

Abstract
Emerging evidence implicates novel roles for post-translational prenylation [i.e., farnesylation and geranylgeranylation] of various signaling proteins in a variety of cellular functions including hormone secretion, survival and apoptosis. In the context of cellular apoptosis, it has been shown previously that caspase-3 activation, a hallmark of mitochondrial dysregulation, promotes hydrolysis of several key cellular proteins. We report herein that exposure of insulin-secreting INS 832/13 cells or normal rat islets to etoposide leads to significant activation of caspase-3 and subsequent degradation of the common α-subunit of farnesyl/geranylgeranyl transferases [FTase/GGTase]. Furthermore, the above stated signaling steps were prevented by Z-DEVD-FMK, a known inhibitor of caspase-3. In addition, treatment of cell lysates with recombinant caspase-3 also caused FTase/GGTase α-subunit degradation. Moreover, nifedipine, a calcium channel blocker, markedly attenuated etoposide-induced caspase-3 activation, FTase/GGTase α-subunit degradation in INS 832/13 cells and normal rat islets. Further, nifedipine significantly restored etoposide-induced loss in metabolic cell viability in INS 832/13 cells. Based on these findings, we conclude that etoposide induces loss in cell viability by inducing mitochondrial dysfunction, caspase-3 activation and degradation of FTase/GGTase α-subunit. Potential significance of these findings in the context of protein prenylation and β-cell survival are discussed.
Keywords: Etoposide, caspase-3, farnesyl transferase, geranylgeranyl transferase, nifedipine
Introduction
A variety of signaling proteins including small G-proteins [e.g., Ras, Cdc42 and Rac1], the γ-subunits of trimeric G-proteins and nuclear lamins [e.g., lamin-B] undergo a series of post-translational modifications at their C-terminal cysteine residues including prenylation [i.e., farnesylation and geranylgeranylation] and carboxylmethylation [1]. Earlier studies have also demonstrated that such modification steps are necessary for the trafficking of prenylated proteins to appropriate cellular compartments for optimal interaction with and activation of their effector proteins leading to cellular activation [2-5]. Prenylation represents the first modification step in which either a 15-carbon [farnesyl] or a 20-carbon [geranylgeranyl] derivatives of mevalonic acid are incorporated into the substrate proteins. The farnesyl transferase [FTase] catalyzes the transfer of farnesyl groups where as the geranylgeranyl transferase [GGTase] facilitates the incorporation of the geranylgeranyl groups in the presence of farnesyl pyrophosphate and geranylgeranyl pyrophosphate, respectively [6]. Examples of farnesylated proteins include H-Ras, lamin-B and geranylgeranylated proteins include small G-proteins such as Cdc42, Rac1, Rho and Rap1 [1]. FTase and GGTases are heterodimeric in nature comprising of a common α-subunit, but distinct β-subunits, and the later confer the substrate specificity for FTase and GGTases [7]. Using various pharmacological inhibitors of FTase and GGTases [e.g., FTI-277 and GGTI-2147], or dominant negative mutant of the FTase/GGTase-α subunit or siRNAs for FTase or GGTase β subunits, several recent studies have demonstrated novel roles for protein prenylation in nutrient-induced insulin secretion from clonal β-cells and normal rodent islets [8-12].
In the context of prenyltransferase-mediated regulation of cellular function, Kim and associates have reported that the common α-subunit of FTase/GGTase undergoes caspase-3 mediated cleavage into a smaller peptide [~35 kDa] under conditions of cellular apoptosis [13]. For example, they demonstrated caspase-3 activation and FTase/GGTase α-subunit degradation in a mouse lymphoma cell line [W4 cell expressing the Fas receptor] following exposure to anti-Fas antibodies. They also observed caspase-3 activation and FTase/GGTase α-subunit degradation in Rat-2/H-ras cells treated with an FTase inhibitor [LB42708] or in Rat-1 cells treated with etoposide. Based on these observations, these authors implicated key roles for caspase-3 mediated degradation of FTase/GGTase-α-subunit in cellular apoptosis [13].
Given the above mentioned regulatory roles of farnesylation and geranylgeranylation in islet function including insulin secretion and cell survival [1,10], we undertook the current study to determine if FTase/GGTase α-subunit undergoes cleavage under conditions of caspase-3 activation and cellular apoptosis in pancreatic β-cells. Specifically, we have examined the effects of etoposide, a genotoxic agent, known to induce robust activation of caspase-3 in insulin-secreting cells on FTase/GGTase α-subunit cleavage and cell viability in isolated pancreatic β-cells [14]. Lastly, we also investigated potential roles of influx and intracellular accumulation of calcium as an intermediary signaling event in etoposide effects on caspase-3 activation, FTase/GGTase α-subunit degradation steps in the cascade of events leading to loss in cell viability in these cells.
Materials and Methods
Materials
Z-DEVD-FMK, a caspase inhibitor, was from R&D Systems, Inc. [Minneapolis, MN]. Antisera directed against cleaved caspase-3 [active form] and β-actin were from Cell Signalling [Danvers, MA]. The antiserum against the FTase/GGTase-α subunit was from Santa Cruz Biotechnology, Inc. [Santa Cruz, CA]. Anti-mouse or anti-rabbit IgG-horseradish peroxidase conjugates and Enhanced hemiluminescence [ECL] kits were from Amersham Biosciences [Piscataway, NJ]. All other reagents used in these studies were from Sigma Aldrich Co. [St. Louis, MO] unless stated otherwise.
