

Abstract
The planar cell polarity (PCP) pathway is a highly conserved signaling cascade that coordinates both epithelial and axonal morphogenic movements during development. Angiogenesis also involves the growth and migration of polarized cells, although the mechanisms underlying their intercellular communication are poorly understood. Here, using cell culture assays, we demonstrate that inhibition of PCP signaling disrupts endothelial cell growth, polarity, and migration, all of which can be rescued through downstream activation of this pathway by expression of either Daam-1, Diversin or Inversin. Silencing of either Dvl2 or Prickle suppressed endothelial cell proliferation. Moreover, loss of p53 rescues endothelial cell growth arrest but not the migration inhibition caused by PCP disruption. In addition, we show that the zebrafish Wnt5 mutant (pipetail (ppt)), which has impaired PCP signaling, displays vascular developmental defects. These findings reveal a potential role for PCP signaling in the coordinated assembly of endothelial cells into vascular structures and have important implications for vascular remodeling in development and disease.
Keywords: Angiogenesis, Endothelial cells, Proliferation, Migration, Dishevelled, Planar cell polarity, TNP-470, Methionine aminopeptidase-2, Chemical biology, Wnt5, Zebrafish, Pipetail
Introduction
Angiogenesis, the formation of blood vessels from pre-existing vasculature, is a complex and coordinated process. Following stimulation by pro-angiogenic factors, endothelial cells degrade their basement membrane and undergo proliferation, directional migration, remodeling of their extracellular matrix and, ultimately, network formation and stabilization to generate new vasculature [1]. How these myriad events are coordinated among endothelial cells and their environment is not fully understood.
At the cellular level, angiogenesis can be envisioned as the coordinated growth and migration of endothelial cell sheets towards an angiogenic signal. Therefore, we hypothesized that endothelial cells during neovascularization behave in a manner similar to epithelial cells whose polarization is responsible for Drosophila bristle pattern formation, namely, as a polarized endothelial sheet that becomes further organized into more complex vascular networks. This intercellular coordination of Drosophila epithelial cell planar polarity is mediated predominantly by a conserved signaling pathway appropriately called the planar cell polarity (PCP) pathway. In addition to Drosophila bristle and photoreceptor patterning [2], this signaling cascade is responsible for the coordinated organization of cells in different systems from vertebrate convergent extension movements during gastrulation [3–6] to dendritic arborization [7, 8]. The PCP pathway is considered one of the β-catenin-independent (or non-canonical) pathways of Wnt signaling, whereby a ligand Wnt acts though a seven-span transmembrane receptor (frizzled) that in turn activates a mobile scaffolding protein dishevelled [9]. Highly conserved dishevelled proteins transduce several Wnt signaling pathways including the canonical Wnt pathway defined by the stabilization of and signaling by β-catenin, the Wnt/Ca2+ pathway (involving calmodulin-dependent kinase II (Cam-KII) activation), and the PCP pathway that includes downstream activation of Daam-1, RhoA, and JNK [10–12].
In the context of angiogenesis, it is known that endothelial cells express their own Wnt5a [13], which has been shown to promote endothelial cell proliferation and survival [14]. Suppression of Wnt5a by either a neutralizing antibody or RNAi further inhibits endothelial cell proliferation and migration [14, 15]. In addition, we recently reported that inhibition of methionine aminopeptidase 2 (MetAP-2) by the anti-angiogenic drug TNP-470 results in loss of β-catenin-independent Wnt signaling without blocking canonical Wnt/β-catenin signaling [16]. Given that PCP signaling is required for the morphogenic movements during vertebrate gastrulation, this finding is consistent with the early gastrulation defect observed in MetAP-2 knockout mice [17] and zebrafish MetAP-2 knockdown [16].
Although TNP-470 displayed anti-angiogenic properties and inhibited β-catenin-independent Wnt signaling, it was not established whether such non-canonical Wnt signaling was important for angiogenesis. We therefore used this compound in our initial studies of PCP signaling in endothelial cell biology and angiogenesis. Interestingly, we observed by both chemical and genetic methods that PCP inhibition suppressed endothelial cell proliferation, migration and network formation. Although the proliferative suppression by PCP inhibition required p53, the absence of p53 did not protect endothelial cells from the anti-migratory effects of PCP signaling inhibition. However, high expression of known PCP downstream mediators rescued migrational defects induced by β-catenin-independent Wnt signaling inhibition. These results suggest a mechanism whereby PCP signaling can direct endothelial cell growth and coordination.
Materials and methods
Transfection and immunoblotting
Wnt5A conditioned media was generated by treating serum free Dulbecco’s MEM with HEK293 cells transiently transfected with a Wnt5a expression vector (Upstate Bio-technology, Lake Placid, NY) using Lipofectamine™ 2000 (Invitrogen) and thereafter collected and filtered though a 0.45-μm filter.
