received adolescent binge ethanol treatment. The mouse olfactory bulb is a large frontal brain region representing over 5% of mouse brain volume that is embedded in the skull making automated skull striping and segmentation difficult. Olfactory bulb volume was reduced 6.5% by automated analysis (Table 3) and 7.8% (Figure 7, **p<0.005) by manual segmentation. Overlays show overall olfactory bulb reductions (Figure 7), consistent with binge ethanol treatment induced adolescent olfactory bulb and cortex necrosis (Crews et al., 2000a) and inhibition of adolescent frontal neurogenesis(Crews et al., 2006) found in rat studies.
The basal forebrain is a collection of structures located ventrally to the striatum. It is considered to be the major cholinergic output of the CNS. It includes a group of structures that lie near the bottom of the front of the brain, including the nucleus basalis, diagonal band of Broca, ventral pallidum and medial septal nuclei. These structures are important in the production of acetylcholine, which is then distributed widely throughout the brain. The adult basal forebrain/septum region volume was reduced in the AE treated young adults. Automated analysis found a −4.4% (Table 3; p<0.03) and manual analysis −4.5% decrease in forebrain/septal volume (Figure 8, **p<0.01). Forebrain/septum includes the nucleus basalis, diagonal band of Broca, and medial septal nuclei. The basal forebrain, through widespread projections to cortex, particularly prefrontal cortex, plays an important role in the modulation of cortical activity in association with different behavioral states including sleep, learning and memory (Everitt and Robbins, 1997; Jones, 2004; Sarter et al., 2003; Weinberger, 2003). By integrating images into a 3-dimensional rendering and overlaying forebrain (Figure 8), we show that the posterior forebrain (bregma + 0.13 to −0.46) accounted for most (71%) of the regional volume loss (Figure 8C, 11% reduced from controls, ***p<0.001). To further investigate forebrain structure, histological sections were prepared and histochemistry used to determine cellular composition and forebrain/septum volume. Immunohistochemistry for GABA interneuron subtypes markers calretinin, calbindin, and parvalbumin (PV) did not show visible changes in the density of these neurons. PV+IR neuron density is high in forebrain, allowing visualization of basal forebrain boundaries facilitating assessment of basal forebrain histology area. Area assessments of forebrain with PV+IR sections indicated adolescent binge alcohol treatment reduced basal forebrain area (p<0.05, 2-Way ANOVA, Figure 9). Dividing forebrain into bregma regions as done with MRI indicated histological forebrain area was reduced particularly in the more posterior regions of the basal forebrain from bregma + 0.02 to − 0.22 (Figure 9A, *p<0.05, t-test). Examples of two of the measured regions of interest in the posterior basal forebrain are shown (Figure 9B, C). Reduced histological basal forebrain area supports the observed MRI volume reduction in the posterior basal forebrain. Thus, adolescent binge ethanol treatment results in reduced volume of adult olfactory bulb and basal forebrain.
Adolescent Binge Ethanol Causes a Reduction in the Number of Cholinergic Neurons in the Adult Basal Forebrain
Basal forebrain cellular composition was investigated using IHC. As mentioned above, histochemistry for GABA interneuron markers calretinin, calbindin, and parvalbumin found no marked changes after adolescent alcohol treatment. Similarly, astroglial marker glial fibrillary acidic protein was similar between groups (data not shown). Our gene expression studies found ChAT gene expression was decreased 71% at P38 following adolescent binge treatment (Figure 10). The basal forebrain is rich in cholinergic neurons, critical for learning and memory, that project to multiple brain regions to integrate cortical activity. We used ChAT+IR to identify cholinergic neurons (Figure 10). The density of ChAT+IR neurons was decreased 7.5% from 166 to 153 cells/mm2 in the Ch4 region (see Methods) of the basal forebrain (Figure 10A, *p<0.05). This change is similar in magnitude to the observed volumetric reduction in these mice, suggesting that cholinergic neuron loss contributes to the reduction in basal forebrain volume. Thus, adolescent binge drinking reduced density of ChAT+IR neurons in the adult basal forebrain.
Figure 10.
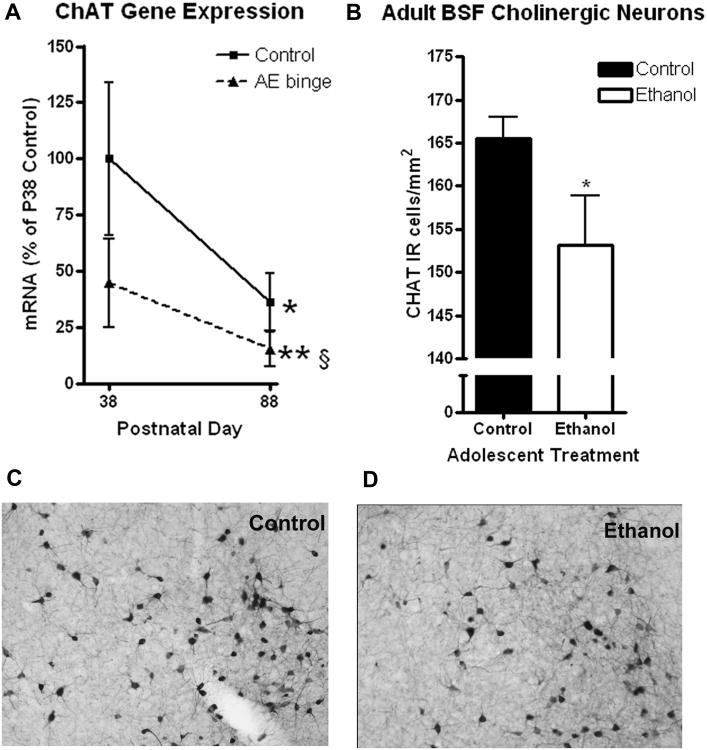
Adolescent Binge Ethanol Treatment Reduces Choline Acetyltransferase (ChAT) gene expression and ChAT-immunoreactivity. Acetylcholine neurons were identified by immunohistochemistry for choline acetyltransferase (ChAT). (A) ChAT mRNA levels changed significantly across development. ChAT mRNA in adolescent mice (P38 – 100%) decreased during maturation to P88 (64% reduction, *p<0.05) in control animals. ChAT mRNA expression 24 hours following adolescent ethanol binge treatment showed a 55% reduction (P38). Fifty days after the adolescent ethanol binge ChAT mRNA levels were reduced 85% (**p<0.01 vs P38 control). (B) Reduced ChAT-immunoreactive (ChAT+IR) neurons in adults. ChAT+IR neurons were counted in the posterior regions of the basal forebrain (bregma 0.14mm to −0.22mm), where the greatest volume changes were observed. This included the horizontal diagonal band, substantia innominata, ventral pallidum, and magnocellular preoptic nucleus. Significant reductions in the number of ChAT+IR neurons per area were observed (7.5% reduction, *p<0.05, N=10 Control, 8 Ethanol). Representative images from an adult controls (C) and adults following adolescent binge ethanol treatment (D) at the same bregma (0.14mm) showing differences in ChAT+IR.
