

Abstract
The pericentriolar stacks of Golgi cisternae are separated from each other in G2 and fragmented extensively during mitosis. MEK1 is required for Golgi fragmentation in G2 and for the entry of cells into mitosis. We now report that Myt1 mediates MEK1’s effects on the Golgi complex. Knockdown of Myt1 by siRNA increased the efficiency of Golgi complex fragmentation by mitotic cytosol in permeabilized and intact HeLa cells. Myt1 knockdown eliminated the requirement of MEK1 in Golgi fragmentation and alleviated the delay in mitotic entry due to MEK1 inhibition. The phosphorylation of Myt1 by MEK1 requires another kinase but is independent of RSK, Plk, and CDK1. Altogether our findings reveal that Myt1 is inactivated by MEK1 mediated phosphorylation to fragment the Golgi complex in G2 and for the entry of cells into mitosis. It is known that Myt1 inactivation is required for CDK1 activation. Myt1 therefore is an important link by which MEK1 dependent fragmentation of the Golgi complex in G2 is connected to the CDK1 mediated breakdown of Golgi into tubules and vesicles in mitosis.
Keywords: CDK1, Golgi membranes, MEK1, Mitosis, Myt1
Introduction
The pericentriolar stacks of Golgi cisternae are separated from each other in G2 (Shima et al, 1998). The Golgi stacks continue to fragment until metaphase and appear to be composed of small tubules and vesicles (Jesch and Linstedt, 1998; Jokitalo et al, 2001; Axelsson and Warren, 2004; Pecot and Malhotra, 2004). In late anaphase/telophase, the fragmented Golgi membranes fuse and assemble into stacks of Golgi cisternae by a process that also involves membrane export from the ER, and finally the stacks are localized to the pericentriolar region of the cells after cytokinesis (Lucocq et al, 1987; Lucocq and Warren, 1987; Colanzi et al, 2007; Persico et al, 2009). This process thus ensures the partitioning of Golgi membranes into daughter cells during cell division (Shorter and Warren, 2002). Surprisingly, inhibiting fragmentation of the Golgi complex in G2 prevents or delays entry of cells into mitosis (Sutterlin et al, 2002; Preisinger et al, 2005; Colanzi et al, 2007; Feinstein and Linstedt, 2007).
What is the role of the pericentriolar stacks of Golgi cisternae and why does inhibiting stack separation and dispersal arrest cells in G2? Incubation of isolated Golgi membranes or permeabilized cells, with mitotic cytosol and an ATP-regenerating system causes extensive Golgi membrane fragmentation, and these procedures have revealed the involvement of three different kinases: the mitogen activated kinase kinase (MEK1), polo like kinase (Plk), and cyclin dependent kinase CDK1 (Misteli and Warren, 1994; Acharya et al, 1998; Lowe et al, 1998; Colanzi et al, 2000, 2003; Sutterlin et al, 2001). MEK1 and polo like kinase (Plk) are required for the dispersal of the pericentriolar Golgi apparatus into smaller stacks and fragments, which then breakdown into small tubules and vesicles by a CDK1 dependent process (Lowe et al, 1998; Kano et al, 2000; Sutterlin et al, 2001; Shorter and Warren, 2002). A protein called Golgi matrix protein 130 (GM130) localized on the cis side of the Golgi membranes is phosphorylated by CDK1 at Ser25 (Lowe et al, 1998). However, new recent evidences suggest that phosphorylation of Ser25 in GM130 is not required for Golgi membrane fragmentation and mitotic progression (Sundaramoorthy et al, 2010). Plk binds to the CDK1 phosphorylated Golgi membrane associated protein GRASP65; however the net effect of this reaction on the Golgi complex during mitosis is poorly understood (Lin et al, 2000).
MEK1 phosphorylates the Golgi membrane protein GRASP55 (Feinstein and Linstedt, 2008). Importantly, depletion of GRASP55 alleviates the requirement of MEK1 in the process of Golgi membrane fragmentation. The data suggests that GRASP55 is required for connecting Golgi stacks laterally; phosphorylation by MEK1/Extracellular signal-regulated kinase (ERK) inactivates the function of GRASP55, which leads to the separation of Golgi stacks from each other in G2 and thus the entry of cell into mitosis (Feinstein and Linstedt, 2008). However, the function of MEK1 in the Golgi membrane fragmentation is also reported to be independent of its well-characterized targets ERK1/ERK2 (Acharya et al, 1998). For example, deletion of the N-terminal residues of MEK1 required for binding ERK1/ERK2 does not affect its ability to fragment Golgi membranes (Colanzi et al, 2000). There are also reports of a specific spliced variant of ERK, ERK1c, as the target of MEK1 dependent Golgi membrane fragmentation (Shaul and Seger, 2006). It is likely that MEK1 has additional targets on the Golgi membranes or that MEK1 does not directly phosphorylate GRASP55 to catalyse Golgi stacks separation.
As mentioned above, the overall breakdown of Golgi complex into tubules and vesicles is mediated sequentially by MEK1 and CDK1, respectively. How are these events connected? We have tested the hypothesis that these sequential events are connected by the ER-Golgi complex associated kinase called Myt1, which is expressed only in the metazoans (Liu et al, 1997). Our assumption is based on the following facts. Myt1 phosphorylates CDK1 on Thr14 and Tyr15 (Mueller et al, 1995) and the Myt1 phosphorylated CDK1 is inactive (Booher et al, 1997). Inactivation of Myt1 is therefore necessary for the activation of CDK1 and entry into mitosis. MEK1 is known to phosphorylate Myt1 via p90RSK (90 kDa ribosomal S6 kinase) and this event is required for the entry of Xenopus oocytes into meiosis (Palmer et al, 1998). We now report that MEK1 inactivates Myt1 in mammalian somatic cells. We show that this reaction is required for the process by which Stacks of Golgi cisternae are separated from each other -independent of CDK1- in G2 and promote entry of cells into mitosis. Surprisingly, the MEK1 dependent inactivation of Myt1 is RSK and CDK1 independent. The description of our findings on the involvement of Myt1 in Golgi membrane reorganization and mitotic entry follows.
Results
Myt1 knockdown promotes early entry into mitosis
HeLa cells were arrested in S-phase or mitosis (pre-metaphase) by incubation for 18 h with thymidine or nocodazole, respectively, and the cell lysates western blotted with an anti-Myt1 antibody. Compared with S-phase cells, Myt1 in mitotic cells migrated as a higher mol.wt polypeptide (Figure 1A). The total cell lysate of cells arrested in mitosis was incubated with λ protein phosphatase and probed by western blotting with an anti-Myt1 antibody. Phosphatase treatment shifted Myt1 to the size observed in non-mitotic cells (Figure 1B). This confirms the mitosis specific phosphorylation of Myt1 reported by Nishida and colleagues (Nakajima et al, 2008). To investigate the kinetics of Myt1 phosphorylation during mitotic progression, HeLa cells were arrested in S-phase with a double thymidine block, thymidine was then removed and the cells placed in normal thymidine free medium. At the indicated times, cells were lysed and the lysates western blotted with an anti-Myt1 antibody. Myt1 phosphorylation peaked at 10 h post release from the double thymidine block, and contrary to a previous report (Nakajima et al, 2008), Myt1 was dephosphorylated and not degraded after M-phase (Figure 1C).
Figure 1.
Myt1 knockdown promotes early entry into mitosis. (A) HeLa cells were incubated for 18 h with DMSO (asynchronized cells), thymidine (S-phase cells) or nocodazole (mitotic cells), and Myt1 expression was analysed by western blotting the total cell lysate. Western blotting with an anti-β-actin antibody was used as a loading control. (B) Lysates from cells synchronized in mitosis were treated with or without λ protein phosphatase and analysed by western blotting with an anti-Myt1 and an anti-β-actin antibody, respectively. (C) HeLa cells were arrested in S-phase with a double thymidine block, the cells were washed to remove thymidine and at the indicated times the total cell lysates were western blotted with an anti-Myt1 antibody. Western blotting with an anti-β-actin antibody was used as a loading control and western blotting with an anti-cyclin-B1 antibody was used to monitor the progression of cells into mitosis. (D) HeLa cells were transfected with control or Myt1 specific siRNA oligos and after 48 and 72 h, respectively, the levels of Myt1 were analysed by western blotting the total cell lysates. Western blotting with an anti-β-actin antibody was used as a loading control. (E) Quantitation of Myt1 protein upon siRNA transfection. (F) The percentage of cells in mitosis (mitotic index) was determined by staining DNA with DAPI and an anti-phospho-histone H3 antibody at the indicated times after thymidine release for control and Myt1 siRNA transfected cells. 400 cells were counted for each time point (mean±s.d., n=3). (G) The percentage of cells in late G2 in the total number of cells in late G2 and all stages of mitosis was determined at the indicated times after thymidine release for control and Myt1 siRNA transfected cells. For each time point, 400 cells were counted (mean±s.d., n=3).
Source data for this figure is available on the online supplementary information page.
