

Abstract
The transcription factor Nrf2 is responsible for regulating a battery of antioxidant and cellular protective genes, primarily in response to oxidative stress. A member of the cap 'n' collar family of transcription factors, Nrf2 activation is tightly controlled by a series of signaling events. These events can be separated into the basal state, a preinduction response, gene induction, and finally a postinduction response, culminating in the restoration of redox homeostasis. However, despite the immensely intricate level of control the cellular environment imposes on Nrf2 activity, there are many opportunities for perturbations to arise in the signaling events that favor carcinogenesis and, therefore, implicate Nrf2 as both a tumor suppressor and a protooncogene. Herein, we highlight the ways in which Nrf2 is regulated, and discuss some of the Nrf2-inducible antioxidant (NQO1, NQO2, HO-1, GCLC), antiapoptotic (Bcl-2), metabolic (G6PD, TKT, PPARγ), and drug efflux transporter (ABCG2, MRP3, MRP4) genes. In addition, we focus on how Nrf2 functions as a tumor suppressor under normal conditions and how its ability to detoxify the cellular environment makes it an attractive target for other oncogenes either via stabilization or degradation of the transcription factor. Finally, we discuss some of the ways in which Nrf2 is being considered as a therapeutic target for cancer treatment.—Shelton, P., Jaiswal, A. K. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene?
Keywords: cancer, signal transduction, oxidative stress, chemoprevention, targeted therapy
The accumulation of reactive oxygen species (ROS) in the cellular environment is considered to result in a state of oxidative stress (1). Left unbalanced the ROS can interact with and damage DNA, RNA, and proteins. Spontaneous mutations that arise from the oxidative stress can lead to the initiation of cancer, and various cancers have been found to be in a constant state of oxidative stress, which suggests a role for oxidative stress in cancer promotion as well (2–4). Thus, this well-established link between oxidative stress and cancer has attracted a great amount of focus in the cancer biology field. As we attempt to seek out better treatments and early diagnostic tools, it is important that we continue to refine our understanding of how not only to combat but also how to prevent cancer.
Fortunately, the cell itself possesses its own internal defense mechanism to fight against oxidative stress. Chiefly, this is mediated by the transcription factor NF-E2-related factor 2 (Nrf2; refs. 5, 6 and Fig. 1). A master regulator of a battery of defensive and detoxification genes, Nrf2 becomes stabilized and activated when the ROS levels and electrophiles accumulate. Specifically, oxidative stress leads to oxidation of key cysteine residues on the inhibitor of Nrf2 [INrf2; Kelch-like ECH-associated protein 1 (Keap1)] (Fig. 1) and phosphorylation of Nrf2, which collectively disrupt the interaction of the two proteins and allow Nrf2 to become stabilized (7–10). Through a series of signaling events, Nrf2 can then induce transcription of genes responsible for detoxifying the ROS, removing damaged proteins and promoting the overall survival of the cell. Accordingly, the Nrf2 pathway has garnered attention in order to elucidate a preventive mechanism in the oxidative stress-induced carcinogenesis model.
Figure 1.
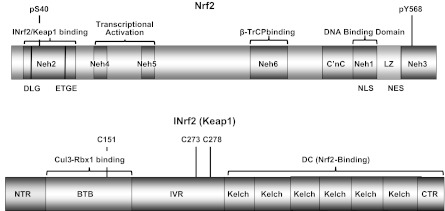
Protein domains of Nrf2 and INrf2/Keap1. Nrf2 domains: Nrf2 possesses 6 highly conserved domains called Nrf2-ECH homology (Neh) domains. The Neh1 domain includes C′nC-bZIP(LZ) regions that heterodimerize with small Maf or Jun proteins. The Neh2 domain binds with INrf2 (Keap1). The Neh3 domain contributes to stabilization of the Nrf2 protein. Neh4 and Neh5 are transcriptional activation domains. The Neh6 domain binds to β-TrCP. LZ, feucine zipper or BZIP region; NLS, nuclear localization signal; NES, nuclear export signal. INrf2 (Keap1) domains: NTR, N-terminal region; BTB, bric-á-brac domain that binds with Nrf2 ubiquitin factors Cul3-Rbx1; IVR, intervening region; DC, DGR/CTR region that binds with Nrf2; DGR, double glycine repeat; CTR, C-terminal region. C151 is important for stress sensing, and C273/C278 is required for Nrf2 suppression.
Interestingly, and somewhat paradoxically, recent research has highlighted that persistent accumulation of Nrf2 is harmful, since it can promote the survival and proliferation of cells that have acquired cancer-promoting mutations (11–14). This has been viewed as too much of a good thing, and active investigation into the negative role of Nrf2 in cancer has led some to characterize it as a cancer promoter.
As we move forward with our understanding of Nrf2 in cancer, it is important that we review and highlight the molecular mechanisms that control this transcription factor and the genes that it regulates. Perhaps then we can identify steps that will allow us to intervene to prevent malignancy secondary to oxidative stress.
CAP 'N' COLLAR (CNC) NUCLEAR TRANSCRIPTION FACTORS
NF-E2-related factors
NF-E2-related factors are a conserved family of vertebrate CNC nuclear transcription factors that comprise 4 members, namely, p45 NF-E2, Nrf1, Nrf2, and Nrf3 (15, 16). These proteins are basic leucine zipper (bZIP) transcription factors that have a conserved 43-aa CNC domain located N-terminally to the DNA binding domain, and they function in response to environmental stress, as well as during development (ref. 17 and Fig. 1). Nrf1 and Nrf2 are ubiquitously expressed, whereas the expression of Nrf3 is restricted to the placenta and liver, and NF-E2 is restricted to erythrocytes, hematopoietic progenitor, mast, and megakaryocytic cells (16–18). Knockout studies reveal that despite their overlapping expression patterns, Nrf1 and Nrf2 have distinct phenotypes and different roles (19, 20). The Nrf1 gene is essential for embryonic development, as Nrf1-knockout mice are embryonically lethal, and liver-specific Nrf1-knockout mice develop nonalcoholic steatohepatitis (21, 22). In contrast, Nrf2-knockout mice are viable and exhibit no obvious phenotypic defects, but are nevertheless sensitive to oxidative stress and neurodegeneration (16, 23–26).