Discussion
Changes in adult neurotransmitter-specific gene expression, postmortem MRI brain region volumes, forebrain histological area, cholinergic neuron density and reversal learning converge to support the conclusion that adolescent binge drinking in mice persistently alters the adult brain. We found greater levels of neurotransmitter-specific gene expression in adolescent brain than adult brain, consistent with other studies finding that dopaminergic, cholinergic and other key neurotransmitters peak during adolescence (Andersen et al., 2000; Crews et al., 2007; Spear, 2000; Tarazi and Baldessarini, 2000). Adulthood changes in neurotransmitter gene expression over 50 days were not as large as adolescent maturational changes in neurotransmitter gene expression, but were statistically evident, suggesting neurotransmitter maturation continues into adulthood. Binge ethanol treatment in adolescence dramatically reduced expression of many neurotransmitter-specific genes early after treatment (P38, Figure 2). None of the neurotransmitter-specific genes showed increased expression following ethanol treatment of adolescents. The greatest reductions were found with the combination of binge AE treatment followed by development to young adulthood, P88. The expression of several genes implicated in mental illnesses were decreased by adolescent binge ethanol treatment. Binge ethanol during adolescence, but not adulthood, decreased DA-D4R mRNA and orbital frontal cortical D4DR+IR in adolescence. D4 receptors bind atypical antipsychotics with higher affinity than other dopamine receptors (Van Tol et al., 1991), and are implicated in novelty seeking, substance abuse (Lusher et al., 2001; Munafo et al., 2008) and attention deficit with hyperactivity disorder (Bellgrove and Mattingley, 2008). Thus, adolescent binge-drinking alters D4 dopamine receptors, particularly in the orbital frontal cortex.
GRPR (gastrin-releasing peptide receptor), COMT, dopamine receptors D1A, and D2 were not altered in adolescence immediately following binge treatment (P38), but after treatment, during young adulthood, mRNA expressions were about half as much as their age matched controls (Table 1, P88 Ethanol). Binge ethanol treatment of adolescent rats has been found to reduce D1 and D2 protein levels in frontal cortex and other brain regions (Pascual et al., 2009) and human alcoholics have fewer brain D2 receptors that are suggested to be a predisposing factor due to persistently low levels in both early and late withdrawal (Volkow et al., 2002). Decreases in dopamine receptors could be associated with decreased motivation (Ernst et al., 2009). GRPR expression, a receptor for gastrin releasing peptide and bombesin, was stable across the ages studied, but adolescent binge treatment reduced adult expression 45% without altering adolescent expression. GRPR is enriched in amygdala and GRPR deficient mice show enhanced fear conditioning with normal spatial learning and no anxiety-like behavior in the water maze and elevated plus maze, respectively (Shumyatsky et al., 2002). Environmental enrichment during adolescence increases GRPR expression and reduces fearfulness (Qian et al., 2008), suggesting that environmental amygdala plasticity during adolescence is in part reflected in GRPR expression and lifelong sensitivity to amygdala fear conditioning. COMT polymorphisms have been implicated in many disorders including addiction (Enoch, 2006), schizophrenia (Tan et al., 2007), aggression (Volavka et al., 2004), and Alzheimer's disease (Serretti et al., 2007). These findings indicate that AE exposure alters expression of neurotransmitter genes in the young adult brain that are associated with psychopathology.
