

Abstract
The deleterious effect of superoxide radicals on cell growth and survival is predominately caused by rapid oxidation of labile [Fe-S] clusters in proteins. Oxidation of these clusters releases Fe(II) ions, which participate in Fenton chemistry that damages DNA. Here it is shown that elevated levels of the YggX protein increase the resistance of Salmonella enterica to superoxide stress, reverse enzymatic defects attributed to oxidized [Fe-S] clusters, and decrease the spontaneous mutation frequency. The data are consistent with a model in which YggX protects protein [Fe-S] clusters from oxidation.
Cell physiology is characterized by the interplay of numerous metabolic pathways and processes. Integration is essential to create a metabolism that is robust, yet adaptable to complex environmental conditions, such as growth in the presence or absence of oxygen. Although aerobic respiration provides a substantial energetic advantage, it necessarily generates toxic oxygen species that can damage macromolecules (1, 2). For example, superoxide radicals (O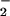 ) can oxidize labile [4Fe-4S] to inactive [3Fe-4S] clusters (3, 4). Such oxidation has at least two detrimental consequences, inactivation of enzymes containing [Fe-S] clusters (3, 5–7), and increased DNA damage (8, 9). DNA damage results from ferrous ions, released during the oxidation of [4Fe-4S] clusters. These ions participate in Fenton chemistry [Fe(II) + H2O2 + H + → Fe(III) + H2O + OH⋅], with the hydroxyl radicals damaging DNA and other macromolecules (9–11). It would not be surprising that many cellular anomalies caused by increased superoxide concentration result from oxidization of [Fe-S] clusters (9).
) can oxidize labile [4Fe-4S] to inactive [3Fe-4S] clusters (3, 4). Such oxidation has at least two detrimental consequences, inactivation of enzymes containing [Fe-S] clusters (3, 5–7), and increased DNA damage (8, 9). DNA damage results from ferrous ions, released during the oxidation of [4Fe-4S] clusters. These ions participate in Fenton chemistry [Fe(II) + H2O2 + H + → Fe(III) + H2O + OH⋅], with the hydroxyl radicals damaging DNA and other macromolecules (9–11). It would not be surprising that many cellular anomalies caused by increased superoxide concentration result from oxidization of [Fe-S] clusters (9).
Several systems exist to reduce the potential for damage by superoxide radicals (12). In general, these systems either prevent the damage from occurring or repair it. The Sox regulon is a good example of the former. This regulon includes a number of genes that are induced under conditions of oxidative stress via the SoxRS regulatory system (13–15). One component of this system is the superoxide dismutase (SOD) enzymes (EC 1.15.1.1) that catalyze the formation of molecular oxygen and hydrogen peroxide from two superoxide radicals (O + O
+ O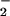 + 2H+ → O2 + H2O2). The resulting hydrogen peroxide (H2O2) is a substrate for catalase (hydroperoxidase) enzymes (EC 1.11.1.6, 1.11.1.7) that convert it to water and molecular oxygen. A distinct way of protecting [Fe-S] clusters is shown by the FeSII protein of Azotobacter vinelandii. The FeSII, or Shetna protein, forms a complex with nitrogenase under periods of high oxygen exposure, thus protecting the essential [Fe-S] cluster from oxidation (16, 17).
+ 2H+ → O2 + H2O2). The resulting hydrogen peroxide (H2O2) is a substrate for catalase (hydroperoxidase) enzymes (EC 1.11.1.6, 1.11.1.7) that convert it to water and molecular oxygen. A distinct way of protecting [Fe-S] clusters is shown by the FeSII protein of Azotobacter vinelandii. The FeSII, or Shetna protein, forms a complex with nitrogenase under periods of high oxygen exposure, thus protecting the essential [Fe-S] cluster from oxidation (16, 17).
In addition to eliminating superoxide per se, mechanisms to repair damage incurred by the superoxide radicals have evolved. This second strategy includes multiple repair systems that are specific for DNA damage (18–20). The DNA glycosylase MutY, which itself contains an [Fe-S] cluster (21, 22), recognizes the mispairing of an oxidized guanine base (8-oxo-guanine) with adenine and cleaves the relevant adenine (23). This cleavage product becomes the target for additional repair enzymes that prevent the generation of a G⋅C to T⋅A transversion mutation.
Another example involves direct repair of oxidized [Fe-S] clusters in vivo. The enzyme paradigm for the majority of studies addressing the in vivo and in vitro reconstitution of [Fe-S] clusters is aconitase (Acn, EC 4.2.1.3) (24–26). Part of the catalytic [4Fe-4S] center in Acn is exposed to the solution and is not sequestered by the enzyme; thus the enzyme is sensitive to attack by superoxide (25, 27). Although extensive work has been performed to characterize in vitro reactivation of oxidized [Fe-S] clusters (24), the participants in [Fe-S] cluster repair in vivo are less well defined (26). The benefit of in vivo repair of [Fe-S] clusters is at least 2-fold, first the restoration of enzyme activity, and second, the decrease of free iron.