Insulin-secreting cell culture and treatments
INS 832/13 cells were cultured in RPMI-1640 medium containing 10 % heat-inactivated fetal bovine serum supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, and 10 mM HEPES [pH 7.4]. The medium was changed twice, and cells were subcloned weekly. Islets from normal 6 week-old male Sprague-Dawley rats [Harlan Laboratories, Oxford, MI] were isolated by collagenase digestion method. All protocols, including isolation of pancreatic islets from rats, were reviewed and approved by our Institutional Animal Care and Use Committee. INS 832/13 cells or rat islets were incubated with diluents [DMSO and/or ethanol] or etoposide [60 μM] in presence or absence of nifedipine [10 μM] for 6-12 h as indicated in the text. In a separate experiment INS 832/13 cells were treated with either diluent or etoposide (60 μM) in the presence or absence of peptide inhibitor Z-DEVD-FMK [25 μM] for 6 h as stated in the text.
Recombinant caspase-3 studies
INS 832/13 cells were cultured to 80 % confluency and washed twice with PBS, harvested and resuspended in sample buffer [0.5 % Nonidet P-40, 20 mM HEPES (pH 7.4), 100 mM NaCl and 20 mM DTT]. Fifty μg of lysate proteins were treated with recombinant caspase-3 [0-0.1 unit/mg protein] and incubated at 25 °C for 1 h. Samples were processed and immunoprobed for caspase-3 and FTase/GGTase α.
Western blotting
Proteins from INS 832/13 cells or rat islets were separated by SDS-PAGE on 10 % [w/v] polyacrylamide mini gels and electrotransferred to nitrocellulose membrane. The membranes were blocked with 5 % non fat dry milk in TBS-T [10 mM Tris-HCl; pH 7.4], 8.8 g/litre NaCl, and 0.1% Tween 20] for 2 h at room temperature. The membranes were then incubated overnight at 4 °C with antisera raised against the cleaved [active fragment] caspase-3 [1:250] or FTase/GGTase-α subunit [1:400) in TBS-T containing 5 % BSA. The membranes were washed 5 times for 5 min each with TBS-T and probed with appropriate horseradish peroxidase-conjugated secondary antibodies in 5 % non fat dry milk in TBS-T at room temperature for 1 h. After washing, the immune complexes comprising of the target proteins were detected using the ECL kit. The membranes then were stripped and reprobed with β-actin. The band intensity was quantified and protein expression levels were calculated relative to β-actin in the same sample.
Metabolic cell viability assays
INS 832/13 cells were seeded in 96-well plates and treated as described above. After 6 h cells viability was determined by using 3-(4, 5 dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide [MTT] assay which was carried out using manufacturer's instructions. The results are shown as relative optical density.
Statistical analysis
Results are expressed as means with their standard errors as indicated. Data are analyzed using one way ANOVA followed by Bonferroni posthoc test [GraphPad Prism 5; GraphPad Software, Inc., La Jolla, CA, USA]. Differences between control and treatment groups were considered significant if p is < 0·05.
Results
Etoposide induces caspase-3 activation in INS 832/13 cells
At the outset, we quantitated etoposide-induced activation of caspase-3 in INS 832/13 cells. This was accomplished by incubating these cells with etoposide [60 μM for 6 hr] followed by identification of the cleaved peptide of caspase-3 [active form] by Western blotting method. Data in Figure 1 [Panel a] show that etoposide significantly increased the emergence of the active form of caspase-3 [lane 3 vs. lane 1]. Further, coprovision of Z-DEDV-FMK, a known inhibitor of caspase-3, markedly attenuated etoposide-induced activation of caspase-3 [lane 4 vs. lane 3]. Data from multiple experiments are provided in Figure 1 [Panel b] suggesting that etoposide significantly augments caspase-3 activity in INS 832/13 cells.
Fig. 1. Etoposide induces caspase-3 activation and FTase/GGTase-α degradation in INS 832/13 cells: Protection of these signaling steps by Z-DEVD-FMK, an inhibitor of caspase 3.