Nrf2 and oxidative stress
Although all three Nrfs play a role in stress response, Nrf2 is the main mediator of cellular adaptation to redox stress (27–33). When the level of ROS and electrophiles become greater than the cell's ability to detoxify them, the result is oxidative stress. This initiates a series of events, detailed in a later section, which results in the activation of Nrf2. As a transcription factor, activated Nrf2 heterodimerizes with small Maf or Jun proteins and binds to the antioxidant response element (ARE) located in the promoter region of Nrf2 target genes (27–34). The binding of Nrf2 to the ARE results in the coordinated activation of a battery of antioxidant (e.g., NQO1, NQO2, HO-1), antiapoptotic (e.g., Bcl-2), metabolic (e.g., G6PD, TKT, PPARγ), and detoxification (e.g., ABCG2, MRP3, MRP4, GST) proteins (35–40). Collectively, the Nrf2 response serves to restore redox homeostasis, detoxify and efflux xenobiotics, remove damaged proteins, and prevent the cell from undergoing apoptosis (17).
NRF2 SIGNALING IN STRESS AND CHEMOPROTECTION
The activity and stabilization of Nrf2 is tightly regulated by various signaling events. In response to acute stress, the Nrf2 response can be viewed more precisely in terms of a basal state, a preinduction response, the gene induction response, and finally, the postinduction response (Fig. 2).
Figure 2.
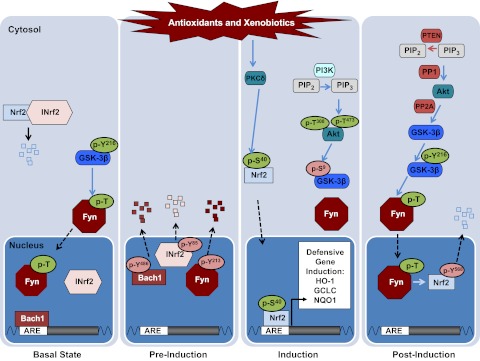
Model depicting the canonical Nrf2 stress response pathway in each of the different phases: basal, preinduction, induction, postinduction.
Basal state
In the absence of a stress signal, Nrf2 is constantly maintained in a repressed, basal state in the cytoplasm. The inhibition of Nrf2 is principally achieved by INrf2 (Keap1), which is an adaptor protein that facilitates ubiquitination and degradation of Nrf2 (8, 41, 42). The interaction between INrf2 and Nrf2 has been proposed to be a 2-site “hinge and latch” model, where an INrf2 homodimer binds to the ETGE (hinge) and DLG (latch) motifs in the Neh2 domain of Nrf2 (43, 44). In addition, the N-terminal BTB domain of INrf2 binds with the ubiquitin ligase complex Cul3-Rbx1, thereby facilitating the ubiquitination of Nrf2 and its degradation by the 26S proteasome (ref. 42 and Fig. 1).
While the INrf2-mediated ubiquitination and degradation of Nrf2 primarily occurs in the cytoplasm, it has been reported that some INrf2 is localized in the nucleus and is able to repress nuclear Nrf2 under basal conditions. Along with INrf2, additional inhibitory proteins converge to prevent Nrf2 from accumulating in the nucleus, binding to the ARE, and inducing defensive gene expression. One of the proteins involved in the basal repression of nuclear Nrf2 is the related transcription factor BTB and CNC homology 1 (Bach1). In the basal state, Bach1 is found in the promoter region of Nrf2 target genes and acts as a transcriptional repressor by competing with Nrf2 for binding to the ARE sequences (45). In addition, Nrf2 has been shown to be a substrate of the Src-A subfamily kinase members Src, Fyn, Yes, and Fgr, which phosphorylate and negatively regulate Nrf2 nuclear localization (46, 47). Moreover, we have recently demonstrated that nuclear translocation of the Src-A subfamily kinases is regulated by glycogen synthase kinase 3β (GSK3β), which is constitutively active in the basal state (46, 48). Therefore, in a resting cell, active GSK3β facilitates the nuclear localization of the Src-A subfamily kinase members, which in turn regulate the nuclear export of Nrf2.
Preinduction
On exposure to oxidative stress, Nrf2 becomes activated and translocates into the nucleus. However, before Nrf2 is able to bind to the ARE and induce gene transcription, the negative factors in the nucleus must be exported out in order to allow for efficient gene induction. Accordingly, the proteins Bach1, INrf2, and Fyn have all been found to be phosphorylated and exported out of the nucleus after a short exposure (1–2 h) to the antioxidant tert-butylhydroquinone (t-BHQ; refs. 49–51). In Bach1, tyrosine 486 has been shown to be responsible for the nuclear export (49). Similarly, Fyn kinase has been found to be phosphorylated at tyrosine 213 within 0.5–1 h exposure to t-BHQ, and this regulates the nuclear export of Fyn (50). It has also been demonstrated that INrf2 becomes phosphorylated at tyrosine 85 during this preinduction response (51). It is noteworthy that all of the negative nuclear factors are phosphorylated at tyrosine residues. While further investigation is necessary, it is tempting to predict that a single tyrosine kinase may regulate the phosphorylation of all of these proteins.
Induction
The induction phase is characterized by stabilization and activation of Nrf2, as well as inactivation of GSK3β-mediated nuclear import of the Src-A subfamily kinases. On exposure to ROS, key cysteine residues in INrf2 (Keap1) become oxidized and disrupt INrf2's interaction with Nrf2 and Cul3-Rbx1, which allows Nrf2 to become phosphorylated at serine 40 by PKCδ (7–9). Together, this allows Nrf2 to be primed for nuclear translocation. Once inside the nucleus, with the negative factors exported out, Nrf2 is able to heterodimerize with small Maf or Jun proteins, recognize and bind to the ARE sequence, and facilitate the recruitment of the transcriptional machinery to induce the expression of proteins capable of combating the oxidative stress (52–53).