Several experiments have suggested that glutathione (GSH) is involved in the in vivo repair and possibly biosynthesis, of the [Fe-S] center in Acn (26). When Escherichia coli strains in vivo were challenged with oxygen total Acn activity decreased, as expected for an enzyme with a labile [Fe-S] cluster. However, when the oxygen challenge was removed, unlike the wild-type strain, gshA (encodes γ-l-glutamyl-l-cysteine synthetase, EC 6.3.2.2) mutants were unable to regain Acn activity in the absence of protein synthesis (26). Further, gshA mutants of E. coli have reduced total Acn activity (26). These results led the authors to suggest that GSH was the cellular reductant involved in repairing the oxidized [Fe-S] cluster of Acn and possibly similar clusters in other proteins.
Here, we show that increased levels of the YggX protein reverse several metabolic defects attributed to a lack of GSH, increase resistance to superoxide stress, and decrease the spontaneous mutation frequency in Salmonella enterica. The phenotypic consequences of increased YggX protein are consistent with a model in which this protein protects labile [Fe-S] clusters from oxidative damage.
Materials and Methods
Strains, Media, and DNA Manipulations.
Strains used were derivatives of S. enterica Serovar Typhimurium strain LT2. Media, antibiotics, and insertion nomenclature have been described (28). All chemicals were purchased from Sigma.
Enzymes for DNA manipulations were purchased from Promega and used as per the manufacturer's instructions. Sequencing was carried out by the University of Wisconsin Biotechnology Center. PCR amplification of S. enterica yggX used E. coli ORFmers (b2962-A, b2962-C) with conditions specified by the manufacturer (Sigma-Genosys).
Genetics.
Transduction.
The methods of transduction using P22 (HT105/1, int-201) (29) and purification of transductants have been described (30).
Isolation of mutants overexpressing YggX (yggX*).
Cells from an overnight nutrient broth culture were pelleted and washed twice with NaCl (85 mM), and aliquots were plated on minimal glucose medium. Colonies arose after 2–3 days of 37°C incubation. A Tn10d(Cm) insertion (31) linked to the causative mutation was identified by standard genetic techniques (32).
Identification of yggX locus.
Genomic DNA from a suppressed gshA strain was partially digested with Sau3A and ligated into vector pSU19(Cm), and the resulting DNAs were electroporated into LT2 cells. Electroporants were selected for CmR and screened for increased resistance to paraquat (PQ) (100 μl of 0.1% PQ spread on a nutrient broth plate). Plasmid DNA was isolated and electroporated into strains DM271 (apbE) and DM4620 (gshA), and prototrophy was scored. One clone, pPQR4 (Fig. 1), satisfied all requirements and was used further.
Figure 1.

Physical parameters of yggX and its gene product. (A) Alignment of YggX homologs. (B) Operon structure of mutY/yggX in E. coli and S. enterica LT2. Promoters were mapped by Gifford and Wallace in E. coli (43).
Generation of chromosomal yggX insertion.
Plasmid pYGGX3A∷Gm was transduced into a polA-deficient strain (DM3961). The transduction was allowed to proceed for 1 h, and cells were then washed twice in LB + 5 mM EGTA and incubated at room temperature overnight before spreading onto nutrient broth/Gm plates. Colonies that arose on nutrient broth/Gm plates were screened for CmS, indicating loss of the parent plasmid by a double crossover event. The GmR cassette from GmRCmS strains was transduced into wild-type LT2; the insertion in yggX was confirmed by PCR amplification.
Strain construction.
A soxR deletion strain of E. coli (DJ901) was obtained, and the marker [zjc-2204∷Tn10 (Km)] linked to the deletion (33) was transduced into S. enterica LT2 via a mutS intermediate as described (34, 35). Transductants were scored for the ΔsoxR901 allele (sensitivity to 4 μM PQ). An isogenic pair of strains with (DM5317) and without (DM5319) the ΔsoxR901 allele was constructed. The presence of the yggX* mutation in relevant strains was confirmed by backcross into a gshA strain (DM4620).
Nutritional requirements.
Nutritional requirements were tested with solid medium, soft agar overlays, and growth curves in microtiter plates (36, 37).
Spontaneous mutation frequency.
Cultures were grown by shaking overnight in LB at 37°C. Aliquots (100–200 μl) were plated on solid LB media containing 100 μg/ml rifampacin and incubated overnight at 37°C. In the case of d-cycloserine resistance, cultures were grown overnight in defined medium. Aliquots (10–100 μl) were plated on minimal glucose plates containing 0.2 mM d-cycloserine (0.2 M stock in phosphate buffer, pH 8.0), and incubated overnight at 37°C. Colony-forming units were determined by plating on nonselective media.
