

Abstract
We have modified the infectious reovirus RNA system so as to generate a reovirus reverse genetics system. The system consists of (i) the plus strands of nine wild-type reovirus genome segments; (ii) transcripts of the genetically modified cDNA form of the tenth genome segment; and (iii) a cell line transformed so as to express the protein normally encoded by the tenth genome segment. In the work described here, we have generated a serotype 3 reovirus into the S2 double-stranded RNA genome segment of which the CAT gene has been cloned. The virus is stable, replicates in cells that have been transformed (so as to express the S2 gene product, protein σ2), and expresses high levels of CAT activity. This technology can be extended to members of the orbivirus and rotavirus genera. This technology provides a powerful system for basic studies of double-stranded RNA virus replication; a nonpathogenic viral vector that replicates to high titers and could be used for clinical applications; and a system for providing nonselectable viral variants (the result of mutations, insertions, and deletions) that could be valuable for the construction of viral vaccine strains against human and animal pathogens.
The reovirus genome comprises 10 segments of double-stranded (ds)RNA that are transcribed nonconservatively within cores into plus strands which are extruded from virus particles by means of 12 icosahedrally distributed projections/spikes. These plus strands exercise two functions: they are translated into the reovirus-encoded proteins and they are transcribed into minus strands with which they remain associated, thereby generating the progeny dsRNA genome segments (1, 2).
Several years ago, we demonstrated that reovirus RNA is infectious (3). The basic features of the infectious reovirus RNA system are as follows. Monolayers of L929 mouse fibroblasts are lipofected with the 10 species of plus strands [single-stranded (ss)RNA] or dsRNA of reovirus serotype (ST)3, with a rabbit reticulocyte lysate in which ssRNA or melted dsRNA has been translated for 60 min. Eight hours later, the cells are infected with reovirus ST1 or ST2 as helper viruses. Virus yields are harvested 24 or 48 h later and plaqued either in the presence of antiserum against ST1 virus if it was the helper virus, or alone if the helper virus was ST2 virus. (ST3 virus forms plaques after 5 days, whereas ST2 virus forms plaques only after 12 days.) No reassortants are formed. All 10 species of RNA are necessary. Virus yields are 20 times higher if ds- rather than ssRNA is used, and if both are used, they are 10 times higher still, because dsRNA increases the infectivity of ssRNA. Lipofection of RNA translation mixtures is not essential but increases virus yields 100-fold.
This system can be used to generate novel viruses by controlling the nature of the 10 RNA species that are lipofected into cells. Thus, we have constructed a double temperature-sensitive (ts) mutant of ST3 virus by using (together with 8 ssRNA species of ST3 virus) ssRNA species that contain ts mutations derived from two ts mutants of ST3 virus (4). We also have lipofected mixtures of ssRNA species of different reovirus STs to generate genome segment reassortants, and have identified the recognition signals that are required for the introduction of heterologous RNA species into the ST3 reovirus genome (2, 5–7). We now have taken a step further in the generation of a reovirus reverse genetics system by introducing into the ST3 virus genome a genome segment the plus strand of which is transcribed from a cDNA template that encompasses the 198 5′-terminal bp of the S2 double-stranded RNA genome segment, the 572-bp long CAT gene, and the 284 3′-terminal bp of the S2 genome segment. The resulting virus is stable, replicates in cells transformed to express the σ2 protein, and expresses the CAT protein fused to the first 60-aa residues of protein σ2. Thus, reovirus now can be used as an expression vector, and reovirus particles that are modified in any desired manner in any genome segment now can be generated. Further, there is no reason why this technology cannot be extended to other members of the Reoviridae family, including orbiviruses and rotaviruses.
Materials and Methods
Virus and Cell Lines.
Reovirus ST3 strain Dearing and reovirus ST2 strain Lang were used. Both were grown in L929 mouse fibroblasts in MEM or RPMI medium 1640 supplemented with 5% (vol/vol) FBS. The recombinant virus containing the CAT gene (ST3.S2.CAT) was grown in L929 cells transformed with pHβAPr-1-neo (8) that contained ST3 S2 cDNA under the control of the human β-actin promoter. The transformed cells, L-ST3.S2 cells, expressed protein σ2 at levels that were sufficient to rescue tsC 447 (9, 10), a ST3 virus mutant with a ts mutation in the S2 genome segment.
Engineering of Reovirus S2 cDNA.
