

ABSTRACT
Streptolysin O is a potent pore-forming toxin produced by group A Streptococcus. The aims of the present study were to dissect the relative contributions of different structural domains of the protein to hemolytic activity, to obtain a detoxified form of streptolysin O amenable to human vaccine formulation, and to investigate the role of streptolysin O-specific antibodies in protection against group A Streptococcus infection. On the basis of in silico structural predictions, we introduced two amino acid substitutions, one in the proline-rich domain 1 and the other in the conserved undecapeptide loop in domain 4. The resulting streptolysin O derivative showed no toxicity, was highly impaired in binding to eukaryotic cells, and was unable to form organized oligomeric structures on the cell surface. However, it was fully capable of conferring consistent protection in a murine model of group A Streptococcus infection. When we engineered a streptococcal strain to express the double-mutated streptolysin O, a drastic reduction in virulence as well as a diminished capacity to kill immune cells recruited at the infection site was observed. Furthermore, when mice immunized with the toxoid were challenged with the wild-type and mutant strains, protection only against the wild-type strain, not against the strain expressing the double-mutated streptolysin O, was obtained. We conclude that protection occurs by antibody-mediated neutralization of active toxin.
IMPORTANCE
We present a novel example of structural design of a vaccine antigen optimized for human vaccine use. Having previously demonstrated that immunization of mice with streptolysin O elicits a protective immune response against infection with group A Streptococcus strains of different serotypes, we developed in this study a double-mutated nontoxic derivative that represents a novel tool for the development of protective vaccine formulations against this important human pathogen. Furthermore, the innovative construction of an isogenic strain expressing a functionally inactive toxin and its use in infection and opsonophagocytosis experiments allowed us to investigate the mechanism by which streptolysin O mediates protection against group A Streptococcus. Finally, the ability of this toxin to directly attack and kill host immune cells during infection was studied in an air pouch model, which allowed parallel quantification of cellular recruitment, vitality, and cytokine release at the infection site.
Introduction
The well-known family of bacterial toxins named cholesterol-dependent cytolysins (CDCs) has the ability to bind cholesterol on eukaryotic cell membranes. Pneumolysin (PLY) from Streptococcus pneumoniae, streptolysin O (SLO) from Streptococcus pyogenes (group A Streptococcus [GAS]), perfringolysin O (PFO) from Clostridium perfringens, and listeriolysin O (LLO) from Listeria monocytogenes are major representatives of the family that are intimately involved in pathogenesis (1–3). The oxygen-labile hemolytic toxin SLO, produced by group A Streptococcus and many group C and G streptococci (4), has been shown to be extremely toxic in vivo (5) and to induce high antibody responses (anti-streptolysin O [ASO titers]), which are instrumental in the diagnosis of streptococcal infection (6, 7). SLO is coexpressed with Streptococcus pyogenes NAD-glycohydrolase (SPN), and SLO-dependent translocation of SPN into the host cell is a second mechanism by which SLO contributes to GAS pathogenesis (8, 9).
We recently demonstrated that immunization of mice with recombinant SLO is a highly effective approach to conferring protection against infection with multiple GAS serotypes (10). However, inclusion of SLO in a vaccine formulation is likely to be hampered by its high toxicity. Here we describe how the analysis of the SLO structure/function relationship led to the development of different variants of the protein impaired in toxicity. Two distinct mutations were combined to attain a SLO derivative that had no detectable toxic activity and was still able to induce highly protective immune responses in animal models of GAS infection. The use of mutated recombinant proteins and of GAS strains harboring the same double amino acid substitution in in vitro and in vivo experiments led to a better understanding of the involvement of SLO in GAS virulence and of the role played by SLO-specific antibodies in protection from GAS infections.
RESULTS
Strategy for SLO genetic detoxification.
Since several members of the CDC family have been well characterized with respect to their structural and functional domains (1–3), we used this information for SLO detoxification by genetic manipulation. As SLO exhibits 67% identity with the conserved core of PFO (11), we initially modeled the three-dimensional structure of SLO protein domains (Fig. 1) by threading the SLO amino acid sequence onto the available PFO X-ray coordinates (12). The first 71 amino acids of SLO are not present in other CDC members and were excluded from the modeling approach.
FIG 1 .
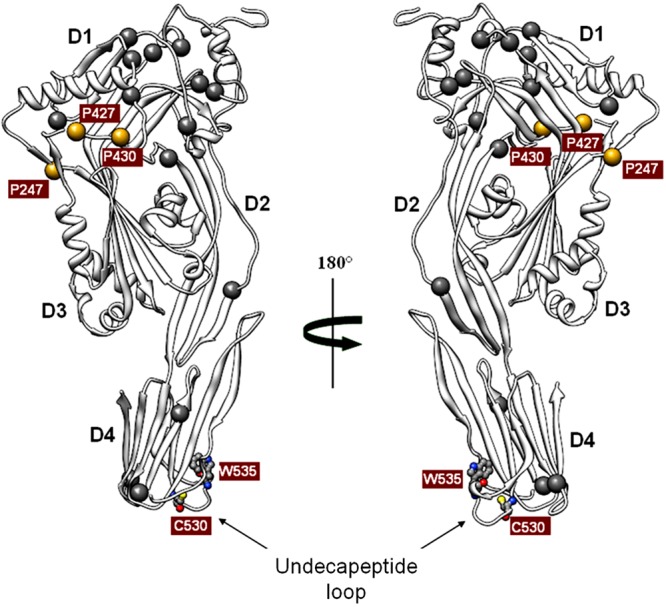
Predicted three-dimensional structure of streptolysin O. The image shows a ribbon representation of the water-soluble SLO monomer lacking the first unfolded 71 amino acids in two orientations rotated 180° relative to each other. D1, D2, D3, and D4 indicate domains 1, 2, 3 and 4, respectively. The undecapeptide loop in domain 4 is indicated by the arrows in both views. The mutagenized residues in this loop, tryptophan 535 (W535) and cysteine 530 (C530), are reported in ball-and-sticks. Golden spheres highlight the three prolines (P247, P427, and P430) located at the interfaces between two adjacent protein domains as indicated by in silico prediction analysis. Gray spheres indicate the additional 14 proline residues (4 prolines in the N-terminal region are not represented).