Enzyme Assays.
Acn.
Acn activity was assayed in cell-free crude extracts by the protocol of Gruer and Guest (38), as modified by Skovran and Downs (39). Specific activity was described in units/mg protein where a unit was the change in absorbance at 240 nm per min. Protein concentration was determined by the Bradford assay (40).
SOD (EC 1.15.1.1).
SOD assays were modified from McCord and Fridovich (41). Cultures (5 ml of LB grown overnight at 37°C) were washed once with 3 ml of 50 mM KH2PO4/0.1 mM EDTA, then resuspended in 1 ml of this buffer. Cells were kept on ice and sonicated three times for 10 sec (0.5-sec pulses, power set to 3) by using a Sonic Dismembrator 550 (Fischer Scientific). Extracts were centrifuged to remove cell debris and unbroken cells and kept on ice until assayed. A unit of SOD activity was as described (41).
YggX Overexpression and Purification.
The yggX gene was cloned into the NdeI and SmaI sites of the pTYB2 expression vector (New England Biolabs) contained in the IMPACT T7 Kit. The resulting plasmid, pJAG100, was electroporated into strain BL21(λDE3). Overexpression and purification were performed per the manufacturer's recommendations, with the exception that the DTT concentration used during the on-column cleavage step was 50 mM. Protein was concentrated by using an Ultrafree-15 centrifugal filter device (Millipore) with a 5-K molecular weight cutoff. Anti-YggX polyclonal rabbit antibodies against purified YggX were generated at the University of Wisconsin Animal Care Unit.
Results
A Suppressor of gshA Mutant Phenotypes.
We recently demonstrated that gshA mutants of S. enterica Serovar Typhimurium strain LT2 are thiamine auxotrophs (28). When a gshA mutant strain was incubated on minimal glucose plates for 2–3 days, colonies arose at a frequency of ≈10−5. Genetic analyses of 10 independent colonies demonstrated that prototrophic growth resulted from a single lesion. An insertion [zgf-8077∷Tn10d(Cm)] was 80% linked by P22 transduction to the causative mutation in each of the 10 revertants. The suppressing allele was designated yggX* to be consistent with annotation of the E. coli genome.
An Intact yggX Locus Is Required for Suppression.
A plasmid library was generated by using genomic DNA from a gshA yggX* double-mutant strain (DM5015). Assuming the yggX* mutation was dominant, clones were screened for ability to confer PQ resistance (see below) and prototrophic growth to strain DM4620 (gshA). One such plasmid (pPQR4) is diagrammed in Fig. 1 and was further characterized. Sequence analysis determined that plasmid pPQR4 contained two full genes (yggX, mltC) and part of a third (mutY). Because additional independent clones also carried yggX, the involvement of this gene in prototrophic growth was pursued. A DNA fragment containing yggX and reduced flanking sequences was PCR-amplified from pPQR4 and used to generate plasmid pYGGX3A (Fig. 1). This plasmid conferred the same growth phenotype as pPQR4, establishing the sufficiency of the yggX gene for suppression.
To investigate the role of yggX in the growth phenotype, a targeted null mutation was generated. A cassette encoding gentamycin resistance (42) was engineered into a unique BglII site in the yggX coding sequence on plasmid pYGGX3A. The resulting plasmid, pYGGX3A∷Gm, failed to restore growth of strain DM4620 (gshA) on minimal glucose medium. When the chromosomal yggX∷Gm insertion was transduced into strain DM5015 (yggX* gshA), the suppression of the thiamine requirement was lost. We concluded that an intact yggX locus was required for the phenotypic suppression caused by a yggX* mutation. No nutritional requirement was detected for the single yggX null mutant (data not shown).
Increased Expression of yggX Is Sufficient for Phenotypic Suppression of gshA Mutants.
Three results led to the conclusion that the yggX* mutation results in increased levels of YggX protein that cause the phenotypes attributed to this mutation. First, there were no differences in the yggX coding sequence between wild-type and yggX* strains. Second, ORFmers were used to amplify the yggX coding sequence from wild-type and yggX* mutant strains and generate plasmids containing only the yggX coding region in each of two orientations. Only the two plasmids with inserts properly oriented with respect to the plasmid-encoded lac promoter restored prototrophic growth of the gshA mutant.
Third, Western blot analyses of cell-free extracts showed that strain DM5105 (yggX*) had increased levels of YggX protein (11 kDa) compared with the isogenic strain DM5104 (Fig. 2). In fact, YggX was not detectable in the wild-type strain by this assay. The above results demonstrated that increasing the levels of YggX was sufficient to cause the phenotypes associated with the yggX* mutation and they were consistent with the yggX* mutation affecting expression of yggX.
Figure 2.