The gene selected to be inserted into the S2 genome segment of ST3 virus was the CAT gene. Scheme 1 is a diagram of the S2-CAT cDNA template (pS2-CAT13) that was inserted into pUC18.
 |
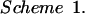 |
This template was transcribed by T7 RNA polymerase to yield an RNA transcript that possessed the same 5′- and 3′-terminal sequences as authentic s2 RNA. The 5′-terminal S2 cDNA sequence ending at bp 198 was fused, in frame, to the CAT gene-coding sequence (752 bp). The 3′ terminus of the CAT sequence was fused to the 3′ S2 cDNA sequence beginning at bp 1,047, and the 3′ terminus of the S2 cDNA sequence, including the untranslated sequence, was fused to the hepatitis delta virus (HDV) ribozyme cDNA sequence. Transcription by T7 RNA polymerase was terminated with the T7 terminator sequence located 3′ of the construct. Transcription of this construction yielded an RNA that contained the 198 5′ nucleotides of the s2 ssRNA genome segment fused in-frame to the CAT mRNA sequence followed by the 284 3′-terminal nucleotides of s2 RNA. The clone was constructed by inserting the HDV ribozyme sequence in such a way that when the ribozyme underwent autocleavage, it left a terminal C at the 3′ terminus; as a result, the 3′ terminal sequence of the transcript was the authentic reovirus RNA 3′-terminal sequence −UCAUC. Recloning and subsequent sequencing and cleavage analysis confirmed the authenticity of the 5′- and 3′-terminal sequences.
The pS2CAT13 construct was transcribed in vitro by using T7 RNA polymerase, and the transcript was capped using (m)7 GpppG (Promega) to yield s2-CAT mRNA. It was translated in vitro by using a rabbit reticulocyte lysate system (Promega), and the lysate was found to contain CAT activity [chloramphenicol transacetylase (CAT-ELISA), Roche Molecular Biochemicals] (Table 1).
Table 1.
Demonstration that the translation product of s2-CAT mRNA possesses CAT activity
| mRNA | CAT activity, pg/ml |
|---|---|
| s2 | <4 |
| s2-CAT | 64 |
Both mRNAs were translated in a rabbit reticulocyte lysate system. For experimental details, see text.
The Reovirus Infectious RNA System.
The system was used as described by Roner et al. (3), using L-ST3.S2 cells. Reovirus ST2 was used as the helper virus.
Removal of Wild-Type (wt) s2 RNA.
wt s2 RNA was removed from the mixture of 10 ssRNA species as described (4). The oligodeoxyribonucleotide (ODRN) selected for this purpose was complementary to nucleotides 937–949, and 10 pmols of it were added to 2 pmols of ST3 RNA. After hybridization, the mixture was treated with RNaseH for 20 min. The RNA was extracted 3 times with phenol/chloroform and precipitated with sodium acetate. Degradation of s2 RNA was confirmed by gel electrophoresis of the RNA (Fig. 1) and of its translation products.
Figure 1.

(Lane A) 32P-labeled RNA transcribed by T7 RNA polymerase from the pS2-CAT13 template lacking the CAT gene insert, that is, authentic s2 RNA. (Lane B) Cleavage products after hybridization to the ODRN complementary to residues 937–949 and digestion with RNaseH. Electrophoresis was carried out in an SDS/PAGE.
The set of nine ssRNAs was supplemented with s2-CAT RNA, and the resulting mixture was lipofected into L-ST3.S2 cells that then were infected with ST2 virus (3). As a control, the set of nine ssRNAs was supplemented with the transcripts shown in lane A of Fig. 1 which were identical with s2 RNA.
Virus Titration/Determination of CAT Activity.
Monolayers of lipofected and infected L-ST3.S2 cells were incubated at 37°C for 5 days. Neutral red was added 24 h before counting plaques (4). CAT activity in cell lysates was assayed by using CAT-ELISA (Roche Molecular Biochemicals).
Conditions for Determining Virus Growth Curves.
The growth characteristics of wt reovirus ST3 and ST3.S2.CAT virus in L929 and L.ST3.S2 cell monolayers (1 × 106 cells) were determined. Virus was allowed to adsorb; then the monolayers were washed, more medium was added, and incubation was continued. At the indicated times, triplicate samples of cells were harvested, sonicated, and stored at −20°C before titration, as described previously.
Measurement of RNA Synthesis.