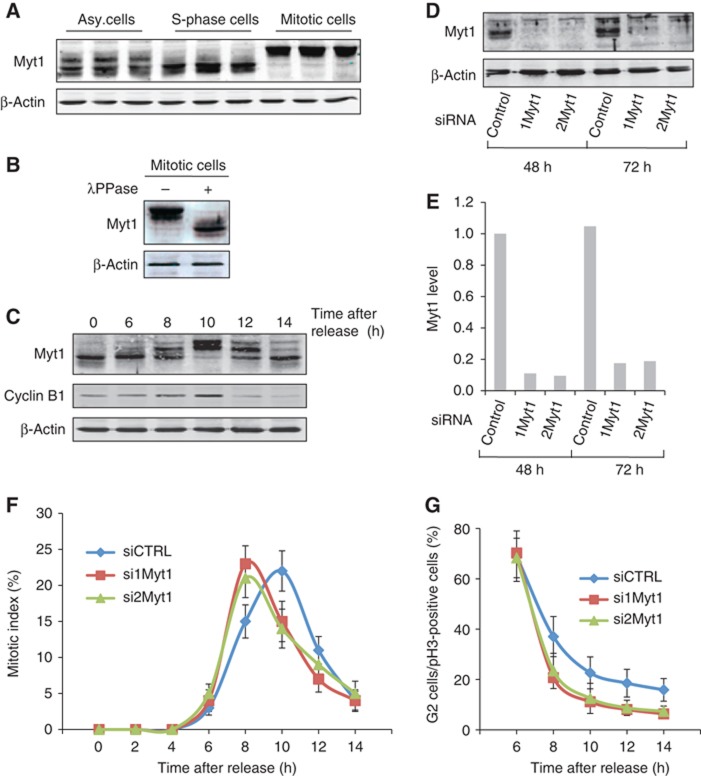
Is Myt1 required for mitotic entry and progression under our experimental conditions? HeLa cells were transfected with control and Myt1 specific siRNA oligos, and the cell lysates western blotted with an anti-Myt1 antibody. Transfection with Myt1 specific siRNA oligo reduced Myt1 level by 80% compared to control cells (Figures 1D and E). The same procedure of Myt1 knockdown was repeated in cells arrested in S-phase with a double thymidine block, the cells were then washed to remove thymidine and incubated in normal medium. At the indicated times, the cells were stained with a DNA binding dye DAPI to identify mitotic cells, and with an anti-phospho-histone H3 antibody to identify cells in late G2-phase and in mitosis (Colanzi et al, 2007). In control cells, a mitotic index (cells in mitosis/total number of cells) of 25% was observed at 10 h after removal of the cells from S-phase block. Knockdown of Myt1 did not increase the mitotic index, however, the peak of mitotic index was observed at 8 h post removal from the S-phase block (Figure 1F). In other words, Myt1 knockdown changed the kinetics of entry into mitosis, and the peak of mitotic index was observed at 8 h instead of 10 h. To further test the involvement of Myt1 in G2 to M-phase transition, we counted the number of cells in late G2-phase (uncondensed DNA and specific punctate phospho-histone H3 staining, as also shown by Corda and colleagues (Colanzi et al, 2007)) and the total number of cells that were in late G2 and M-phase (all the cells that are phospho-histone H3 positive from late G2 to all the mitotic stages). In control and Myt1 siRNA transfected cells, 6 h after thymidine release, 70% of phospho-histone H3 positive cells were in G2. Interestingly, 8 h after thymidine release, 40% of phospho-histone H3 positive control cells were still in G2 whereas in Myt1 knockdown cells, only 20% of phospho-histone H3 positive cells were in G2 (Figure 1G). These results suggested that Myt1 knockdown increases the kinetics of G2-M transition.
The role of Myt1 in the organization of Golgi complex
We investigated the role of Myt1 in Golgi complex organization and function in interphase cells. HeLa cells were transfected with control or Myt1 specific siRNA oligos and after 48 h, the cells were fixed and visualized by fluorescence microscopy with an anti-TGN46 (of the Trans Golgi Network) and anti-GM130 (of the early Golgi cisternae) antibody, respectively. Myt1 knockdown did not affect the overall organization of the Golgi complex (Figure 2A). Moreover, fluorescence recovery after photobleaching an area of the Golgi membranes revealed no obvious kinetic difference between control and Myt1 specific siRNA transfected cells. Thus unlike the dissociation of Golgi stacks from each other upon GRASP55 knockdown in HeLa cells (Feinstein and Linstedt, 2008), Myt1 knockdown does not affect the lateral connexions between Golgi stacks (Figure 2B). HeLa cells stably expressing signal sequence and V5 tagged horseradish peroxidase (ss-HRP) were transfected with control, Myt1, or SCFD1 (a Sec family domain containing 1) specific siRNA oligos and the quantity of HRP secreted by the cells was monitored by chemiluminescence as described previously (von Blume et al, 2009). Knockdown of SCFD1, which is involved in vesicular transport between the ER and the Golgi apparatus, significantly inhibited HRP secretion, whereas knockdown of Myt1 was without any obvious effect (Figure 2C). Altogether, these data reveal that Myt1 knockdown does not affect Golgi membrane organization and protein secretion in non-mitotic HeLa cells.
Figure 2.
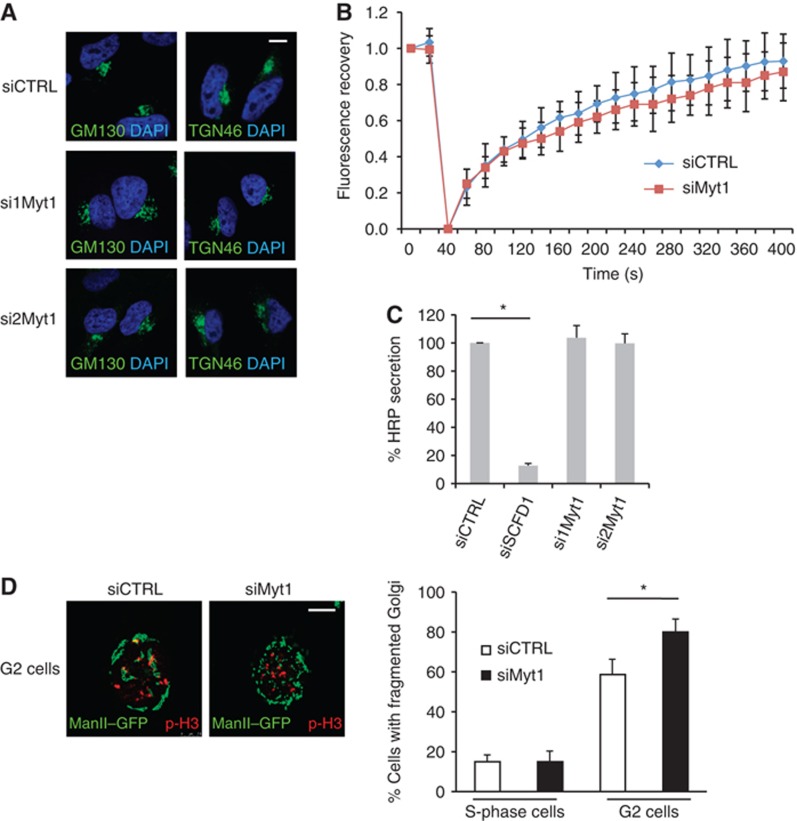
Myt1 knockdown does not modify Golgi organization and function in S-phase but promotes fragmentation of the Golgi complex in late G2. (A) HeLa cells grown on coverslips were transfected with control or Myt1 specific siRNA oligos. 48 h after transfection, cells were fixed and processed for immunofluorescence microscopy with DAPI and antibodies to GM130 and TGN46, respectively. Scale bar is 10 μm. (B) HeLa cells expressing ManII-GFP were transfected with control or Myt1 specific siRNA oligo. 48 h after transfection, the central area of the Golgi complex was bleached and the recovery of fluorescence monitored for 400 s. The fluorescence recovery was quantified as the ratio of GFP fluorescence of the bleached and the unbleached Golgi membrane area and normalized. The rate of recovery in control and Myt1 siRNA transfected cells was plotted and shown (mean±s.d., n=3, >10 cells each). (C) The media from control, SCFD1 and Myt1 knockdown HeLa cells stably expressing ss-HRP were used to detect HRP secretion by chemiluminescence. HRP activity in the medium was normalized to the total HRP activity in the cell lysates (mean±s.d., n=3, *P<0.05). (D) Left panel. Control and Myt1 siRNA transfected HeLa cells stably expressing ManII-GFP were arrested in S-phase with a double thymidine block. Cells were washed to remove thymidine, incubated for 8 h in thymidine free medium, fixed and stained with an anti-phospho-histone H3 antibody and visualized by fluorescence microsopy. The images show cells in G2. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi in S-phase and G2 in control and in Myt1 knockdown cells. For each condition, 200 cells on 2 different coverslips were counted (mean±s.d., n=3, *P<0.05).
Preventing fragmentation of the pericentriolar Golgi complex in G2 is known to prevent or delay entry of the cells into mitosis (Sutterlin et al, 2002; Preisinger et al, 2005; Colanzi et al, 2007; Feinstein and Linstedt, 2007). Is Myt1 required for the fragmentaion of Golgi complex in G2? Control and Myt1 siRNA transfected cells stably expressing Mannosidase II (ManII)-GFP were arrested in S-phase with a double thymidine block. The cells were then washed to remove thymidine and incubated in normal medium. After 8 h, the cells were stained with an anti-phospho-histone H3 antibody to identify the cells in G2. We visualized the organization of Golgi membranes in these cells stably expressing ManII-GFP by fluorescence microscopy. Quantitation of this data revealed that the Golgi complex was fragmented in 60% of the control cells in G2. However, 80% of the cells transfected with Myt1 siRNA had fragmented Golgi complex in G2 (Figure 2D). Myt1 knockdown did not affect the Golgi membrane organization in cells arrested in S-phase by thymidine treatment (Figure 2D). Taken together these findings indicate that Myt1 knockdown by siRNA promotes Golgi membrane fragmentation in G2.
We then tested whether Myt1 was required for fragmentation of the Golgi complex in permeabilized HeLa cells incubated with mitotic cytosol (Acharya et al, 1998). Briefly, permeabilized HeLa cells stably expressing ManII-GFP were incubated with cytosol prepared from HeLa cells treated with thymidine (S-phase block) or nocodazole (pre-metaphase block), and an ATP-regenerating system at 32°C for 1 h. The cells were visualized by fluorescence microscopy and revealed extensive fragmentation of the Golgi complex in 65% of permeabilized cells incubated with mitotic cytosol (Figure 3A).
Figure 3.