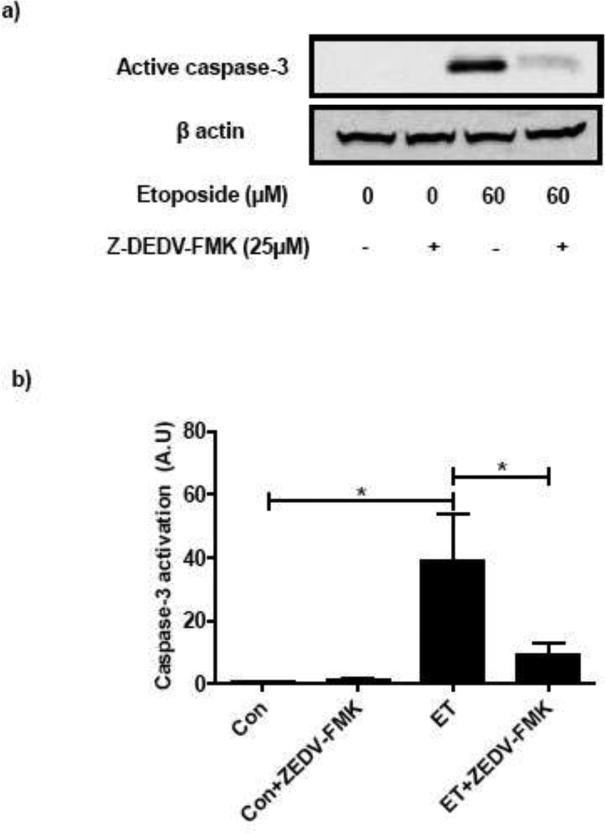
INS 832/13 cells were treated with either diluents (DMSO and/or ethanol) or etoposide (60 μM) in the presence or absence of peptide inhibitor Z-DEVD-FMK (25 μM; 6 h). Caspase-3 activation and FTase/GGTase-α degradation were determined by Western blotting. Equal amount of lysates protein were resolved by SDS-PAGE (10 %). Protein loading was determined by actin content in individual lanes. A representative blot indicating the caspase-3 activation (a) and FTase/GGTase-α degradation (c) is provided. Quantitative analysis of data obtained from three independent experiments for caspase-3 activation (b) and FTase/GGTase-α degradation (d) was carried out by densitometry. Results are shown as means ± SEM. *p < 0.05, ***p < 0.001
Etoposide induces FTase/GGTase α-subunit degradation in INS 832/13 cells and normal rat islets
As stated above, earlier observations from Kim and associates have shown that the α-subunit of FTase/GGTase undergoes cleavage by caspase-3. We next determined if etoposide-induced caspase-3 activation culminates in the degradation of the FTase/GGTase α-subunit. To examine this, lysates from studies described under Figure 1 [Panels a and b] were subjected to SDS-PAGE and relative abundance of the degraded product of FTase/GGTase α-subunit [~ 35 kDa] was determined by Western blotting. As depicted in Figure 1 [Panels c and d], a significant increase in the degradation of FTase/GGTase α-subunit was noticed in cells incubated with etoposide [Figure 1; Panel c; lane 3 vs. lane 1]. In a manner akin to caspase-3 activation [Figure 1; Panels a and b], incubation of INS 832/13 cells with an inhibitor of caspase-3 markedly reduced etoposide-induced FTase/GGTase α-subunit degradation [Figure 1; Panel c; lane 4 vs. lane 3]. Data from multiple experiments are included in Figure 1 [Panel d].
Recombinant caspase-3 degrades FTase/GGTase α-subunit in INS 832/13 cells
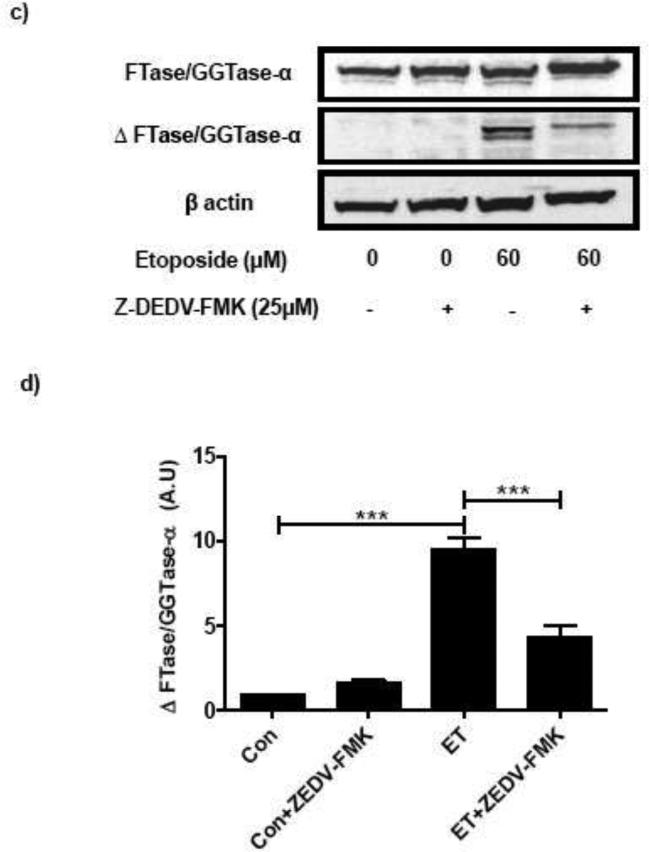
To further demonstrate that caspase-3 mediates the hydrolysis of FTase/GGTase α-subunit, INS 832/13 cell lysates were incubated with human recombinant caspase-3 and the relative abundance of cleaved product of FTase/GGTase α-subunit was determined as above. Indeed, data is Figure 2 show the emergence of the degradative product of FTase/GGTase α-subunit under conditions of caspase-3 activation. These data further support our observations in Figure 1 suggesting that etoposide induces caspase-3 activation leading to cleavage of FTase/GGTase α-subunit in pancreatic β-cells.
Fig. 2. Incubation of INS 832/13 cell lysates with human recombinant caspase-3 (CPP32) results in FTase/GGTase-α degradation in INS 832/13 cells.