L929 or L-ST3.S2 cells were pelleted and resuspended at a concentration of 1 × 107 cells per ml in MEM (phosphate-free) supplemented with 1% dialyzed FBS. The cells were infected with wt ST3 or ST3.S2.CAT virus at multiplicity of infection optimized for each virus [10–50 plaque-forming units per cell]. After 1 h at 4°C, the cells were diluted 10-fold in the same medium supplemented with actinomycin D (5 μg/ml) to inhibit cellular RNA synthesis, and [32P]orthophosphate was added at 50 μCi/ml (1 Ci = 37 GBq).
Measurement of Reovirus Protein and RNA Synthesis.
L929 or L-ST3.S2 cells were pelleted and resuspended at a concentration of 1 × 107 cells per ml in MEM (methionine- and phosphate-free) supplemented with 1% dialyzed FBS. The cells were infected with wt ST3 or ST3.S2.CAT virus, as described previously. After 1 h at 4°C, the cells were diluted 10-fold into the same medium supplemented with [35S]methionine at 1.5 μCi/ml and [32P]orthophosphate at 50 μCi/ml.
Immunoprecipitation.
Immunoprecipitations were performed on ice or in a cold room at 4°C. Antibody–antigen reactions were allowed to proceed for 45 min. Antibody–antigen complexes were recovered by incubation for 10 min in the presence of Pansorbin immunoadsorbant (Calbiochem) prepared according to Kessler (11), with the exception that Pansorbin was resuspended at a concentration of 10% in Tris/magnesium/NP-40 (TMN)-0.1% TX-100 instead of in NP-40/EDTA/Tris (NET) buffer. Pansorbin was pelleted from the immunoprecipitation reactions by centrifuging at 12,000 × g for 1 min. Pellets were washed with three cycles of resuspension in TMN-0.1% TX-100 and pelleting. Then immune complexes were released from the immunoadsorbent for analysis as described below.
Electrophoresis of Proteins.
Proteins were analyzed in SDS/PAGE as described by Gaillard and Joklik (12).
Results
Construction of s2-CAT RNA.
There are four prerequisites for developing a reverse genetic system for dsRNA viruses with segmented genomes. (i) The RNA must be infectious; that is, insertion of the complement of RNA genome segments into cells must lead to formation of infectious virus particles. (ii) Techniques must be available for removing any particular genome segment (this is not a general prerequisite, but it is for the construction of variant viruses with nonselectable phenotypes). (iii) The cDNA form of any particular genome segment, modified by mutation, insertion, or deletion, must be transcribed into an RNA that can be incorporated into the viral genome. (iv) If the modified genome segments do not encode proteins that can exercise the wt protein functions, cell lines must be constructed in which these proteins are expressed in amounts sufficient to enable the variant viruses to replicate.
The first prerequisite was met for orthoreoviruses some time ago; the infectious reovirus RNA system has not only been optimized, but it also has been used to identify the signals required for the introduction of genome segments into the orthoreovirus genome (3, 5). The second prerequisite also has been solved for the construction of a nonselectable reovirus double-ts mutant (4). It involves the use of short (12–15-residues-long) ODRNs that are hybridized to the RNA to be eliminated, and then the annealed complexes are digested with RNaseH. The choice of the ODRN is of great importance for avoiding incomplete digestion of the target RNA on the one hand, and for avoiding excessive breakdown of nontarget RNAs on the other. The ODRN chosen here differs from that used previously (4), because although the latter removed s2 RNA efficiently from a mixture of the four s-size class RNAs, it caused excessive degradation of m-size class RNAs when it was used for removing s2 RNA from the complete set of reovirus ssRNAs. It is therefore best to test a series of ODRNs for efficiency of target RNA removal without excessive loss of nontarget RNAs. The efficiency of target RNA removal was usually about 90% (Fig. 1) with efficiencies occasionally as high as 98% and as low as 70% for reasons yet to be identified. Removal of target RNAs also can be confirmed by electrophoresis of in vitro translation products.