Assuming that SLO could induce pore formation by a mechanism similar to that proposed for PLY, which seems to primarily involve domains 1 and 4 (13), we focused our attention on these two regions. According to the proposed model, domain 1 is supposedly unaffected by the dramatic conformational rearrangements occurring during pore formation. We realized that this domain spans 30% of the entire SLO protein sequence and contains 12 of the 21 proline residues present in its primary sequence. It was therefore conceivable that, due to their intrinsic torsional rigidity, prolines could be instrumental in ensuring the conformational integrity of this critical region of the protein during its rearrangements on the cell membrane. We thus reasoned that these residues could represent good candidates for generating SLO derivatives with impaired toxicity and unaltered immunological properties. In particular, we selected proline residues interfacing adjacent domains, assuming that these regions were subjected to conformational stress and likely involved in intramolecular interactions. These criteria were met by prolines 247, 427, and 430 (P247, P427, and P430) (Fig. 1). We finally concentrated on P427 for two main reasons: first, it was predicted to be located at the interface between domains 1 and 3, a region considered critical for CDC oligomerization and insertion (14); second, the corresponding P356 in PFO had the highest B-factor (15) among the three prolines, suggesting that this amino acid residue could be particularly stressed during SLO structural rearrangements and therefore crucial for maintaining the local conformational integrity required for oligomer pore formation.
The second protein region taken into consideration was located in SLO domain 4, between residues 529 and 539 (Fig. 1), corresponding to the highly conserved protein undecapeptide loop responsible for eukaryotic cell membrane binding in almost all CDCs (1). Cysteine 530 (C530) and tryptophan 535 (W535) were chosen for modification, since changes of the corresponding residues (C428 and W433) were previously shown to reduce PLY toxicity while retaining its protective properties (16).
Generation of SLO single-mutation protein derivatives.
Six His-tagged SLO derivatives were generated. Proline 427 was replaced with glycine, leucine, glutamic acid, or lysine. While the first mutation (P427G) was intended to relax the intrinsic conformational rigidity conferred by the proline residue, the other mutations were chosen to allow us to evaluate the effects of replacing the small hydrophobic side chain with larger hydrophobic (P427L), basic (P427K), or acidic (P427E) residues. In domain 4, C530 and W535 were replaced with glycine (C530G) and phenylalanine (W535F), respectively.
The six recombinant proteins were tested for hemolytic activity on sheep blood cells and for their capacity to bind to A549 human pulmonary epithelial cells, in comparison to wild-type SLO (Fig. 2A). A 3-log reduction in hemolysis was attained with the two domain 4 mutants, while partial (P427G), negligible (P427K), or no (P427E and P427L) detectable hemolysis was observed in the case of P427 mutants. Interestingly, while the reduction in the hemolytic capacity of SLO mutants in the domain 4 undecapeptide loop coincided with a sharp reduction in their capability to bind eukaryotic cells, a lower cell binding capacity could not account for the huge toxicity impairment of the P427 mutants.
FIG 2 .
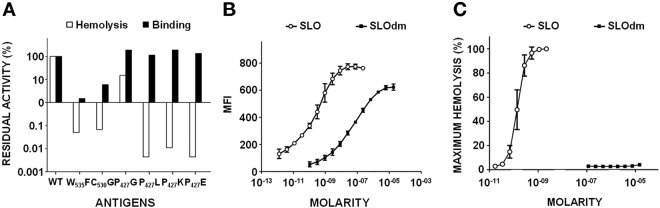
Hemolytic activity and binding properties of SLO single and double mutants. (A) Percent residual binding to A549 human lung epithelial cells measured by FACS analysis and hemolytic activity on sheep blood cells of SLO (WT) and of various SLO single mutants. (B) Binding curves of SLO and SLO(P427L/W535F) (SLOdm) on A549 cells as determined by correlating the MFI detected by FACS analysis with the different amounts of protein tested, expressed as molarities. (C) Hemolytic curves of SLO and SLOdm reported as percentages of maximum hemolysis of sheep blood cells obtained using different antigen doses expressed as molarities.
A SLO double mutant (SLOdm) is highly impaired in toxicity.
On the basis of the above results, a P427L/W535F SLO double mutant (SLOdm) was generated. In domain 4, the W535F substitution was selected over C530G, since the corresponding mutation in PLY (W433F) had been described as the most effective in reducing hemolytic activity (16–18). In silico predictions suggested that, among the three P427 mutations, P427L had the hydrophobic profile most similar to that of the wild-type toxin, reducing the potential to introduce conformational changes that could alter the overall protein stability.
SLOdm was characterized with respect to cell binding and hemolysis. We observed a reduction in binding to A549 cells that was 2.5 to 3 orders of magnitude lower than that of SLO (Fig. 2B), while hemolytic activity was undetectable (Fig. 2C), in agreement with the results obtained for the related W535F and P427L single mutants. The highly reduced toxicity of SLOdm was confirmed in vivo, as 5 µg of recombinant SLO injected intravenously was lethal in mice (5), while a 20-fold-higher dose of SLOdm did not affect mouse survival (see Table S1 in the supplemental material).
Remarkably, despite the extremely high impairment of in vitro and in vivo toxicity, the double mutant displayed only partial reduction in its capacity to bind eukaryotic cells, which could be attributed entirely to the W535F, not to the P427L, mutation (Fig. 2A). To better assess the contribution of the latter mutation to the overall toxicity reduction, we evaluated the oligomerization capability of wild-type SLO, the P427L single mutant (SLOsm), and SLOdm by SDS-AGE (19) and found that only SLO was able to form stable high-molecular-weight oligomers (Fig. 3A). These data suggest that the P427L substitution interferes with oligomerization, a precondition for pore formation. To further support this hypothesis, we performed transmission electron microscopy (TEM) of eukaryotic cell membranes treated with SLO, SLOsm, or SLOdm (Fig. 3B), demonstrating that SLOsm assembled into long linear structures, as opposed to the circular pores obtained with SLO. No macromolecular protein structures were detected in association with the SLOdm-treated cell membranes, even when the assay was repeated using a 100-fold-increased concentration of the double mutant. Finally, to exclude major structural alterations caused by the P427 mutation, recombinant tagless SLO, SLOsm, and SLOdm were subjected to circular dichroism (CD) and differential scanning calorimetry (DSC) analysis, showing that the three recombinant proteins had comparable profiles and melting temperatures (see Fig. S1 in the supplemental material).
FIG 3 .

Oligomerization on eukaryotic cell membranes of SLO and mutant derivatives. (A) SDS agarose gel Western blot analysis of SLO (wt), SLOP427L (sm) and SLOdm (dm) performed either in the absence (proteins only) or in the presence (proteins and cells) of sheep blood cells. Putative monomeric (M), dimeric (D), and oligomeric (O) forms are indicated. Sheep blood cells treated with buffer alone were used as negative control (NC). (B) TEM analysis of sheep blood cell ghost membranes alone or incubated with SLO, SLOsm, or SLOdm. Black arrows indicate typical structured pores on the membrane. White asterisks indicate long and linear structures.
GAS expressing SLOdm displays attenuated virulence in vivo.