Genetic constructs consisting of pCMV5 vectors expressing HA-tagged mouse Dvl2 (and mutations) were constructed as described previously [18]. K446M-Dvl2 was generated by introducing a point mutation into HA-Dvl2 using the QuikChange II XL Site-Directed Mutagenesis Kit (Cat#: 200521, Stratagene). Co-transfections with a given construct of interest and GFP-expressing construct were done at a respective 10:1 ratio (total, 6 μg DNA per 10-cm plate) using Lipofectamine 2000 (Invitrogen). A similar approach was used to generate MPE cell populations with various Dvl2 mutants, whereby co-transfections with a given construct of interest were introduced with a plasmid conferring puromycin-resistance (10:1 ratio, 6 μg DNA per 10-cm plate) followed by selection (1 μg/ml for 2 days). Transfection of MPE cells was typically observed to occur at approximately 70–80% efficiency as determined by scoring GFP-positive cells.
Western blots were performed by routine and standard methods. Whole cell MPE lysates were collected using RIPA buffer (50 mM Tris pH 7.5, 0.1% SDS, 1% NP-40 and 0.5% deoxycholic acid) at 500 ml/10-cm plate. Cells were sonicated and the supernatant was collected after a 5-min spin at 1,000 rpm in a microcentrifuge. Primary antibodies utilized against the hemagglutnin-tag (Y11, Santa Cruz Biotechnology), FLAG-tag (M2, Sigma), and α-tubulin (DM1A, Sigma) as recommended by the respective suppliers. Secondary antibodies utilized (after 3 washes in TBST) included the ECL™ sheep anti-mouse or anti-rabbit (as appropriate) conjugated to HRP (GE Healthcare), which were diluted 1:2,000 in TBST supplemented with 5% powdered milk and incubated for 1 h. Blots were thereafter washed three times with TBST, washed 2 min in Western Blotting Luminol Reagents A and B (sc-2048, Santa Cruz) and exposed to film for image acquisition.
Reagents and cell lines
MPE cells, including those deficient in p53, were harvested from mice by standard methods [19]. Adult normal human dermal fibroblasts (NHDFs) were acquired from Clonetics (Cat#: CC2511).
Image acquisition and processing
Images from polarity, migration and Matrigel™ assays (described below) were acquired using an Olympus CK40 fluorescent microscope and an Olympus C-3040ZOOM digital camera. Confocal images were acquired using a Zeiss Inverted Axiovert confocal microscope with a Bio-Rad MRC 1040 laser source. Images were process with LaserSharp Image J software (NIH) and merged artificially using Adobe Photoshop CS. Objective magnification and fluorochromes used are as described per individual experiment. In all cases, slides were mounted with Vectashield mounting medium (Vector Laboratories Inc., Burlington, CA). Figure presentations were prepared utilizing Adobe Illustrator 10.0.3.
Assessing cellular polarity by use of Dunn chemotaxis chambers
Endothelial cells were seeded on gelatin coated coverslips and serum starved for 24 h prior to assay. Dunn chemotaxis chambers were used per established methods [20]. Outer concentric wells contained media that included serum free DMEM treated with HEK293 cells transiently transfected with Wnt5a or control vectors. This chamber establishes a radially directed linear diffusion gradient in 10–30 min, which is maintained for 10–30 h [21]. Cells were maintained within this gradient for 9 h (37°C), after which the coverslips were labeled to note the bridge and wells of the chemotaxis chamber. The resulting cells were then fixed and processed for immunohistochemistry for caveolin-1 [20]. Cells were considered polar if they displayed asymmetrical localization of caveolin-1.
Migration assays
Migration assays were performed in 24-well plate, BD Biocoat™ Control Inserts (Cat#: 354578, BD Biosciences, Bedford, MA) with 2 ng/ml VEGF (±TNP-470, 10 nM) for 6 h after a 2-h incubation with drug or vehicle [22, 23]. Respective test cells were harvested, diluted to 5 × 104 cells/ml and preincubated 2 h in serum-free medium supplemented with vehicle (0.1% DMSO) or TNP-470 (10 nM). Then cells in 0.5-ml aliquots were added to the upper chambers of each well. Lower chambers were filled with 0.75 ml of serum free media supplemented with VEGF (0.2 μg/ml, Sigma) and either DMSO or TNP-470 as described above. The assembled chemotaxis chamber was incubated for 6 h at 37°C with 5% CO2 to allow cells to migrate through the filter. Non-migrated cells on the upper surface of the filter were removed by gentle scraping with a PBS-doused sterile cotton tipped applicator. Cells were thereafter fixed and stained with 100% methanol (2 min) and 1% Toluidine blue (2 min, Sigma), respectively. Cells were quantified in units of migrated cells per 100× field of view (FOV) (5 FOV/well, N = 3 wells per test condition). In migration assays involving GFP-expressing cells, the assay was performed as above. In place of staining, after the removal of non-migrated cells, cells were viewed directly under a fluorescent microscope (200× FOV, 5 FOV/well, N = 3 wells per test condition). The migration index (MI), where applicable, was determined as the number of migrating cells with drug compared to their vehicle-treated counterparts.