Fragmentation of the Golgi complex with mitotic cytosol in permeabilized cells. (A) Left panel. HeLa cells stably expressing ManII-GFP were grown on coverslips and incubated with thymidine for 12 h before permeabilization. Permeabilized cells were incubated with an ATP-regenerating system and either KHM buffer (top panel), S-phase cytosol (centre panel), or mitotic cytosol (bottom panel), at 32°C for 1 h. Cells were fixed and visualized by fluorescence microscopy. Scale bar is 10 μm. Right panel. Quantitation of the experimental data. 200 cells on 2 different coverslips were counted (mean±s.d., n=3, *P<0.05) for each experimental condition to obtain the percentage of cells with fragmented Golgi complex. (B) Left panel. Total cell lysate, S-phase, and mitotic cytosol were western blotted with the antibodies shown. Right panel. The level of the same (indicated) proteins was analysed by western blotting the total cell lysate of non-permeabilized cells and cells permeabilized for 1, 2 and 3 min with digitonin and washed with 1M KCl in KHM buffer.
Source data for this figure is available on the online supplementary information page.
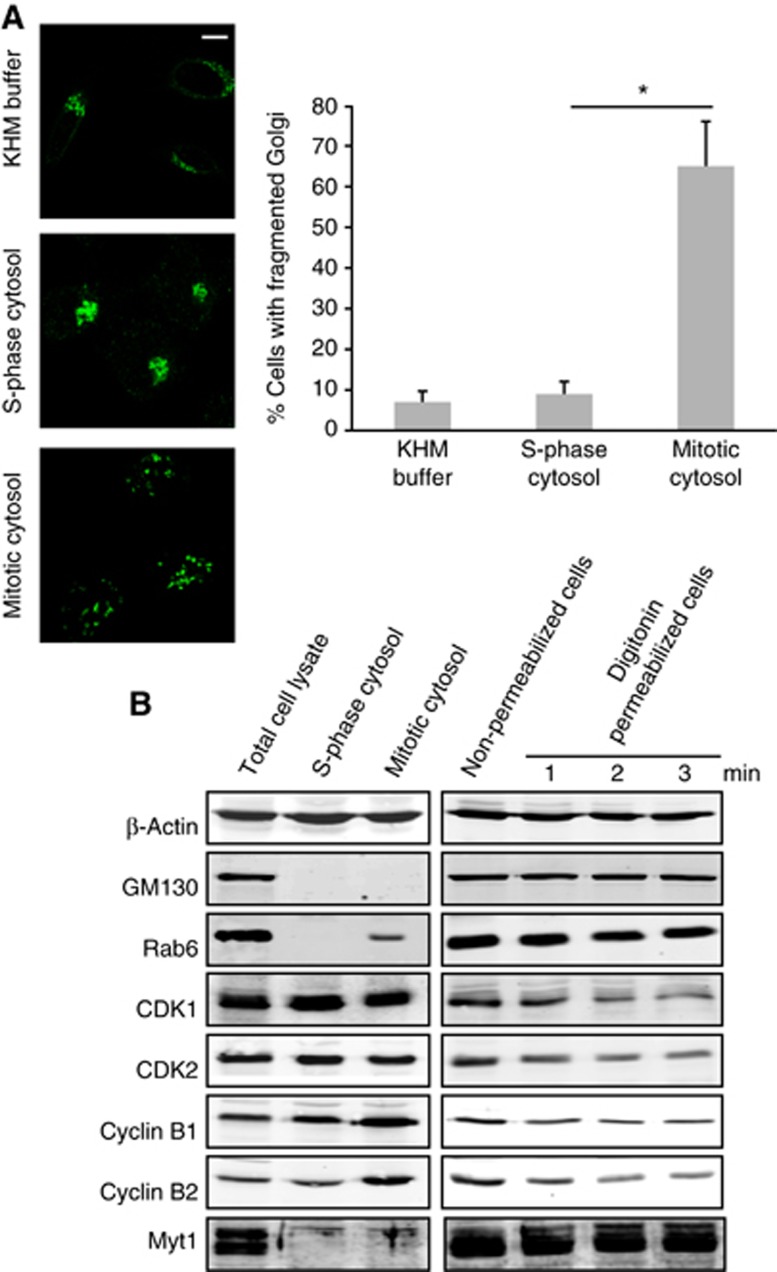
Myt1 is anchored to the cytoplasmic surface of the Golgi membranes and the ER and we tested whether it was contained in the mitotic cytosol preparation used for the fragmentation of the pericentriolar Golgi complex in permeabilized HeLa cells. Permeabilized HeLa cells and the cytosol prepared from S-phase (thymidine treated cells) and mitotic cytosol (nocodazole treated cells) were analysed by western blotting. Myt1 is detected in the permeabilized HeLa cells but not in the S-phase or in the mitotic cytosol preparations (Figure 3B). Therefore, Myt1 is not contained in the S-phase or the mitotic cytosol used in our experimental procedures. We then transfected HeLa cells stably expressing ManII-GFP with control and Myt1 specific siRNA oligos for 48 h. Cells were permeabilized, washed to remove cytoplasmic proteins, incubated with S-phase or mitotic cytosol, and the organization of the Golgi complex monitored by fluorescence microscopy. Incubation of HeLa cells transfected with control siRNA oligo and mitotic cytosol revealed fragmented Golgi complex in 60% of the cells. The percentage of HeLa cells with fragmented Golgi complex increased to almost 100% in cells transfected with Myt1 specific siRNA oligos (Figure 4A). Thus knockdown of Myt1 potentiated the ability of mitotic cytosol to fragment Golgi complex. However, this effect was mitotic cytosol specific as there was no change in the organization of the Golgi complex in Myt1 knockdown cells incubated with S-phase cytosol (Figure 4A). To further ascertain the effect of Myt1 knockdown on fragmentation of the Golgi complex by mitotic cytosol, we incubated HeLa cells transfected with control or Myt1 specific siRNA oligos with serial dilutions of mitotic cytosol. After 1 h at 32°C, the organization of the Golgi membranes was monitored by fluorescence microscopy. We found that 2-fold diluted mitotic cytosol was ineffective in fragmenting Golgi complex in cells transfected with control siRNA oligo. However, 4-fold diluted mitotic cytosol fragmented Golgi membranes in cells transfected with Myt1 specific siRNA oligos (Figure 4B). This further suggests that depletion of Myt1 increases the potency of mitotic cytosol to fragment Golgi complex.
Figure 4.
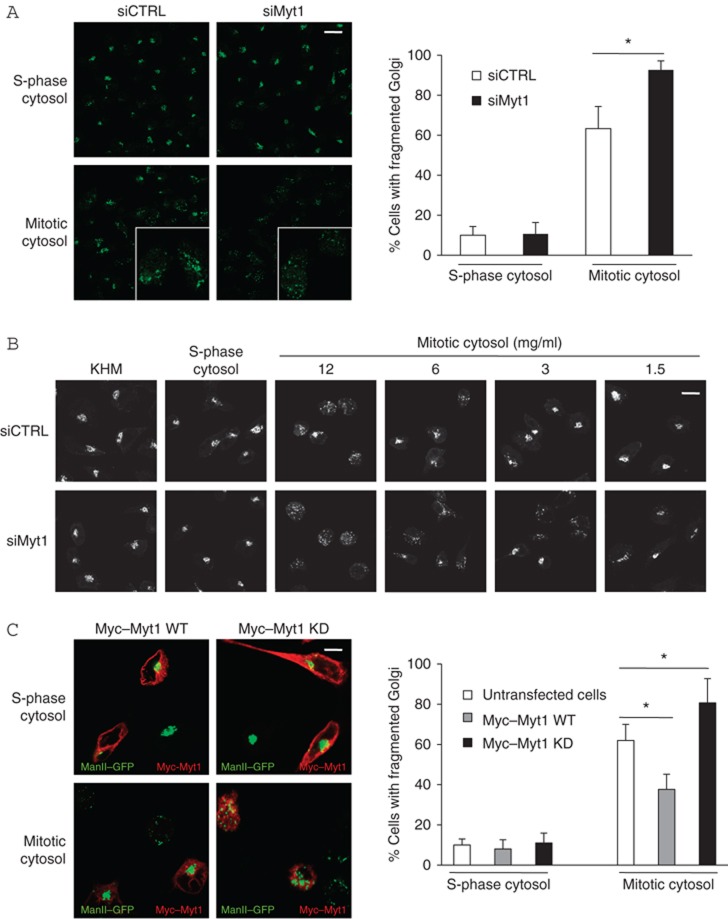
Myt1 inactivation promotes fragmentation of the Golgi complex. (A) Left panel. HeLa cells stably expressing ManII-GFP were transfected with control or Myt1 specific siRNA oligo. After incubation with thymidine for 12 h, cells were permeabilized, salt-washed and incubated with S-phase cytosol (top panel) or mitotic cytosol (bottom panel), and visualized by fluorescence microscopy. Scale bar is 40 μm. Right panel. Percentage of cells with fragmented Golgi upon incubation with S-phase or mitotic cytosol in control and in Myt1 knockdown cells. For each condition, 200 cells on 2 different coverslips were counted (mean±s.d., n=3, *P<0.05). (B) HeLa cells stably expressing ManII-GFP were transfected with control or Myt1 specific siRNA oligo. After incubation with thymidine for 12 h, permeabilized and salt-washed cells were incubated with KHM buffer, S-phase cytosol, or serial dilutions of mitotic cytosol. The organization of Golgi membranes was analysed by fluorescence microscopy. Scale bar is 20 μm. (C) Left panel. HeLa cells stably expressing ManII-GFP were transfected with Myc-Myt1 wild type (WT) plasmid and Myc-Myt1 kinase dead (KD) plasmid. After incubation with thymidine for 12 h, permeabilized and salt-washed cells were incubated with S-phase cytosol (top panel) or with mitotic cytosol (bottom panel) and visualized by fluorescence microscopy. Transfected cells were identified by staining with an anti-Myc antibody. Scale bar is 15 μm. Right panel. Percentage of cells with fragmented Golgi upon incubation with S-phase or mitotic cytosol in untransfected cells, cells expressing Myc-Myt1 WT, or Myc-Myt1 KD. For each condition, 200 cells on 2 different coverslips were counted (mean±s.d., n=3, *P<0.05).