INS 832/13 cell lysates were treated with CPP32 (0-0.1 unit/mg protein) for 1 hr as indicated in the figure followed by separation on SDS-PAGE (10 %). Separated proteins were transferred onto a nitrocellulose membrane and immunoblotted for Caspase-3 and FTase/GGTase-α. A representative Western blot for caspase-3 activation (a) and FTase/GGTase-α degradation (b) is shown here
Nifedipine protects etoposide-induced caspase-3-mediated degradation of FTase/GGTase α-subunit in INS 832/13 cells and normal rodent islets
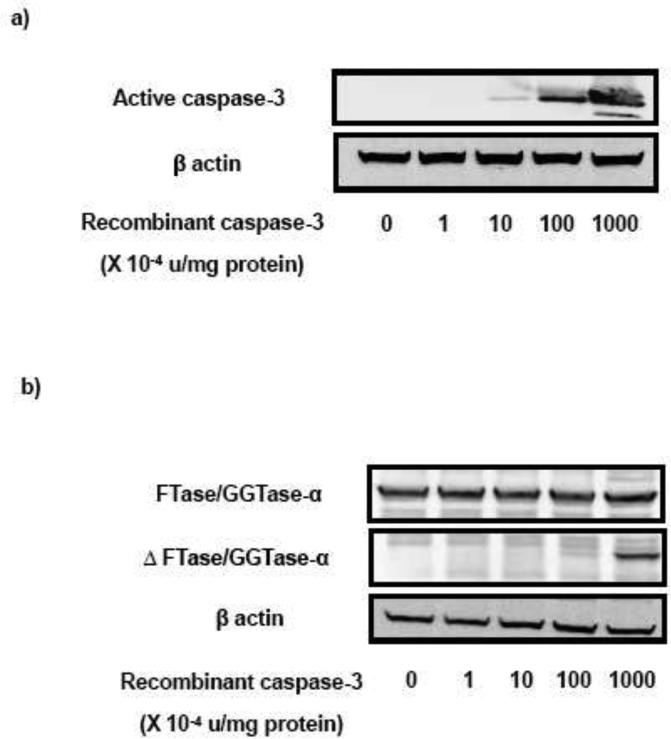
Existing evidence in other cell types implicates critical roles for intracellularly accumulated calcium in eliciting mitochondrial dysfunction and caspase-3 activation [15-18]. In addition, available data in many cell types indicated that etoposide-induced caspase activation may, in part, be due to accumulation of calcium intracellularly [19,20]. Therefore, in the next of experiments, we investigated if blocking the influx of extracellular calcium by nifedipine, a known blocker of voltage-gated calcium channels [21,22], prevents etoposide-induced caspase-3 activation and FTase/GGTase α-subunit cleavage in INS 832/13 cells. Data depicted in Figure 3 [Panel a] demonstrates a significant reduction in etoposide-induced caspase-3 activation in INS 832/13 cells following coprovision with nifedipine. Data from multiple experiments revealed ~ 70 % inhibition in etoposide-induced caspase-3 activation following incubation with nifedipine [Figure 3; Panel b]. We then investigated if nifedipine-treatment of pancreatic β-cells prevents etoposide-induced degradation of FTase/GGTase-α-subunit. Data in Figure 3 [Panel c and d] demonstrate a marked reduction in etoposide-induced FTase/GGTase α-subunit hydrolysis by nifedipine.
Fig. 3. Nifedipine prevents etoposide induced caspase-3 activation and FTase/GGTase-α degradation in INS 832/13 cells.
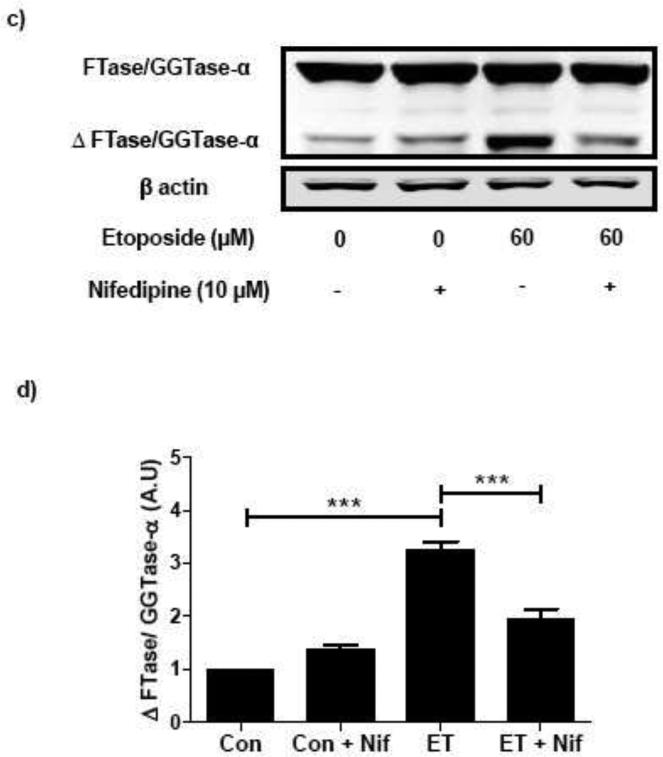
INS 832/13 cells were treated with either diluents (DMSO and/or ethanol) or etoposide (ET) (60 μM) in the presence or absence of Nifedipine (Nif; 10 μM; 6 h). Caspase-3 activation and FTase/GGTase-α degradation were determined by Western blot analysis. Protein loading was determined by actin content in individual lanes. Data are representative of four independent experiments. A representative blot indicating the caspase-3 activation (a) and FTase/GGTase-α degradation (c) is shown here. Quantitation of the active caspase-3 (b) and FTase/GGTase-α degradation (d) was carried out by densitometry. Results are shown as means ± SEM. ***p < 0.001
In a manner akin to INS 832/13 cells, we also observed protective effects of nifedipine against etoposide induced caspase 3 activation and FTase/GGTase α-subunit degradation in normal rat islets. As shown in Figure 4 [Panel a and b] coprovision of nifedipine markedly attenuated etoposide-induced caspase-3 activation in rat islets. Furthermore, nifedipine-treatment of rat islets resulted in marked reduction in etoposide-induced FTase/GGTase α-subunit hydrolysis [Figure 4; Panel c and d].
Fig. 4. Nifedipine prevents etoposide induced caspase-3 activation and FTase/GGTase-α degradation in rat islets.
