

Summary
Mixed Lineage Leukemia (MLL) fusion proteins cause oncogenic transformation of hematopoietic cells by constitutive recruitment of elongation factors to HOX promoters, resulting in over-expression of target genes. The structural basis of transactivation by MLL fusion partners remains undetermined. We show that the ANC1 Homology Domain (AHD) of AF9, one of the most common MLL translocation partners, is intrinsically disordered and recruits multiple transcription factors through coupled folding and binding. We determined the structure of the AF9 AHD in complex with the elongation factor AF4, and show that aliphatic residues which are conserved in each of the AF9 binding partners form an integral part of the hydrophobic core of the complex. NMR relaxation measurements show AF9 retains significant dynamic behavior which may facilitate exchange between disordered partners. We propose that AF9 functions as a signaling hub which regulates transcription through dynamic recruitment of co-factors in normal hematopoiesis and in acute leukemia.
Introduction
Chromosomal translocations involving the Mixed Lineage Leukemia (MLL) gene are responsible for a subset of acute leukemias characterized by poor prognosis and early relapse (Dimartino and Cleary, 1999). The majority result in expression of oncogenic fusion proteins which include the N-terminal chromatin targeting domains of the MLL protein fused to one of over sixty translocation partners (Meyer et al., 2009). Through constitutive recruitment of elongation factors, these proteins upregulate transcription of MLL target genes, including HOXA9 and MEIS1, resulting in oncogenic transformation of hematopoietic cells. Despite the large number of fusion partners, the five most common account for ~80% of cases and are thought to share a common mechanism of transactivation.
The normal MLL protein regulates transcription of many genes, but is specifically required for H3K4 trimethylation and transcriptional initiation at a subset of HOX promoters (Guenther et al., 2005). Following initiation at these promoters, RNA Polymerase II (PolII) is subject to promoter proximal pausing. The major regulatory step in HOX gene expression is through release of PolII from this paused state (Chopra et al., 2009). This requires coordinated chromatin remodeling, phosphorylation of pause factors, and phosphorylation of serine 2 of the RNA PolII C-terminal domain by P-TEFb (Marshall et al., 1996; O’Brien et al., 1994).
Among the most common MLL translocation partners are AF9 and ENL, closely related members of the YEATS domain superfamily. Both proteins have previously been identified as components of biochemically isolated complexes with functions in transcriptional elongation such as SEC (Super Elongation Complex)(Lin et al., 2010), AEP (AF4, ENL, P-TEFb)(Yokoyama et al., 2010), and Dot-Com (Dot1 Complex)(Mohan et al., 2010). AF9 and ENL are able to recruit and activate P-TEFb through recruitment of AF4 family members via their ANC1 homology domain (AHD). Disruption of the AF4-AF9 interaction results in necrotic cell death in several cell lines harboring MLL translocations suggesting that the AF9 AHD may be an attractive pharmacological target (Palermo et al., 2008). In addition to AF4, the AHD also binds to Dot1L, a histone methyltransferase which methylates H3K79, a histone modification associated with transcriptional elongation (Steger et al., 2008). Paradoxically, this domain was also shown to recruit the BCL6 Corepressor (BCoR) and Polycomb 3 (hPC3/CBX8), which are involved in transcriptional repression (Bardos et al., 2000; Huynh et al., 2000).
It was recently demonstrated that AEP components colocalize with MLL at a subset of promoters, suggesting that AF9 or ENL may be recruited to these loci in a context dependent manner to regulate transcriptional elongation downstream of MLL. This hypothesis is supported by recently identified interactions between the AF9 and ENL YEATS domains and the PAF elongation complex (PAF-c) (He et al., 2011), which in turn interacts with the MLL CXXC domain and flanking regions (Milne et al., 2010; Muntean et al., 2010). These results raise the possibility that AF9 or ENL may be recruited by both normal MLL and MLL fusion proteins through interactions with PAF-c, and transcriptional elongation may be regulated through interactions of transcription factors with the AHD.
The importance of the AHD in both transcriptional regulation and in acute leukemia has been established, however the structural basis of its function has not been demonstrated. Here we present the first structure of the AF9 AHD, in complex with a peptide from the elongation factor AF4. We show that this domain is intrinsically disordered in isolation and interacts with each of its binding partners through mutual synergistic folding. The relatively extensive intermolecular interface generated in this complex maintains high affinity binding, while accommodating significant conformational entropy. This may allow exchange between binding partners in response to changes in local concentrations or post-translational modifications, which may be essential to dynamic transcriptional control.