Increased levels of YggX protein in yggX* mutant. Western blot analysis was performed according to Harlow and Lane (59). Proteins were visualized by using alkaline phosphatase conjugated to anti-rabbit secondary antibody (Promega). Lanes A–C were loaded with crude cell-free extracts (1 μg protein) from strains DM5104, DM5105 (yggX*), and DM5647 (yggX∷Gm), respectively. Lane D was loaded with 1 ng purified YggX.
The yggX gene is located at minute 66 on the E. coli and S. enterica chromosomes. In a number of organisms, yggX is located adjacent to mutY (encoding adenine DNA glycosylase), and at least in E. coli, these genes appear to be cotranscribed (43). The gene organization of mutY and yggX appears to be conserved in at least 17 of the 23 eubacteria. We have not found yggX sequences in any archeal or eukaryotic genome sequences available in the GenBank Database at the National Center for Biotechnology Information.
Increased Level of YggX Does Not Act by Increasing the Cellular Levels of Free Thiols.
Inactivation of gshA results in loss of GSH, the predominant free thiol in the cell (44). Because the phenotypes of a gshA mutant must be explained in the context of this loss, it was conceivable that the phenotypic suppression by yggX* could be due to either gshA-independent formation of GSH or elevation of a distinct free-thiol pool. The results of two experiments eliminated both of these possibilities. First, GSH levels of 14.0 pmol/mg wet weight were detected in wild-type strain (LT2) by using a GSH cycling assay (45), yet no GSH (<0.1 pmol) was detectable in either gshA or gshA yggX* mutant strains (DM5014 and 5015, respectively). Second, the yggX* mutation did not alter the sensitivity of a gshA mutant to N-methyl-N′-nitro-N-nitrosguanidine (MNNG). MNNG is a common mutagen whose toxicity is accelerated by the presence of free thiols in the cell (46). Growth analyses were preformed in the presence of 60 μM MNNG, and the results are presented in Fig. 3. As expected, strain DM5014 (gshA) was significantly more resistant to MNNG than wild-type strain LT2 (47), and the yggX* mutation had no deleterious affect on this resistance. In fact, the gshA yggX* double mutant (DM5015) appeared to have a slightly increased growth rate. A general stimulation of growth rate was observed in several strains containing the yggX* mutation or the overexpression plasmid and was attributed to the general effect of increased levels of YggX on distinct areas of metabolism described below. The resistance of gshA yggX* double mutants to MNNG suggested that an increased level of YggX did not elevate the pool size of a free thiol.
Figure 3.

The yggX* mutation does not increase MNNG resistance of gshA mutants. Strain LT2 was grown in LB with (▴) and without (▵) 60 μM MNNG. Both gshA (○) and gshA yggX* (●) mutant strains were grown in LB with 60 μM MNNG.
The Breadth of Phenotypes Suppressed by Increased Levels of YggX Suggests a Role for This Protein in Protecting [Fe-S] Clusters.
Mutants defective in gshA belong to a recently defined class of thiamine auxotrophs that share several phenotypic similarities (28, 39) including a requirement for the thiazole moiety of thiamine that can be eliminated by anaerobic growth. It has been proposed that this defect reflects an inability to repair the oxidized [Fe-S] cluster in the ThiH biosynthetic enzyme (28). Although the function of their gene products has not been determined, lesions in apbC (48) and apbE (49, 50) result in a thiamine phenotype similar to that caused by a gshA mutation. The effect of the yggX* mutation on thiamine-independent growth in these mutant strains was quantified, and data from representative experiments are shown in Table 1. The data showed that the requirement for thiamine was eliminated by a yggX* mutation in a strain defective in gshA, apbC, or apbE (Table 1, lines 2–7). These results were consistent with thiamine synthesis in these mutant strains being disrupted by a similar mechanism.
Table 1.
yggX* mutation eliminates thiamine requirement of gshA mutants
| Line | Strain | Relevant genotype | Growth rate, μ
|
|
|---|---|---|---|---|
| Minimal | Min + Thi | |||
| 1 | LT2 | Wild type | 0.45 | 0.47 |
| 2 | DM5014 | gshA | 0.11 | 0.32 |
| 3 | DM5015 | gshA yggX* | 0.46 | 0.35 |
| 4 | DM5784 | apbE | 0.09 | 0.31 |
| 5 | DM5783 | apbE yggX* | 0.44 | 0.42 |
| 6 | DM1774 | apbC | 0.20 | 0.37 |
| 7 | DM1773 | apbC yggX* | 0.46 | 0.45 |
Specific growth rate was determined by using μ = ln(X/X0)/T, where X is Abs650 during the log portion of the growth curve and T is time. Numbers shown are representative of at least two experiments.