Heavy episodic drinking within the past 2 weeks is reported by 12% of 8th graders, 22% of 10th graders, 28% of 12th grade seniors and 44% of college students (Johnston et al., 2004; Masten et al., 2008; Wechsler et al., 1995; Windle et al., 2008). In our model of adolescent binge drinking in C57BL/6 mice the 5 gm/kg/day dose is less than mice from this strain drink by choice (>15 gm/kg/day) (Crews et al., 2004). The high peak blood ethanol levels (288 mg/dL) in this model are appropriate since alcohol dependent human adolescents report approximately 13 drinks per drinking episode (Deas et al., 2000), consistent with blood ethanol levels of 250-299 mg/dL, 50-65 mM (Jones and Holmgren, 2009). C57BL/6 mice metabolize ethanol approximately 3.5 times faster than humans (≈9.7mmole/kg/hr in mice vs ≈2.7mmole/kg/hr in humans), suggesting that elevated blood alcohol levels persist longer in humans than mice (Thurman et al., 1982; Bradford et al., 2007). Our adolescent binge ethanol treatment followed by assessments in young adults mimics the “young adult alcohol dependent subtype,” the most common alcohol dependence subtype characterized by heavily drinking during adolescence -young adulthood, and maturation out of dependence (Jacob et al., 2005; Moss et al., 2007). Although it is of interest to distinguish adolescent from adult responses, since human adolescents represent the majority of binge drinkers, understanding the persistent effects of adolescent binge ethanol on the adult brain may be the most important for human health considerations. Therefore, though we compared the effects of binge ethanol during adolescence with that of adults on neurotransmitter receptor genes, we focused the remainder of our analyses on the persistent effects of the adolescent binge. Our model replicates rat models of binge drinking that did not demonstrate changes in locomotor activity (Slawecki et al., 2001), water maze spatial learning (Schulteis et al., 2008; White et al., 2002) or elevated plus maze performance (White et al., 2002). We found alterations in reversal learning in young adult mice after adolescent binge treatment, consistent with an adult rat binge drinking model (Obernier et al., 2002). Reversal learning deficits have been observed in human alcoholics (C. B. Fortier et al., 2008), cocaine addicts (Fortier et al., 2008; Stalnaker et al., 2009) and neurodegenerative diseases (Freedman and Oscar-Berman, 1989; Oscar-Berman and Zola-Morgan, 1980). Frontal cortex, where we found altered adolescent D4DR expression, and marked rat adolescent binge brain damage (Crews et al., 2000a), is implicated in drug dependence and reversal learning deficits (Schoenbaum et al., 2009). Reversal learning deficits have also been observed in rats and marmosets following lesions of the basal forebrain (Cabrera et al., 2006; Roberts et al., 1992; Tait and Brown, 2008). Our finding suggests that individuals who drink heavily during adolescence may be more likely to have reversal learning deficits, possibly mediated by chemical or structural changes in frontal cortex or basal forebrain.
Alcohol is known to have differential effects on adolescents than adults. Developing neurocircuitry in adolescents likely underlies the increased sensitivity to disruption of working memory and hippocampal function (White and Swartzwelder, 2004), reduced ethanol sedative response compared to adults (Silveri and Spear, 1998), increased neurotoxicity (Crews et al., 2000b) and exaggerated thrill seeking, social and motivational behaviors (Ernst et al., 2006). Human studies using in vivo brain imaging (Pfefferbaum et al., 2009) and post-mortem histology (Harper, 2009) find alcoholics have brain volume reductions compared to age matched controls, including adolescents (De Bellis et al., 2005). Pre-existing conditions (Prescott and Kendler, 1999) and/or binge drinking induced pathology (Stephens and Duka, 2008) could contribute to differences between binge and non-binge drinkers.
To our knowledge, this is the first report of young adult mouse brain MRI following adolescent binge treatment. We found reduced olfactory bulb and basal forebrain volume. Alcoholics have persistent olfactory deficits (Kesslak et al., 1991; Rupp et al., 2003), associated with loss of brain volume and ventricular expansion (Shear et al., 1992). We found a 4-10% volume reduction across basal forebrain regions, comparable in magnitude to human MRI brain regional volume reductions in alcohol use disorder (Makris et al., 2008; Sullivan et al., 2005). Human MRI medial septal/diagonal band volume is negatively correlated with age in alcoholic individuals, with volume reductions being associated with deficits in verbal working memory (Sullivan et al., 2005). Basal forebrain cholinergic neurons project to frontal cortex, hippocampus, and amygdala. These neurons modulate information processing, allowing contextual associated information to exist concurrently in memory with little interference to facilitate reversal plasticity. Proactive interference, previous learning disrupting later learning, is minimized by forebrain acetylcholine. Human fMRI shows basal forebrain activation during resolution of proactive interference tasks in normal individuals but not in alcoholics (De Rosa et al., 2004). Damage to the basal forebrain could alter an individual's ability to resolve proactive interference, altering reversal learning ability. These findings, taken together with our findings suggest persistent basal forebrain dysfunction following adolescent binge drinking that continues through life.
We found decreases in the expression of many cholinergic-specific genes including ChAT, as well as all 5 subtypes of the muscarinic cholinergic receptors measured in young adult mice following adolescent binge treatment. Post-mortem brains from humans with alcohol use disorder have fewer muscarinic receptors and other cholinergic markers in hippocampus (Freund and Ballinger, 1989a; Nordberg et al., 1983) and cerebral cortex (Freund and Ballinger, 1989b). Adolescent binge treatment of rats has been shown to disrupt adult sleep and electrophysiology consistent with altered cholinergic systems (Ehlers and Criado, 2010). In adult rats, 28 weeks of alcohol treatment results in a progressive and persistent loss of 60-80% of multiple induces of forebrain cholinergic neurons (Arendt et al., 1995; Arendt et al., 1988b) that are associated with memory impairments (Arendt et al., 1988a). After 8 weeks of ethanol treatment in adult rats, full recovery of reductions in ChAT activity was observed in the basal forebrain during 4 weeks of abstinence (Arendt et al., 1989). We found reduced forebrain histological area and cholinergic neuron density in young adult mice following adolescent binge treatment for only 10 days, with no evidence of recovery from gene array or immunohistochemistry.
In summary, adolescent binge ethanol treatment of mice reduces young adult neurotransmitter gene expression, particularly cholinergic genes, as well as forebrain MRI volume, histologic area, cholinergic neuron immunohistochemistry, and reversal learning performance.
Supplementary Material
Supplemental Figure 1: Adolescent binge ethanol treatment delayed the adolescent growth spurt. Mice received either water or ethanol (5g/kg) once a day for 10 days during adolescence (P28-37) Mice were weighed on multiple days during the adolescent binge treatment and several days after the treatment. In control mice, an increase in weight occurred from P28 to P32 (15.8 g to 17.5 g, 11%) followed by a plateau in weight from P32-P36 and another increase in weight from P36 to P49 (3.5 g, 19% increase). The adolescent growth spurt was delayed in adolescent ethanol treated mice until P36, resulting in an average weight less than controls from P32 to P36 (*p<0.05 ANOVA with Bonferroni post-tests). By P42, ethanol treated mice recovered and underwent a greater weight gain than controls from P36 to P49 (4.73 g, 31% increase) arriving at weights by P49 that were not significantly different (Control: 21.7 ± 0.4, Ethanol: 20.23 ± 0.4, p>0.05).