In addition, stress activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling cascade has been reported to be influential in Nrf2-mediated gene induction and, more generally, in cancer (54–60). Recent evidence suggests that one of the potential mechanisms involved in the PI3K/Akt cascade's ability to regulate Nrf2 is its ability to regulate the nuclear localization of the Src-A subfamily kinases Src, Fyn, Yes, and Fgr (46–47). Specifically, it is thought that oxidative stress presumably activates the canonical PI3K/Akt pathway to convert phosphatidylinositol-(4,5)-bisphosphate (PIP2) into phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) and recruit Akt to the cellular membrane via its pleckstrin homology (PH) domain. Akt is then phosphorylated at residues T308 and S473 by PDK1 and PDK2, respectively, which fully activates Akt. When activated, Akt initiates the phosphorylation of GSK3β at S9, which causes GSK3β to shift into an inactive conformation (61). As was noted previously, GSK3β is responsible for the activation and nuclear translocation of the Src-A subfamily kinases Src, Fyn, Yes, and Fgr (46–47). Therefore, the PI3K/Akt cascade's inactivation of GSK3β during the induction phase facilitates unimpeded entry of Nrf2 into the nucleus by preventing the Src-A subfamily kinases to localize in the nucleus and cause Nrf2's nuclear export.
Postinduction
Following the induction phase, the cell responds by exporting Nrf2 out of the nucleus and restoring the accumulation of the negative regulators Bach1, INrf2, and the Src-A subfamily kinases (49–51). The effective result is a turning off of the Nrf2 response and a return to the basal state.
In part, this is achieved by an Nrf2-mediated negative feedback loop. Specifically, the negative transcriptional regulator Bach1 has been reported to be regulated by Nrf2, and its de novo synthesis is induced in response to oxidative stress. The de novo protein then translocates into the nucleus during the postinduction response, where it decreases Nrf2 binding to the ARE (49). In addition, INrf2 also reaccumulates in the nucleus during the postinduction response (51).
More important, the nuclear accumulation of the Src-A subfamily kinases is instrumental in the nuclear export of Nrf2. As it has been previously outlined, the Src-A subfamily kinases phosphorylate Nrf2 at Y568, which triggers its nuclear export (47, 62). The reaccumulation of the Src-A subfamily kinases is a result of GSK3β S9 dephosphorylation and subsequent reactivation of GSK3β through phosphorylation of Y216. Preliminary evidence, along with established roles of protein phosphatases involved in the dephosphorylation of the inhibitory S9 phosphorylation, has suggested a role for protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) (63–64). Although there is still need for further investigation into how these phosphatases are regulated, it is tempting to speculate that Nrf2 itself may regulate their activation, thereby initiating another negative feedback mechanism to turn itself off after it has finished with the induction phase.
The coordination of these stress response phases allows the cell to effectively respond to the cellular stress by activating Nrf2 to increase defensive gene expression. On successful induction of the detoxification enzymes, the cell initiates a negative feedback loop whereby it shuts off the response by phosphorylating Nrf2 and exporting it out of the nucleus for degradation, thus returning homeostasis to the basal state. It is important to note that although the responses are separated into different phases in this characterization, which implies a linear series of events, these responses are, in fact, not static and will have interplay between the different phases.
NRF2 IN CANCER: A PROTO-ONCOGENE?
The hallmarks of cancer include resisting cell death, inducing angiogenesis, activating invasion and metastasis, enabling replicative immortality, evading growth suppressors, and sustaining proliferative signaling that allow cancer cells to survive, proliferate, and spread. More recently, the deregulation of cellular energetics and the avoidance of immune destruction have been identified as emerging hallmarks, and genomic instability and tumor-promoting inflammation have been acknowledged as enabling characteristics that allow oncogenesis to occur (65). Surprisingly, Nrf2 regulates or is targeted by many of the proteins involved in both the prevention and the direction of these oncogenic processes. As such, the precise role that Nrf2 plays in cancer remains somewhat controversial. It is considered to be both a tumor suppressor, as well as a tumor promoter protein, with some postulating that its precise role is dependent on the stage of tumorigenesis (66). Therefore, it has been termed a double-edged sword with dual roles in cancer (11, 14). Herein we will highlight some aspects that characterize Nrf2 as both a chemopreventive target and an oncogenic factor.
Nrf2 as a tumor suppressor
As we have previously detailed, Nrf2 is the primary regulator of the antioxidant response. By way of inducing the antioxidant enzymes, the cell is able to combat ROS and electrophiles that may otherwise cause genomic instability and induce inflammation, which are conducive to initiate oncogenesis. In addition to antioxidant enzymes, Nrf2 plays a role in regulating other detoxifying and defensive genes, such as anti-inflammatory genes, as well as components of the proteasome that help rid the cell of damaged proteins (17). Recent evidence has also revealed a role for Nrf2 in the regulation of PPARγ, a protein known to inhibit cell proliferation and induce apoptosis and cell differentiation (67, 68). In particular, one study found that persistent activation of Nrf2 in A549 non-small cell lung cancer cells led to increased expression of PPARγ and attenuation of oncogenic-cyclin D and cancer stem cell markers (68). Therefore, the intrinsic ability of Nrf2 to regulate enzymes that confer defense against oncogenesis characterizes it as an anticancer protein.
In vivo data using Nrf2-knockout mice has also highlighted the tumor-suppressor nature of Nrf2, as knockout mice have been shown to display increased sensitivity to carcinogens and toxicants. Specifically, Nrf2−/− mice are prone to acute damages induced by acetaminophen, ovalbumin, pentachlorophenol, and 4-vinylcyclohexene diepoxide (69–73). Nrf2−/− mice showed increased pulmonary DNA adducts and bladder tumors when exposed to diesel exhaust and N-nitrosobutyl (4-hydroxybutyl) amine, respectively (26, 74–75). Moreover, loss of Nrf2 in lung cancer cells has been associated with providing a microenvironment that favors metastasis (76).