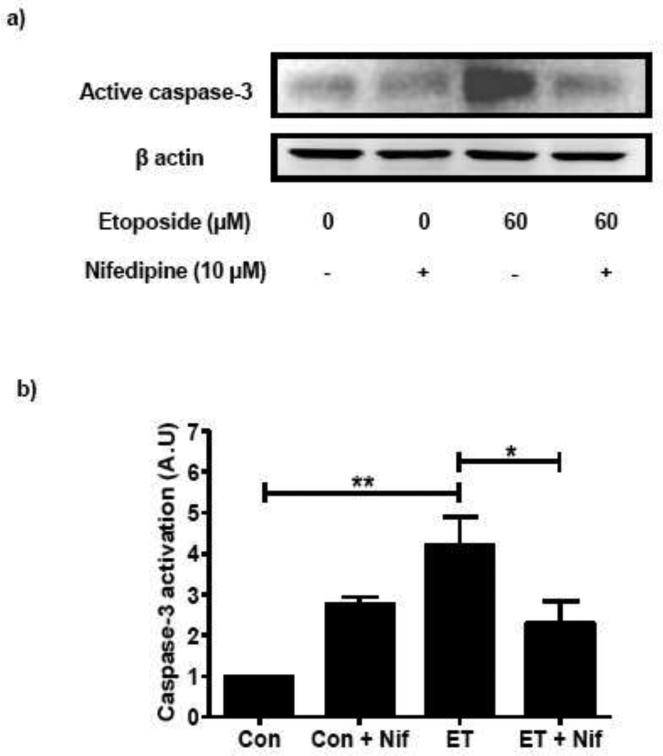
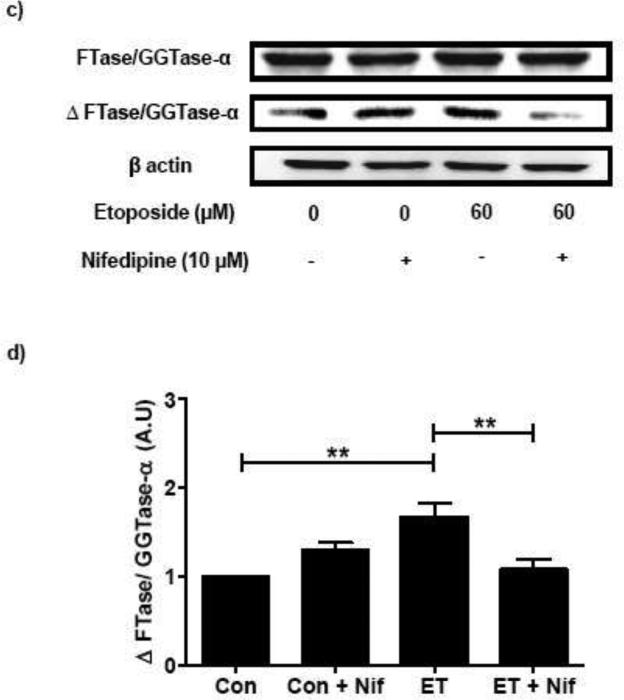
Rat islets were treated with either diluents (DMSO and/or ethanol) or etoposide (ET) (60 μM) in the presence or absence of Nifedipine (Nif; 10 μM; 12 h). Caspase-3 activation and FTase/GGTase-α degradation were determined by Western blot analysis. Protein loading was determined by actin content in individual lanes. Data are representative of four independent experiments. A representative blot indicating the caspase-3 activation (a) and FTase/GGTase-α degradation (c) is shown here. Quantitation of the active caspase-3 (b) and FTase/GGTase-α degradation (d) was carried out by densitometry. Results are shown as means ± SEM. *p < 0.05, **p < 0.01
Nifedipine protects etoposide-induced loss in cell viability in INS 832/13 cells
As mentioned above, several lines of evidence in multiple cell types including the islet β-cell implicate roles for prenylated G-proteins in cell survival [1,10]. Therefore, in the last series of studies, we have examined if: etoposide induces loss in metabolic cell viability under conditions it induced caspase-3 activation and FTase/GGTase α-subunit degradation, and if so, whether nifedipine prevents such an effect. To test this, cells were incubated with diluents or etoposide and/or nifedipine in different combinations followed by quantitation of viability of these cells using the MTT method [see Methods for additional details]. Data in Figure 5 indicate no significant effects of nifedipine on cell viability [bar 1 vs bar 2]; these data are in agreement with lack of effects of nifedipine on caspase-3 activation and FTase/GGTase α-subunit degradation in these cells. In a manner akin to its effects on caspase-3 activation and FTase/GGTase α-subunit degradation, etoposide significantly reduced cell viability in INS 832/13 cells [bar 3 vs. bar 1]. Coprovision of nifedipine to these cells alongside etoposide markedly prevented loss in cell viability [bar 4 vs bar 3]. Together, these data suggest that nifedipine affords protective effects against etoposide-induced caspase-3 activation, FTase/GGTase α-subunit degradation and loss in cell viability in pancreatic β-cells.
Fig. 5. Nifedipine prevents etoposide induced reduction in metabolic cell viability in INS 832/13 cells.