Results
The AF9 AHD is intrinsically disordered and interacts with AF4 by mutual synergistic folding
We expressed the C-terminal 79 amino acids of AF9, which includes the minimal portion of AF9 required for oncogenic activity of an MLL-AF9 fusion protein in colony forming assays (Prasad et al., 1995) and roughly corresponds to the minimal domain observed in a clinical case of leukemia with an MLL-AF9 translocation (Mitterbauer et al., 1999). During initial efforts to express and purify the AHD, we found that it had limited solubility and that the domain was prone to proteolytic degradation. 15N-1H HSQC spectra showed extreme broadening, with only a small number of peaks with narrow 1H chemical shift dispersion and an insufficient number of peaks for the amino acids in the domain (Figure 1a). Since the interacting regions between AF4 and AF9 had been finely mapped (Srinivasan et al., 2004), we titrated a peptide derived from the AF9 interaction motif of AF4 (residues 761 to 774) into the AF9 AHD. This resulted in dramatically improved chemical shift dispersion and an increased number of observable peaks in the HSQC spectrum (Figure 1b). In order to understand the structural changes induced by peptide binding, we conducted circular dichroism (CD) experiments on the AF9 AHD alone and in complex with the AF4 peptide. Surprisingly, these showed that in isolation the AHD is almost entirely random coil with only a small amount of beta structure predicted using K2D (Andrade et al., 1993)(Figure 1c). Upon addition of the AF4 peptide, the domain undergoes a structural rearrangement to form a mixed alpha-beta structure consistent with the dramatic change in the HSQC spectrum (Figure 1b). These data show that the AF9 AHD is intrinsically disordered, and recruits AF4 through mutual synergistic folding (Demarest et al., 2002).
Figure 1. The AF9 AHD is intrinsically disordered and folds upon binding to AF4.
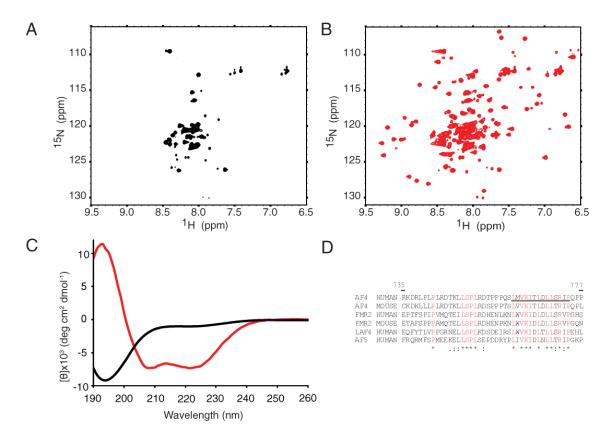
A 15N-1H HSQC of AF9 490-568 B 15N-1H HSQC of AF9 490-568 saturated with unlabeled AF4 761-774. C Far-UV Circular dichroism spectrum of the constructs shown in A and B, AF9 only (black), AF9 with AF4 peptide (red) D alignment of AF4 family members, conserved residues are in red, the AF4 peptide is underlined.
Structure of the AF4-AF9 complex
In order to understand the structural basis for AF9 AHD function, we determined the structure of the AF4-AF9 complex using NMR spectroscopy (PDB 2LM0, BMRB 18094 and Table 1). Due to the problems encountered in expression and purification of the AF9 AHD, we generated a construct which included 43 amino acids of AF4 fused to the flexible residues immediately preceding the AF9 AHD. This approach resulted in expression of a monomeric homogeneous complex and yielded high quality spectra suitable for structure determination.
Table 1.