Mutations in the isc gene cluster of S. enterica (39) and E. coli (51, 52) cause a number of metabolic phenotypes, two of which are relevant here. A polar mutation in iscA caused a requirement for thiazole similar to that described for the class of mutants discussed above (39). This requirement was eliminated by the presence of either the yggX* mutation or plasmid pYGGX3A (data not shown). Further, the nicotinic acid requirement generated by lack of the iscS gene was eliminated by the overexpression of YggX (ref. 39 and unpublished results). The nicotinic acid requirement can be traced back to a reduced activity of NadA (quinolinic synthetase) (39, 53), an enzyme that also contains an oxygen-labile [Fe-S] center (5).
The emerging correlation between increased YggX levels and activity of [Fe-S] proteins prompted us to address Acn activity. In both E. coli (26) and S. enterica (28), gshA mutants have reduced total Acn activity. This loss in activity was suggested to reflect an inability to repair the oxidized [Fe-S] center of Acn in the absence of GSH (26). The specific activity of Acn in cell-free extracts of wild-type, gshA and gshA yggX* mutant strains was 3.50 ± 0.32, 1.23 ± 0.22, and 3.66 ± 0.23 units/mg protein, respectively.
Increased levels of YggX restored activity of at least two enzymes when assayed nutritionally (ThiH, NadA) and one when assayed biochemically (Acn). The ability of the yggX* mutation to completely restore Acn activity makes it feasible that suppression of the nutritional requirements reflects a significant change in the relevant enzyme activities. Experiments below identified additional metabolic consequences of increased levels of YggX, all of which could be accounted for by a model in which YggX was either limiting oxidation of [Fe-S] centers and/or facilitating their repair.
Increased Levels of YggX Result in soxR-Independent Resistance to Superoxide.
Strains carrying the yggX* mutation, or the expression plasmids described above, displayed increased resistance to superoxide. Supplementing the growth medium with the redox-cyling herbicide PQ increased the concentration of superoxide (54). Fig. 4 illustrates the effect of the yggX* mutation on the growth of four strains in the presence of PQ. Data in Fig. 4B show that wild-type S. enterica grew slowly in the presence of 40 μM PQ and that a yggX* mutation restored rapid growth. A gshA mutant was sensitive to the presence of 4 μM PQ, as shown in Fig. 4A (28), and the yggX* mutation improved growth, restoring it to a wild-type rate. In other experiments using phenazine methosulfate (16 μM) as the generator of superoxide, similar trends were seen. In a representative experiment, the specific growth rates of a gshA (DM5014) and a gshA yggX* mutant strain (DM5015) in LB containing phenazine methosulfate were 0.15 and 0.51, respectively.
Figure 4.

The yggX* mutation increases resistance of S. enterica to PQ. (A) Growth of gshA (○) and gshA yggX* (●) mutant strains in LB with 4 μM PQ. (B) Growth of LT2 (▵) and yggX* (▴) strains in LB with 40 μM PQ.
Growth in PQ induces expression of genes in the soxRS regulon, currently the best understood system to combat superoxide stress (15). To test whether the increased resistance of the yggX* mutants to PQ was mediated through the soxRS regulon, various strains with lesions in soxR were constructed and analyzed. Some of the data from these experiments are shown in Fig. 5. In our system, as in others, a soxR mutant (DM5317) was more sensitive to PQ than the isogenic soxR+ strain (DM5319). The growth data showed that a yggX* mutation significantly increased the resistance of the soxR strain to PQ (0.4 μM), but was unable to restore resistance to the wild-type level. We observed that a yggX* mutation restored prototrophic growth to a gshA mutant strain, even in the presence of the soxR mutation (data not shown). Together, these results indicated that the resistance to PQ allowed by increased levels of YggX was not mediated through the soxRS system. Because inactivation of enzymes containing labile [Fe-S] centers contributes to the lethality of PQ, these results were also consistent with a model in which YggX protects [Fe-S] centers from oxidation.
Figure 5.

yggX* does not require soxR to mediate resistance to PQ. Strains LT2 (♦), soxR (◊), and soxR yggX* (▾) were grown in LB with 4.0 μM PQ.
It was formally possible that YggX overexpression increased the cellular level of SOD activity independent of the soxRS system. When SOD activity of the wild-type (DM5104) and yggX* mutant strain (DM5105) were measured to address this possibility, they were found to be 6.78 ± 0.49 and 6.61 ± 0.49 units, respectively.
Increased Levels of YggX Result in a Decreased Frequency of Spontaneous Mutations.