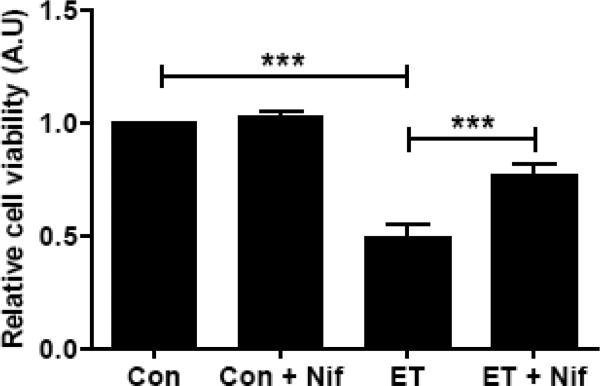
INS 832/13 cells were incubated with either diluents or etoposide (ET; 60 μM) in the presence or absence of Nifedipine (Nif; 10 μM; 6 h) as indicated in the figure. Cell viability was measured by MTT assay. Data represent mean ± SEM from four independent experiments. *** p < 0.001
Discussion
It is well established that small molecular mass and heterotrimeric G-proteins play important roles in cellular signaling events leading to glucose-stimulated insulin secretion [5,12]. A growing body of evidence is also suggestive of novel roles for these signaling proteins in other β-cell functions including cell cycle progression, survival and apoptosis [1]. It is noteworthy that the majority of these proteins undergo post-translational modifications at their c-terminal cysteine residues [e.g., prenylation, methylation and acylation], which are essential for their trafficking to relevant cell membranes for optimal interaction with their respective effector proteins culminating in optimal regulation of β-cell function [1,4,5]. Despite the compelling evidence that FTase and GGTases are acutely regulated by glucose in the β-cell [11,12], and that they are critical for insulin secretion [1,8,9], very little is known with regard to potential alterations in these enzymes under conditions of cellular apoptosis. Along these lines, studies by Kim and associates have demonstrated degradation of the FTase/GGTase α-subunit under conditions of caspase-3 activation [13]. With this in mind, we quantitated caspase-3 activation, FTase/GGTase degradation and cellular dysfunction in isolated β-cells exposed to etoposide, a known inducer apoptosis in pancreatic β-cells. Our findings suggested that: [i] etoposide-induces caspase-3 activation, which in turn, degrades the common α-subunit of FTase/GGTase; these signaling events are blocked by a peptide inhibitor of caspase-3; [ii] nifedipine, a known inhibitor of plasma membrane-associated calcium channels, markedly attenuates etoposide-mediated effects on caspase-3 activation and FTase/GGTase α-subunit degradation in INS 832/13 cells and normal rat islets; and [iii] nifedipine protects pancreatic β-cells against etoposide-induced loss in metabolic viability. Based on these findings we speculate that influx and intracellular accumulation of calcium as one of the signaling events leading to etoposide-induced metabolic dysfunction of the islet β-cell, leading to the activation of caspase-3 and FTase/GGTase α-subunit degradation [see below]. Given our observations, one might ask the question about the relevance and implications of these current findings in the context of β-cell survival and function. These aspects are discussed below.
Existing evidence suggests that inhibition of protein prenylation culminates in growth arrest, loss in cell viability and apoptosis, thus implicating regulatory roles for prenylated proteins, including small molecular mass and trimeric G-proteins as well as nuclear lamins in cell survival pathways [23,24]. Furthermore, it has been demonstrated that functional inactivation of G-proteins or GTP-requiring signaling steps by depletion of intracellular GTP results in cell demise. For example, original studies by Li and associates [25] have suggested that prolonged depletion of intracellular GTP via inhibition of inosine monophosphate dehydrogenase using mycophenolic acid, results in the apoptotic demise of the islet β-cell. These authors have also demonstrated that functional inactivation of Rho G-proteins by clostridial toxins further potentiated mycophenolic acid-induced β-cell apoptosis. These data raise an interesting possibility that functional inactivation of islet endogenous G-proteins either by GTP-depletion or functional inactivation via Clostridial toxins or FTase/GGTase inhibitors results in impaired cell viability. Lastly, it was also demonstrated that inhibition of protein farnesylation using selective inhibitors of FTases [FTI-277 and FTI-2628] or siRNA-mediated knockdown of the β-subunit of FTase, markedly reduced glucose-induced ERK1/2 phosphorylation, Rac1 activation and insulin secretion in clonal β-cells and normal rat islets [11]. Our current findings indicating a potential causal relationship between the degradation of FTase/GGTase α-subunit and loss in cell viability further validates the above experimental models and postulations.
It is also noteworthy that our current observations also suggested regulatory roles for accumulation of cytosolic calcium in etoposide-mediated effects on caspase-3 activation and FTase/GGTase α-subunit degradation. These findings are in concert with recent observations by Wang and associates [26] who reported cytoprotective effects, by nifedipine, against endoplasmic reticulum stress and apoptosis induced by glucotoxic conditions in pancreatic β-cells. Therefore, it is likely that such conditions could also promote β-cell dysfunction via a calcium-mediated mitochondrial dysregulation, caspase-3 activation and FTase/GGTase α-subunit degradation pathways leading to loss in cell viability. This possibility needs to be verified experimentally. Indeed, recent observations from Shalev's laboratory [27] have provided compelling evidence to suggest a marked reduction by verapamil, a calcium channel blocker, of increased expression of pro-apototic thioredoxin-interacting protein expression and restoration of normal β-cell survival and function in the BTBR ob/ob mice. Our current findings provide further evidence in support of the formulation that influx of extracellular calcium and subsequent overload of mitochondrial calcium leads to dysregulation of cellular function. Thus, calcium channel blockers such as verapamil [27] and nifedipine [current study] appear to exert cytoprotective effects in β-cells against noxious stimuli and improve β-cell survival and function.