NMR and refinement statistics for the AF4-AF9 complex
| NMR distance and dihedral constraints | |
|---|---|
| Distance constraints | |
| Total NOE | 2168 |
| Intra-residue | 1042 |
| Inter-residue | 1126 |
| Sequential (∣i – j∣ = 1) | 529 |
| Medium-range (∣i – j∣ < 4) | 291 |
| Long-range (∣i – j∣ > 5) | 306 |
| Intermolecular | 76 |
| Hydrogen bonds | 22 |
| Total dihedral angle restraints | 116 |
| ϕ | 59 |
| Ψ | 57 |
| Structure statistics | |
| Violations (mean and s.d.) | |
| Distance constraints (Å) | 0.024 ± 0.004 |
| Dihedral angle constraints (°) | 0.91 ± 0.167 |
| Max. dihedral angle violation (°) | 7.5 |
| Max. distance constraint violation (Å) | 0.517 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.003 ± 0.000 |
| Bond angles (°) | q0.47 ± 0.012 |
| Impropers (°) | 0.36 ± 0.03 |
| Average pairwise r.m.s. deviation** (Å) | |
| Heavy | 1.171 |
| Backbone | 0.572 |
| Total RDCs | |
| HN | 49 |
| Cα-C | 41 |
| Cα-Hα | 32 |
| RDCs used for validation but not for structure calculation | |
| HN | 34 |
| Qfree (%) | 29.2 |
The AF4-AF9 complex forms a mixed alpha-beta structure with the AF4 residues forming an integral part of the hydrophobic core of the complex (Figure 2a-d). A DALI search indicated that this synergistically folded complex represents a novel fold. 1H-15N heteronuclear NOE experiments show that AF4 residues 761-775 are ordered in the complex whereas the remainder is flexible (Figure 4b). Importantly, the linker region between AF4 and AF9 is also flexible. The beta structure evident in the CD spectrum of the isolated AHD appears to be due to the presence of a beta-hairpin formed by AF9 residues 535 to 546. AF4 residues 761-766 form a β-strand which extends the AF9 β-hairpin to a 3-stranded antiparallel β-sheet. The rest of the AF9 domain folds around the AF4 peptide in the form of 3 α-helices, with five C-terminal residues remaining flexible. Immediately C-terminal to the AF4 strand is a conserved LXXL motif (L767-L770)(Figure 2c,d), which folds into a turn and packs behind the end of the hairpin to form a key hydrophobic cluster. The last four structured residues (771-775) of AF4 make contacts with AF9, although these are not as intimate as in the remainder of the peptide. The interface between AF4 and AF9 is extensive and hydrophobic, with a number of aliphatic residues from AF4 deeply buried in the hydrophobic core (Figures 2c and d). Specifically, V763 and I765 pack into the interface of the hairpin and the α1 and α3 helices, and appear to stabilize the tertiary fold of the complex. Specificity of AF4 binding also appears to involve at least one electrostatic interaction, between AF4 K764 and AF9 D544. The extensive hydrophobic interface between AF4 and AF9 may explain how the significant entropic cost of folding of the complex can be overcome upon binding. Clearly, the hydrophobic core of AF9 is not sufficiently extensive to maintain an independently folded structure.
Figure 2. Structure of the AF4-AF9 complex.
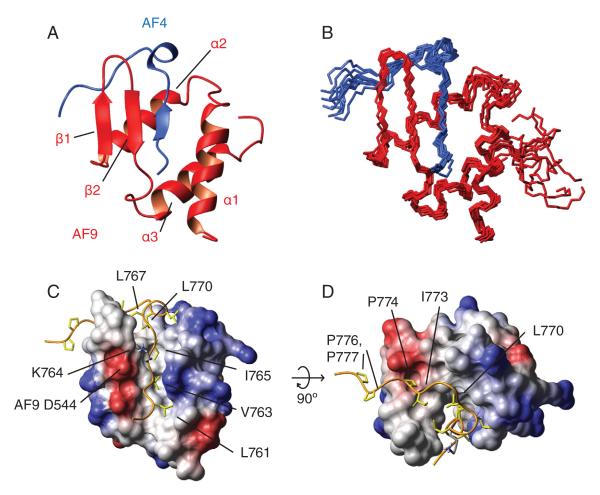
A Cartoon representation of the AF4-AF9 complex. B Ensemble of the 10 lowest energy conformers. AF4 is shown in blue, AF9 in red. The AF4 peptide forms a third strand with the beta hairpin from AF9, which folds around the AF4 peptide. C and D: The AF4-AF9 interface. The Cα trace of AF4 is shown as an orange ribbon. Hydrophobic residues from AF4 are in khaki. AF9 surface is colored according to electrostatic potential. Hydrophobic residues from the AF4 strand (L761, V763, I765) penetrate deeply into the AF9 core. Asp 544 makes an electrostatic interaction with Lys 764, which is shown in blue.
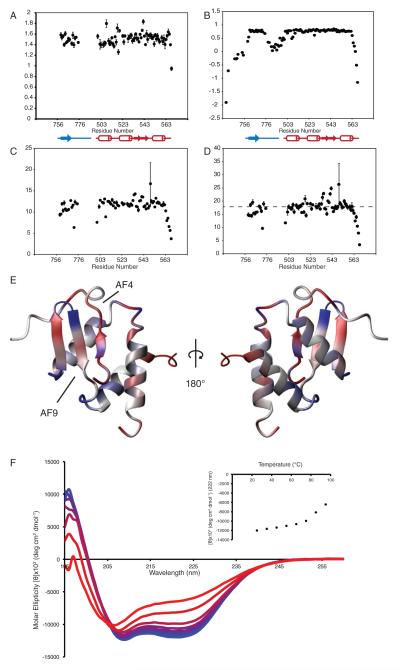
Figure 4. Dynamics of the AF4-AF9 complex.