Research into the role of oxidative stress in aging, along with the observation that cancer incidence increases with age, has provided another intriguing hypothesis that Nrf2 is a defender against oncogenesis. At the center of this theory is the notion that aging is the result of progressive oxidative damage to cells that is caused by free radicals (17). Although Nrf2 protects the cells from oxidative stress, in vivo studies with rats has shown that the basal Nrf2 protein level declines with age, and correlates with lower expression of Nrf2 target genes (17). Therefore, it is plausible that Nrf2 functions to defend against the free radical-induced aging process and that this progressively declines over time, which leads to the accumulation of free radicals that can promote the formation of cancer.
A further line of evidence that demonstrates the tumor suppressor nature of Nrf2 is that one of the principal actions of known chemopreventive compounds, such as sulforaphane, curcumin, EGCG (green tea), oltipraz, and others, is their ability to activate Nrf2 (30, 75). These compounds activate Nrf2 by reacting with the cysteine residues of INrf2 (Keap1) (66). Clinical trials investigating these compounds are ongoing in various tissues, as both chemopreventive and tumor suppressive agents. It is important to note that these drugs can also target other proteins besides INrf2 (Keap1) and may exhibit their anticancer activity via pathways other than Nrf2 (66).
Interestingly, Nrf2 has been found to be both up-regulated by tumor suppressor proteins, as well as targeted for degradation by oncoproteins (Table 1). Therefore, this indirect evidence also highlights Nrf2's action as a tumor suppressor protein. More specifically, a recent study demonstrated that under moderate oxidative stress conditions the tumor suppressor protein p21 is up-regulated and acts to stabilize Nrf2 by preventing INrf2 (Keap1)-mediated ubiquitination and degradation of Nrf2 (77). Similarly, a separate study has found partner and localizer of BRCA2 (PALB2), another tumor suppressor protein that is chiefly responsible for BRCA function, to interact with the ETGE domain of Nrf2 and prevent its interaction with INrf2 (Keap1) in the nucleus (78). In addition, there is growing evidence that has shown that oncoproteins target Nrf2 for degradation, either directly or indirectly. These oncoproteins include the Src-A subfamily kinases, the ubiquitin ligase complex SCFβ-TrCP, and GSK3β (47, 79, 80). Interestingly, the mechanism proposed for SCFβ-TrCP-mediated degradation is INrf2 (Keap1) independent and involves Nrf2 being directly phosphorylated by GSK3β in its Neh6 domain, which serves as a motif for ubiquitination and degradation by SCFβ-TrCP (80). Taken together, Nrf2's ability to provide cellular protection against cancer promoting carcinogens, become activated by chemopreventive compounds, and also be stabilized by tumor suppressor proteins or degraded by oncoproteins highlight its ability to act as an anticancer protein, particularly during acute stress and premalignant states.
Table 1.
Proteins affecting the expression levels of Nrf2
| Proteins increasing Nrf2 accumulation |
Proteins decreasing Nrf2 accumulation |
||||
|---|---|---|---|---|---|
| Protein | Function | Ref. | Protein | Function | Ref. |
| p21 | 154KRR motif in p21 directly interacts with the 29DLG and 79ETGE motifs in Nrf2 | 77 | INrf2/Keap1 | Facilitates Cul3-Rbx1-mediated ubiquitination of Nrf2 via Neh2 domain of Nrf2 | 8, 10, 41–44 |
| p62 | Sequesters Keap1 into aggregates | 89, 90 | Bach1 | Inhibits binding of Nrf2 to ARE | 45 |
| PKCδ | Phosphorylates Nrf2 at S40 and releases Nrf2 from INrf2 | 7 | Src-A subfamily kinases | Phosphorylates Nrf2 at Y568, triggering its nuclear export and degradation | 46, 47 |
| PALB2 | Direct interaction between the ETGE motif of PALB2 and the Kelch domain of INrf2/Keap1 | 78 | GSK3β | Directly phosphorylates a cluster of serines in the ETGE domain of Nrf2, targeting it for degradation by SCFβTrCP; indirect via Src-A subfamily kinases | 79, 47 |
| K-Ras, B-Raf, and Myc | Increase Nrf2 transcription and basal expression | 81 | SCF/β-TrCP | Adaptor protein for Skp1-Cul1-Rbx1-mediated ubiquitination of Nrf2 via Neh6 domain of Nrf2 | 79 |
Nrf2 as a tumor promoter
Conflicting with the antitumorigenic characteristics of Nrf2 is its opposing characterization as a tumor promoter. Nrf2 regulates many genes that control the hallmarks of cancer. Specifically, Nrf2 has been shown to regulate the antiapoptotic protein Bcl-2, which helps evade cell death (39). More recently, a seminal paper highlighted the ability of Nrf2 to regulate many of the enzymes responsible for reprogramming energy metabolism and de novo nucleotide synthesis, such as glucose-6-phosphate dehydrogenase (G6PD) and transketolase (TKT), and that these enzymes are required to regulate tumor growth (40). While it has long been known that cancer cells can reprogram their glucose metabolism toward glycolysis, this study demonstrates Nrf2 as a principal regulator in the process and provides evidence that this glycolytic switch generates de novo nucleotide synthesis to drive proliferation. The researchers in this study also found that the oncogenic PI3K-Akt pathway contributes to the Nrf2-mediated activation of metabolic genes. Yet another key mechanism recently reported that implicates a protumorigenic role for Nrf2 is that the oncogenes K-Ras, B-Raf, and Myc specifically target the transcription and amplification of Nrf2 in cancer cells (81). The researchers in the study noted that primary cells and tissues of mice overexpressing the oncogenes K-Ras, B-Raf, or Myc all had decreased ROS levels due to increased transcription and basal expression of Nrf2, and genetic targeting of Nrf2 in vivo decreased the ability of K-Ras to induce oncogenesis, implicating Nrf2 in providing an oncogenic environment for cells with mutations in those genes. In effect, this finding further supports a role for Nrf2 in sustained proliferating signaling in cancer cells and also suggested a novel role whereby the increased expression of Nrf2 helps to maintain ROS levels below a toxic threshold to escape death in the cancer cells (82). Together, the researchers conclude that Nrf2 is a mediator of oncogenesis. This is also supported by the fact that the basal activity of Nrf2 has been found to be increased in neoplasia, and that deletion of Nrf2 is able to reduce carcinogen-induced lung tumor development in mice (81, 83). However, in vivo studies using INrf2 (Keap1)-knockout mice in which Nrf2 is constitutively active showed no increase in cancer, which suggests that Nrf2 may not initiate oncogenesis but rather plays a role in selection during tumor development (84).