A role for YggX in mutagenesis was explored for two reasons. First, the conserved location of yggX adjacent to mutY raised the possibility that YggX was associated with MutY function. It was intriguing that MutY itself contains an [Fe-S] center, while it functions under conditions of oxidative stress in the repair of oxidatively damaged DNA (20, 21, 23). In a more general context, our working model suggests that YggX reduces the oxidation of [Fe-S] clusters (see below). Thus, YggX would reduce the loss of Fe(II) ions from clusters. The resulting decrease in free-iron levels would generate fewer hydroxyl radicals and thus reduce DNA damage (9). As an initial test of this aspect of the model, the frequency of spontaneous mutants acquiring resistance to rifampicin or d-cycloserine was determined in several strains. Representative data for these two assays of mutation frequency are shown in Table 2. As shown by the data in Table 2, in an otherwise wild-type background, the yggX* mutation reduced the number of spontaneous mutations by more than 10-fold. As predicted by our working model, a gshA mutant displayed an increased mutation frequency. When the yggX* mutation was present in the gshA mutant background, the frequency of Rfr colonies was decreased from 176 to a background level of 1–2/108. A similar trend was noted in the frequency of spontaneous mutants resistant to d-cycloserine.
Table 2.
Frequency of rifampacin and d-cycloserine resistant colonies is reduced by yggX*
| Strain | Genotype | CFU/108 cells
|
|
|---|---|---|---|
| Rifampacin | d-cycloserine | ||
| DM5104 | Wild type | 40 | 1,600 |
| DM5105 | yggX* | 1–2 | 420 |
| DM5014 | gshA | 176 | ND |
| DM5015 | gshA yggX* | 1–2 | ND |
Numbers presented are representative of three experiments. See Materials and Methods for description of technique. ND, not determined. CFU, colony-forming units.
Discussion
This work was initiated to characterize a frequent mutation that suppresses the requirement of a class of thiamine auxotrophs (28). Molecular analysis found the causative mutation, yggX*, increased the level of the YggX protein. Overexpression of the yggX gene was found to alter several metabolic processes “unrelated” to thiamine synthesis. The phenotypes resulting from YggX overexpression are broad enough to suggest a role for this protein in a central metabolic process. Our working model holds that YggX protects labile [Fe-S] clusters from attack by oxygen species, including superoxide.
Fig. 6 depicts the consequences of superoxide radicals relevant to our model for the function of YggX. Superoxide (and/or other oxygen species) attack the labile [Fe-S] centers in a number of proteins (e.g., Acn) (3, 5, 6, 25, 55). This molecular attack results in inactivation of the respective enzymes and release of both free-iron and hydrogen peroxide that generates DNA damaging hydroxyl radicals via Fenton chemistry (9–11). It was suggested that in a wild-type cell, GSH minimizes the effects of oxidation damage by providing reductant to facilitate reconstitution of the [Fe-S] clusters (26), completing a cycle of damage and repair to the [Fe-S] clusters that remains in equilibrium under normal growth conditions. When GSH is absent (e.g., a gshA mutant), the effects of these oxygen species are exacerbated and the resulting phenotypes include, reduced activity of enzymes with labile [Fe-S] centers (i.e., ThiH, Acn), increased sensitivity to the superoxide (e.g., growth in PQ), and increased mutation frequency. Increasing the level of YggX reversed each of these phenotypes. One interpretation of these results is that YggX acts before the damage and protects labile [Fe-S] clusters from oxidation. In this scenario, blocking the initial attack on the [Fe-S] clusters would abrogate the above phenotypes (Fig. 6). It is formally possible the YggX acts to remove superoxide or to facilitate GSH independent repair of the oxidized clusters. We observed no increased SOD activity in yggX* mutant extracts, and elevated levels of YggX increased the resistance of a wild-type strain (i.e., not limited for GSH) to superoxide, suggesting that cluster repair is not the affected step.
Figure 6.

Model showing how YggX protects S. enterica from oxidative damage. The result of superoxide attack on [Fe-S] clusters is depicted. We hypothesize that YggX is able to block oxidative damage to labile clusters and thus prevent the normal downstream consequences of such oxidation.
This work and the model described above are consistent with the suggestion that the requirement of gshA mutants for the thiazole moiety of thiamine was caused by the oxygen lability of the ThiH enzyme (28). The recent identification of ThiH as a member of a SAM radical protein family is consistent with this notion because members of this family share a motif that is indicative of an oxygen labile [Fe-S] cluster (56, 57). Thus, the characterization of YggX presented here supports our hypothesis that the role of GSH in thiamine synthesis is in repair of the oxidzed [Fe-S] cluster in ThiH (28).
This work raises a number of provocative questions for future studies. The phenotypes characterized here were the result of relatively high levels of YggX. The conserved location of yggX adjacent to mutY is intriguing. MutY contains an [Fe-S] center and must function under conditions of oxidative stress to perform its role in repairing oxidatively damaged DNA. Considering results herein, we suggest that YggX protects the [Fe-S] cluster of MutY under conditions of oxidative stress. Although in vitro studies on the homolgous enzyme endonuclease III suggest the [Fe-S] cluster in MutY is not accessible to oxidation (58), the need for protection in vivo or perhaps during protein folding after synthesis, maturation, and/or conformation changes associated with function are not ruled out.