R1, R2, and R1 × R2 values for co-expressed AF4-AF9 are shown in A, C and D, Heteronuclear NOE for the AF4-AF9 fusion used for structure determination is shown in B. Dashed line in D shows a 10% trimmed mean for R1×R2 after exclusion of values < 0.65. E shows deviation of R1R2 products from the trimmed mean. Depressed values (fast exchange) are in red, elevated values (slow exchange) are in blue. F shows far-UV CD spectra of coexpressed AF4-AF9 recorded at 10°C increments from 25°C to 95°C. Insert: average molar ellipticity at 222 nm as a function of temperature.
The AF9 AHD recruits multiple partners through coupled folding and binding
In addition to AF4 family members, the AF9 AHD has previously been shown to interact with hPC3, BCoR and Dot1L. Having established that AF4 and AF9 interact through mutual synergistic folding, we hypothesized that other binding partners may interact with AF9 in a similar manner.
The interactions of AF9 with its other binding partners have been identified through a combination of co-immunoprecipitation (Co-IP) and two hybrid experiments (Erfurth et al., 2004; Hemenway et al., 2001; Srinivasan et al., 2003; Zhang et al., 2006). Interestingly, all of the sequences which interact with AF9 are in regions of their respective proteins which were predicted to be disordered by IUPRED (Dosztanyi et al., 2005)(Figure S1). We postulated that co-expression of these sequences with AF9 using a bicistronic vector may allow us to validate previous interaction studies and to finely map these interactions. To check the feasibility of this approach, we co-expressed the interacting regions of AF9 and AF4. This resulted in high expression of the AF4-AF9 complex, which was stable for extended periods and produced high quality spectra (Fig. 3a).
Figure 3. Comparison of AF9 in complex with four binding partners.
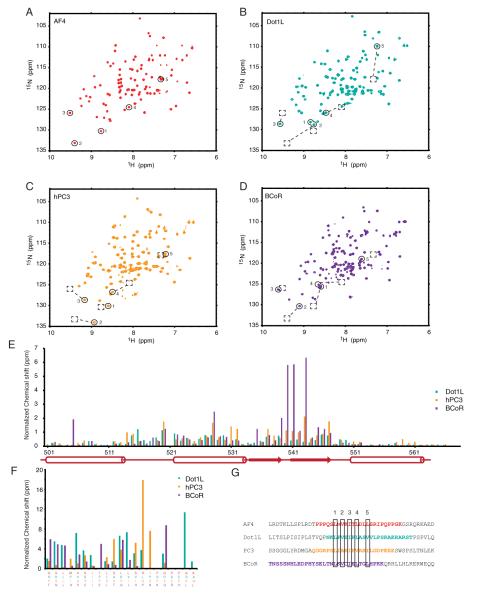
HSQC spectra of A AF4-AF9, B Dot1L-AF9, C hPC3-AF9, D BCoR-AF9. Circles show peaks corresponding to conserved residues in G, dashed boxes show position of peaks from AF4-AF9. Normalized chemical shift differences from AF4-AF9 are shown in colors corresponding to spectra, in E for AF9 and F for binding partners. Aligned sequences of interacting peptides are shown in G.