It is noteworthy that many cancers have been found to up-regulate Nrf2. This is thought to provide an advantageous environment for the cancer cells to evade apoptosis, proliferate, and even metastasize (13, 85–86). One of the mechanisms in which cells up-regulate Nrf2 is through mutations that disrupt the interaction between Nrf2 and INrf2 (Keap1). For example, it has been reported that various cancer cells have loss of function mutations in INrf2 (Keap1), primarily in the IVR (29%) and DC (65%) domains of INrf2 (Keap1), and hypermethylation in its promoter region has also been reported (89). Similarly, Nrf2 has also been reported to be susceptible to mutations in the DLG (43%) and ETGE (57%) motifs where it interacts with INrf2 (Keap1) (88). In addition to mutations that prevent the interaction of Nrf2 and INrf2, certain cancers have been found to have proteins that interact with Nrf2 or INrf2 (Keap1) in their respective interacting domains, disrupting INrf2's ability to bind Nrf2, which also leads to persistent activation of Nrf2. For example, the autophagy-associated protein p62 has been found to be a target gene for Nrf2, and it directly interacts with INrf2 (Keap1), thereby facilitating increased Nrf2 activity (89–91). Under these circumstances the end result is a persistent activation of Nrf2 in the cells.
The persistent activation of Nrf2 not only provides a microenvironment that is conducive for the tumor cells to survive, but also helps protect the cells from therapeutic intervention. As has been previously highlighted, Nrf2 up-regulates many genes that are involved in the efflux of drugs, as well as genes that can defend against radiation (12, 38, 92–94). Consequently, the increased activity of Nrf2 in cancer cells provides a mechanism for resistance against drug therapy and radiation therapy. Taken together, the detection of increased Nrf2 expression and activity in many cancers, along with its inherent ability to provide defense to tumor cells against treatment, suggest that Nrf2 also acts as a tumor promoter protein.
By definition, this would suggest that Nrf2 is a protooncogene. First described by Varmus, Bishop, and colleagues (95) in 1976 with c-Src, a protooncogene is a normal gene that typically controls cell growth and division, but when it obtains a gain of function mutation, or its expression is altered, it becomes oncogenic (95, 96). When expressed at normal levels, Nrf2 is a beneficial tool to help detoxify and defend against oxidative stress conditions. However, when Nrf2 becomes overexpressed it provides a prosurvival benefit that tilts a mutated, precancerous cell into becoming a malignancy that can resist apoptosis, chemotherapeutics, and radiation.
NRF2 AS A THERAPEUTIC TARGET
Despite the beneficial ability of Nrf2 to help maintain the redox balance in the cellular environment by detoxifying harmful ROS, it is still an attractive therapeutic target in the treatment of Nrf2-addicted cancer cells. As has been previously discussed, many cancer cells have been found to have an increased expression and activity of Nrf2. These cells are able to drive the progression of carcinogenesis and even avoid standard radio- and chemotherapies. Therefore, it is tempting to target this addiction that cancer cells have to Nrf2, possibly at a subclinical level and in combination with standard therapies to increase their effectiveness. Ideally, this would increase the effectiveness of the targeted therapies specifically in the cancer cells, without drastically affecting the normal cellular environment. One compound that has recently been described to have such an effect on Nrf2 is brusatol, a plant extract from Brucea javanica (97). In this study, the researchers found brusatol to increase the ubiquitination and degradation of Nrf2, resulting in reduction of the Nrf2-downstream genes and sensitization to chemotherapy in cancer cell lines and a mouse xenograft model. Similarly, procyanidins from cinnamomi cortex extract (CCE) have also been found to suppress Nrf2-mediated transcription, and enhance A549 lung cancer cells to chemotherapy (98). In addition to these natural compounds, one can envision developing inhibitors of Nrf2 that are domain specific and target the Neh1 domain to disrupt nuclear import and DNA binding, or the transactivation domain.
CONCLUSIONS AND FUTURE VISION
Nrf2 is regulated at multiple levels as the cell responds to the oxidative stress. Under normal circumstances, this is an acute response that is helpful in preventing cellular damage that could lead to genomic instability. However, persistent activation of Nrf2 can confer an advantageous environment for a cancer cell to survive and proliferate with an inherent ability to resist traditional cancer therapies. As we move forward in our approach to treat cancer, it is important to be mindful of this dual nature of Nrf2. Perhaps results from the cancer genome project could yield information detailing what cancers and in what particular tumor stages Nrf2 tends to exhibit persistent activation, allowing clinicians to identify which patients would best benefit from Nrf2 targeted therapy. In turn, by targeting Nrf2 in the cancer specific cell we could improve the efficacy of traditional chemotherapeutic drugs, thereby effectively lowering their cytotoxic side effects, while still maintaining Nrf2 levels in normal cells so as to retain its beneficial power. With antibody-directed therapy to cancer cells and improvements in drug delivery, one can envision a treatment regimen whereby Nrf2 can be specifically targeted either by restoring INrf2 (Keap1) levels, or disrupting Nrf2's ability to initiate transcription. Hopefully, as our understanding of Nrf2 continues to improve, so too will our ability to include it as a cancer target.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) grants RO1GM047466 and RO1ES012265.
The helpful suggestions from Dr. Suresh Niture (University of Maryland, Baltimore) are greatly appreciated.