To determine whether the kinase activity of Myt1 was required for fragmentation of the Golgi complex, HeLa cells were transfected with a wild type (WT) or a kinase dead (KD) variant of Myc-Tagged Myt1. HeLa cells stably expressing ManII-GFP were permeabilized and incubated in the presence of an ATP-regenerating system at 32°C, for 1 h with S-phase or mitotic cytosol, respectively. The organization of the Golgi membranes was monitored by fluorescence microscopy. Transfection of the WT or the KD form of Myt1 had no effect on the organization of Golgi complex in permeabilized cells incubated with S-phase cytosol. However, incubation of cell expressing Myt1-WT with mitotic cytosol decreased the number of cells with fragmented Golgi complex (from 60–65% to under 40%) whereas the expression of the KD form of Myt1 increased the number of cells with fragmented Golgi complex to 80% (Figure 4C).
Taken together, these results strongly suggest that Myt1 inhibits fragmentation of the Golgi complex by mitotic cytosol; its knockdown, or overexpression of a KD form, increases the efficiency of this reaction.
MEK1 regulates Golgi membrane fragmentation via Myt1
Plk is known to phosphorylate and inactivate Myt1 (Nakajima et al, 2003; Inoue and Sagata, 2005). Is Myt1 involved in Plk dependent Golgi complex fragmentation by mitotic cytosol? We depleted Plk from mitotic cytosol by immunoadsorption with an anti-Plk antibody. This procedure resulted in greater than 70% depletion of Plk from the mitotic cytosol (Figure 5A and B). Permeabilized HeLa cells were incubated with control or Plk-depleted mitotic cytosol and after 1 h incubation at 32°C, in the presence of an ATP- regenerating system visualized by fluorescence microscopy. Incubation of permeabilized cells with Plk-depleted mitotic cytosol significantly inhibited fragmentation of the Golgi complex (Figure 5C). We then tested the effect of Myt1 knockdown on Plk dependent Golgi complex fragmentation. HeLa cells were transfected with control or Myt1 specific siRNA oligos as described above and the cells were incubated with either control or Plk-depleted mitotic cytosol. Fluorescence microscopy revealed that knockdown of Myt1 did not alleviate the requirement of Plk for the mitotic cytosol dependent Golgi complex fragmentation (Figure 5D). This suggests that Myt 1 is not downstream of the Plk mediated Golgi fragmentation process.
Figure 5.
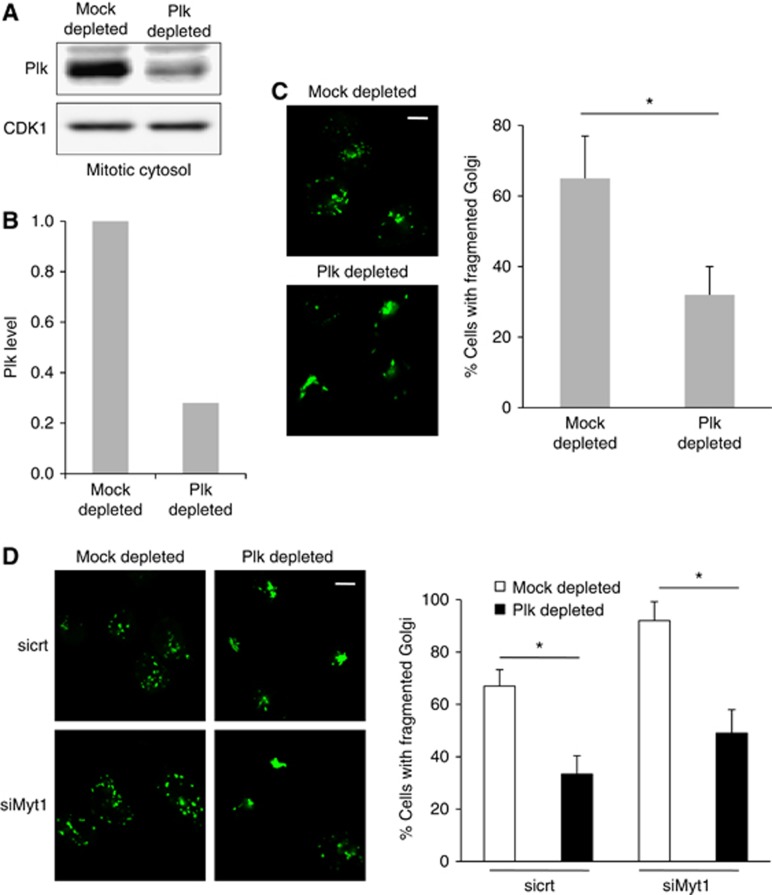
Plk does not regulate fragmentation of the Golgi complex via Myt1. (A) Mock and Plk-depleted mitotic cytosol were western blotted with an anti-Plk antibody. Western blotting with an anti-CDK1 antibody was used as a loading control. (B) Quantification of Plk levels in mitotic cytosol upon immunodepletion. (C) Left panel. After incubation with thymidine for 12 h, permeabilized and salt-washed cells were incubated with mock or Plk-depleted mitotic cytosol, and an ATP-regenerating system for 1 h at 32°C. The organization of the Golgi membranes was visualized by fluorescence microscopy. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi upon incubation with mock or Plk-depleted mitotic cytosol. For each condition, 200 cells were counted on 2 different coverslips (mean±s.d., n=3, *P<0.05). (D) Left panel. HeLa cells stably expressing ManII-GFP were transfected with control or Myt1 specific siRNA oligo, and after incubation with thymidine for 12 h, permeabilized and salt-washed cells were incubated with mock or Plk-depleted mitotic cytosol and an ATP-regenerating system. The organization of the Golgi membranes was visualized by fluorescence microscopy. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi in the experimental conditions describe above. For each condition, 200 cells were counted on 2 different coverslips (mean±s.d., n=3, *P<0.05).
Is Myt1 required for MEK1 dependent Golgi complex fragmentation by mitotic cytosol? Permeabilized HeLa cells were incubated with mitotic cytosol in the presence of DMSO (control), the MEK1 inhibitor PD98058 (PD) or U0126, the RSK inhibitor SL0101 or BI-D1870, and the CDK1 inhibitor olomoucine or RO-3306. The inhibitory activity of these chemicals on their respective kinases was confirmed and shown in Supplementary Figure S1. After 1 h of incubation at 32°C, in the presence of an ATP-regenerating system, the cells were visualized by fluorescence microscopy. PD or U0126 treatment inhibited fragmentation of the Golgi complex in 75% of the cells (Figure 6A and Supplementary Figure S2). This confirms our previous findings that MEK1 is required for mitotic cytosol dependent Golgi complex fragmentation (Acharya et al, 1998; Colanzi et al, 2000, 2003). RSK inhibitor SL0101 or BI-D1870 did not block Golgi complex fragmentation by mitotic cytosol. This suggests that RSK, which is required for MEK1 dependent Myt1 phosphorylation (and its inactivation in Xenopus egg extracts (Palmer et al, 1998)) is not important for the fragmentation of Golgi complex (Figure 6A and Supplementary Figure S2). The CDK1 inhibitor olomoucine or RO-3306 was ineffective in mitotic cytosol dependent fragmentation of Golgi complex to the extent reconstituted in our assay as also reported previously (Figure 6A and Supplementary Figure S2) (Colanzi et al, 2000).
Figure 6.
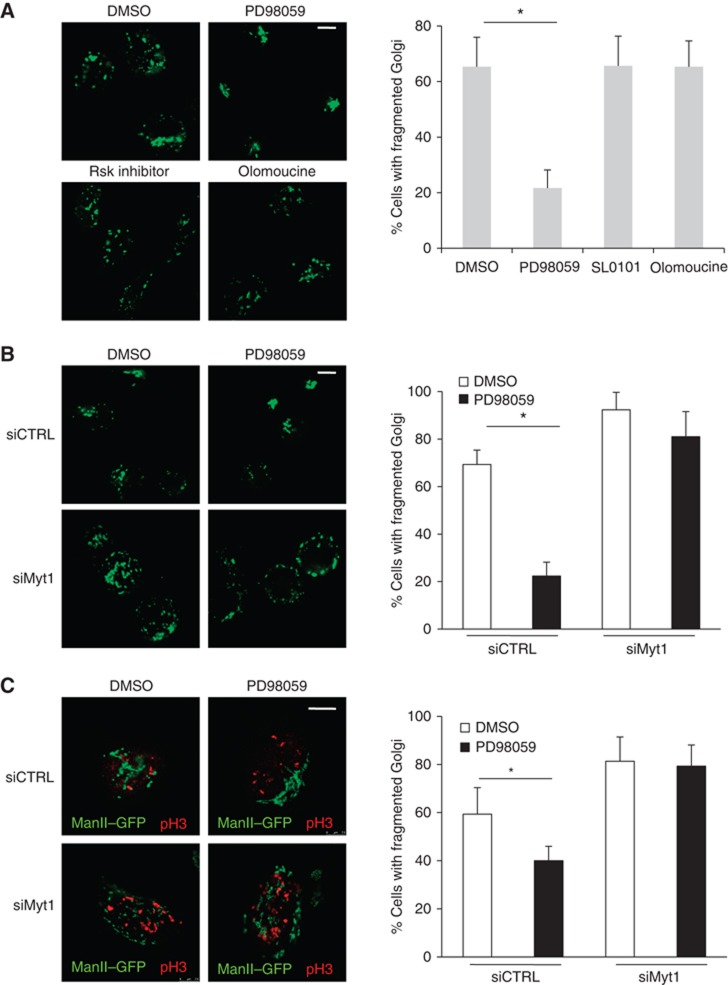
MEK1 regulates fragmentation of the Golgi complex through Myt1. (A) Left panel. After incubation with thymidine for 12 h, ManII-GFP expressing HeLa cells were permeabilized, salt-washed and incubated with an ATP-regenerating system and mitotic cytosol that was pretreated with DMSO, PD, SL0101 or olomoucine. The organization of the Golgi membranes was visualized by fluorescence microscopy. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi under the experimental conditions describe above. For each condition, 200 cells were counted on 2 different coverslips (mean±s.d., n=3, *P<0.05). (B) Left panel. HeLa cells stably expressing ManII-GFP were transfected with control or Myt1 specific siRNA oligo and after incubation with thymidine for 12 h, permeabilized and salt-washed cells were incubated with mitotic cytosol preincubated with DMSO or PD, and an ATP-regenerating system. The organization of the Golgi membranes was monitored by fluorescence microscopy. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi under the experimental conditions describe above. For each condition, 200 cells were counted on 2 different coverslips (mean±s.d., n=3, *P<0.05). (C) Left panel. Control and Myt1 siRNA transfected HeLa cells stably expressing ManII-GFP were arrested in S-phase with a double thymidine block. Cells were washed to remove thymidine, incubated 8 h in thymidine free medium with either DMSO or PD. Then, cells were fixed and stained with an anti-phospho-histone H3 antibody and analysed by fluorescence microscopy. The images show cells in G2. Scale bar is 10 μm. Right panel. Percentage of cells with fragmented Golgi in G2-phase under the experimental conditions describe above. For each condition, 200 cells on 2 different coverslips were counted (mean±s.d., n=3, *P<0.05).