Supplemental Figure 2: Morris Water Maze Learning. There were no differences in the ability of both groups of mice to learn the platform location. (A) Mice were first trained to locate the platform with a visual cue placed on the platform. There were no significant differences in learning acquisition between the two treatment groups on any testing day (Day 2, p=0.09 t test) or across days (2-Way ANOVA: p=0.6, F=0.28), however there was significant learning across treatment days in both groups (2-Way ANOVA: p<0.0001, F=55.85; N=10 Control, 8 Ethanol). (B) Initial learning with the hidden platform location over four days. Both treatment groups significantly improved in learning of the location of the hidden platform across four days (2-Way ANOVA: p=0.0006, F=6.82). There were no significant differences between treatment groups (2-Way ANOVA p=0.89, F=0.19; N=10 Control, 8 Ethanol) (C) Learning probe trial. Mice were placed in the tank for 60 seconds with no platform to measure which quadrant in the tank they preferred. Both groups of mice spent significantly more time in Quadrant 1 (*p<0.05 one way-ANOVA, quadrant 1 versus each other quadrant), where the platform was during the hidden platform trials. (2-Way ANOVA: p=0.0001, F=10.43; N=10 Control, 8 Ethanol) (D) Reversal learning acquisition. The hidden platform was moved to Quadrant 3. Both groups of mice significantly improved in learning the new platform location over three days (2-Way ANOVA: p=0.029 F=4.027, N=9 Control, 8 Ethanol). There were no differences during reversal training between the two groups. (2-Way ANOVA: p=0.90, F=0.017).
Supplemental Figure 3: Adult behaviors that were mostly unchanged by adolescent binge ethanol (A) There was a nonsignificant reduction in prepulse inhibition, a measure of sensorimotor gating, across all decibels in adult mice that received alcohol during adolescence (2-way ANOVA: p=0.11, F=2.93). Reductions in prepulse inhibition reached significance at prepulse decibels of 78 and 90dB (*p<0.05, t-test) (B) Locomotor behavior. There were no differences between groups in total distance traveled across the 1 hour testing period (2-way ANOVA: p =0.27, F=1.304) (C) Elevated Plus Maze. There were no differences in time spent in the open arms (control: 35.9 sec ± 8.3, ethanol 39.4 sec ± 4.5, p=0.73, t test) or closed arms (control: 203.6 ± 16.1, ethanol 210.7 ± 7.1, p=0.72, t test) of the elevated plus maze. (D) Center time in the open field, a measure of anxiety-like behavior showed no differences between the two groups (2-way ANOVA: p=0.94, F=0.005). N=10 Control, 8 Ethanol.
Supplemental Figure 4: Comparison of automatic and manual assessments of anterior commissure, hippocampus and olfactory bulb volumes. Mice were treated with either water or alcohol (5g/kg) for ten days during adolescence (P28-37). Postmortem MRI was performed on adults (P79). Automatic segmentation of brain regions and volume analysis reported possible volume reductions in the hippocampus, anterior commissure and olfactory bulb. These changes not confirmed when the brain regions were manually segmented by blinded investigators using ITK-SNAP software. (A) Hippocampal volumes. Automatic segmentation showed a nonsignificant reduction (4.5%) in adult mice that received ethanol during adolescence (Control: 24.64 ± 0.51mm3, N=10; Ethanol: 23.52 ± 0.39mm3, N=8, p=0.11). There was no significant reduction found following manual segmentation (Control: 26.87 ± 0.38 mm3; Ethanol: 26.22 ± 0.40 mm3, p = 0.26) (B) Anterior Commissure volume automatic segmentation showed a 5.1% reduction in adult mice that received ethanol during adolescence that reached significance (Control: 1.168 ± 0.018mm3; Ethanol: 1.113 ± 0.018mm3,* p<0.05). This was not confirmed by automatic segmentation (p=0.31) (C) Olfactory Bulb volume automatic segmentation showed a 6.5% reduction that was not confirmed by automatic segmentation. N=10 Control, 8 Ethanol.
Acknowledgments
The authors wish to thank the National Institute of Alcohol Abuse and Alcoholism (AA06069; AA011605, AA018051, AA020023, AA020022), the UNC-Bowles Center for Alcohol Studies and the UNC Neurodevelopmental Disorders Research Center (NINDS R42 NS059095, NIDA P01 DA022446-01) for support. Behavioral tests were conducted in the Mouse Behavioral Phenotyping Laboratory of the Neurodevelopmental Disorders Research Center with Sherl Moy, supported by NICHD grant P30 HD03110. We also thank Shriya Soora and Diana Lotito for technical support.