Footnotes
- Akt
- protein kinase B
- ARE
- antioxidant response element
- Bach1
- BTB and CNC homology 1
- bZIP
- basic leucine zipper
- CNC
- cap 'n' collar
- G6PE
- glucose-6-phosphate dehydrogenase
- GSK3β
- glycogen synthase kinase 3β
- INrf2
- inhibitor of Nrf2
- Keap1
- Kelch-like ECH-associated protein 1
- Nrf2
- NF-E2-related factor 2
- PH
- pleckstrin homology
- PI3K
- phosphoinositide 3-kinase
- PIP2
- phosphatidylinositol-(4,5)-bisphosphate
- PIP3
- phosphatidylinositol-(3,4,5)-trisphosphate
- PP1
- protein phosphatase 1
- PP2A
- protein phosphatase 2A
- ROS
- reactive oxygen species
- t-BHQ
- tert-butylhydroquinone
- TKT
- transketolase
REFERENCES
- 1. Sies H. (1997) Oxidative stress: oxidants and antioxidants. Exp. Physiol. 82, 291–295 [DOI] [PubMed] [Google Scholar]
- 2. Klaunig J. E., Kamendulis L. M., Hocevar B. A. (2010) Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Path. 38, 96–109 [DOI] [PubMed] [Google Scholar]
- 3. Tudek B., Winczura A., Janik J., Siomek A., Foksinski M., Oliński R. (2010) Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am. J. Transl. Res. 2, 254–284 [PMC free article] [PubMed] [Google Scholar]
- 4. Wu R. P., Hayashi T., Cottam H. B., Jin G., Yao S., Wu C. C., Rosenbach M. D., Corr M., Schwab R. B., Carson D. A. (2010) Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 107, 7479–7484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nguyen T., Huang H. C., Pickett C. B. (2000) Transcriptional regulation of the antioxidant response element-activation by Nrf2 and repression by MafK. J. Biol. Chem. 275, 15466–15473 [DOI] [PubMed] [Google Scholar]
- 6. Jaiswal A. K. (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 36, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 7. Niture S. K., Jain A. K., Jaiswal A. K. (2009) Antioxidant induced modification of INrf2 cysteine151 and PKCd-mediated phosphorylation of Nrf2 serine40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J. Cell Sci. 122, 4452–4464 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Zhang D. D., Hannink M. (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bloom D. A., Jaiswal A. K. (2003) Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H: quinone oxidoreductase1 gene expression. J. Biol. Chem. 278, 44675–44682 [DOI] [PubMed] [Google Scholar]
- 10. Eggler A. L., Liu G., Pezzuto J. M., van Breemen R. B., Mesecar A. D. (2005) Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. U. S. A. 102, 10070–10075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lau A., Villeneuve N. F., Sun Z., Wong P. K., Zhang D. D. (2008) Dual roles of Nrf2 in cancer. Pharmacol. Res. 58, 262–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X. J., Sun Z., Villeneuve N. F., Zhang S., Zhao F., Li Y., Chen W., Yi X., Zheng Y. X., Wondrack G. T., Wong P. K., Zhang D. D. (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Homma S., Ishii Y., Morishima Y., Yamadori T., Matsuno Y., Haraguchi N., Kikuchi N., Satoh H., Sakamoto T., Hizawa N., Itoh K., Yamamoto M. (2009) Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423–3432 [DOI] [PubMed] [Google Scholar]
- 14. Kensler T. W., Wakabayashi N. (2010) Nrf2: friend or foe for chemoprevention? Carcinogenesis 31, 90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan J. Y., Cheung M. C., Moi P., Chan K., Kan Y. W. (1995) Chromosomal localization of the human NF-E2 family of bZIP transcription factors by fluorescence in situ hybridization. Hum. Genet. 95, 265–269 [DOI] [PubMed] [Google Scholar]
- 16. Motohashi H., O'Connor T., Katsuoka F., Engel J. D., Yamamoto M. (2002) Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 294, 1–12 [DOI] [PubMed] [Google Scholar]
- 17. Sykiotis G. P., Bohmann D. (2010) Stress-activated cap ‘n’ collar transcription factors in aging and human disease. Sci. Signal. 3(112), re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sankaranarayanan K., Jaiswal A. K. (2004) Nrf3 negatively regulates antioxidant-response element-mediated expression and antioxidant induction of NAD(P)H: quinone oxidoreductase1 gene. J. Biol. Chem. 279, 50810–50817 [DOI] [PubMed] [Google Scholar]
- 19. Leug L., Kwong M., Hou S., Lee C., Chan J. Y. (2003) Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 278, 48021–48029 [DOI] [PubMed] [Google Scholar]
- 20. Ohtsuji M., Katsuoka F., Kobayashi A., Aburatani H., Hayes J. D., Yamamoto M. (2008) Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 283, 33554–33562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chan J. Y., Kwong M., Lu R., Chang J., Wang B., Yen T. S., Kan Y. W. (1998) Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 17, 1779–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu Z. R., Chen L., Leung L., Yen. T. S. B., Lee C., Chan J. Y. (2005) Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. U. S. A. A102, 4120–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan K., Lu R., Chang J. C., Kan Y. T. (1996) Nrf2, a member of the NF-E2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U. S. A. 93, 13943–13948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan J. Y., Kwong M. (2000) Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta 1517, 19–26 [DOI] [PubMed] [Google Scholar]
- 25. Kwak M. K., Egner P. A., Dolan P. M., Ramos-Gomez M., Groopman J. D., Itoh K., Yamamoto M., Kensler T. W. (2001) Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat. Res. 480-481, 305–315 [DOI] [PubMed] [Google Scholar]
- 26. Ramos-Gomez M., Kwak M. K., Dolan P. M., Itoh K., Yamamoto M., Talalay P., Kensler T. W. (2001) Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 98, 3410–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaspar J. W., Niture S. K., Jaiswal A. K. (2009) Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dhakshinamoorthy S., Long D. J., 2nd, Jaiswal A. K. (2000) Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Curr. Top. Cell Regul. 36, 201–206 [DOI] [PubMed] [Google Scholar]
- 29. Kobayashi M., Yamamoto M. (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 46, 113–140 [DOI] [PubMed] [Google Scholar]
- 30. Zhang D. D. (2006) Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 38, 769–789 [DOI] [PubMed] [Google Scholar]