The third prerequisite now has been addressed by the construction of a reovirus genome segment into which a foreign gene, the CAT gene, was incorporated. The genome segment that we chose was S2 [for which the RNaseH removal procedure had been worked out (4)]. The CAT mRNA is only about one-half as long as s2 RNA (752 and 1,331 nucleotides, respectively) and it was thought desirable for the recombinant genome segment to be approximately the same length as the wt S2 genome segment (the exact locations of encapsidation signals are not known), significant portions of the wt S2 genome segment were retained at the 5′ end (198 bp) and at the 3′ end (284 bp). The T7 polymerase promoter was placed at the 5′ end of the construct and the T7 terminator at the 3′ end; and to achieve the appropriate cleavage of the modified s2 from the transcript, the hepatitis delta virus ribozyme sequence was placed so as to ensure the presence of the universal reovirus RNA 3′-terminal sequence, −UCAUC, at the 3′ terminus of the s2-CAT RNA. This RNA was 1,234-nucleotides-long, 97 nucleotides shorter than the wt s2 RNA. It encoded a σ2-CAT fusion protein that possessed 60 σ2 amino acids at its N terminus and did not exercise protein σ2 function, but possessed CAT catalytic activity (Table 1). It is conceivable that the σ2 sequence preceding that of the protein to be expressed can be shortened, or eliminated, by mutating all AUG start codons in the s2 sequence required for the introduction of the recombinant genome segment in the reovirus genome.
Construction of a Protein σ2-Expressing Cell Line.
Because the σ2-CAT protein does not exercise protein σ2 function, it was necessary to construct a cell line that expressed this protein.
To create this cell line, mouse L929 fibroblasts were transformed with pβAPr-1-neo that contained the S2 cDNA coding sequence under the control of the β-actin promoter. To demonstrate that the σ2 protein that this cell line produced (Fig. 2) was functional, one-step growth curves at permissive (30°C) and nonpermissive (39°C) temperatures were constructed for wt ST3 virus and tsC447, a mutant with a ts lesion on genome segment S2 (8, 9). At the permissive temperature, both viruses replicated equally well in L929 and L-ST3.S2 cells, but at the nonpermissive temperature, tsC447 failed to replicate in L929 cells but replicated well in L-ST3.S2 cells (Fig. 3). Wt ST3 virus replicated equally well in both cell lines at the nonpermissive temperature.
Figure 2.

Demonstration that protein σ2 is expressed in L-ST3.S2 cells. (Lane A) Proteins formed in L929 cells infected with ST3 virus that are precipitated with antiserum against ST3 virus. (Lane B) As in lane A, for mock-infected L929 cells. (Lane C) As in lane A, for uninfected L-ST3.S2 cells. Electrophoresis was carried out in SDS/PAGE.
Figure 3.

Growth curves of ST3 virus and ts447 in L929 cells and L-ST3.S2 cells at 39°C. For experimental details, see Materials and Methods and text. (▪) ST3 virus in L929 cells. (▴) ts447 in L929 cells. (□) ST3 virus in L-ST3.S2 cells. (▵) ts447 in L-ST3.S2 cells.
Construction of ST3.S2.CAT Reovirus.
To generate a CAT-expressing reovirus, the s2 RNA-depleted set of nine ST3 virus ssRNA species was supplemented with s2-CAT RNA. One-half of the mixture was translated in a rabbit reticulocyte lysate for 60 min, and both it and the untranslated one-half were lipofected into L-ST3.S2 cells. Recombinant virus was isolated by using the optimal conditions as described (3) and in Materials and Methods. Virus in individual plaques was tested for ability to express CAT; virus in 50% of plaques was CAT+. Five clones were selected for characterization and further study.
Characterization of ST3.S2.CAT Reovirus.
We characterized the novel ST3.S2.CAT virus.
(i) The S2-CAT genome segments of all five clones were sequenced and found to conform exactly to the sequence outlined in the diagram of pS2-CAT13 in Materials and Methods. The electrophoretic migration patterns of the genome segments of ST3 virus and ST3.S2.CAT virus are shown in Fig. 4. Interestingly, the S2-CAT genome segment migrates more slowly than S2 despite being 97 nucleotides shorter. Other examples of such lack of correlation between the lengths of dsRNA segments and their electrophoretic migration rates are known, such as the case of the ST1 and ST3 S1 genome segments that are the same size (1,463 and 1,461 bp, respectively), but the ST3 S1 segment migrates considerably more slowly than the ST1 virus.
Figure 4.

Electropherograms of the genome segments of ST1, ST2, ST3 virus, and ST3.S2.CAT virus. Electrophoresis was carried out in an SDS/PAGE. Lane 1, ST1 virus; lane 2, ST2 virus; lane 3, ST3 virus; lane 4, ST3.S2.CAT virus.
(ii) The rate and extent of replication of ST3.S2.CAT virus in L929 and L-ST3.S2 cells were determined (Fig. 5). The virus failed to replicate in L929 cells, and replicated somewhat more slowly in L-ST3.S2 cells than ST3 virus but as extensively.
Figure 5.