References
- Andersen SL, Thompson AT, Rutstein M, Hostetter JC, Teicher MH. Dopamine receptor pruning in prefrontal cortex during the periadolescent period in rats. Synapse. 2000;37(2):167–9. doi: 10.1002/1098-2396(200008)37:2<167::AID-SYN11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Marchbanks RM, Schugens MM, Sinden J, Lantos PL, Gray JA. Cholinergic system and memory in the rat: Effects of chronic ethanol, embryonic basal forebrain projection system. Neuroscience. 1989;33(4):435–462. doi: 10.1016/0306-4522(89)90397-7. [DOI] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Sinden J, Schugens MM, Marchbanks RM, Lantos PL, Gray JA. Cholinergic-rich brain transplants reverse alcohol-induced memory deficits. Nature. 1988a;332(6163):448–50. doi: 10.1038/332448a0. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bruckner MK, Magliusi S, Krell T. Degeneration of rat cholinergic basal forebrain neurons and reactive changes in nerve growth factor expression after chronic neurotoxic injury-I. Degeneration and plastic response of basal forebrain neurons. Neuroscience. 1995;65(3):633–645. doi: 10.1016/0306-4522(94)00526-b. [DOI] [PubMed] [Google Scholar]
- Arendt T, Henning D, Gray JA, Marchbanks R. Loss of neurons in the rat basal forebrain cholinergic projection system after prolonged intake of ethanol. Brain Res Bull. 1988b;21(4):563–9. doi: 10.1016/0361-9230(88)90193-1. [DOI] [PubMed] [Google Scholar]
- Bellgrove MA, Mattingley JB. Molecular genetics of attention. Ann N Y Acad Sci. 2008;1129:200–12. doi: 10.1196/annals.1417.013. [DOI] [PubMed] [Google Scholar]
- Blakemore SJ, Choudhury S. Development of the adolescent brain: implications for executive function and social cognition. J Child Psychol Psychiatry. 2006;47(3-4):296–312. doi: 10.1111/j.1469-7610.2006.01611.x. [DOI] [PubMed] [Google Scholar]
- Cabrera SM, Chavez CM, Corley SR, Kitto MR, Butt AE. Selective lesions of the nucleus basalis magnocellularis impair cognitive flexibility. Behav Neurosci. 2006;120(2):298–306. doi: 10.1037/0735-7044.120.2.298. [DOI] [PubMed] [Google Scholar]
- Coleman LG, Jr, Jarskog LF, Moy SS, Crews FT. Deficits in adult prefrontal cortex neurons and behavior following early post-natal NMDA antagonist treatment. Pharmacol Biochem Behav. 2009;93(3):322–30. doi: 10.1016/j.pbb.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835(1):18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav. 2007;86(2):189–99. doi: 10.1016/j.pbb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, 3rd, Knapp DJ. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res. 2000a;24(11):1712–23. [PubMed] [Google Scholar]
- Crews FT, Braun CJ, S RC, III, Knapp DJ. Binge ethanol causes differential brain damage in young adolescent rats compared to adult rats. Alcoholism: Clinical and Experimental Research. 2000b;24(11):1712–1723. [PubMed] [Google Scholar]
- Crews FT, Mdzinarishvili A, Kim D, He J, Nixon K. Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience. 2006;137(2):437–45. doi: 10.1016/j.neuroscience.2005.08.090. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME. Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol. 2004;33(1):63–71. doi: 10.1016/j.alcohol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Narasimhan A, Thatcher DL, Keshavan MS, Soloff P, Clark DB. Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol Clin Exp Res. 2005;29(9):1590–600. doi: 10.1097/01.alc.0000179368.87886.76. [DOI] [PubMed] [Google Scholar]
- De Rosa E, Desmond JE, Anderson AK, Pfefferbaum A, Sullivan EV. The human basal forebrain integrates the old and the new. Neuron. 2004;41(5):825–37. doi: 10.1016/s0896-6273(04)00080-7. [DOI] [PubMed] [Google Scholar]
- Deas D, Riggs P, Langenbucher J, Goldman M, Brown S. Adolescents are not adults: developmental considerations in alcohol users. Alcohol Clin Exp Res. 2000;24(2):232–7. [PubMed] [Google Scholar]
- Ehlers CL, Criado JR. Adolescent ethanol exposure: does it produce long-lasting electrophysiological effects? Alcohol. 2010;44(1):27–37. doi: 10.1016/j.alcohol.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch MA. Genetic and environmental influences on the development of alcoholism: resilience vs. risk. Ann N Y Acad Sci. 2006;1094:193–201. doi: 10.1196/annals.1376.019. [DOI] [PubMed] [Google Scholar]
- Ernst M, Pine DS, Hardin M. Triadic model of the neurobiology of motivated behavior in adolescence. Psychol Med. 2006;36(3):299–312. doi: 10.1017/S0033291705005891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Romeo RD, Andersen SL. Neurobiology of the development of motivated behaviors in adolescence: a window into a neural systems model. Pharmacol Biochem Behav. 2009;93(3):199–211. doi: 10.1016/j.pbb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–84. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Fortier CB, Steffen EM, Lafleche G, Venne JR, Disterhoft JF, McGlinchey RE. Delay discrimination and reversal eyeblink classical conditioning in abstinent chronic alcoholics. Neuropsychology. 2008;22(2):196–208. doi: 10.1037/0894-4105.22.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin, Paxinos The Mouse Brain in Stereotaxic Coordinates 2001 [Google Scholar]
- Freedman M, Oscar-Berman M. Spatial and visual learning deficits in Alzheimer's and Parkinson's disease. Brain Cogn. 1989;11(1):114–26. doi: 10.1016/0278-2626(89)90009-2. [DOI] [PubMed] [Google Scholar]
- Freund G, Ballinger WE., Jr Loss of muscarinic and benzodiazepine neuroreceptors from hippocampus of alcohol abusers. Alcohol. 1989a;6(1):23–31. doi: 10.1016/0741-8329(89)90069-4. [DOI] [PubMed] [Google Scholar]
- Freund G, Ballinger WE., Jr Loss of muscarinic cholinergic receptors from the temporal cortex of alcohol abusers. Metab Brain Dis. 1989b;4(2):121–41. doi: 10.1007/BF00999390. [DOI] [PubMed] [Google Scholar]