- 31. Aleksunes L. M., Manautou J. E. (2007) Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Path. 35, 459–473 [DOI] [PubMed] [Google Scholar]
- 32. Hayes J. D., McMahon M. (2009) NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 34, 176–188 [DOI] [PubMed] [Google Scholar]
- 33. Kwak M. K., Cho J. M., Huang B., Shin S., Kensler T. W. (2007) Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 43, 809–817 [DOI] [PubMed] [Google Scholar]
- 34. Blank V. (2008) Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J. Mol. Biol. 376, 913–925 [DOI] [PubMed] [Google Scholar]
- 35. Venugopal R., Jaiswal A. K. (1996) Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: quinone oxidoreductase1gene. Proc. Natl. Acad. Sci. U. S. A. 93, 14960–14965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alam J., Stewart D., Touchard C., Boinapally S., Choi A. M., Cook J. L. (1999) Nrf2, a cap 'n' collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078 [DOI] [PubMed] [Google Scholar]
- 37. Wang W., Jaiswal A. K. (2006) Nuclear factor Nrf2 and antioxidant response element regulate NRH: quinone oxidoreductase 2 (NQO2) gene expression and antioxidant induction. Free Radic. Biol. Med. 40, 1119–1130 [DOI] [PubMed] [Google Scholar]
- 38. Maher J. M., Dieter M. Z., Aleksunes L. M., Slitt A. L., Guo G., Tanaka Y., Scheffer G. L., Chan J. Y., Manautou J. E., Chen Y., Dalton T. P., Yamamoto M., Klaassen C. D. (2007) Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2–related factor-2 transcriptional pathway. Hepatology 46, 1597–1610 [DOI] [PubMed] [Google Scholar]
- 39. Niture S. K., Jaiswal A. K. (2012) Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873–9886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 [DOI] [PubMed] [Google Scholar]
- 41. Cullinan S. B., Gordan J. D., Jin J., Harper J. W., Diehl J. A. (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24, 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang D. D., Lo S. C., Cross J. V., Templeton D. J., Hannink M. (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tong K. I., Katoh Y., Kusunoki H., Itoh K., Tanaka T., Yamamoto M. (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 26, 2887–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tong K. I., Padmanabhan B., Kobayashi A., Shang C., Hirotsu Y., Yokoyama S., Yamamoto M. (2007) Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 27, 7511–7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dhakshinamoorthy S., Jain A. K., Bloom D. A., Jaiswal A. K. (2005) Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H: quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 280, 16891–16900 [DOI] [PubMed] [Google Scholar]
- 46. Jain A. K., Jaiswal A. K. (2007) GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 282, 16502–16510 [DOI] [PubMed] [Google Scholar]
- 47. Niture S. K., Jain A. K., Shelton P. M., Jaiswal A. K. (2011) Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J. Biol. Chem. 286, 28821–28832 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Doble B. W., Woodgett J. R. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaspar J., Jaiswal A. K. (2010) Antioxidant induced phosphorylation of tyrosine486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to ARE and activate defensive genes expression. J. Biol. Chem. 285, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaspar J. W., Jaiswal A. K. (2011) Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 25, 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaspar J. W., Niture S. K., Jaiswal A. K. (2012) Antioxidant-induced INrf2 (Keap1) tyrosine 85 phosphorylation controls the nuclear export and degradation of the INrf2-Cul3-Rbx1 complex to allow normal Nrf2 activation and repression. J. Cell Sci. 125, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322 [DOI] [PubMed] [Google Scholar]
- 53. Marini M. G., Chan K., Casula L., Kan Y. W., Cao A., Moi P. (1997) hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J. Biol. Chem. 16490–16497 [DOI] [PubMed] [Google Scholar]
- 54. Lee J. M., Hanson J. M., Chu W. A., Johnson J. A. (2001) Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant responsive element in IMR-32 human neuroblastoma cells. J. Biol. Chem. 276, 20011–20016 [DOI] [PubMed] [Google Scholar]
- 55. Vivanco I., Sawyers C. L. (2002) The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 [DOI] [PubMed] [Google Scholar]
- 56. Nakaso K., Yano H., Fukuhara Y., Takeshima T., Wada-Isoe K., Nakashima K. (2003) PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 546, 181–184 [DOI] [PubMed] [Google Scholar]
- 57. Martin D., Rojo A. I., Salinas M., Diaz R., Gallardo G., Alam J., De Galarreta C. M., Cuadrado A. (2004) Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 279, 8919–8929 [DOI] [PubMed] [Google Scholar]
- 58. Papaiahgari S., Zhang Q., Kleeberger S. R., Cho H. Y., Reddy S. P. (2006) Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Signal. 8, 43–52 [DOI] [PubMed] [Google Scholar]
- 59. Lim J. H., Kim K. M., Kim S. W., Hwang O., Choi H. J. (2008) Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: novel cytoprotective mechanism against oxidative damage. Pharmacol. Res. 57, 325–331 [DOI] [PubMed] [Google Scholar]
- 60. Liu P., Cheng H., Roberts T. M., Zhao J. J. (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Disc. 8, 627–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cohen P., Frame S. (2001) The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2, 769–776 [DOI] [PubMed] [Google Scholar]
- 62. Jain A. K., Jaiswal A. K. (2006) Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 281, 12132–12142 [DOI] [PubMed] [Google Scholar]
- 63. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 64. Hernández F., Langa E., Cuadros R., Avila J., Villaneuva N. (2010) Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol. Cell. Biochem. 344, 211–215 [DOI] [PubMed] [Google Scholar]