One-step growth curves of ST3 virus and ST3.S2.CAT virus in L929 and L-ST3.S2 cells. For experimental details, see Materials and Methods and text. (▪) ST3 virus in L929 cells. (▴) ST3.S2.CAT virus in L929 cells. (□) ST3 virus in ST3.S2 cells. (▵) L-ST3.S2.CAT virus in L-ST3.S2 cells.
(iii) Examination of the ssRNAs transcribed in L-ST3.S2 cells infected with ST3.S2.CAT revealed the presence of an RNA consistent with a length of 1,234 nucleotides that hybridized to a probe that hybridized to the 18 5′-terminal residues of CAT mRNA (Fig. 6). Transcription of a similar RNA was detected in infected L929 cells in which no progeny virus was formed.
Figure 6.

Demonstration of s2.CAT RNA in L-ST3.S2.CAT virus-infected L-ST3.S2 cells. (Lane A) s2 RNA revealed in L929 cells infected with ST3 virus by hybridization with a probe that hybridized to the 18 5′-terminal nucleotides of s2 RNA. (Lane B) s2.CAT RNA revealed in L-ST3.S2 cells infected with ST3.S2.CAT virus by hybridization to a probe that hybridized to the 18 5′-terminal nucleotides of CAT mRNA. For experimental details, see Materials and Methods and text.
(iv) The amount of CAT formed was measured in L929 and L-ST3.S2 cells infected with ST3.S2.CAT virus infected at a multiplicity of infection of 20 plaque-forming units per cell (Table 2). As expected from the relative amounts of viral mRNA formed, some CAT was formed soon after infection in L929 cells, and much larger amounts were formed in L-ST3.S2 cells, maximum amounts accumulating by 20 h after infection. ST3.S2.CAT virus was an excellent vector; the amount of CAT formed in cells infected for 48 h amounted to 0.73% of total cell protein.
Table 2.
Kinetics of CAT formation in ST3.S2.CAT virus-infected L929 and L-ST3.S2 cells
| Time, h | A | B | C | D |
|---|---|---|---|---|
| 0 | <4 | <4 | <4 | <4 |
| 4 | <4 | 16 | <4 | 16 |
| 8 | <4 | 64 | <4 | 500 |
| 12 | <4 | 64 | <4 | 500 |
| 16 | <4 | 8 | <4 | 1200 |
| 24 | <4 | 4 | <4 | 1800 |
| 36 | <4 | 4 | <4 | 2000 |
| 48 | <4 | 4 | <4 | 2000 |
A, ST3 virus in L929 cells; B, ST3.S2.CAT virus in L929 cells; C, ST3 virus in ST3.S2 cells; D, ST3.S2.CAT virus in ST3.S2 cells. Numbers refer to CAT enzyme levels in terms of pg/ml. For experimental details, see Materials and Methods and text.
Discussion
The results presented in this paper complete the work required to devise a reverse genetic system for members of the Reoviridae family. The fact that the complete complement of reovirus genome segments is infectious has been known for some time (3). A technique for removing most if not all of the ssRNA form of specific genome segments and the advantages of the availability of such a technique has been described already also (4). Now, we demonstrate that genetically manipulated genome segments, that is, genome segments the plus strands of which are transcripts of their genetically manipulated cDNA versions, can be inserted into the reovirus genome. In the particular case reported here, the manipulation was the removal of a portion of the S2 genome segment and the insertion of a foreign gene, the CAT gene. As a result, the genome segment no longer encoded a functional reovirus protein, which was supplied by specially constructed host cells, and the foreign protein encoded by the genome segment was expressed abundantly when the virus replicated; that is, the virus was a vector. In other words, lethal modifications now can be incorporated in the reovirus genome, selected, and expressed.