In order to investigate the other interactions with the AF9 AHD, we cloned DNA coding for the sequences derived from hPC3, Dot1L and BCoR into our coexpression vector and found that all of these co-purified with AF9. Using triple resonance based backbone assignments and 1H 15N heteronuclear NOE experiments, we were able to map the residues of these proteins involved in the interaction with the AF9 AHD. Immediately apparent is the similarity in the chemical shifts of the AF9 NH moieties observed in the HSQC spectra of these complexes. As chemical shifts are highly sensitive to structure, this similarity indicates that AF9 retains a similar fold in all of the complexes (Figure 3a-d). Alignment of the sequences of AF9 binding partners shows that although there is little sequence conservation between the binding partners, there is a consensus sequence which includes the buried aliphatic residues from AF4, corresponding to L761, V763 and I765 (Figure 3g). Additionally, each of the binding partners has two aliphatic residues in the positions occupied by leucines 767 and 770 of AF4, which form a turn in the AF4-AF9 complex. Comparison of these spectra show that the characteristically downfield shifted peaks corresponding to the beta strand in the AF4-AF9 spectra are also present in the other complexes (labeled 1-3), suggesting that the beta strand is common to each of the different complexes. Interestingly, the plot of normalized chemical shift differences in comparison to AF4-AF9 shows that the chemical shifts of peaks from AF9 are remarkably similar in each of the complexes, with the largest deviations in residues adjacent to the AF4 binding site. These are along the β2 strand, along loop 1 of AF9, and also in the α2 helix, which contains a number of aliphatic residues which are involved in packing with the buried residues of AF4. Despite having significantly different sequences, the AF9 binding partners also share similar chemical shifts, particularly through the conserved beta strand. Together, these data strongly suggest that AF9 retains a similar fold in each of the complexes, and that each of its binding partners share a common binding site on AF9. This is consistent with previous data showing that AF9 binding partners interact with the AF9 AHD in a mutually exclusive manner (Srinivasan et al., 2003; Yokoyama et al., 2010). It was previously proposed that where IDPs compete for binding to a common site, coupled folding and binding may allow one IDP to displace another without its prior dissociation, facilitating rapid exchange between high affinity partners (De Guzman et al., 2004). The differing functions of the AF9 binding partners suggest that the function of the AHD may be to regulate gene expression through alternate binding to its partners, and intrinsic disorder may facilitate their exchange as a function of local concentration and affinity.
The differences in sequence between the AF9 binding partners prompted us to determine how this affects their affinities for AF9, and presumably their binding in vivo. Our efforts were severely hampered by the hydrophobic nature of the isolated AHD and the propensity of the peptides to aggregate in a number of conditions tested. Expression of the AF9 AHD as a fusion to the Maltose Binding Protein (MBP) dramatically increased the solubility of the domain and was critical for determining binding affinities by fluorescence anisotropy. The affinity for AF4 is extremely high (KD = 0.17 ± 0.05 nM)(Figure S2). A similarly high affinity was observed for Dot1L (1.6 ± 0.3 nM ) and BCoR (32 ± 20 nM), however the peptide from hPC3 binds with much lower affinity (KD > 0.9 μM). The hPC3 sequence lacks a buried valine residue which is part of the consensus sequence (Fig. 3g), and which we hypothesized may be responsible for the lower affinity. Introduction of a valine at position 335 in the hPC3 peptide decreased the dissociation constant between AF9 and hPC3 to 8.7 ± 0.7 nM (Figure S2) demonstrating that this is indeed the case.
Dynamics of the AF9-AF4 complex assessed by 15N backbone relaxation measurements
The AF4 peptide is integral to the fold of the AF4-AF9 complex, so exchange between binding partners would require significant structural rearrangements. We therefore hypothesized that the AF4-AF9 complex may retain some conformational entropy in order to reduce the activation energy for exchange, and that this may be reflected in the backbone dynamics of the AF4-AF9 complex. To investigate this, we measured 15N longitudinal (R1) and transverse (R2) relaxation rates for the coexpressed AF4-AF9 complex as well as 1H-15N heteronuclear NOE values for the AF4-AF9 fusion (Figure 4a, b, and c). In order to analyze backbone dynamics, we calculated the product of R1 and R2, which largely results in cancellation of the effects of anisotropic motions on nuclear relaxation, and allows direct evaluation of both slow and fast timescale motions (Figure 4d) (Kneller et al., 2002). Conformational exchange on the ms-μs timescale results in elevation of the R1R2 product, whereas fast exchange (ps-ns), results in decreased R1R2. Deviation of R1R2 products from a trimmed mean are shown on the AF4-AF9 structure in figure 4E, with depressed values, corresponding to ps-ns timescale motions in red, and elevated values, corresponding to ms-μs timescale motions, in blue.
Several loops, particularly those near the AF4 peptide, are involved in conformational exchange. One of these is the loop formed by residues 539-542 at the tip of the hairpin, which is adjacent to the turn formed by AF4 residues 767-770. Another is the preceding loop formed by residues 531-535 which makes contact with the C-terminal residues of AF4. More importantly, the strand formed by AF9 residues 761-765, and parts of the helix adjacent to it exhibit dynamic behavior on a fast (ns-ps) timescale, which have been shown to correlate with sidechain conformational entropy (Gagne et al., 1998). Interestingly, the methyl resonances in the core of the protein, particularly those adjacent to the aromatic rings of AF9 F543 and F545, show surprisingly poor chemical shift dispersion, suggestive of internal dynamics within the hydrophobic core. Further support for this comes from thermal equilibrium unfolding measurements monitored by far UV CD (Figure 4). We were unable to achieve complete unfolding, even at 95°C, consistent with the high affinity of the AF4 peptide for AF9. Importantly, AF4-AF9 unfolds with a broad transition, quite different from the highly cooperative transitions observed for rigid domains with stably packed hydrophobic cores. These data suggest that aliphatic residues at the AF4-AF9 interface retain a significant amount of conformational entropy, which may partially compensate for the loss of conformational entropy during coupled folding and binding, and may be required fordynamic exchange between binding partners.