To assess whether SLO genetic detoxification was associated with changes in GAS virulence, we generated an M1-3348 GAS strain derivative in which the slo gene was replaced by the double-mutant counterpart (strain 3348slodm). The virulence of this mutant was compared with that of the wild-type M1-3348 strain and of an additional mutant strain containing a complete in-frame deletion of the slo gene (strain 3348Δslo). Both mutants were confirmed to have growth rates similar to that of the wild-type strain (data not shown). Moreover, replacement of slo with the double-mutant gene did not affect toxin expression levels in culture supernatants (see Fig. S2A in the supplemental material), nor surface exposure, as detected by fluorescence-activated cell sorting (FACS) analysis (data not shown). SLO-dependent hemolysis of sheep blood cells was examined using spent broth from bacterial cultures. Compared with the wild-type strain, 3348slodm and 3348Δslo were unable to induce hemolysis (see Fig. S2B in the supplemental material). To rule out any polar effect on the adjacent spn gene, we performed a Western blot analysis, which showed that expression of SPN appeared to be unaffected in the two mutants (see Fig. S2A in the supplemental material).
The slo mutants were tested in a recently reported air pouch model of GAS infection, which has the advantage of allowing evaluation of multiple parameters related to the host immune response (10, 20). Figure 4A shows that naïve mice infected with either 3348slodm or 3348Δslo were able to control bacterial growth in situ. Conversely, infection with the same number of wild-type bacteria resulted in considerable bacterial proliferation over the course of the experiment. Successful containment of infection with the mutant strains was also reflected by a milder inflammation in situ, as confirmed by the significantly lower levels of the proinflammatory cytokines IL-1α, IL-1β, and IL-6 (Fig. 4B). Remarkably, infection with either 3348slodm or 3348Δslo resulted in recruitment and survival of a large number of immune cells, mostly neutrophils, which accumulated in the pouch and presumably were able to trigger bacterial killing. On the other hand, very low numbers of live immune cells were recovered from the air pouches of mice infected with the wild-type strain, while the total number of cells (including dead cells) was comparable to or higher than that obtained from mice infected with the mutant strains (Fig. 4C). These data suggested that the presence of SLO was instrumental for lysis/killing of host immune cells.
FIG 4 .
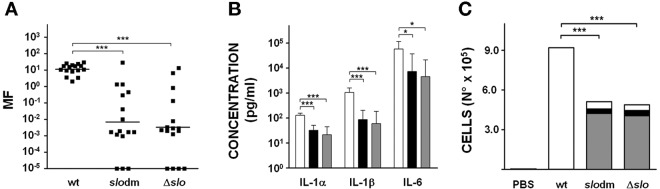
Functional comparison of wild-type and mutant M1-3348 strains. (A) CFU number determination in lavage from the air pouch of mice infected with M1-3348 (wt), 3348slodm (slodm), or 3348Δslo (Δslo). Black squares indicate the multiplication factors as determined for each individual mouse, with the bar in each group reporting the geometric mean value obtained. Asterisks indicate statistical significance, as calculated using the Kruskal-Wallis test followed by Dunn’s test (***, P < 0.001). (B) IL-1α, IL-1β, and IL-6 quantification in lavage from the air pouches of mice infected with M1-3348 (white bars), 3348slodm (black bars), and 3348Δslo (gray bars). Eight to 16 animals per group were assessed, and geometric mean with 95% confidence interval is reported. Statistically significant differences are indicated by the asterisks (***, P < 0.001; *, P < 0.05) as calculated using the Kruskal-Wallis test followed by Dunn’s test. (C) Quantification of dead cells (white), live neutrophils (gray), and live monocytes/macrophages (black) in the air pouch following infection with the indicated strains or phosphate-buffered saline (PBS) used as negative control. Sixteen animals per group were evaluated for cellular recruitment and geometric means for each single subpopulation are reported. Kruskal-Wallis test followed by Dunn’s test was used to perform statistical analysis on each subpopulation of cells retrieved from 3348slodm and 3348Δslo infected mice versus the cell population from the wild-type-infected control group. In all cases, a P value <0.001 (***) was obtained.
Virulence of mutant strains also appeared to be severely affected in mice infected intraperitoneally with 80% lethal doses (LD80s) of 3.6 × 106, 2 × 108, and 2.5 × 108 for M1-3348, 3348slodm, and 3348Δslo, respectively.
Mouse immunization with detoxified SLOdm confers protection similar to that conferred by wild-type SLO.
To evaluate the in vivo protective efficacy of SLOdm, CD1 mice were immunized either with alum adjuvant alone or with SLOdm plus adjuvant and challenged intranasally with three GAS strains of different epidemiologically relevant serotypes. Mice immunized with the homologous recombinant M proteins were used as positive controls. Infected mice were observed for 14 days. In our experience with this model, most animals that succumb to infection do so within the first 5 to 6 days after the inoculum. In this period, the survival curves of mice immunized with SLOdm fully overlapped those obtained with the homologous M protein, our standard protection benchmark, and protection was significantly higher than that in negative-control groups. After 14 days, consistent protection against the M1-3348 strain was obtained, while protection against the M6-S43 and M12-2728 strains was less striking but still statistically significant in both SLOdm- and M-protein-immunized mice (Fig. 5A to C). This outcome closely resembles what we previously reported for the wild-type SLO (SLOwt) (10). Likewise, SLOdm could effectively replace the wild-type antigen in the previously described “Combo” formulation with SPy0416 and SPy0269 (10), producing comparable protection against infection with the three strains (data not shown).
Protective immunity is mediated by neutralization of active SLO.
SLO is a secreted toxin that can also be found on the bacterial cell surface (10). To investigate the mechanism of protection induced by SLO-specific antibodies, we immunized mice with SLOdm and challenged them intraperitoneally (i.p.) with an LD80 of 3348, 3348slodm, or 3348Δslo. Consistent protection was obtained only against the strain producing wild-type SLO, while mice infected with 3348slodm showed a level of survival that was indistinguishable from that of the control group infected with 3348Δslo (Fig. 5D), suggesting that toxin inactivation is the only mechanism of antibody-mediated protection. To further investigate whether in vivo protection could in part be exerted through opsonization of bacteria exposing SLO on their surfaces, rabbit SLOdm antisera were used in opsonophagocytosis experiments with the wild-type M1-3348 and 3348slodm. Consistent killing was observed with the positive-control M1 antisera but not with SLO antisera (Fig. 5E). Overall, these results confirm inactivation of the secreted toxin as the major mechanism in SLO mediated protection.
FIG 5 .