- Giedd JN. Structural magnetic resonance imaging of the adolescent brain. Ann N Y Acad Sci. 2004;1021:77–85. doi: 10.1196/annals.1308.009. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Lenroot RK, Shaw P, Lalonde F, Celano M, White S, Tossell J, Addington A, Gogtay N. Trajectories of anatomic brain development as a phenotype. Novartis Found Symp. 2008;289:101–12. doi: 10.1002/9780470751251.ch9. discussion 112-8, 193-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Woolf NJ, Butcher LL. Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res Bull. 1991;27(6):767–89. doi: 10.1016/0361-9230(91)90209-3. [DOI] [PubMed] [Google Scholar]
- Harper C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009;44(2):136–40. doi: 10.1093/alcalc/agn102. [DOI] [PubMed] [Google Scholar]
- Jacob T, Bucholz KK, Sartor CE, Howell DN, Wood PK. Drinking trajectories from adolescence to the mid-forties among alcohol dependent males. J Stud Alcohol. 2005;66(6):745–55. doi: 10.15288/jsa.2005.66.745. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O'Malley PM, Bachman JG, Schulenberg JE. Monitoring the Future, National Survey Results on Drug Use, 1975-2004. Secondary School Students 2004;1 NIH Pub No. 05-5727. [Google Scholar]
- Jones BE. Activity, modulation and role of basal forebrain cholinergic neurons innervating the cerebral cortex. Prog Brain Res. 2004;145:157–69. doi: 10.1016/S0079-6123(03)45011-5. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Voorn P, Buijs RM, Pool CW, Uylings HB. Development of the dopaminergic innervation in the prefrontal cortex of the rat. J Comp Neurol. 1988;269(1):58–72. doi: 10.1002/cne.902690105. [DOI] [PubMed] [Google Scholar]
- Kesslak JP, Profitt BF, Criswell P. Olfactory function in chronic alcoholics. Percept Mot Skills. 1991;73(2):551–4. doi: 10.2466/pms.1991.73.2.551. [DOI] [PubMed] [Google Scholar]
- Kostovic I. Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life. Prog Brain Res. 1990;85:223–39. doi: 10.1016/s0079-6123(08)62682-5. discussion 239-40. [DOI] [PubMed] [Google Scholar]
- Lee J, Jomier J, Aylward S, Tyszka M, Moy S, Lauder J, Styner M. Evaluation of atlas based mouse brain segmentation. SPIE Medical Imaging 2009 Image Processing. 2009;7259:137–146. doi: 10.1117/12.812762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, McCoy MK, Harms AS, Ruhn KA, Gold SJ, Tansey MG. Regulator of G-protein signaling 10 promotes dopaminergic neuron survival via regulation of the microglial inflammatory response. J Neurosci. 2008;28(34):8517–28. doi: 10.1523/JNEUROSCI.1806-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusher JM, Chandler C, Ball D. Dopamine D4 receptor gene (DRD4) is associated with Novelty Seeking (NS) and substance abuse: the saga continues. Mol Psychiatry. 2001;6(5):497–9. doi: 10.1038/sj.mp.4000918. [DOI] [PubMed] [Google Scholar]
- Makris N, Oscar-Berman M, Jaffin SK, Hodge SM, Kennedy DN, Caviness VS, Marinkovic K, Breiter HC, Gasic GP, Harris GJ. Decreased volume of the brain reward system in alcoholism. Biol Psychiatry. 2008;64(3):192–202. doi: 10.1016/j.biopsych.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masten AS, Faden VB, Zucker RA, Spear LP. Underage drinking: a developmental framework. Pediatrics. 2008;121(4):S235–51. doi: 10.1542/peds.2007-2243A. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6) Neuroscience. 1983;10(4):1185–201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Moss HB, Chen CM, Yi HY. DSM-IV criteria endorsement patterns in alcohol dependence: relationship to severity. Alcohol Clin Exp Res. 2008;32(2):306–13. doi: 10.1111/j.1530-0277.2007.00582.x. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, Magnuson TR, Crawley JN. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res. 2007;176(1):4–20. doi: 10.1016/j.bbr.2006.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 2006;1089(1):186–94. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Munafo MR, Yalcin B, Willis-Owen SA, Flint J. Association of the dopamine D4 receptor (DRD4) gene and approach-related personality traits: meta-analysis and new data. Biol Psychiatry. 2008;63(2):197–206. doi: 10.1016/j.biopsych.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Larsson C, Perdahl E, Winblad B. Changes in cholinergic activity in human hippocampus following chronic alcohol abuse. Pharmacology, Biochemistry and Behavior. 1983;18:397–400. doi: 10.1016/0091-3057(83)90206-x. [DOI] [PubMed] [Google Scholar]
- O'Malley PM, Johnston LD, Bachman JG. Alcohol use among adolescents. Alcohol Health and Research. 1998;22:85–93. [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, Crews FT. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav. 2002;72(3):521–32. doi: 10.1016/s0091-3057(02)00715-3. [DOI] [PubMed] [Google Scholar]
- Oscar-Berman M, Zola-Morgan SM. Comparative neuropsychology and Korsakoff's syndrome. I.--Spatial and visual reversal learning. Neuropsychologia. 1980;18(4-5):499–512. doi: 10.1016/0028-3932(80)90152-9. [DOI] [PubMed] [Google Scholar]
- Pascual M, Boix J, Felipo V, Guerri C. Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J Neurochem. 2009;108(4):920–31. doi: 10.1111/j.1471-4159.2008.05835.x. [DOI] [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132(2):169–80. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Rosenbloom M, Rohlfing T, Sullivan EV. Degradation of association and projection white matter systems in alcoholism detected with quantitative fiber tracking. Biol Psychiatry. 2009;65(8):680–90. doi: 10.1016/j.biopsych.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, Kendler KS. Age at first drink and risk for alcoholism: a noncausal association. Alcohol Clin Exp Res. 1999;23(1):101–7. [PubMed] [Google Scholar]