There is one feature of reovirus genetics that emerged during the course of this work that is worth commenting on because it seems to apply generally. We pointed out that it is rarely possible to remove 100% of a particular ssRNA species, even after testing numerous ODRNs; usually about 10% of the target RNA was not degraded. Even though the RNA species that was to be incorporated into the genome then was added to the mixture of nine RNA species in at least a 10-fold excess over the small amount not degraded, usually about 50% of the progeny virus generated by the infectious reovirus RNA technology was wt virus. Screening did not present a problem—testing very few plaques always turned up the desired novel viral construct. However, it did highlight the fact that some genome segments are inserted or accepted into the viral genome much more efficiently than others, and this has nothing to do with the acceptance signals that we identified (5). In the case encountered in this work, the S2.CAT genome segment was accepted about five times less efficiently into the ST3 virus genome than the wt S2 genome segment despite possessing the latter's 198 bp at the 5′ end and its 284 bp at the 3′ end. We encountered a related situation in our previous work identifying the signals required for acceptance into the ST3 virus genome (5), which indicated that there are “sets” of reovirus genome segments that are preferred over others. Under normal conditions these are the wt genome segment sets. However, when one of the genome segments is replaced by a heterologous one, variants of some of the other nine homologous genome segments, variants that are normally minor population components, are inserted preferentially into the genome, replacing the wt component. Variants containing additional mutations could be very important and should be tested for when devising vaccine strains against pathogens in the Reoviridae family; that is, when replacing one of the pathogen's genome segments with a heterologous one, it is essential to test for the replacement of 1 or more of the 9 or 10 other wt genome segments by a minor population component that may possess an unexpected and undesirable phenotype.
In summary, we have devised a powerful reverse genetics system for members of the Reoviridae family. Reverse genetics for members of the genus orbivirus and rotavirus should be valuable for the construction and identification of vaccine virus strains for use in both humans and animals. The genetic modifications that this system permits are mutations, deletions, and insertions, and it possesses the advantage that the novel variants represent a very high (50% or more) proportion of the infectious reovirus system's virus yields. The technology also permits reovirus to be used as an abundantly replicating nonpathogenic expression vector that could find wide applicability in gene therapy and tumor oncolysis. Finally, the technology provides a powerful system for basic studies on all aspects of reovirus replication, molecular biology, and molecular genetics.
Acknowledgments
We thank Dr. Igor Nepliouev for technical advise and Ms. Kim Van Vliet for expert technical assistance. This work was supported in part by Research Initiation Awards from Florida Atlantic University, and by grants from the Center for Molecular Biology and Biotechnology, Florida Atlantic University.
Abbreviations
- ss
single-stranded
- ds
double-stranded
- ST
serotype
- l
m, and s, large-, medium-, and small-size class ssRNA forms of genome segments
- L
M, and S, large-, medium-, and small-size class dsRNA genome segments
- wt
wild type
- ts
temperature-sensitive
- CAT
chloramphenicol transacetylase
- ODRN
oligodeoxyribonucleotide
References
- 1.Joklik W K. Annu Rev Genet. 1985;19:537–575. doi: 10.1146/annurev.ge.19.120185.002541. [DOI] [PubMed] [Google Scholar]
- 2.Joklik W K, Roner M R. Prog Nucleic Acid Res Mol Biol. 1996;53:249–281. doi: 10.1016/s0079-6603(08)60147-6. [DOI] [PubMed] [Google Scholar]
- 3.Roner M R, Sutphin L A, Joklik W K. Virology. 1990;179:845–852. doi: 10.1016/0042-6822(90)90153-i. [DOI] [PubMed] [Google Scholar]
- 4.Roner M R, Nepliouev I, Sherry B, Joklik W K. Proc Natl Acad Sci USA. 1997;94:6826–6830. doi: 10.1073/pnas.94.13.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roner M R, Lin P-N, Nepluev I, Kong L-J, Joklik W K. Proc Natl Acad Sci USA. 1995;92:12362–12366. doi: 10.1073/pnas.92.26.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joklik W K, Roner M R. J Biol Chem. 1995;270:4181–4184. doi: 10.1074/jbc.270.9.4181. [DOI] [PubMed] [Google Scholar]
- 7.Roner M R. Adv Virus Res. 1999;53:355–367. doi: 10.1016/s0065-3527(08)60356-5. [DOI] [PubMed] [Google Scholar]
- 8.Gunning P, Leavitt J, Muscat G, Ng S Y, Kedes L. Proc Natl Acad Sci USA. 1987;84:4831–4835. doi: 10.1073/pnas.84.14.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito Y, Joklik W K. Virology. 1972;50:189–202. doi: 10.1016/0042-6822(72)90359-5. [DOI] [PubMed] [Google Scholar]
- 10.Wiener J R, McLaughlan T, Joklik W K. Virology. 1989;170:340–341. doi: 10.1016/0042-6822(89)90392-9. [DOI] [PubMed] [Google Scholar]
- 11.Kessler S W. J Immunol. 1975;115:1617–1624. [PubMed] [Google Scholar]
- 12.Gaillard R K, Jr, Joklik W K. Virology. 1985;147:336–348. doi: 10.1016/0042-6822(85)90136-9. [DOI] [PubMed] [Google Scholar]