The model proposed for the function of YggX in vivo encourages us to develop an in vitro assay for protection of oxygen labile [Fe-S] clusters. Such in vitro experiments may distinguish between various mechanisms that could explain the in vivo results and also help frame questions to dissect the possible connection between MutY and YggX functions.
In summary, our work has provided insight on the function of a previously uncharacterized ORF in S. enterica. By the serendipitous use of a strain that was sensitive to the lack of GSH we were able to identify a phenotype associated with increased cellular levels of YggX and offer a plausible model for the role of YggX in cellular metabolism.
Acknowledgments
The first yggX* mutation was isolated by L. Petersen, our associate. We thank J. Imlay for the sox mutant strains and advice on the SOD assays. We acknowledge the discussion and helpful comments of W. McClain and J. C. Escalante-Semerena. This work was supported by the competitive grants program of the National Science Foundation (MCB9723830) and the Shaw Scientist program of the Milwaukee Foundation.
Abbreviations
- Acn
aconitase
- SOD
superoxide dismutase
- GSH
glutathione
- PQ
paraquat, MNNG, N-methyl-N′-nitro-N-nitrosoguanidine
References
- 1.Gonzalez-Flecha B, Demple B. J Biol Chem. 1995;270:13681–13687. doi: 10.1074/jbc.270.23.13681. [DOI] [PubMed] [Google Scholar]
- 2.Imlay J A, Fridovich I. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- 3.Flint D H, Tuminello J F, Emptage M H. J Biol Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 4.Kuo C F, Mashino T, Fridovich I. J Biol Chem. 1987;262:4724–4727. [PubMed] [Google Scholar]
- 5.Gardner P R, Fridovich I. Arch Biochem Biophys. 1991;284:106–111. doi: 10.1016/0003-9861(91)90270-s. [DOI] [PubMed] [Google Scholar]
- 6.Gardner P R, Fridovich I. J Biol Chem. 1991;266:1478–1483. [PubMed] [Google Scholar]
- 7.Gardner P R, Fridovich I. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 8.Imlay J A, Linn S. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 9.Keyer K, Imlay J A. Proc Natl Acad Sci USA. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liochev S I, Fridovich I. Free Radical Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasan C, Liba A, Imlay J A, Valentine J S, Gralla E B. J Biol Chem. 2000;275:29187–29192. doi: 10.1074/jbc.M004239200. [DOI] [PubMed] [Google Scholar]
- 12.Storz G, Imlay J A. Curr Opin Microbiol. 1999;2:188–194. doi: 10.1016/s1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- 13.Hidalgo E, Demple B. EMBO J. 1997;16:1056–1065. doi: 10.1093/emboj/16.5.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaudu P, Moon N, Weiss B. J Biol Chem. 1997;272:5082–5086. doi: 10.1074/jbc.272.8.5082. [DOI] [PubMed] [Google Scholar]
- 15.Liochev S I, Benov L, Touati D, Fridovich I. J Biol Chem. 1999;274:9479–9481. doi: 10.1074/jbc.274.14.9479. [DOI] [PubMed] [Google Scholar]
- 16.Lou J, Moshiri F, Johnson M K, Lafferty M E, Sorkin D L, Miller A, Maier R J. Biochemistry. 1999;38:5563–5571. doi: 10.1021/bi9827823. [DOI] [PubMed] [Google Scholar]
- 17.Shethna Y I, DerVartanian D V, Beinert H. Biochem Biophys Res Commun. 1968;31:862–868. doi: 10.1016/0006-291x(68)90531-7. [DOI] [PubMed] [Google Scholar]
- 18.McCullough A K, Dodson M L, Lloyd R S. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 19.Cadet J, Bourdat A G, D'Ham C, Duarte V, Gasparutto D, Romieu A, Ravanat J L. Mutat Res. 2000;462:121–128. doi: 10.1016/s1383-5742(00)00022-3. [DOI] [PubMed] [Google Scholar]
- 20.Boiteux S, Radicella J P. Biochimie. 1999;81:59–67. doi: 10.1016/s0300-9084(99)80039-x. [DOI] [PubMed] [Google Scholar]
- 21.Michaels M L, Pham L, Nghiem Y, Cruz C, Miller J H. Nucleic Acids Res. 1990;18:3841–3845. doi: 10.1093/nar/18.13.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porello S L, Cannon M J, David S S. Biochemistry. 1998;37:6465–6475. doi: 10.1021/bi972433t. [DOI] [PubMed] [Google Scholar]
- 23.Michaels M L, Tchou J, Grollman A P, Miller J H. Biochemistry. 1992;31:10964–10968. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy M C, Beinert H. J Biol Chem. 1988;263:8194–8198. [PubMed] [Google Scholar]