Discussion
AF9 has previously been shown to regulate a number of genes with roles in development and lineage determination. Previous studies suggest that AF9 and ENL function downstream of MLL to regulate transcriptional elongation, and the distinct functions of the AF9 binding partners suggest a direct role of AF9 in regulation of elongation by promoter proximally paused RNA PolII. We propose that the intrinsically disordered nature of the AF9 AHD and its binding partners is pivotal for integration of signaling mechanisms and transcriptional control at developmentally regulated loci.
Intrinsic disorder conveys a number of important kinetic advantages for protein interactions in signaling networks and in transcriptional regulation (Pontius, 1993; Shoemaker et al., 2000). In addition to providing fast association kinetics, it was also proposed that intrinsic disorder may facilitate displacement of one IDP by another from an overlapping binding site without its complete prior dissociation, and as such binding would be controlled by mass action (De Guzman et al., 2004).
For AF9, where the binding partners have discrete functions, this may facilitate integration of signaling networks through response to local concentrations of proteins, post translational modifications and alternative splicing. Of the AF9 binding partners, the AF9 interaction motif from BCoR is only present in specific isoforms. This suggests that alternative splicing may be a mechanism for regulation of AF9 mediated transcriptional control in a tissue specific or developmentally regulated manner. Similarly, AF4 and hPC3 contain phosphorylation sites within the AF9 consensus binding sequence, which may allow transient and reversible regulation of these interactions. For instance, phosphorylation of T766 of AF4 results in an ~ 30-fold reduction of its affinity for AF9 (Figure S2E). Interestingly, AF4 is also targeted for proteosomal degradation in the absence of binding to AF9 or ENL which would reduce the global concentration of AF4 and downregulate transcriptional elongation (Bitoun et al., 2007). Posttranslational modifications may also increase the avidity of these interactions and stabilize binding of a partner in a regulated manner. The interaction of hPC3 with AF9 is relatively weak, however the increased avidity imparted through binding of the hPC3 chromodomain to H3K27me3 may be sufficient to prevent displacement of hPC3 by other partners, resulting in stable transcriptional silencing.
The potential role for intrinsic disorder of the AF9 binding partners in enhancing exchange is clear, but the reason for the intrinsically disordered nature of AF9 is less clear. A frequently observed facet of molecular recognition through coupled folding and binding is the high specificity afforded through the extensive interface formed between the IDP and its interaction partner (Spolar and Record, 1994).The aliphatic side chains of AF4 form an integral part of the hydrophobic core of the AF4-AF9 complex, generating an extensive hydrophobic interface. However, globular proteins are largely stabilized by van der Waals contacts from close packing of hydrophobic side chains, and their unfolding initially requires unlocking of this core with an associated increase in side chain conformational entropy to form a dry molten globule intermediate (Baldwin et al., 2010). Therefore if the AF4-AF9 complex exhibited the same close packing interactions as globular proteins, unlocking of the protein core would represent a significant free energy barrier to the exchange between binding partners. It is likely that retention of conformational entropy in the core of the AHD, reflected in the 15N backbone relaxation data, may reduce the free energy barrier for exchange between high affinity binding partners. Correspondingly, the lack of an optimally packed core may result in the absence of independent folding of the AHD. Through formation of a slightly ‘Fuzzy’ complex (Tompa and Fuxreiter, 2008), it may therefore be possible to retain both high affinity binding, coupled with a low energy barrier for exchange in response to changes in protein concentrations or modifications. Such behavior is likely to be important for regulation of developmentally critical genes which must be turned on or off with high fidelity, but in a rapid and synchronous manner.