Our current findings form the basis for future investigations including determination of overall impact of degradation of FTase/GGTase α-subunit in the functional regulation of other key cellular proteins such as Probin, a novel farnesylated protein that has been shown to suppress the activation of survival proteins [e.g., Akt] in the islet β-cell [28]. Second, potential implications of our current findings on nuclear lamins and the assembly of nuclear envelope remains to be verified since inhibition of post-translational prenylation of lamins is known to increase their susceptibility to degradation leading to the collapse of nuclear assembly [29,30]. Along these lines, recent studies by Chang and associates have demonstrated that inhibition of farnesylation using FTIs led to a marked reduction in the biogenesis of mature lamin A culminating in the accumulation of prelamin-A intracellularly. Further, their data suggested a critical requirement for a geranylgeranylation signaling step in the conversion of farnesylated lamin A to its mature form mediated by a zinc metalloprotease [31]. Together, these findings implicate novel roles for post-translational prenylation steps in the functional regulation of lamins [23,24]. Lastly, it is likely that the cleavage product of FTase/GGTase α-subunit could have direct effects on the induction of cell death. Indeed, findings by Kim and associates further substantiate this viewpoint. These investigators provided direct evidence to suggest that expression of p35, the cleavage product of FTase/GGTase α markedly increased cell death probably by interfering with the prenylenzyme activities [13]. This remains to be confirmed in the β-cell.
In summary, we propose [Figure 6] that etoposide-induced metabolic dysregulation and loss in cell viability may, in part, be due to calcium-mediated, caspase-3-induced degradation of FTase/GGTase α-subunit and associated downstream signaling events, which remain to be determined in the context of prenylation-dependent regulation of survival and function of the islet β-cell.
Fig. 6. Proposed model for etoposide-induced loss in viability of the β-cell.
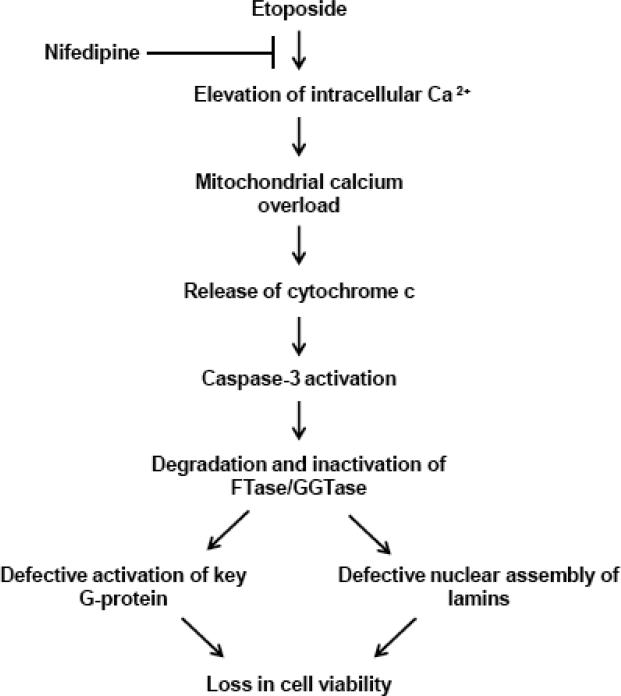
Based on the data accrued in the current study we speculate that exposure of pancreatic β-cells to etoposide leads to mitochondrial dysfunction mediated via intracellular accumulation of calcium, which is sensitive to nifedipine. Such a signaling step leads to increased activation of caspase-3, and peptidic cleavage of FTase/GGTase α-subunit. While our current data do not provide direct evidence, existing data from multiple laboratories including our own suggests that pharmacological and molecular biological inhibition of FTase/GGTases lead to functional inactivation of G-proteins and abnormal lamin metabolism leading to defective nuclear assembly and cell demise [see text for additional details]. Lastly, we cannot rule out the potential possibility that the cleavage product of FTase/GGTase α-subunit could exert direct effects on inducing loss in metabolic cell viability in INS 832/13 cells. This remains to be verified experimentally.
Abbreviations
- CSP3
caspase-3
- ET
etoposide
- FTase α
farnesyltransferase α subunit
- GGTase α
Geranylgeranyltransferase α subunit
- Δ FTase/GGTase
hydrolytic product of FTase/GGTase α-subunit
- MTT
3-(4, 5 dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- Nif
nifedipine
References
- 1.Kowluru A. Small G proteins in islet beta-cell function. Endocr.Rev. 2010;31:52–78. doi: 10.1210/er.2009-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson RP, Seaquist ER, Walseth TF. G proteins and modulation of insulin secretion. Diabetes. 1991;40:1–6. doi: 10.2337/diab.40.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Cox AD, Der CJ. Protein prenylation: more than just glue? Current Opinion in Cell Biology. 1992;4:1008–1016. doi: 10.1016/0955-0674(92)90133-w. [DOI] [PubMed] [Google Scholar]
- 4.Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem J. 1993;295:31–40. doi: 10.1042/bj2950031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kowluru A. Regulatory roles for small G proteins in the pancreatic beta-cell: lessons from models of impaired insulin secretion. American Journal of Physiology - Endocrinology and Metabolism. 2003;285:E669–684. doi: 10.1152/ajpendo.00196.2003. [DOI] [PubMed] [Google Scholar]
- 6.Casey PJ, Seabra MC. Protein Prenyltransferases. Journal of Biological Chemistry. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 7.Lane KT, Beese LS. Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J Lipid Res. 2006;47:681–699. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Amin R, Chen HQ, Tannous M, Gibbs R, Kowluru A. Inhibition of glucose- and calcium-induced insulin secretion from betaTC3 cells by novel inhibitors of protein isoprenylation. J Pharmacol.Exp.Ther. 2002;303:82–88. doi: 10.1124/jpet.102.036160. [DOI] [PubMed] [Google Scholar]