What is the effect of Myt1 knockdown on PD dependent inhibition of Golgi complex fragmentation? HeLa cells were transfected with control or Myt1 specific siRNA oligos as describe above. These cells were then permeabilized, washed and incubated with mitotic cytosol and either DMSO or PD for 1 h at 32°C and visualized by fluorescence microscopy. In cells transfected with control siRNA oligo, PD inhibited Golgi complex fragmentation by mitotic cytosol. Myt1 knockdown, on the other hand, annulled the inhibitory effect of PD on Golgi complex fragmentation (Figure 6B). Does Myt1 knockdown affect PD dependent inhibition of Golgi complex fragmentation in intact cells in G2? Control and Myt1 siRNA transfected cells were arrested in S-phase with a double thymidine block. The cells were then washed to remove thymidine and incubated in normal medium with either DMSO or PD. Eight hours after release, cells were stained with an anti-phospho-histone H3 antibody to identify and quantitate cells in G2. In these cells, PD inhibited Golgi complex fragmentation in cells transfected with control siRNA oligo. However, Myt1 knockdown abrogated the inhibitory effect of PD on Golgi complex fragmentation (Figure 6C). Altogther the data derived from permeabilized and intact cells implies that Myt1 depletion alleviates the need of MEK1 in fragmentation of Golgi complex.
MEK1 regulates entry into the mitosis through Myt1
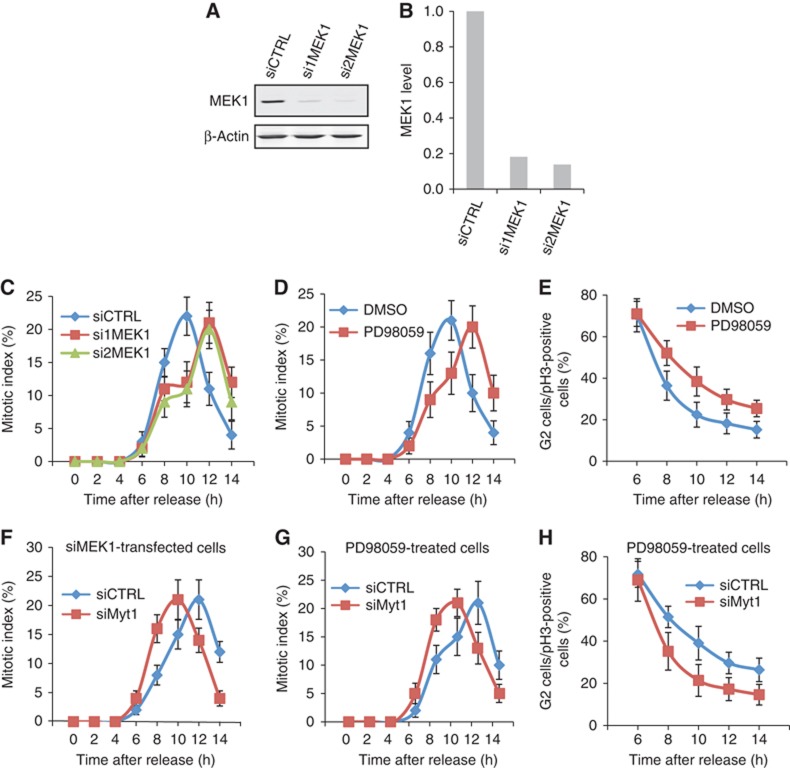
Transfection of HeLa cells with MEK1 specific siRNA oligos reduced MEK1 levels by 80% compared with control siRNA transfected cells (Figures 7A and B). We used this procedure to knockdown MEK1 in HeLa cells arrested in S-phase with a double thymidine block. In a parallel experiment, HeLa cells arrested in S-phase were treated with DMSO or PD. The cells were washed to remove thymidine and incubated in normal medium. At the indicated times the cells were stained with DAPI (to identify mitotic cells) and with an anti-phospho-histone H3 antibody to identify cells in late G2-phase and in mitosis (Colanzi et al, 2007). In control cells, cells transfected with control siRNA, or treated with DMSO, a mitotic index (cells in mitosis/total number of cells) of 25% was observed at 10 h after removal of the cells from the S-phase block. Knockdown of MEK1 or its inhibition with PD delayed the peak mitotic index by 2 h compared with control cells (Figures 7C and D). Under these experimental conditions, the analysis of cells in G2 showed that in PD treated cells, the kinetic of transition from G2 to M-phase was delayed compared with DMSO treated cells (Figure 7E). As described above, knockdown of Myt1 accelerates the peak of mitotic index by 2 h (from 10 to 8 h. Figure 1F). We tested the effect of Myt1 knockdown on the delay in mitotic index observed upon MEK1 knockdown or its inhibition by PD. HeLa cells were arrested in S-phase and transfected with MEK1 specific siRNA oligos or incubated with PD to inactivate MEK1. The cells were then transfected with control or Myt1 specific siRNA oligos. These cells were then washed to relieve the S-phase block and incubated in normal medium. At the indicated times, the mitotic index was determined by staining cells with DAPI and an anti-phospho-histone H3 antibody. The results show that the delay of entry into mitosis due to MEK1 inhibition was annulled by the knockdown of Myt1 (Figures 7F and G). Similarly, knockdown of Myt1 alleviated the G2/M-phase transition delay induced by PD treatment (Figure 7H). Altogether, these results provide strong evidence that Myt1 is downstream in the MEK1 dependent fragmentation of the Golgi complex and the entry of cells into mitosis.
Figure 7.
MEK1 regulates mitotic entry via Myt1. (A) HeLa cells were transfected with control or MEK1 specific siRNA oligos and after 48 h the level of MEK1 depletion was evaluated by western blotting the total cell lysates. Western blotting with an anti-β-actin antibody was used as a loading control. (B) Quantification of the MEK1 levels upon siRNA transfection. (C and D) The mitotic index was determined at the indicated times after thymidine release for control and MEK1 siRNA transfected cells (C) and DMSO and PD treated cells (D). (E) The percentage of cells in late G2 in the total number of cells in late G2 and all stages of mitosis was determined at the indicated times after thymidine release for DMSO and PD treated cells. For each time point, 400 cells were counted (mean±s.d., n=3). (F and G) To test the effect of Myt1 knockdown on the delay in mitotic entry induced by MEK1 downregulation, the mitotic index was calculated at the indicated times after thymidine release for control and Myt1 siRNA transfected cells, in cells transfected with MEK1 siRNA (F) or treated with PD (G). (H) The percentage of cells in late G2 in the total number of cells in late G2 and all stages of mitosis was determined at the indicated times after thymidine release for control and Myt1 siRNA transfected cells, in cells treated with PD. For each time point, 400 cells were counted (mean±s.d., n=3).
Source data for this figure is available on the online supplementary information page.
MEK1 is required for Myt1 phosphorylation
HeLa cells arrested in S-phase or mitosis by incubation for 18 h with thymidine or nocodazole, respectively, were incubated with either DMSO (control) or PD (MEK1 inhibitor). HeLa cells were lysed and the cell lysates western blotted with an anti-Myt1 antibody. The mitosis specific Myt1 phosphorylation was inhibited by PD treatment indicating the involvement of MEK1 in this process (Figure 8A). To further ascertain the role of MEK1 in Myt1 phosphorylation, HeLa cells were arrested in S-phase, then washed to remove thymidine and cultured in thymidine free medium in the absence or the presence of PD. The cells were then collected at the indicated times and western blotted with an anti-Myt1, anti-phospho-CDK1, and anti-phospho-histone H3 specific antibody, respectively. In DMSO treated cells, Myt1 phosphorylation peaked at 10 h post thymidine release. In PD treated cells, Myt1 phosphorylation peaked at 12 h post thymidine release (Figure 8B). We repeated the same experiment but instead of inhibiting MEK1 with PD, the cells were transfected with MEK1 specific siRNA oligos to knockdown MEK1. The cells were released from the S-phase and at the indicated times collected, lysed, and the cells lysates western blotted with an anti-Myt1, anti-phospho-CDK1, and anti-phospho-histone H3 specific antibody, respectively. Knockdown of MEK1 caused a 2 h delay in the peak of Myt1 phosphorylation during mitotic progression (Figure 8C). Myt1 is known to inactivate CDK1 and we therefore tested whether the delay in Myt1 phosphorylation affected CDK1 activation. In DMSO treated cells or control siRNA transfected cells, the peak of Myt1 phosphorylation coincided with CDK1 activation as monitored by dephosphorylation of Tyr15 and the phosphorylation of Histone H3. In PD treated cells or MEK1 siRNA transfected cells, the delay in Myt1 phosphorylation coincided with a delay in CDK1 inactivation and phosphorylation of Histone H3 (Figures 8B and C). These results indicate that MEK1 mediated Myt1 phosphorylation affects CDK1 activation, and therefore likely to inhibit CDK1 dependent downstream mitotic events.