Control of transcriptional elongation through release of promoter proximally paused RNA PolII allows rapid and highly synchronous induction of transcription in processes such as cellular differentiation and embryonic patterning as well as in the heat shock response (Boettiger and Levine, 2009). At HSP70 loci, this is stimulated through sequential recruitment of transcription factors (Zobeck et al., 2010). Intrinsic disorder in the AF9 AHD and its partners may facilitate this sequential recruitment to repress or activate transcription in a highly regulated manner. For instance, MLL regulates transcriptional initiation through methylation of H3K4 and recruitment of CBP/p300. AF9 or ENL may then stabilize promoter proximal pausing through recruitment of BCoR to deacetylate H3K27. Trimethylation of H3K27 would increase binding avidity of hPC3 resulting in a stably repressed but poised promoter. Conversely, H3K79 methylation has been proposed to antagonize epigenetic silencing, and so we envisage that Dot1L may be recruited to inhibit polycomb mediated silencing, either before or after release of RNA PolII pausing by recruitment of P-TEFb by AF4. Direct fusion of AF9 to MLL results in loss of context dependence of AF9 recruitment, and the high affinity of AF4 for AF9 suggests that this may dominate binding to MLL-AF9 leading to upregulation of HOXA9 and MEIS1 and ultimately to leukemia.
Experimental procedures
Protein preparation and construct design
For initial studies of the AF9 AHD, we expressed residues 490-568 of AF9 from pET 32a with the region coding for thioredoxin removed. Proteins were purified by Ni-NTA and size exclusion chromatography. To prepare a saturated sample of AF9 bound to the minimal AF4 peptide, the peptide was added to a dilute sample of purified AF9 AHD and incubated at room temperature overnight. The sample was concentrated by ultrafiltration and the complex purified by size exclusion chromatography.
The AF4-AF9 fusion included an N-terminal 6-his tag and TEV protease site, with the coding sequence for AF4 residues 738-779 directly fused to AF9 residues 490-568 in pET 32a (Novagen). Proteins were expressed in Rosetta2DE3 cells (Novagen) in EMBL medium supplemented with 10ml/L of Bioexpress (Cambridge Isotopes), where necessary substituting with isotopically labeled bioexpress, 15N labeled ammonium sulfate or 13C labeled glucose. Proteins were purified by Ni-NTA chromatography, purification tags were removed by digestion with Enterokinase (AF9 AHD)(NEB) or TEV protease (AF4-AF9) and aggregates and impurities removed by Gel Filtration chromatography using a Superdex 75 column (GE life sciences).
Co-expression constructs were generated by cloning DNA coding for AF9 residues 500-568 and for binding partners into pET Duet 1 modified by addition of a TEV cleavage site between the His-tag and the fusion partner. Proteins were expressed and purified as for the AF4-AF9 fusion. Residues that remained rigid in the complex were identified using assignments from HNCACB and CBCA(CO)NH experiments and heteronuclear NOE experiments performed as detailed in supplementary data.
Circular Dichroism experiments
Circular dichroism samples were exchanged into 10mM potassium phosphate, 10 mM NaCl, 0.5 mM DTT at the gel filtration step. CD experiments were conducted with 3.7 μM AF9 or 9.4 μM AF4-AF9, data collected at 1 nm intervals with 3 s averaging time. Melting experiments were conducted with 20 μM coexpressed AF4-AF9. Spectra are shown as an average of 16 scans.
All data was collected on an Aviv 410 spectropolarimeter.
Fluorescence Anisotropy binding experiments
DNA coding for AF9 residues 475-568 of mouse AF9 were cloned into pMAL C2 (NEB) and the protein expressed in Rosetta2DE3 cells in LB medium. MBP-AF9 was purified on Amyloseresin (NEB) and impurities and aggregates removed immediately by size exclusion chromatography. Proteins were titrated into 0.5 nM (AF4) or 1nM (BCoR, hPC3 or Dot1L) peptide in 50 mM Tris-HCl, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 8.5 and incubated for 1 hour at room temperature. Anisotropy measurements were recorded on a Pherastar plate reader. Data were fitted to a single site binding model accounting for ligand depletion (Veiksina et al., 2010).
NMR methods and structure determination
Full details of NMR methods are included in the supplementary data.
All NMR experiments for initial structure determination were conducted with a 0.6 mM sample of AF4-AF9 in 25 mM Bis-Tris/MES pH 6.0, 100 mM NaCl, 1 mM DTT at 25°C.
Supplementary Material
Highlights.