- 25.Gardner P R, Fridovich I. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- 26.Gardner P R, Fridovich I. Arch Biochem Biophys. 1993;301:98–102. doi: 10.1006/abbi.1993.1120. [DOI] [PubMed] [Google Scholar]
- 27.Beinert H, Kennedy M C, Stout C D. Chem Rev. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 28.Gralnick J, Webb E, Beck B, Downs D. J Bacteriol. 2000;182:5180–5187. doi: 10.1128/jb.182.18.5180-5187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmieger H. Mol Gen Genet. 1972;119:75–88. doi: 10.1007/BF00270447. [DOI] [PubMed] [Google Scholar]
- 30.Downs D M. J Bacteriol. 1992;174:1515–1521. doi: 10.1128/jb.174.5.1515-1521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Way J C, Davis M A, Morisato D, Roberts D E, Kleckner N. Gene. 1984;32:369–379. doi: 10.1016/0378-1119(84)90012-x. [DOI] [PubMed] [Google Scholar]
- 32.Kleckner N, Roth J, Botstein D. J Mol Biol. 1977;116:125–159. doi: 10.1016/0022-2836(77)90123-1. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg J T, Monach P, Chou J H, Josephy P D, Demple B. Proc Natl Acad Sci USA. 1990;87:6181–6185. doi: 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien K, Deno G, Ostrovsky de Spicer P, Gardner J F, Maloy S R. Gene. 1992;118:13–19. doi: 10.1016/0378-1119(92)90243-i. [DOI] [PubMed] [Google Scholar]
- 35.Beck B J, Connolly L E, De Las Penas A, Downs D M. J Bacteriol. 1997;179:6504–6508. doi: 10.1128/jb.179.20.6504-6508.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen L, Enos-Berlage J, Downs D M. Genetics. 1996;143:37–44. doi: 10.1093/genetics/143.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christian T, Downs D M. Can J Microbiol. 1999;45:565–572. [PubMed] [Google Scholar]
- 38.Gruer M J, Guest J R. Microbiology. 1994;140:2531–2541. doi: 10.1099/00221287-140-10-2531. [DOI] [PubMed] [Google Scholar]
- 39.Skovran E, Downs D M. J Bacteriol. 2000;182:3896–3903. doi: 10.1128/jb.182.14.3896-3903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 41.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 42.Schweizer H D. BioTechniques. 1993;15:831–834. [PubMed] [Google Scholar]
- 43.Gifford C M, Wallace S S. J Bacteriol. 1999;181:4223–4236. doi: 10.1128/jb.181.14.4223-4236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apontoweil P, Berends W. Biochim Biophys Acta. 1975;399:10–22. doi: 10.1016/0304-4165(75)90206-8. [DOI] [PubMed] [Google Scholar]
- 45.Anderson M E. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- 46.Lawley P D, Thatcher C J. Biochem J. 1970;116:693–707. doi: 10.1042/bj1160693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerklaan P, Bouter S, Mohn G. Mutat Res. 1983;122:257–266. doi: 10.1016/0165-7992(83)90004-0. [DOI] [PubMed] [Google Scholar]
- 48.Petersen L A, Downs D M. J Bacteriol. 1997;179:4894–4900. doi: 10.1128/jb.179.15.4894-4900.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beck B J, Downs D M. J Bacteriol. 1998;180:885–891. doi: 10.1128/jb.180.4.885-891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beck B J, Downs D M. J Bacteriol. 1999;181:7285–7290. doi: 10.1128/jb.181.23.7285-7290.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwartz C J, Djaman O, Imlay J A, Kiley P J. Proc Natl Acad Sci USA. 2000;97:9009–9014. doi: 10.1073/pnas.160261497. . (First Published July 25, 2000; 10.1073/pnas.160261497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lauhon C T, Kambampati R. J Biol Chem. 2000;275:20096–20103. doi: 10.1074/jbc.M002680200. [DOI] [PubMed] [Google Scholar]
- 53.Zhu N. Thesis. Salt Lake City: Univ. of Utah; 1990. [Google Scholar]
- 54.Hassan H M. Methods Enzymol. 1984;105:523–532. doi: 10.1016/s0076-6879(84)05072-2. [DOI] [PubMed] [Google Scholar]
- 55.Flint D H, Emptage M H, Finnegan M G, Fu W, Johnson M K. J Biol Chem. 1993;268:14732–14742. [PubMed] [Google Scholar]
- 56.Sofia H J, Chen G, Hetzler B G, Reyes-Spindola J F, Miller N E. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frey P A, Booker S. Adv Free Radical Chem. 1999;2:1–43. [Google Scholar]
- 58.Cunningham R P, Asahara H, Bank J F, Scholes C P, Salerno J C, Surerus K, Munck E, McCracken J, Peisach J, Emptage M H. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 59.Harlow E, Lane D. Antibodies. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]