Figure 8.
MEK1 is required for Myt1 phosphorylation. (A) HeLa cells were incubated 18 h with DMSO (asynchronized cells), thymidine (S-phase cells) or nocodazole (mitotic cells), with or without PD, and Myt1 phosphorylation monitored by western blotting the total cell lysates. Western blotting with an anti-β-actin antibody was used as a loading control. (B) HeLa cells were arrested in S-phase with double thymidine block, washed to remove thymidine, and incubated with DMSO or PD. At the indicated times, the cell lysates were western blotted with antibodies to Myt1, CDK1 (pY15), Histone H3 (pSer10) and β-actin. (C) HeLa cells transfected with control or MEK1 specific siRNA oligos were arrested in S-phase with double thymidine block, washed to remove thymidine, and at the indicated times, the cell lysates were western blotted with antibodies to Myt1, CDK1 (pY15), Histone H3 (pSer10) and β-actin. (D) Total cell lysate, S-phase and mitotic cytosol, and Golgi membrane preparation was western blotted with antibodies to TGN46, Myt1 and MEK1. (E) Isolated Golgi membranes were incubated for 1 h at 37 °C with S-phase or mitotic cytosol preincubated with DMSO, PD, SL0101 or olomoucine, and an ATP-regenerating system. The reaction was terminated by the addition of an excess of cold KHM buffer supplemented with phosphatase inhibitors. The Golgi membranes were collected by ultracentrifugation, resuspended in the SDS sample buffer and western blotted with an anti-Myt1 antibody. Western blotting of the same preparation with an anti-TGN46 antibody was used as a loading control.
Source data for this figure is available on the online supplementary information page.
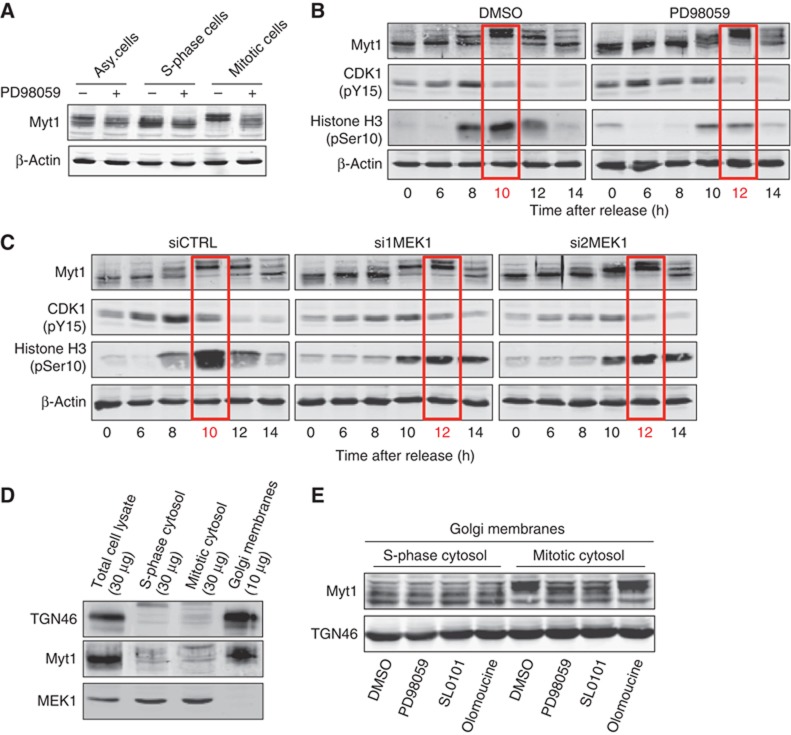
We tested the requirement of MEK1 for Myt1 phosphorylation in vitro. Western blotting isolated Golgi membranes (from interphase cells), S-phase and mitotic cytosol with an anti-Myt1 and MEK1 antibody revealed the presence of Myt1 but not MEK1 in the Golgi membrane fraction. MEK1 on the other hand was present only in the cytosol preparations (Figure 8D). Golgi membranes isolated from HeLa cells were incubated with MEK1 containing S-phase or mitotic cytosol in the presence of DMSO, PD, RSK inhibitor SL0101, or the CDK1 inhibitor olomoucine, and an ATP-regenerating system for 1 h at 37°C. The reactions were terminated by the addition of excess of cold buffer containing phosphatase inhibitor, the membranes collected by centrifugation and western blotted with an anti-Myt1 antibody. The results reveal that Myt1 in the Golgi membrane fraction is phosphorylated by mitotic cytosol and this event is inhibited by treatment with PD and RSK inhibitor (Figure 8E). While RSK inhibitor blocked Myt1 phosphorylation, it had no effect on the fragmentatoin of the Golgi complex by mitotic cytosol (Figure 6A and Supplementary Figure S2). It is therefore unlikely that RSK is involved in mitotic cytosol dependent Myt1 phosphorylation in the events leading to the fragmentation of the Golgi complex.
The finding that PD inhibited Myt1 phosphorylation prompted us to test whether MEK1 was directly involved in this reaction. We purified recombinant wild type (WT) and kinase dead (KD) Myt1, and constitutively active (CA) and kinase dead (KD) MEK1. The activity of recombinant MEK1 proteins was tested by their ability to phosphorylate ERK2 (Figure 9A, left panel). Myt1 undergoes autophosphorylation (Figure 9B), and in silico analysis has revealed that Myt1 contains 32 potential phosphorylation sites. Recombinant WT or KD-Myt1 was incubated with MEK1-CA, or MEK1-KD with γ-(32P) ATP at 30°C for 30 min. The samples were then analysed by SDS–PAGE followed by autoradiography and reveal that recombinant MEK1 does not directly phosphorylate Myt1 (Figure 9B, Left panel). It is reported that the activity is MEK1 is specifically regulated during mitosis (Colanzi et al, 2003; Harding et al, 2003). We therefore tested whether MEK1 isolated from mitotic cytosol phosphorylates Myt1 directly. MEK1 was immunoprecipitated from S-phase and mitotic cytosol and incubated with ERK2 (Figure 9A, right panel) or with recombinant WT or KD-Myt1 in the presence of γ-(32P)ATP at 30°C for 30 min (Figure 9B, Right panel). The reaction mixtures were analysed by SDS–PAGE and the radiolabeled polypeptides visualized by autoradiography. We did not observe any obvious shift or an increase in the amount of radioactivity incorporated into Myt1 by mitotic MEK1 compared with S-phase MEK1 (Figure 9B, Right panel). We then tested whether MEK1 required ERK2 to phosphorylate Myt1. MEK1 was immunoprecipitated from mitotic cytosol and incubated with recombinant pure ERK2 and recombinant KD-Myt1 in the presence of γ-(32P)ATP at 30°C for 30 min. The samples were then analysed by SDS–PAGE followed by autoradiography and our results show that ERK2 does not phosphorylate Myt1 (Supplementary Figure S3). Altogether, these results suggest that MEK1 mediated phosphorylation of Myt1 is not via ERK2, Plk, RSK or CDK1.
Figure 9.
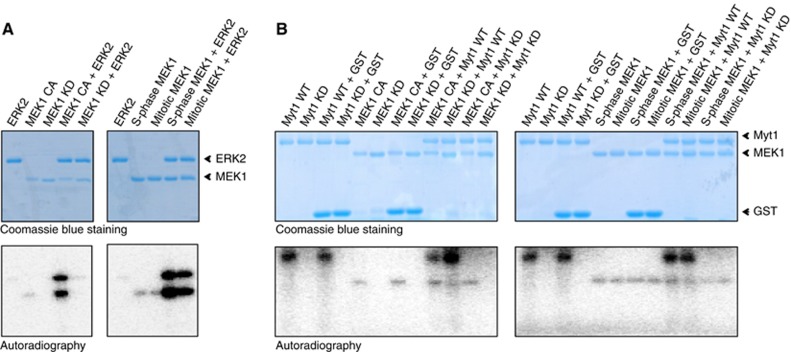
Myt1 is not directly phosphorylated by MEK1 in vitro. (A) 500 ng of recombinant 6-His-tagged MEK1 proteins (constitutively active, CA; kinase dead, KD) (Left panels), and 500 ng of FLAG-tagged MEK1 proteins purified from S-phase and mitotic cells (Right panels) were incubated with 1 μg of recombinant ERK2 protein in the presence of γ-(32P)ATP for 30 min at 30°C. The reactions were stopped by the addition of SDS sample buffer and analysed by SDS/PAGE. Coomassie blue staining and the autoradiogram of the gels are shown. (B) 500 ng recombinant 6-His-tagged MEK1 proteins (CA and KD form) (Left panels) and 500 ng of FLAG-tagged MEK1 proteins purified from S-phase and mitotic cells (Right panels) were incubated with 1 μg of GST-tagged Myt1 proteins (wild type, WT; kinase dead, KD) in the presence of γ-(32P)ATP for 30 min at 30°C. The reactions were stopped by the addition of SDS sample buffer and analysed by SDS/PAGE. Coomassie blue staining and the autoradiogram of the gels are shown.