The AF9 AHD is intrinsically disordered
The AHD recruits AF4, BCoR, Dot1L and hPC3 by coupled folding and binding
AF9 binding partners compete for binding to a common site
Dynamics of the AF4-AF9 complex may facilitate exchange between partners
Acknowledgements
We thank N. Zeleznik-Le for and C. Hemenway for critical discussions and reading the manuscript. JHB was supported by the William Lawrence and Blanche Hughes Foundation and the National Cancer Institute (RO1 CA155328)
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- Baldwin RL, Frieden C, Rose GD. Dry molten globule intermediates and the mechanism of protein unfolding. Proteins. 2010;78:2725–2737. doi: 10.1002/prot.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardos JI, Saurin AJ, Tissot C, Duprez E, Freemont PS. HPC3 is a new human polycomb orthologue that interacts and associates with RING1 and Bmi1 and has transcriptional repression properties. J Biol Chem. 2000;275:28785–28792. doi: 10.1074/jbc.M001835200. [DOI] [PubMed] [Google Scholar]
- Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- Boettiger AN, Levine M. Synchronous and stochastic patterns of gene activation in the Drosophila embryo. Science. 2009;325:471–473. doi: 10.1126/science.1173976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra VS, Hong JW, Levine M. Regulation of Hox gene activity by transcriptional elongation in Drosophila. Curr Biol. 2009;19:688–693. doi: 10.1016/j.cub.2009.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman RN, Martinez-Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J Biol Chem. 2004;279:3042–3049. doi: 10.1074/jbc.M310348200. [DOI] [PubMed] [Google Scholar]
- Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–626. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Erfurth F, Hemenway CS, de Erkenez AC, Domer PH. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia. 2004;18:92–102. doi: 10.1038/sj.leu.2403200. [DOI] [PubMed] [Google Scholar]
- Gagne SM, Tsuda S, Spyracopoulos L, Kay LE, Sykes BD. Backbone and methyl dynamics of the regulatory domain of troponin C: anisotropic rotational diffusion and contribution of conformational entropy to calcium affinity. J Mol Biol. 1998;278:667–686. doi: 10.1006/jmbi.1998.1723. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex(SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemenway CS, de Erkenez AC, Gould GC. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene. 2001;20:3798–3805. doi: 10.1038/sj.onc.1204478. [DOI] [PubMed] [Google Scholar]
- Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000;14:1810–1823. [PMC free article] [PubMed] [Google Scholar]
- Kneller JM, Lu M, Bracken C. An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc. 2002;124:1852–1853. doi: 10.1021/ja017461k. [DOI] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–1499. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterbauer G, Zimmer C, Fonatsch C, Haas O, Thalhammer-Scherrer R, Schwarzinger I, Kalhs P, Jaeger U, Lechner K, Mannhalter C. Monitoring of minimal residual leukemia in patients with MLL-AF9 positive acute myeloid leukemia by RT-PCR. Leukemia. 1999;13:1519–1524. doi: 10.1038/sj.leu.2401542. [DOI] [PubMed] [Google Scholar]
- Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, Shilatifard A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom) Genes Dev. 2010;24:574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, Basrur V, Elenitoba-Johnson KS, Hess JL. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17:609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T, Hardin S, Greenleaf A, Lis JT. Phosphorylation of RNA polymerase II Cterminal domain and transcriptional elongation. Nature. 1994;370:75–77. doi: 10.1038/370075a0. [DOI] [PubMed] [Google Scholar]
- Palermo CM, Bennett CA, Winters AC, Hemenway CS. The AF4-mimetic peptide, PFWT, induces necrotic cell death in MV4-11 leukemia cells. Leuk Res. 2008;32:633–642. doi: 10.1016/j.leukres.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius BW. Close encounters: why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem Sci. 1993;18:181–186. doi: 10.1016/0968-0004(93)90111-y. [DOI] [PubMed] [Google Scholar]
- Prasad R, Yano T, Sorio C, Nakamura T, Rallapalli R, Gu Y, Leshkowitz D, Croce CM, Canaani E. Domains with transcriptional regulatory activity within the ALL1 and AF4 proteins involved in acute leukemia. Proc Natl Acad Sci U S A. 1995;92:12160–12164. doi: 10.1073/pnas.92.26.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci U S A. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolar RS, Record MT., Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- Srinivasan RS, de Erkenez AC, Hemenway CS. The mixed lineage leukemia fusion partner AF9 binds specific isoforms of the BCL-6 corepressor. Oncogene. 2003;22:3395–3406. doi: 10.1038/sj.onc.1206361. [DOI] [PubMed] [Google Scholar]
- Srinivasan RS, Nesbit JB, Marrero L, Erfurth F, LaRussa VF, Hemenway CS. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia. 2004;18:1364–1372. doi: 10.1038/sj.leu.2403415. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in proteinprotein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Veiksina S, Kopanchuk S, Rinken A. Fluorescence anisotropy assay for pharmacological characterization of ligand binding dynamics to melanocortin 4 receptors. Anal Biochem. 2010;402:32–39. doi: 10.1016/j.ab.2010.03.022. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLLdependent transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–18068. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobeck KL, Buckley MS, Zipfel WR, Lis JT. Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol Cell. 2010;40:965–975. doi: 10.1016/j.molcel.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.