Survival analysis of SLOdm-immunized mice infected with different GAS strains and opsonophagocytosis assay. (A to C) Kaplan-Meier survival curves of CD1 mice immunized with SLOdm and infected intranasally with M1-3348, M6-S43, or M12-2728 strain, respectively. Up to 32 or 48 mice were used for each study group. Mice immunized either with adjuvant alone (alum) or with the homologous M protein were used as negative and positive controls, respectively. (D) Survival percentage of CD1 mice immunized with SLOdm and infected intraperitoneally with M1-3348 (wt), M1-3348slodm (slodm), or M1-3348Δslo (Δslo) strains. Infectious doses used for all strains corresponded to an LD80. Asterisks indicate statistically significant protection, as calculated using the log-rank (Mantel-Cox) test (***, P < 0.001; *, P < 0.05). (E) Whole-blood assay with M1-3348 (wt) and M1-3348slodm (slodm) strains using SLOdm rabbit antisera. Freund’s and anti-M1 protein antisera were used as negative and positive controls, respectively. The MF compared to the starting inoculum is reported on the y axis. Each dot reflects the result of an independent serum evaluation, and bars indicate medians. Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn’s test (ns, P > 0.05).
In this context, in vivo challenge experiments by intravenous injection of SLO showed that toxicity was efficiently countered both by preincubating the toxin with SLOdm antisera (Fig. 6A) and by actively immunizing mice with SLOdm prior to treatment with doses of up to 20 µg of wild-type toxin (Fig. 6B), indicating that specific antibodies can completely neutralize the toxin in vivo. Functional inactivation of the toxin by specific antibodies was further examined by in vitro neutralization experiments, demonstrating that preincubation of SLO with sera from mice immunized with SLOdm either alone or in the Combo formulation could efficiently inhibit hemolysis (Fig. 6C). No differences in immunogenicity were observed following immunization of mice with SLO or SLOdm, as measured by enzyme-linked immunosorbent assay (ELISA) of specific IgG titers (data not shown) and by a functional assay (Fig. 6C).
FIG 6 .
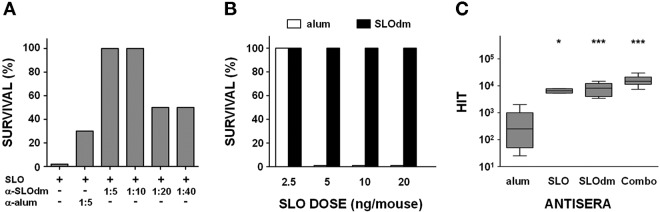
SLOdm antibodies mediate in vivo and in vitro neutralization of SLO toxicity. (A) Survival analysis of CD1 mice (8 to 12 mice/group) injected intravenously with 5 µg of SLO toxin preincubated either with serial 2-fold dilutions (from 1:5 to 1:40) of SLOdm antiserum or with a 1:5 dilution of a serum raised against alum alone as negative control. (B) Survival analysis of CD1 mice (4 to 12 mice/group) actively immunized with SLOdm and intravenously injected with increasing doses of SLO toxin. Animals immunized with alum alone were used as control group. (C) Sheep blood cells hemolysis inhibition titers (HIT) of sera from mice immunized with SLO, SLOdm, or SLOdm in combination with SPy0416 and SPy0269 GAS antigens (Combo). Sera of mice immunized with adjuvant alone (alum) were used as negative controls. Data are represented by box-and-whisker plots, showing the 0-to-90th-percentile ranges together with the 25th-to-75th-percentile ranges and medians. Asterisks indicate statistically significant protection compared to the negative control, as calculated using the Kruskal-Wallis test followed by Dunn’s test (***, P < 0.001; *, P < 0.05).
DISCUSSION
In the last decade, the advent of genomics led to a rapid acceleration of vaccine discovery (21). We recently showed proof of concept for a novel strategy which integrates the classical genomics reverse vaccinology approach with proteomics and protein array analysis of immunogenic antigens. The combined technologies were applied to GAS, resulting in a highly selective identification of few protective antigens, including SLO (10). Further support for vaccine development is derived from the application of structural biology to the rational design of optimized antigens (22, 23).
In this study, we provide an example of how protein structural predictions can be used to design a detoxified vaccine antigen optimized for human vaccine use. This strategy allowed generating a derivative of the GAS streptolysin O antigen which (i) was highly affected in host cell binding, (ii) did not form any detectable organized structure on the cell surface, (iii) preserved the overall structure/conformation, and (iv) was fully equivalent to wild-type SLO in terms of immunogenicity and protection against multiple GAS serotypes. The study also resulted in the collection of relevant data on SLO structure-function relationships, on the contribution of the toxin to GAS virulence, and on the mechanism by which the SLO antigen confers protection against GAS infection.
SLO belongs to the well-known family of CDC toxins secreted by Gram-positive pathogens (1, 11, 24). The 28 members of the family share high sequence homology, resulting in almost identical three-dimensional structures (12, 25–27). The structure-function relationship of CDCs has been extensively studied (12, 14, 28–30). Tilley et al. proposed a scheme for PLY pore formation that has been used as prototype for almost all CDCs members. According to this and other models, the conserved tryptophan-rich undecapeptide loop present at the bottom of domain 4 plays a central role in binding of toxin monomers to cholesterol in eukaryotic cell membranes (1, 11, 13). Oligomerization of SLO monomers is then triggered by significant structural changes in domains 2 and 4, allowing domains 1 and 3 of close monomers to interact and form prepore complexes. As a consequence, dramatic conformational changes allow the free domain 3 to insert into the cell membrane, resulting in pore formation.
Based on this background, the undecapeptide loop of SLO domain 4 was selected for our site-directed mutagenesis approach. The mutant protein SLO(W535F), which was subsequently chosen for the generation of the SLOdm derivative, exhibited highly reduced hemolytic and binding properties, consistent with previous data reported for PLY and PFO (16, 31). The choice of SLO domain 1 as an additional target for modifications was based on the recognition that this domain, in combination with domain 3, plays a central role in the earlier phases of pore formation, in particular in monomer oligomerization, as suggested by data obtained with several members of the CDC family, including listeriolysin (LLO) and PLY (12, 13, 28, 32, 33). Importantly, the conformation of domain 1 is supposedly unchanged during the process of pore formation (13). In this context, our original observation that SLO domain 1 is particularly rich in prolines (12 of the 21 Pro residues in the full protein sequence) suggested that conformational rigidity of this domain could be a prerequisite for SLO activity and prompted us to target this particular region.