GRASP55, a Golgi membrane associated protein is phosphorylated by MEK1/ERK2 (Jesch et al, 2001). We tested whether MEK1/ERK2-mediated GRASP55 phosphorylation during mitosis required Myt1. GRASP55 was immunoprecipitated from HeLa cells synchronized in S-phase (thymidine-treated cells) or mitosis (nocodazole-treated cells). Then, using the mitotic phosphoprotein monoclonal-2 (MPM-2) antibody, which recognizes a Ser/Thr-phospho epitope, we tested whether GRASP55 reacted with the MPM-2 antibody as reported by Linstedt and colleagues (Jesch et al, 2001). As shown in Supplementary Figure S4A, GRASP55 immunoprecipitated from mitotic cells was strongly MPM-2 reactive. GRASP55 immunoprecipitated from S-phase synchronized cells did not show a detectable signal with MPM-2 antibody. Treatment with the MEK1 inhibitor PD reduced the phosphorylation of GRASP55 in mitotic cells as revealed by the decreased reactivity with MPM-2 antibody (Supplementary Figure S4A). In order to determine whether Myt1 activity was required for the GRASP55 phosphorylation by MEK1, HeLa cells were transfected with control or Myt1 specific siRNA oligos and synchronized in mitosis by nocodazole treatment in the presence or absence of PD. The downregulation of Myt1 did not affect the MPM-2 reactivity of GRASP55 immunoprecipitated from mitotic cells (Supplementary Figure S4B), suggesting that MEK1 mediated GRASP55 phosphorylation in mitotic cells is not mediated by Myt1.
Discussion
In the mammalian cells, the pericentriolar stacks of Golgi cisternae are separated from each other and dispersed in the cytoplasm. This change is evident in G2 and first reported by Warren and colleagues (Shima et al, 1998). The cells then enter in mitosis and the stacks are broken into small tubules and vesicles. Understanding the mechanism and the reason for the G2 and mitosis specific changes in Golgi organization are of fundamental importance, and our new findings provide important insights into these two challenges as discussed below.
The first step (separation of Golgi stacks from each other and their dispersal from the pericentriolar region) is mediated by MEK1 and Plk. CDK1 is required for the second step by which the stacks are converted into small tubules and vesicles (Kano et al, 2000). Our new findings reveal the involvement of Myt1 in the MEK1 dependent event, which occurs in G2. Loss of Myt1 promotes the fragmentation of the Golgi complex in G2 and the entry of cells into mitosis. Depletion of Myt1 alleviates the dependence on MEK1 for the purpose of Golgi fragmentation and it also abrogates the delay in mitotic entry produced by the knockdown or inactivation of MEK1. Myt1 therefore is a negative regulator of Golgi fragmentation in G2 and of the process by which the pericentriolar organization of the Golgi membranes controls entry of cell into mitosis.
It is important to note that the MEK1-Myt1 effects on the Golgi fragmentation are independent of CDK1 and the challenge now is to identify the target(s) of Myt1 on Golgi membranes. It is known that inactivation of Myt1 is required for the activation of CDK1. The identification of Myt1 in the MEK1 pathway is therefore important because it connects two sequential steps by which the pericentriolar Golgi stacks are first converted into small stacks by MEK1 in G2 and then into small tubules and vesicles by CDK1 in mitosis.
MEK1 dependent Myt1 phosphorylation is not by RSK
During meiosis, in Xenopus oocytes, inactivation of Myt1 by MEK1 (for the subsequent activation of CDK1) requires RSK (Palmer et al, 1998). We have found that in HeLa cells, inactivation of RSK (by chemical inhibitors) inhibited phosphorylation of Myt1 without affecting mitosis specific Golgi membrane fragmentation. Therefore, RSK is unlikely to be the intermediary kinase in the MEK1 dependent Myt1 phosphorylation. Myt1 is heavily phosphorylated during mitosis as evident by its phosphorylation dependent (approximately) 10 kD increase in mol.wt. These observations are strengthened by in silico analysis, which has revealed 32 potential phosphorylation sites, which include phosphorylation by Plk, RSK and CDK1. Mapping the residue(s) phosphorylated in the presence of MEK1 will reveal the elusive kinase(s) and the mechanism by which Myt1 controls Golgi membrane organization in association with the cell cycle events.
Resolving the controversy on the role of MEK1 in Golgi membranes fragmentation during mitosis
Warren and colleagues have claimed that fragmentation of isolated Golgi enriched fractions with mitotic cytosol is independent of the activity of MEK1 (Lowe et al, 1998). However, their starting material is the end product of a MEK1 dependent reaction; so this is not really a concern. They reported that NRK cells arrested in S-phase with aphidicolin for 14 h then washed and incubated with MEK1 inhibitors for 4 h, were not defective in mitotic entry. As shown here, loss or inactivation of MEK1 causes a 2 h delay but not a strict block in entry of cells into mitosis. This would not be evident based on a single time point that was used to monitor the mitotic index by Warren and colleagues (Lowe et al, 1998). It was also reported that treatment with lethal factor (which inactivates MEK1) did not affect the overall organization of the Golgi membranes in mitotic cells (Lowe et al, 1998). As shown here, MEK1 is required for the early stages of the disassembly of the Golgi complex and the entry of cells into mitosis. Loss of MEK1 affects the kinetics of mitotic entry and not the overall fate (breakdown into mitotic Golgi fragments: tubules and vesicles) of the Golgi complex per se during mitosis. Pines and colleagues reported that overexpression of cyclin B2 (the Golgi localized cyclin) and CDK1 in non-mitotic cells affected the organization of the Golgi complex, which was unaffected by treatment with MEK1 inhibitor PD or U0126 (Draviam et al, 2001). However, MEK1 is required for very early changes in Golgi complex in late G2 and for the subsequent entry of cell into mitosis, and neither MEK1 nor Myt1 have a role in the organization of Golgi membranes in non-mitotic cells in S-phase. Additionally, it has been reported that CHO cells released 3 h post G2 block in the presence of PD were found in all stages of mitosis (Draviam et al, 2001). However, as described above, MEK1 dependent Golgi membrane fragmentation affects the kinetics of mitotic entry: a single point 3 h after release from G2 would not have uncovered the effects on the kinetics of this process.
Fragmentation of the Golgi complex and mitotic entry: a checkpoint or a kinetic timer?
Inhibiting fragmentation of the pericentriolar Golgi apparatus was found to block entry of NRK cells into mitosis (Sutterlin et al, 2002; Colanzi et al, 2007; Feinstein and Linstedt, 2007). Barr and colleagues found that preventing fragmentation of the Golgi complex did not block cells in G2 per se but the entry into mitosis was delayed (Preisinger et al, 2005). Our new findings also reveal that MEK1 knockdown in intact HeLa cells does not arrest cells in G2 but the peak of mitotic index is delayed by 2 h. A plausible explanation for this discrepancy is the involvement of parallel pathways that play a role in the pericentriolar organization of the Golgi membranes. We suggest that a block in Golgi complex fragmentation is a signal to arrest cells in G2. However, the cytoplasmic microtubules that connect Golgi stacks laterally and hold them in the pericentriolar region start depolymerizing, which triggers the separation of Golgi stacks. In fact, it is known that treatment with nocodazole overrides the block in mitotic entry created by a defect in Golgi fragmentation (Sutterlin et al, 2002). In other words, there are two distinct pathways that fragment the Golgi complex in G2: a microtubule dependent and a microtubule independent pathway. A defect in mitotic entry induced by a block in Golgi complex fragmentation through the MEK1-Myt1 pathway is compensated by the microtubule dependent separation of Golgi membranes. The contribution of the microtubule dependent Golgi stack separation could be cell type and might explain the difference in the kinetic of the mitotic entry due to the defect on pericentriolar Golgi fragmentation reported by differents groups. It is also important to map the timing of microtubule dependent stack separation with respect to MEK1-Myt1 process for a better understanding of the significance and the mechanism of Golgi apparatus mediated control of mitotic entry.
Materials and methods
Reagents, antibodies and plasmids
Reagents used in this study were obtained from the following sources: Phorbol-12-myristate-13-acetate (PMA), nocodazole, thymidine and digitonin were from Sigma Aldrich. PD98059, SL0101 and olomoucine were from Calbiochem. U0126 was from Promega. BI-D1870 was from Symansis. Lambda protein phosphatase was from New England Biolabs. RO-3306 and recombinant human protein ERK2 were from Millipore. Antibodies used in this study were purchased from the following sources: Mouse monoclonal antibodies against β-actin and c-Myc (clone 9E10) were from Sigma Aldrich. Mouse monoclonal antibodies against GM130 and MEK1 were from BD Biosciences. The mouse monoclonal antibody against Plk (clone F-8) was from Santa Cruz Biotechnology, the sheep antibody against TGN46 was from AbD Serotec, and the rabbit polyclonal antibody against phospho-Histone H3 was from Upstate Biotechnology. Rabbit polyclonal antibodies against phospho-GSK3α/β (Ser21/9) and phospho-CDK1 (Tyr15) were from Cell signaling Technology. Rabbit polyclonal antibodies against Cyclin B1 (clone H-433), Cyclin B2 (clone H-105), Rab6 (clone C-19), CDK1 (clone H-297) and CDK2 (clone M2) were from Santa Cruz Biotechnology. Rabbit monoclonal antibodies against ERK1/2 and phospho-ERK1/2 (Thr202/Tyr204) were from Cell Signaling Technology. Secondary antibodies for immunofluorescence microscopy and western blotting were from Invitrogen. The mouse IgG2a and rabbit IgG control isotypes were from BD Biosciences and Sigma Aldrich, respectively. We generated the rabbit polyclonal antibody against GRASP55 as described previously (Duran et al, 2008). We kindly thank Dr Angel R Nebrada (Institute for Research in Biomedicine (IRB), Barcelona, Spain) for the generous gift of the mouse monoclonal antibodies against MPM-2 and GSK3α/β, and Dr Helen Piwnica-Worms (Washington University School of Medicine, St Louis, MO) for the generous gift of the rabbit polyclonal antibody against Myt1, and c-Myc-Myt1 wild type (WT) and c-Myc-Myt1 kinase dead (KD) plasmids. The cDNA encoding full-length human Myt1 was inserted into pGEX4T1 to express protein with a N-terminal GST-tag. The kinase dead Myt1 N238A was generated by mutagenesis using the Quick Change II site directed mutagenesis Kit (Stratagene) following the manufacturer’s recommendations. The cDNA encoding full-length human MEK1 was inserted into pRSETA to express protein with an N-terminal His-tag. The constitutively active (CA) form of MEK1 S128E S222E, and KD form of MEK1 K97M, was generated by mutagenesis using the Quick Change II site directed mutagenesis Kit (Stratagene) following the manufacturer’s recommendations. The cDNA encoding full-length human MEK1 was inserted into pcDNA3 to express protein with a C-terminal FLAG-tag.