- 9.Veluthakal R, Kaur H, Goalstone M, Kowluru A. Dominant-negative alpha-subunit of farnesyl- and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes. 2007;56:204–210. doi: 10.2337/db06-0668. [DOI] [PubMed] [Google Scholar]
- 10.Kowluru A. Protein prenylation in glucose-induced insulin secretion from the pancreatic islet beta-cell: a perspective. Journal of Cellular and Molecular Medicine. 2008;12:164–173. doi: 10.1111/j.1582-4934.2007.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta-cells. Diabetes. 2010;59:967–977. doi: 10.2337/db09-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goalstone M, Kamath V, Kowluru A. Glucose activates prenyltransferases in pancreatic islet beta-cells. Biochem Biophys.Res.Commun. 2010;391:895–898. doi: 10.1016/j.bbrc.2009.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KW, Chung HH, Chung CW, et al. Inactivation of farnesyltransferase and geranylgeranyltransferase I by caspase-3: cleavage of the common alpha subunit during apoptosis. Oncogene. 2001;20:358–366. doi: 10.1038/sj.onc.1204099. [DOI] [PubMed] [Google Scholar]
- 14.Collier JJ, Fueger PT, Hohmeier HE, Newgard CB. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes. 2006;55:1398–1406. doi: 10.2337/db05-1000. [DOI] [PubMed] [Google Scholar]
- 15.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma AK, Rohrer B. Calcium-induced Calpain Mediates Apoptosis via Caspase-3 in a Mouse Photoreceptor Cell Line. Journal of Biological Chemistry. 2004;279:35564–35572. doi: 10.1074/jbc.M401037200. [DOI] [PubMed] [Google Scholar]
- 17.Tripathi A, Chaube SK. High cytosolic free calcium level signals apoptosis through mitochondria-caspase mediated pathway in rat eggs cultured in vitro. Apoptosis. 2012;17:439–448. doi: 10.1007/s10495-012-0702-9. [DOI] [PubMed] [Google Scholar]
- 18.Smith MA, Schnellmann RG. Calpains, Mitochondria and Apoptosis. Cardiovasc.Res. 2012 doi: 10.1093/cvr/cvs163. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Distinct pathways for stimulation of cytochrome c release by etoposide. J Biol.Chem. 2000;275:32438–32443. doi: 10.1074/jbc.C000518200. [DOI] [PubMed] [Google Scholar]
- 20.Parihar A, Parihar MS, Ghafourifar P. Significance of mitochondrial calcium and nitric oxide for apoptosis of human breast cancer cells induced by tamoxifen and etoposide. Int.J Mol Med. 2008;21:317–324. [PubMed] [Google Scholar]
- 21.Sorkin EM, Clissold SP, Brogden RN. Nifedipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy, in ischaemic heart disease, hypertension and related cardiovascular disorders. Drugs. 1985;30:182–274. doi: 10.2165/00003495-198530030-00002. [DOI] [PubMed] [Google Scholar]
- 22.Bean BP. Classes of calcium channels in vertebrate cells. Annu.Rev.Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- 23.Sane KM, Mynderse M, LaLonde DT, et al. A Novel Geranylgeranyl Transferase Inhibitor in Combination with Lovastatin Inhibits Proliferation and Induces Autophagy in STS-26T MPNST Cells. Journal of Pharmacology and Experimental Therapeutics. 2010;333:23–33. doi: 10.1124/jpet.109.160192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geryk-Hall M, Yang Y, Hughes DP. Driven to death: inhibition of farnesylation increases Ras activity in osteosarcoma and promotes growth arrest and cell death. Mol Cancer Ther. 2010;9:1111–1119. doi: 10.1158/1535-7163.MCT-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Segu VB, Rabaglia ME, Luo RH, Kowluru A, Metz SA. Prolonged Depletion of Guanosine Triphosphate Induces Death of Insulin-Secreting Cells by Apoptosis. Endocrinology. 1998;139:3752–3762. doi: 10.1210/endo.139.9.6207. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Gao L, Li Y, Chen H, Sun Z. Nifedipine Protects INS-1 beta-Cell from High Glucose-Induced ER Stress and Apoptosis. Int.J Mol Sci. 2011;12:7569–7580. doi: 10.3390/ijms12117569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu G, Chen J, Jing G, Shalev A. Preventing β-Cell Loss and Diabetes With Calcium Channel Blockers. Diabetes. 2012;61:848–856. doi: 10.2337/db11-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyathanahalli CN, Kowluru A. A farnesylated G-protein suppresses Akt phosphorylation in INS 832/13 cells and normal rat islets: regulation by pertussis toxin and PGE(2). Biochem Pharmacol. 2011;81:1237–1247. doi: 10.1016/j.bcp.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutz RJ, Trujillo MA, Denham KS, Wenger L, Sinensky M. Nucleoplasmic localization of prelamin A: implications for prenylation-dependent lamin A assembly into the nuclear lamina. Proc.Natl.Acad.Sci U.S.A. 1992;89:3000–3004. doi: 10.1073/pnas.89.7.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prokocimer M, Davidovich M, Nissim-Rafinia M, et al. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med. 2009;13:1059–1085. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang SY, Hudon-Miller SE, Yang SH, et al. Inhibitors of protein geranylgeranyltransferase-I lead to prelamin A accumulation in cells by inhibiting ZMPSTE24. Journal of Lipid Research. 2012;53:1176–1182. doi: 10.1194/jlr.M026161. [DOI] [PMC free article] [PubMed] [Google Scholar]