Mutation of P427 significantly reduced SLO hemolysis. Unexpectedly, the replacement of P427 with a residue that should increase flexibility (P427G) had only a mild effect on SLO hemolytic activity. This could possibly be due to a conformational compensation effect exerted by nearby proline residues (i.e., P430). In contrast, the other three changes, which introduced amino acids with charged or uncharged longer lateral chains, had a clear effect on hemolytic activity, possibly due to local alteration of essential steric features of the molecule. The double mutant which we finally generated showed no detectable toxicity. This result was achieved by interfering with SLO activity both at the very early step of cell binding (W535F mutation) and during the more advanced process of pore organization (P427L mutation). The CD and DSC profiles of SLO and SLOdm were very similar, and thus it is unlikely that the two mutations disrupted the overall protein structure.
The ability of SLOdm to confer protection against GAS infection in immunized mice is the other key feature of this molecule. SLOdm induced protective immunity against infection with three different epidemiologically relevant GAS strains, at levels very similar to those obtained with the homologous M protein protection benchmark. Protection rates were particularly high when immunized mice were challenged with strain M1-3348. This clinical isolate contains an inactivating mutation in the covRS locus (as confirmed by sequence analysis), which we predict will result in a hypervirulent phenotype and overexpression of virulence factors, including SLO (34, 35). High expression levels of SLO and SPN and hemolytic activity in culture media were confirmed (see Fig. S2 in the supplemental material).
In a previous study, FACS and surfome analysis of bacterial cells revealed that SLO is not only secreted by the bacteria but also surface exposed (10). Anti-SLO antibodies similarly recognized SLO and SLOdm on the surfaces of M1-3348 and 3348slodm cells (data not shown), raising the possibility that anti-SLO-mediated opsonophagocytosis of bacteria could account for bacterial protection. Our in vitro and in vivo results suggest that neutralization of secreted/circulating toxin is possibly the only protective mechanism exerted by anti-SLO antibodies, in agreement with data recently reported for PLY (36). However, we cannot exclude the possibility that, due to the high 3348slodm and 3348Δslo bacterial numbers needed to reach LD80, other virulence factors might come into play, which could mask the SLO protection effect.
In vivo challenge with the M1-3348 double mutant strain and 3348Δslo in the murine models also helped define the role of SLO as a virulence factor. SLO toxicity in vitro and in vivo has been widely documented and acknowledged, but infection models have yielded conflicting results regarding its contribution to virulence (37, 38). Using the air pouch model, we observed a dramatic reduction in the virulence of M1-3348 when SLO was absent or functionally compromised. Such bacteria failed to establish an infection in situ and, concomitantly, live immune host cells were present in high numbers. To our knowledge, this is the first observation which directly correlates in vivo streptolysin O expression with killing of host recruited immune cells and bacterial replication at the infection site. Overall inflammation was also significantly reduced during infection with these strains. Levels of IL-6, a cytokine with predictive value for disease outcome (39), were 3-fold lower in these cases (Fig. 5C). We hypothesize that the use of SLOdm in immunization contributes to protection through two main mechanisms. First, the functional interference of SLO toxicity by specific antibodies reduces bacterial virulence and host cell lysis. Second, the increased survival of host immune cells promotes killing of the bacteria. Our genetically inactivated SLO toxin should thus be of value for the development of a vaccine against GAS infections. Finally, the strategy of genetically constructing a detoxified variant of a virulence factor (functional knockout) and comparing the strain that has this variant with a strain where the gene has been deleted (structural knockout) allows the dissection of the relative contribution to virulence of selected functional/structural properties. In combination with immunization experiments (e.g., antigen combinations), such a strategy can guide future studies in which the relative contribution of single antigens to virulence and protection can be approached.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and plasmids.
M1-SF370, M1-3348, M6-S43, and M12-2728 strains, as well as conditions used to grow them were previously reported (10). The M1-3348 isogenic mutants 3348Δslo and 3348slodm are described below. pET21b(+), pET24b(+) (Novagen), and pJRS233 (40) plasmids were used, respectively, to clone and express recombinant proteins and to generate the GAS M1-3348 isogenic mutants.
Generation of SLO protein mutants.
The SF370-derived His-tagged fusion SLO gene previously reported (10) was used as the template to obtain all the mutant derivatives described in this study. Tagless derivatives were obtained by cloning the SLO sequence of interest in the pET24b+ Escherichia coli expression vector. Splicing by overlapping extension-PCR (SOE-PCR) (41) was used to carry out site-directed mutagenesis, using the oligonucleotides listed in Table S2 in the supplemental material for PCR amplification. Plasmid DNA from engineered E. coli was isolated with either Qiagen plasmid mini- or maxiprep kits (Qiagen). Restriction enzymes and DNA ligase were from Biolabs, and PFU Ultra II was from Stratagene. All the enzymes and kits were used according to the manufacturer’s protocols.
Construction of M1-3348 mutant strains.
The plasmid pJRS233 is a temperature-sensitive shuttle vector (40). The vector pJRS233slodm was obtained by transferring the SLOdm gene from the E. coli expression plasmid pET24b+(slodm) using the restriction enzymes BamHI and XhoI. The vector pJRS233Δslo contains the slo locus (from 649 bp upstream to 593 bp downstream of slo) with an in-frame deletion in slo (total of 1,293 bp) and was constructed using a SOE strategy with primers FsloSOE1, RsloSOE1, FsloSOE2, and RsloSOE2 (see Table S3 in the supplemental material) and subsequent cloning into pJRS233 with the restriction enzymes BamHI and XhoI (42). For E. coli cloning, we used maximum-efficiency competent DH5α cells (Invitrogen) according to the manufacturer’s protocol. An insertion/duplication and excision mutagenesis strategy was used to obtain the strains with the slo chromosomal replacement (3348slodm) and the in-frame deletion slo knockout (3348Δslo). Briefly, pJRS233slodm and pJRS233Δslo were purified from E. coli and introduced into strain M1-3348 by electroporation (43). Transformants were selected by growth on Todd-Hewitt--yeast extract (THYe) agar containing erythromycin (Erm) at 30°C. Integration was performed by growth of transformants at 37°C with Erm selection. Excision of the integrated plasmid was performed by serial passages in THYe at 30°C. Replacement of the slo gene with the deletion construct or the double mutant was verified by PCR sequencing (primers FsloEXT and RsloEXT; see Table S3 in the supplemental material).
Protein expression and purification.