- Qian J, Zhou D, Pan F, Liu CX, Wang YW. Effect of environmental enrichment on fearful behavior and gastrin-releasing peptide receptor expression in the amygdala of prenatal stressed rats. J Neurosci Res. 2008;86(13):3011–7. doi: 10.1002/jnr.21736. [DOI] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Robbins TW, Everitt BJ, Muir JL. A specific form of cognitive rigidity following excitotoxic lesions of the basal forebrain in marmosets. Neuroscience. 1992;47(2):251–64. doi: 10.1016/0306-4522(92)90241-s. [DOI] [PubMed] [Google Scholar]
- Rosenberg DR, Lewis DA. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study. Biol Psychiatry. 1994;36(4):272–7. doi: 10.1016/0006-3223(94)90610-6. [DOI] [PubMed] [Google Scholar]
- Rupp CI, Kurz M, Kemmler G, Mair D, Hausmann A, Hinterhuber H, Fleischhacker WW. Reduced olfactory sensitivity, discrimination, and identification in patients with alcohol dependence. Alcohol Clin Exp Res. 2003;27(3):432–9. doi: 10.1097/01.ALC.0000057945.57330.2C. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP, Givens B. Attentional functions of cortical cholinergic inputs: what does it mean for learning and memory? Neurobiol Learn Mem. 2003;80(3):245–56. doi: 10.1016/s1074-7427(03)00070-4. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Roesch MR, Stalnaker TA, Takahashi YK. A new perspective on the role of the orbitofrontal cortex in adaptive behaviour. Nat Rev Neurosci. 2009;10(12):885–92. doi: 10.1038/nrn2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulteis G, Archer C, Tapert SF, Frank LR. Intermittent binge alcohol exposure during the periadolescent period induces spatial working memory deficits in young adult rats. Alcohol. 2008;42(6):459–67. doi: 10.1016/j.alcohol.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serretti A, Olgiati P, De Ronchi D. Genetics of Alzheimer's disease. A rapidly evolving field. J Alzheimers Dis. 2007;12(1):73–92. doi: 10.3233/jad-2007-12108. [DOI] [PubMed] [Google Scholar]
- Shaw P, Greenstein D, Lerch J, Clasen L, Lenroot R, Gogtay N, Evans A, Rapoport J, Giedd J. Intellectual ability and cortical development in children and adolescents. Nature. 2006;440(7084):676–9. doi: 10.1038/nature04513. [DOI] [PubMed] [Google Scholar]
- Shear PK, Butters N, Jernigan TL, DiTraglia GM, Irwin M, Schuckit MA, Cermak LS. Olfactory loss in alcoholics: correlations with cortical and subcortical MRI indices. Alcohol. 1992;9(3):247–255. doi: 10.1016/0741-8329(92)90061-e. [DOI] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, Battey JF, Dulac C, Kandel ER, Bolshakov VY. Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell. 2002;111(6):905–18. doi: 10.1016/s0092-8674(02)01116-9. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. Decreased sensitivity to the hypnotic effects of ethanol early in ontogeny. Alcohol Clin Exp Res. 1998;22(3):670–6. doi: 10.1111/j.1530-0277.1998.tb04310.x. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Betancourt M, Cole M, Ehlers CL. Periadolescent alcohol exposure has lasting effects on adult neurophysiological function in rats. Brain Res Dev Brain Res. 2001;128(1):63–72. doi: 10.1016/s0165-3806(01)00150-x. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24(4):417–63. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Takahashi Y, Roesch MR, Schoenbaum G. Neural substrates of cognitive inflexibility after chronic cocaine exposure. Neuropharmacology. 2009;56(1):63–72. doi: 10.1016/j.neuropharm.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DN, Duka T. Review. Cognitive and emotional consequences of binge drinking: role of amygdala and prefrontal cortex. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3169–79. doi: 10.1098/rstb.2008.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Deshmukh A, De Rosa E, Rosenbloom MJ, Pfefferbaum A. Striatal and forebrain nuclei volumes: contribution to motor function and working memory deficits in alcoholism. Biol Psychiatry. 2005;57(7):768–76. doi: 10.1016/j.biopsych.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Tait DS, Brown VJ. Lesions of the basal forebrain impair reversal learning but not shifting of attentional set in rats. Behav Brain Res. 2008;187(1):100–8. doi: 10.1016/j.bbr.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Tan HY, Callicott JH, Weinberger DR. Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia. Cereb Cortex. 2007;17(1):i171–81. doi: 10.1093/cercor/bhm069. [DOI] [PubMed] [Google Scholar]
- Tarazi FI, Baldessarini RJ. Comparative postnatal development of dopamine D(1), D(2) and D(4) receptors in rat forebrain. Int J Dev Neurosci. 2000;18(1):29–37. doi: 10.1016/s0736-5748(99)00108-2. [DOI] [PubMed] [Google Scholar]
- Van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, Civelli O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature. 1991;350(6319):610–4. doi: 10.1038/350610a0. [DOI] [PubMed] [Google Scholar]
- Volavka J, Bilder R, Nolan K. Catecholamines and aggression: the role of COMT and MAO polymorphisms. Ann N Y Acad Sci. 2004;1036:393–8. doi: 10.1196/annals.1330.023. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Maynard L, Fowler JS, Jayne B, Telang F, Logan J, Ding YS, Gatley SJ, Hitzemann R, Wong C, Pappas N. Effects of alcohol detoxification on dopamine D2 receptors in alcoholics: a preliminary study. Psychiatry Res. 2002;116(3):163–72. doi: 10.1016/s0925-4927(02)00087-2. [DOI] [PubMed] [Google Scholar]
- Wechsler H, Dowdall GW, Davenport A, Castillo S. Correlates of college student binge drinking. Am J Public Health. 1995;85(7):921–6. doi: 10.2105/ajph.85.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger NM. The nucleus basalis and memory codes: auditory cortical plasticity and the induction of specific, associative behavioral memory. Neurobiol Learn Mem. 2003;80(3):268–84. doi: 10.1016/s1074-7427(03)00072-8. [DOI] [PubMed] [Google Scholar]