- 65. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 66. Sporn M. B., Liby K. T. (2012) NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer 12, 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cho H.-Y., Gladwell W., Wang X., Chorley B., Bell D. S., Reddy P., Kleeberger S. R. (2010) Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 182, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhan L., Zhang H., Zhang Q., Woods C. G., Chen Y., Xue P., Dong J., Tokar E. J., Xu Y., Hou Y., Fu J., Yarborough K., Wang A., Qu W., Waalkes M. P., Andersen M. E., Pi J. (2012) Regulatory role of KEAP1 and NRF2 in PPARγ expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radic. Biol. Med. 53, 758–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O'Connor T., Harada T., Yamamoto M. (2001) High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 59, 169–177 [DOI] [PubMed] [Google Scholar]
- 70. Chan K., Han X. D., Kan Y. W. (2001) An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc. Natl. Acad. Sci. U. S. A. 98, 4611–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rangasamy T., Guo J., Mitzner W. A., Roman J., Singh A., Fryer A. D., Yamamoto M., Kensler T. W., Tuder R. M., Georas S. N., Biswal S. (2005) Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 202, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Iizuka T., Ishii Y., Itoh K., Kiwamoto T., Kimura T., Matsuno Y., Morishima Y., Hegab A. E., Homma S., Nomura A., Sakamoto T., Shimura M., Yoshida A., Yamamoto M., Sekizawa K. (2005) Nrf2 deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10, 1113–1125 [DOI] [PubMed] [Google Scholar]
- 73. Hu X., Roberts J. R., Apopa P. L., Kan Y. W., Ma Q. (2006) Accelerated ovarian failure induced by 4-Vinyl Cyclohexane diepoxide in Nrf2-null mice. Mol. Cell. Biol. 26, 940–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aoki Y., Sato H., Nishimura N., Takahashi S., Itoh K., Yamamoto M. (2001) Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharm. 173, 154–160 [DOI] [PubMed] [Google Scholar]
- 75. Iida K., Itoh K., Kumagai Y., Oyasu R., Hattori K., Kawai K., Shimazui T., Akaza H., Yamamoto M. (2004) Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 64, 6424–6431 [DOI] [PubMed] [Google Scholar]
- 76. Satoh H., Moriguchi T., Taguchi K., Takai J., Maher J., Suzuki T., Winnard P. T., Raman V., Ebina M., Nukiwa T., Yamamoto M. (2010) Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 31, 1833–1843 [DOI] [PubMed] [Google Scholar]
- 77. Chen W., Sun Z., Wang X. J., Jiang T., Huang Z., Fang D., Zhang D. D. (2009) Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell. 34, 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma J., Cai H., Wu T., Sobhian B., Huo Y., Alcivar A., Mehta M., Cheung K. L., Ganesan S., Kong A. N., Zhang D. D., Xia B. (2012) PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 32, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rada P., Rojo A. I., Chowdhry S., McMahon M., Hayes J. D., Cuadrado. A. (2011) SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 31, 1121–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Salazar M., Rojo A. I., Velasco D., de Sagarra R. M., Cuadrado A. (2006) Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281, 14841–14851 [DOI] [PubMed] [Google Scholar]
- 81. DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Perera R. M., Bardeesy N. (2011) Cancer: when antioxidants are bad. Nature 475, 43–44 [DOI] [PubMed] [Google Scholar]
- 83. Bauer A. K., Cho H.-Y., Miller-DeGraff L., Walker C., Helms K., Fostel J., Yamamoto M., Kleeberger S. R. (2011) Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS ONE 6, e26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ma Q., He X. (2012) Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol. Rev. 64, 1055–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Singh A., Misra V., Thimmulappa R. K., Lee H., Ames S., Hoque M. O., Herman J. G., Baylin S. B., Sidransky D., Gabrielson E., Brock M. V., Biswal S. (2006) Dysfunctional Keap1-Nrf2 interaction in non-small-cell lung cancer. PLoS Med. 3, e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Padmanabhan B., Tong K. I., Ohta T., Nakamura Y., Scharlock M., Ohtsuji M., Kang M. I., Kobayashi A., Yokoyama S., Yamamoto M. (2006) Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 21, 689–700 [DOI] [PubMed] [Google Scholar]
- 87. Kweon M. H., Adhami V. M., Lee J. S., Mukhtar H. (2006) Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J. Biol. Chem. 281, 33761–33772 [DOI] [PubMed] [Google Scholar]
- 88. Taguchi K., Motohashi H., Yamamoto M. (2011) Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140 [DOI] [PubMed] [Google Scholar]
- 89. Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y. S., Ueno I., Sakamoto A., Tong K. I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 [DOI] [PubMed] [Google Scholar]
- 90. Lau A., Wang X. J., Zhao F., Villeneuve N. F., Wu T., Jiang T., Sun Z., White E., Zhang D. D. (2010) A Noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Inami Y., Waguri S., Sakamoto A., Kouno T., Nakada K., Hino O., Watanabe S., Ando J., Iwadate M., Yamamoto M., Lee M. S., Tanaka K., Komatsu M. (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vollrath V., Wielandt A. M., Iruretagoyena M., Chianale M. (2006) Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 395, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim Y. J., Ahn J. Y., Liang P., Ip C., Zhang Y., Park Y. M. (2007) Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 67, 546–554 [DOI] [PubMed] [Google Scholar]
- 94. Okawa H., Motohashi H., Kobayashi A., Aburatani H., Kensler T. W., Yamamoto M. (2006) Hepatocyte-specific deletion of the Keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 339, 79–88 [DOI] [PubMed] [Google Scholar]
- 95. Stehelin D., Varmus H. E., Bishop J. M., Vogt P. K. (1976) DNA related to the transforming gene(s) of avian sarcomaviruses is present in normal avian DNA. Nature 260, 170–173 [DOI] [PubMed] [Google Scholar]
- 96. Bishop J. M. (1988) Oncogenes and proto-oncogenes. J. Cell. Physiol. 129(S4), 1–5 [DOI] [PubMed] [Google Scholar]
- 97. Ren D., Villeneuve N. F., Jiang T., Wu T., Lau A., Toppin H. A., Zhang D. D. (2011) Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. U. S. A. 108, 1433–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ohnuma T., Matsumoto T., Itoi A., Kawana A., Nishiyama T., Ogura K., Hiratsuka A. (2011) Enhanced sensitivity of A549 cells to the cytotoxic action of anticancer drugs via suppression of Nrf2 by procyanidins from cinnamomi cortex extract. Biochem. Biophys. Res. Commun. 413, 623–629 [DOI] [PubMed] [Google Scholar]