Recombinant proteins were expressed as His-tagged or tagless derivatives. His-tagged proteins were expressed and purified as described elsewhere (44). For tagless proteins, bacterial culture pellets were suspended in 30 mM Tris-HCl (pH 8.0) and lysed by 3 cycles in an Emulsiflex C5 (Avestin) high-pressure homogenizer at 12,000 to 14,000 lb/in2. Following centrifugation for 60 min at 16,900 × g, the supernatant was salted by addition of NaCl to a final concentration of 2 M and filtered (0.45 to 0.22 µm) using a Sartobran 300 capsule (Sartorius). The clarified lysate was then loaded on a phenyl Sepharose 6 Fast Flow HS (GE Healthcare) column, previously equilibrated with 30 mM Tris, 2 M NaCl (pH 8.0). The protein was eluted with Tris 30 mM pH 8.0, salted again to a final conductivity of 12 to 15 mS/cm with 30 mM Tris, 1 M NaCl (pH 8.0), and loaded on a Capto Q (GE Healthcare) column. The protein was recovered in the flowthrough fraction and loaded directly on a 40-μm hydroxyapatite column (Bio-Rad). After a washing with 150 mM sodium phosphate, pH 6.8, the protein was eluted with 400 mM sodium phosphate (pH 6.8), the fractions containing SLO proteins were pooled, the buffer was exchanged by dialysis, and the protein concentration was determined by the bicinchoninic acid (BCA) method (Pierce).
CDs and DSCs.
Protein samples for CD and DSC analysis were dialyzed against 10 mM KH2PO4/K2HPO4 (pH 7.4) before analysis. The potassium phosphate used for dialysis was the reference solution in both the experiments. For DC analysis, protein concentration was 10–4 M and the assay was performed at a temperature of 20.0 ± 0.5°C. CD spectra were obtained using a Jasco J-810 spectropolarimeter (Easton, MD). Samples were analyzed in a quartz cell with a 1-mm optical path length, and the scan range was in the far-UV region, covering wavelengths between 190 and 260 nm. Scan parameters were a response time of 8 s and a scan speed of 5 nm/min. CD Multivariate SSE software was used to estimate the second structure composition of the sample. A VP-DSC microcalorimeter (Microcal, Northampton, MA) was employed for DSC analysis using a scan rate of 90°C/h and the middle feedback setting. Data were analyzed using Origin software, v. 7.0 (Microcal), which was used to subtract baseline scans, normalize the data to the protein concentration, and fit the data to one unfolding transition. Normalized data were exported, and the thermograms were plotted using Prism 5.0b (GraphPad, La Jolla, CA).
Hemolytic activity titration and hemolysis inhibition assay.
Hemolysis and hemolysis inhibition assays were performed as previously described (10), yielding complete dose-response curves for SLO and SLOdm. When the hemolytic activities of a mutated toxin derivative were to be compared to that of SLO, a hemolytic titer was calculated for the protein as the dose required to obtain 50% of maximum hemolysis. SLO titer was eventually divided by the titer of the mutated derivative, yielding a value which represented residual hemolytic activity.
Binding assay.
A549 human epithelial cells (American Type Culture Collection) were grown in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 250 mM HEPES, 200 U/ml ampicillin and streptomycin, and complemented fetal bovine serum (10% [vol/vol]; Sigma). Confluent cells were detached with 0.25% (w/vol) trypsin plus 1 mM EDTA (GIBCO), and incubated for 20 min with agitation at room temperature (RT) in phosphate-buffered saline (PBS) plus Live/Dead-AquaFluor (Invitrogen) at a final concentration of 1:1,000 (vol/vol). Cells were then fixed in 2% (vol/vol) formaldehyde (Sigma) for 20 min under the same conditions. Serial dilutions of SLO in PBS plus 1% BSA (PBSA) were then added to the fixed cells and incubated for 45 min at 4°C. A 1:1,000 (vol/vol) dilution in PBSA of a mouse polyclonal anti-SLO serum was used to label bound SLO, washed, and then incubated for 30 min at 4°C with 1:100 (vol/vol) α-mouse phycoerythrin-conjugated antibody (Jackson ImmunoResearch Laboratories) in PBSA. Cells were finally suspended in PBS plus 5 mM EDTA, and the amount of bound protein was detected by flow cytometry and analyzed with FlowJo software. Mean fluorescence intensity (MFI) values obtained were correlated to the different amounts of SLO used in the assay to obtain binding curves. When the ability of a single mutant derivative to bind A549 cells was to be compared to that of SLO, a binding titer for the mutated protein was calculated as the dose required to obtain a pre-established MFI value. Finally, SLO binding titer was divided by the titer of the mutated derivative obtaining the percent residual binding activity. When SLOdm was compared to SLO, the dose-response curves of both the proteins were entirely calculated.
SDS-agarose gel electrophoresis and transmission electron microscopy analysis.
Denaturing agarose gel electrophoresis (SDS-AGE) was carried out as previously described (19), and Western blot analysis using a mouse polyclonal SLO antiserum was performed according to standard procedures. For TEM analysis, doses ranging from 1 to 100 µg of each protein were incubated at 37°C for 30 min with ghost membranes from sheep blood cells prepared as previously reported (45). Samples were then adsorbed onto 200 mesh Formvar-carbon coated copper grids for 5 min. Negative staining was performed by placing the specimen grid on a drop of negative stain (2% uranyl acetate in distilled water) and repeating this step once. Samples were observed with a JEOL 1200 EX II electron microscope at 100 kV, and micrographs were acquired with an Olympus SIS Veleta charge-coupled device (CCD) camera equipped with iTEM software.
In vivo protection assays.
Immunization with SLO, intranasal (i.n.) infection with GAS, and intravenous (i.v.) injection of SLO proteins were carried out as previously described (10). When a preincubation with mouse sera was required prior to i.v. administration of SLO, serially diluted immune sera were incubated with the wild-type toxin for 20 min at RT. Prior to i.p. challenge of immunized mice, dose-ranging challenge experiments (data not shown) were performed to establish lethal doses. The slodm and Δslo mutants were severely attenuated in virulence compared to the wt strain, and even at the highest doses, lethality did not consistently exceed 80%. The use of extremely high inocula (needed to reach the LD90) risked confounding the effects of an infection with unspecific toxicity due to proinflammatory bacterial components. We thus chose to perform the experiment at the LD80 level. Animal protocols were approved by the Novartis Animal Ethical Committee, Siena, Italy, and the Italian Ministry of Health, Rome, Italy. Mice were monitored on a daily basis and euthanized for humane reasons when they exhibited defined endpoints that had been pre-established for these studies in agreement with the Novartis Animal Welfare Policies.
Whole-blood assay.
The whole-blood assay was performed as previously described (10), and the multiplication factor (MF) was calculated as the ratio between the number of CFU after 5 h incubation and the number at time zero. Sera from rabbits immunized with SLOdm were used to assess the ability of specific antibodies to induce opsonophagocytosis. Three sera raised against SLOdm were tested either 3 times or twice, for a total of 8 independent replicas. Sera from rabbit immunized with adjuvant alone (Freund’s) and M1 protein were used as negative and positive controls, respectively.
Mouse air pouch infection and sample analysis.