- White AM, Bae JG, Truesdale MC, Ahmad S, Wilson WA, Swartzwelder HS. Chronic-intermittent ethanol exposure during adolescence prevents normal developmental changes in sensitivity to ethanol-induced motor impairments. Alcohol Clin Exp Res. 2002;26(7):960–8. doi: 10.1097/01.ALC.0000021334.47130.F9. [DOI] [PubMed] [Google Scholar]
- White AM, Swartzwelder HS. Hippocampal function during adolescence: a unique target of ethanol effects. Ann N Y Acad Sci. 2004;1021:206–20. doi: 10.1196/annals.1308.026. [DOI] [PubMed] [Google Scholar]
- Windle M, Spear LP, Fuligni AJ, Angold A, Brown JD, Pine D, Smith GT, Giedd J, Dahl RE. Transitions into underage and problem drinking: developmental processes and mechanisms between 10 and 15 years of age. Pediatrics. 2008;121(4):S273–89. doi: 10.1542/peds.2007-2243C. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Adolescent binge ethanol treatment delayed the adolescent growth spurt. Mice received either water or ethanol (5g/kg) once a day for 10 days during adolescence (P28-37) Mice were weighed on multiple days during the adolescent binge treatment and several days after the treatment. In control mice, an increase in weight occurred from P28 to P32 (15.8 g to 17.5 g, 11%) followed by a plateau in weight from P32-P36 and another increase in weight from P36 to P49 (3.5 g, 19% increase). The adolescent growth spurt was delayed in adolescent ethanol treated mice until P36, resulting in an average weight less than controls from P32 to P36 (*p<0.05 ANOVA with Bonferroni post-tests). By P42, ethanol treated mice recovered and underwent a greater weight gain than controls from P36 to P49 (4.73 g, 31% increase) arriving at weights by P49 that were not significantly different (Control: 21.7 ± 0.4, Ethanol: 20.23 ± 0.4, p>0.05).
Supplemental Figure 2: Morris Water Maze Learning. There were no differences in the ability of both groups of mice to learn the platform location. (A) Mice were first trained to locate the platform with a visual cue placed on the platform. There were no significant differences in learning acquisition between the two treatment groups on any testing day (Day 2, p=0.09 t test) or across days (2-Way ANOVA: p=0.6, F=0.28), however there was significant learning across treatment days in both groups (2-Way ANOVA: p<0.0001, F=55.85; N=10 Control, 8 Ethanol). (B) Initial learning with the hidden platform location over four days. Both treatment groups significantly improved in learning of the location of the hidden platform across four days (2-Way ANOVA: p=0.0006, F=6.82). There were no significant differences between treatment groups (2-Way ANOVA p=0.89, F=0.19; N=10 Control, 8 Ethanol) (C) Learning probe trial. Mice were placed in the tank for 60 seconds with no platform to measure which quadrant in the tank they preferred. Both groups of mice spent significantly more time in Quadrant 1 (*p<0.05 one way-ANOVA, quadrant 1 versus each other quadrant), where the platform was during the hidden platform trials. (2-Way ANOVA: p=0.0001, F=10.43; N=10 Control, 8 Ethanol) (D) Reversal learning acquisition. The hidden platform was moved to Quadrant 3. Both groups of mice significantly improved in learning the new platform location over three days (2-Way ANOVA: p=0.029 F=4.027, N=9 Control, 8 Ethanol). There were no differences during reversal training between the two groups. (2-Way ANOVA: p=0.90, F=0.017).
Supplemental Figure 3: Adult behaviors that were mostly unchanged by adolescent binge ethanol (A) There was a nonsignificant reduction in prepulse inhibition, a measure of sensorimotor gating, across all decibels in adult mice that received alcohol during adolescence (2-way ANOVA: p=0.11, F=2.93). Reductions in prepulse inhibition reached significance at prepulse decibels of 78 and 90dB (*p<0.05, t-test) (B) Locomotor behavior. There were no differences between groups in total distance traveled across the 1 hour testing period (2-way ANOVA: p =0.27, F=1.304) (C) Elevated Plus Maze. There were no differences in time spent in the open arms (control: 35.9 sec ± 8.3, ethanol 39.4 sec ± 4.5, p=0.73, t test) or closed arms (control: 203.6 ± 16.1, ethanol 210.7 ± 7.1, p=0.72, t test) of the elevated plus maze. (D) Center time in the open field, a measure of anxiety-like behavior showed no differences between the two groups (2-way ANOVA: p=0.94, F=0.005). N=10 Control, 8 Ethanol.
Supplemental Figure 4: Comparison of automatic and manual assessments of anterior commissure, hippocampus and olfactory bulb volumes. Mice were treated with either water or alcohol (5g/kg) for ten days during adolescence (P28-37). Postmortem MRI was performed on adults (P79). Automatic segmentation of brain regions and volume analysis reported possible volume reductions in the hippocampus, anterior commissure and olfactory bulb. These changes not confirmed when the brain regions were manually segmented by blinded investigators using ITK-SNAP software. (A) Hippocampal volumes. Automatic segmentation showed a nonsignificant reduction (4.5%) in adult mice that received ethanol during adolescence (Control: 24.64 ± 0.51mm3, N=10; Ethanol: 23.52 ± 0.39mm3, N=8, p=0.11). There was no significant reduction found following manual segmentation (Control: 26.87 ± 0.38 mm3; Ethanol: 26.22 ± 0.40 mm3, p = 0.26) (B) Anterior Commissure volume automatic segmentation showed a 5.1% reduction in adult mice that received ethanol during adolescence that reached significance (Control: 1.168 ± 0.018mm3; Ethanol: 1.113 ± 0.018mm3,* p<0.05). This was not confirmed by automatic segmentation (p=0.31) (C) Olfactory Bulb volume automatic segmentation showed a 6.5% reduction that was not confirmed by automatic segmentation. N=10 Control, 8 Ethanol.