Preparation of S-phase and mitotic cytosols and incubation with permeabilized cells
HeLa cells were grown in complete medium consisting of DMEM containing 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 7% CO2. Cytosol from HeLa cells, arrested in S-phase (thymidine treated cells) or mitosis (nocodazole treated cells) at a concentration of 12–14 mg/ml was prepared as described previously (Acharya et al, 1998). Permeabilization of cells grown on coverslips and the Golgi membrane fragmentation assay was performed as described previously (Acharya et al, 1998). In brief, HeLa cells stably expressing ManII-GFP were treated with 2 mM thymidine for 12 h and permeabilized on ice with 30 μg/ml digitonin in KHM buffer (25 mM HEPES-KOH, pH 7.4, 125 mM potassium acetate and 2.5 mM magnesium acetate). Permeabilized cells were washed with 1M KCl-containing KHM and incubated with S-phase or mitotic cytosol and an ATP-regenerating system. After 1 h of incubation at 32°C, cells were fixed and processed for immunofluorescence microscopy. When chemical inhibitors were used, cytosolic extracts were pretreated with the corresponding inhibitors for 10 min at 32°C, and intact cells were preincubated with the corresponding inhibitors for 30 min at 37°C and permeabilized as describe above. PD was used at 75 μM, olomoucine and SL0101 at 20 μM, U0126 and BI-D1870 at 10 μM, and RO-3306 at 5 μM.
Statistical analysis
All data are presented as mean±s.d. Differences in the mean values between 2 groups were assessed by 2-tailed Student t-test. P<0.05 was taken to imply statistical significance.
Supplementary Material
Acknowledgments
We thank all members of the Malhotra Laboratory for valuable discussions, Arrate Mallabiabarrena and Raquel García for help with live cell imaging and fluorescence microscopy. Protein technologies Unit in the Centre for Genomic Regulation are thanked for help with the expression and purification of recombinant proteins. We thank SnapGene: http://www.snapgene.com for the software for molecular biology procedures. Julien Villeneuve is funded by a long-term EMBO postdoctoral fellowship. Vivek Malhotra is an Institució Catalana de Recerca i Estudis Avançats (ICREA) professor at the Centre for Genomic Regulation, and the work in his laboratory is funded by grants from Plan Nacional (BFU2008-00414), Consolider (CSD2009-00016), Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) Grups de Recerca Emergents (SGR2009-1488; AGAUR-Catalan Government), and European Research Council (268692). The project has received research funding from the European Union. This paper reflects only the author’s views. The Union is not liable for any use that may be made of the information contained therein.
Author contributions: JV and VM designed the experiments, analysed the data and wrote the manuscript. JV, MS and MOB performed the experiments. All authors discussed the results and contributed to the final manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V (1998) Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 92: 183–192 [DOI] [PubMed] [Google Scholar]
- Axelsson MA, Warren G (2004) Rapid, endoplasmic reticulum-independent diffusion of the mitotic Golgi haze. Mol Biol Cell 15: 1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Holman PS, Fattaey A (1997) Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem 272: 22300–22306 [DOI] [PubMed] [Google Scholar]
- Colanzi A, Deerinck TJ, Ellisman MH, Malhotra V (2000) A specific activation of the mitogen-activated protein kinase kinase 1 (MEK1) is required for Golgi fragmentation during mitosis. J Cell Biol 149: 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Hidalgo Carcedo C, Persico A, Cericola C, Turacchio G, Bonazzi M, Luini A, Corda D (2007) The Golgi mitotic checkpoint is controlled by BARS-dependent fission of the Golgi ribbon into separate stacks in G2. EMBO J 26: 2465–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Sutterlin C, Malhotra V (2003) RAF1-activated MEK1 is found on the Golgi apparatus in late prophase and is required for Golgi complex fragmentation in mitosis. J Cell Biol 161: 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draviam VM, Orrechia S, Lowe M, Pardi R, Pines J (2001) The localization of human cyclins B1 and B2 determines CDK1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J Cell Biol 152: 945–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran JM, Kinseth M, Bossard C, Rose DW, Polishchuk R, Wu CC, Yates J, Zimmerman T, Malhotra V (2008) The role of GRASP55 in Golgi fragmentation and entry of cells into mitosis. Mol Biol Cell 19: 2579–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Linstedt AD (2007) Mitogen-activated protein kinase kinase 1-dependent Golgi unlinking occurs in G2 phase and promotes the G2/M cell cycle transition. Mol Biol Cell 18: 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Linstedt AD (2008) GRASP55 regulates Golgi ribbon formation. Mol Biol Cell 19: 2696–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding A, Giles N, Burgess A, Hancock JF, Gabrielli BG (2003) Mechanism of mitosis-specific activation of MEK1. J Biol Chem 278: 16747–16754 [DOI] [PubMed] [Google Scholar]
- Inoue D, Sagata N (2005) The Polo-like kinase Plx1 interacts with and inhibits Myt1 after fertilization of Xenopus eggs. EMBO J 24: 1057–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesch SA, Lewis TS, Ahn NG, Linstedt AD (2001) Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol Biol Cell 12: 1811–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesch SA, Linstedt AD (1998) The Golgi and endoplasmic reticulum remain independent during mitosis in HeLa cells. Mol Biol Cell 9: 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokitalo E, Cabrera-Poch N, Warren G, Shima DT (2001) Golgi clusters and vesicles mediate mitotic inheritance independently of the endoplasmic reticulum. J Cell Biol 154: 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F, Takenaka K, Yamamoto A, Nagayama K, Nishida E, Murata M (2000) MEK and Cdc2 kinase are sequentially required for Golgi disassembly in MDCK cells by the mitotic Xenopus extracts. J Cell Biol 149: 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Madsen ML, Yarm FR, Jang YJ, Liu X, Erikson RL (2000) Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2. Proc Natl Acad Sci USA 97: 12589–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Stanton JJ, Wu Z, Piwnica-Worms H (1997) The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol 17: 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jamsa E, Rahman D, Pappin DJ, Warren G (1998) Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94: 783–793 [DOI] [PubMed] [Google Scholar]
- Lucocq JM, Pryde JG, Berger EG, Warren G (1987) A mitotic form of the Golgi apparatus in HeLa cells. J Cell Biol 104: 865–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucocq JM, Warren G (1987) Fragmentation and partitioning of the Golgi apparatus during mitosis in HeLa cells. EMBO J 6: 3239–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Warren G (1994) COP-coated vesicles are involved in the mitotic fragmentation of Golgi stacks in a cell-free system. J Cell Biol 125: 269–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG (1995) Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 270: 86–90 [DOI] [PubMed] [Google Scholar]
- Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E (2003) Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem 278: 25277–25280 [DOI] [PubMed] [Google Scholar]
- Nakajima H, Yonemura S, Murata M, Nakamura N, Piwnica-Worms H, Nishida E (2008) Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J Cell Biol 181: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A, Gavin AC, Nebreda AR (1998) A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J 17: 5037–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot MY, Malhotra V (2004) Golgi membranes remain segregated from the endoplasmic reticulum during mitosis in mammalian cells. Cell 116: 99–107 [DOI] [PubMed] [Google Scholar]
- Persico A, Cervigni RI, Barretta ML, Colanzi A (2009) Mitotic inheritance of the Golgi complex. FEBS Lett 583: 3857–3862 [DOI] [PubMed] [Google Scholar]
- Preisinger C, Korner R, Wind M, Lehmann WD, Kopajtich R, Barr FA (2005) Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J 24: 753–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul YD, Seger R (2006) ERK1c regulates Golgi fragmentation during mitosis. J Cell Biol 172: 885–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Cabrera-Poch N, Pepperkok R, Warren G (1998) An ordered inheritance strategy for the Golgi apparatus: visualization of mitotic disassembly reveals a role for the mitotic spindle. J Cell Biol 141: 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Warren G (2002) Golgi architecture and inheritance. Annu Rev Cell Dev Biol 18: 379–420 [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy S, Goh JB, Rafee S, Murata-Hori M (2010) Mitotic Golgi vesiculation involves mechanisms independent of Ser25 phosphorylation of GM130. Cell Cycle 9: 3100–3105 [DOI] [PubMed] [Google Scholar]
- Sutterlin C, Hsu P, Mallabiabarrena A, Malhotra V (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109: 359–369 [DOI] [PubMed] [Google Scholar]
- Sutterlin C, Lin CY, Feng Y, Ferris DK, Erikson RL, Malhotra V (2001) Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc Natl Acad Sci USA 98: 9128–9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Blume J, Duran JM, Forlanelli E, Alleaume AM, Egorov M, Polishchuk R, Molina H, Malhotra V (2009) Actin remodeling by ADF/cofilin is required for cargo sorting at the trans-Golgi network. J Cell Biol 187: 1055–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.