Formation of the air pouches, infection, and MF were assessed as previously described (10). The cellular fraction was obtained from the lavage samples by centrifugation (320 × g, 4°C). Cells were suspended in 1 ml of PBS with 1:1,000 of Live/Dead-AquaFluor (Invitrogen) for 30 min at 4°C. Cells were fixed in 2% (vol/vol) formaldehyde (Sigma), centrifuged as described above, and resuspended in 25 µl staining solution for 30 min at 4°C with a combination of the following cell marker antibodies: anti-Ly6C conjugated to fluorescein isothiocyanate (FITC), anti-CD11b conjugated to phycoerythrin-Cy7 (PE-Cy7), anti-Ly6G-PE, anti-Ly6C plus anti-Ly6G (GR1) conjugated to peridinin-chlorophyll (PerCP) (BD Pharmingen), and anti-F4/80-PacificBlue (eBioscience). The stained cells were analyzed using an LSR II SOS flow cytometer and BD DIVA software (BD Bioscience). For chemokine analysis, lavage material from the air pouch was centrifuged as described above, and supernatants were filtered through 0.22-µm filters to remove bacteria. Chemokine concentrations were measured with a Milliplex Map kit (Millipore) according to the manufacturer’s instructions.
In silico analysis.
The crystal structures and B factors of perfringolysin O (code 1PFO) were downloaded from Protein Brookhaven Database website (http://www.pdb.org). The SLO sequence was threaded onto the PFO and coordinated using Swiss PDB Viewer (http://www.expasy.org/spdbv/), as described elsewhere (46). The same software was used for molecular rendering.
Statistical analysis.
For the SLO i.v. injection model, the two-tailed Fisher exact test was used. For the GAS i.n. and i.p. models of infection, the log-rank (Mantel-Cox) test was used. For the GAS air pouch infection model, cytokine levels, hemolysis inhibition, and whole blood assays, the Kruskal-Wallis test followed by Dunn’s test were used. In all cases, GraphPad Prism 5 software was used to calculate statistical significance. P values lower than 0.05 were considered significant.
SUPPLEMENTAL MATERIAL
Structural analysis of SLO and mutant derivatives. Circular dichroism profiles of SLO, SLO(P427L) (SLOsm), and SLO(P427L-W535F) (SLOdm). The percentage of protein regions with different secondary structures and the melting temperature (Tm) obtained by differential scanning calorimetry (DSC) are reported in the lower table. Download
Phenotypic characterization of M1-3348 isogenic mutants. (A) Western blot analysis of secreted SLO and NAD-glycohydrolase (SPN) in the culture medium of 3348 strains expressing SLO (wt) or SLOdm (slodm) or not expressing the toxin (Δslo).(B) Hemolysis test performed on sheep blood cells using supernatants from GAS cultures of wild-type M1-3348 (wt), 3348slodm (slodm), or 3348Δslo (Δslo). Download
SLO(P427L/W535F) (SLOdm) is not toxic in a mouse model of intravenous injection.
Oligonucleotides used to clone SLO mutant derivatives.
Oligonucleotides used to generate the 3348slodm and 3348Δslo strains.
ACKNOWLEDGMENTS
We thank Massimo Mariani for support in protein purification, Graziella Orefici, Laboratory of Bacterial Pathogenesis and Immunology, Istituto Superiore di Sanità, Rome, Italy, and John Zabriskie, Rockefeller University, New York, NY, for providing bacterial strains. A special acknowledgment is due to the Novartis Animal Resources Center, Siena, Italy, and in particular to Giacomo Matteucci for the coordination of animal tests and to Marco Vagaggini, Roberto Dini, and Marco Martelli for their excellent technical support.
This work was supported by internal funding from Novartis NVD and by the Italian Ministry of Instruction, University and Research, Rome, Italy (grant MIUR no. 10911).
Footnotes
Citation Chiarot E, et al. 2013. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio 4(1):e00387-12. doi:10.1128/mBio.00387-12.
REFERENCES
- 1. Billington SJ, Jost BH, Songer JG. 2000. Thiol-activated cytolysins: structure, function and role in pathogenesis. FEMS Microbiol. Lett. 182:197–205 [DOI] [PubMed] [Google Scholar]
- 2. Palmer M. 2001. The family of thiol-activated, cholesterol-binding cytolysins. Toxicon 39:1681–1689 [DOI] [PubMed] [Google Scholar]
- 3. Gilbert RJ. 2010. Cholesterol-dependent cytolysins. Adv. Exp. Med. Biol. 677:55–56 [DOI] [PubMed] [Google Scholar]
- 4. Tiesler E, Trinks C. 1982. Release of extracellular metabolic products by streptococci groups C and G. Zentralbl. Bakteriol. Mikrobiol. Hyg. A. 253:81–87 [In German.] [PubMed] [Google Scholar]
- 5. Alouf JE. 1980. Streptococcal toxins (streptolysin O, streptolysin S, erythrogenic toxin). Pharmacol. Ther. 11:661–717 [DOI] [PubMed] [Google Scholar]
- 6. Sheeler RD, Houston MS, Radke S, Dale JC, Adamson SC. 2002. Accuracy of rapid strep testing in patients who have had recent streptococcal pharyngitis. J. Am. Board Fam. Pract. 15:261–265 [PubMed] [Google Scholar]
- 7. Todd EW. 1932. Antigenic streptococcal hemolysin. J. Exp. Med. 55:267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bricker AL, Cywes C, Ashbaugh CD, Wessels MR. 2002. NAD+ glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol. Microbiol. 44:257–269 [DOI] [PubMed] [Google Scholar]
- 9. Madden JC, Ruiz N, Caparon M. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive Bacteria. Cell 104:143–152 [DOI] [PubMed] [Google Scholar]
- 10. Bensi G, et al. 2012. Multi high-throughput approach for highly selective identification of vaccine candidates: the group A streptococcus Case. Mol. Cell. Proteomics 11:M111, 015693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heuck AP, Moe PC, Johnson BB, Harris JR. 2010. The cholesterol-dependent cytolysin family of Gram-positive bacterial toxins, p 551–577 In Harris JR, Cholesterol binding and cholesterol transport proteins: structure and function in health and disease, vol. 51 Springer Verlag, Dordrecht, Netherlands: [DOI] [PubMed] [Google Scholar]
- 12. Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. 1997. Structure of a cholesterol-Binding, thiol-activated cytolysin and a model of its membrane form. Cell 89:685–692 [DOI] [PubMed] [Google Scholar]
- 13. Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. 2005. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 121:247–256 [DOI] [PubMed] [Google Scholar]
- 14. Sekiya K, et al. 2007. Ultrastructural analysis of the membrane insertion of domain 3 of streptolysin O. Microbes Infect. 9:1341–1350 [DOI] [PubMed] [Google Scholar]
- 15. Yuan Z, Bailey TL, Teasdale RD. 2005. Prediction of protein B-factor profiles. Proteins 58:905–912 [DOI] [PubMed] [Google Scholar]
- 16. Paton JC, et al. 1991. Purification and immunogenicity of genetically obtained pneumolysin toxoids and their conjugation to Streptococcus pneumoniae type 19F polysaccharide. Infect. Immun. 59:2297–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander JE, et al. 1994. Immunization of mice with pneumolysin toxoid confers a significant degree of protection against at least nine serotypes of Streptococcus pneumoniae. Infect. Immun. 62:5683–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kirkham LA, et al. 2006. Construction and immunological characterization of a novel nontoxic protective pneumolysin mutant for use in future pneumococcal vaccines. Infect. Immun. 74:586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shepard LA, Shatursky O, Johnson AE, Tweten RK. 2000. The mechanism of pore assembly for a cholesterol-dependent cytolysin: formation of a large prepore complex precedes the insertion of the transmembrane β-hairpins. Biochemistry 39:10284–10293 [DOI] [PubMed] [Google Scholar]
- 20. Chiappini N, et al. 2012. Streptococcus pyogenes SpyCEP influences Host-pathogen interactions during infection in a murine air pouch model. PLoS One 7:e40411 http://dx.doi.org/doi:10.1371/journal.pone.0040411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seib KL, Zhao X, Rappuoli R. 2012. Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin. Microbiol. Infect. 18:1–8 [DOI] [PubMed] [Google Scholar]
- 22. Nuccitelli A, et al. 2011. Structure-based approach to rationally design a chimeric protein for an effective vaccine against group B streptococcus infections. Proc. Natl. Acad. Sci. USA 108:10278–10283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scarselli M, et al. 2011. Rational design of a meningococcal antigen inducing broad protective immunity. Sci. Transl. Med. 3:91ra62. [DOI] [PubMed] [Google Scholar]
- 24. Giddings KS, Zhao J, Sims PJ, Tweten RK. 2004. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 11:1173–1178 [DOI] [PubMed] [Google Scholar]
- 25. Bourdeau RW, et al. 2009. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 284:14645–14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polekhina G, Giddings KS, Tweten RK, Parker MW. 2004. Crystallization and preliminary X-ray analysis of the human-specific toxin intermedilysin. Acta Crystallogr. D Biol. Crystallogr. 60(Pt 2):347–349 [DOI] [PubMed] [Google Scholar]
- 27. Xu L, et al. 2010. Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell. 1:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palmer M, et al. 1996. Membrane-penetrating domain of streptolysin O identified by cysteine scanning mutagenesis. J. Biol. Chem. 271:26664–26667 [DOI] [PubMed] [Google Scholar]
- 29. Palmer M, et al. 1998. Streptolysin O: a proposed model of allosteric interaction between a pore-forming protein and its target lipid bilayer. Biochemistry 37:2378–2383 [DOI] [PubMed] [Google Scholar]
- 30. Shatursky O, et al. 1999. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99:293–299 [DOI] [PubMed] [Google Scholar]
- 31. Polekhina G, Giddings KS, Tweten RK, Parker MW. 2005. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. U. S. A. 102:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Darji A, et al. 1996. Neutralizing monoclonal antibodies against listeriolysin: mapping of epitopes involved in pore formation. Infect. Immun. 64:2356–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de los toyos JR, et al. 1996. Functional analysis of pneumolysin by use of monoclonal antibodies. Infect. Immun. 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ato M, Ikebe T, Kawabata H, Takemori T, Watanabe H. 2008. Incompetence of Neutrophils to Invasive group A streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS One 3:e3455 http://dx.doi.org/doi:10.1371/journal.pone.0003455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-Wide Analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2:e5 http://dx.doi.org/doi:10.1371/journal.ppat.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salha D, et al. 2012. Neutralizing antibodies elicited by a novel detoxified pneumolysin derivative, PlyD1, provide protection against both pneumococcal infection and lung injury. Infect. Immun. 80:2212–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fontaine MC, Lee JJ, Kehoe MA. 2003. Combined contributions of streptolysin O and streptolysin S to virulence of serotype M5 Streptococcus pyogenes Strain Manfredo. Infect. Immun. 71:3857–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Limbago B, Penumalli V, Weinrick B, Scott JR. 2000. Role of streptolysin O in a mouse model of Invasive group A streptococcal disease. Infect. Immun. 68:6384–6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. 2002. Six at six: interleukin-6 measured 6 H after the initiation of sepsis predicts mortality over 3 days. Shock 17:463–467 [DOI] [PubMed] [Google Scholar]
- 40. Perez-Casal J, Price JA, Maguin E, Scott JR. 1993. An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol. Microbiol. 8:809–819 [DOI] [PubMed] [Google Scholar]
- 41. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 42. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8:528–535 [PubMed] [Google Scholar]
- 43. Upton M, Tagg JR, Wescombe P, Jenkinson HF. 2001. Intra- and interspecies signaling betweenStreptococcus salivarius and Streptococcus pyogenes mediated by SalA and SalA1 Lantibiotic peptides. J. Bacteriol. 183:3931–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodríguez-Ortega MJ, et al. 2006. Characterization and identification of vaccine candidate proteins through analysis of the group A streptococcus surface proteome. Nat. Biotechnol. 24:191–197 [DOI] [PubMed] [Google Scholar]
- 45. Burton GW, Ingold KU, Thompson KE. 1981. An improved procedure for the isolation of ghost membranes from human red blood cells. Lipids 16:946 [DOI] [PubMed] [Google Scholar]
- 46. Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural analysis of SLO and mutant derivatives. Circular dichroism profiles of SLO, SLO(P427L) (SLOsm), and SLO(P427L-W535F) (SLOdm). The percentage of protein regions with different secondary structures and the melting temperature (Tm) obtained by differential scanning calorimetry (DSC) are reported in the lower table. Download
Phenotypic characterization of M1-3348 isogenic mutants. (A) Western blot analysis of secreted SLO and NAD-glycohydrolase (SPN) in the culture medium of 3348 strains expressing SLO (wt) or SLOdm (slodm) or not expressing the toxin (Δslo).(B) Hemolysis test performed on sheep blood cells using supernatants from GAS cultures of wild-type M1-3348 (wt), 3348slodm (slodm), or 3348Δslo (Δslo). Download
SLO(P427L/W535F) (SLOdm) is not toxic in a mouse model of intravenous injection.
Oligonucleotides used to clone SLO mutant derivatives.
Oligonucleotides used to generate the 3348slodm and 3348Δslo strains.