Network formation assay on matrigel™
Matrigel assays were performed as described in the product literature (BD BioSciences, with reference to [24]. Briefly, 20,000 MPE cells in 50 μL EGM-2 MV media (Clonetics) were applied to each well of a BD Matrigel™ basement membrane (BD Biosciences) coating in 96-well plates. Media were supplemented with DMSO or 10 nM TNP-470 as appropriate prior to application to cells. Digital acquisition was performed 16–18 h thereafter. In the case of the mosaic analysis, MPE cells co-expressing a given Dvl2 construct and GFP mixed with an equal volume of GFP-negative MPE cells prior to plating onto Matrigel (total cell number kept constant). Cell extensions were counted if they consisted of a clear, unbroken extension of cells from one given node of cells to another. Cell extension length (by relative comparison to a micrometer) and quantity were determined directly off the digital images assisted with Adobe Photoshop CS software. Statistical analyses were performed as described below.
Zebrafish
Embryos were obtained by natural crosses. Fli1:EGFP (generously provided by Dr. Weinstein) was crossed into the pptti265 strain. F1 offspring were intercrossed to identify ppt and EGFP carriers. Fli1:EGFP transgenic embryos were microinjected with Wnt5 morpholino (3×10−13 mol, GCAAACACAATAATTTCTTACCACC, Gene Tools LLC),in 1× Danieau solution using a pneumatic injector at the one cell stage.
Embryos were reared at 28°C and assayed between 24 to 48 h post-fertilization (hpf). For in vivo analysis of the vasculature, live embryos (n = 60) were mounted in methylcellulose and fluorescent images captured with a Zeiss axiocam. For whole mount in situ hybridization, embryos were fixed with 4% paraformaldehyde/phosphate buffered saline (PBS) overnight (n = 62). Embryos older than 24 hpf were treated with pigment inhibitor. Sense and antisense digoxigenin-UTP RNA probes (Roche) were synthesized from linearized template and whole mount in situ hybridization was performed as described [25]. Approximately 25% of the embryos observed had vascular defects; n = 15.
TNP-470 treatment
Prior to treatment, embryos were dechorionated using 1 mg/ml pronase. TNP-470 (25 μM) or Control DMSO (25 μM) was added to the embryo medium at 10 hpf and incubated until approximately 30 hpf.
Transplantation
Donor embryos were microinjected with 20 ng lineage marker (dextran-conjugated Texas Red, Molecular Probes) mixed with Wnt5 MO at the 1–2 cell stage. Donor and host Fli1:EGFP embryos were manually dechorionated prior to transplantation. At sphere stage, 20–30 cells were removed from donor embryos and transplanted into the blastodermal margin of each host embryo. After 30 h, host embryos with transplanted cells incorporated into the somite region were fixed in 4% paraformaldehyde and subjected to confocal imaging (n = 25). Grafted cells varied in size and distribution yet vessels in 40% of the mosaic fish were disrupted.
Statistical analyses
All results are expressed as mean ± SEM One-way ANOVA analyses followed by Tukey’s multiple comparison tests were performed for polar versus non-polar assessments, as well as for comparing differences between various mutants of Dvl2. Similar analyses were performed to test differences between autonomous (GFP-positive) extensions between different groups. Two-way ANOVA analyses followed by Bonferroni’s post-tests were performed to determine significant differences within groups between vehicle and drug-treated data as well as for GFP-positive and GFP-negative extensions formed on Matrigel™. *, P <0.05; **, P <0.01; ***, P <0.001. GraphPad Prism v4.0b software was used to perform these analyses.
Results
TNP-470 disrupts caveolin-1 polarity in endothelial cells
While TNP-470 has been shown to inhibit endothelial cell migration in transwell cell culture assays [26], these assays did not allow for endothelial cells to be examined for steady state polarity in the absence of migration. Being able to do so was critical in our early analysis as it was unknown whether PCP inhibition disrupted polarity or rather spared polarity while nonetheless disrupting migration. Recently, caveolin-1 has been shown to regulate cell polarization and directional migration [27], and is localized to the trailing end of migrating endothelial cells [28], thus making it an appropriate marker for endothelial cell polarity. Treatment with TNP-470 resulted in delocalization of caveolin-1 from the trailing end of migrating MPE cells to internal distinct punctae (Fig. 1a–b). This was more informative as a marker than the microtubule organization center (MOTC, γ-tubulin), which localized to the anterior (lamellipodial side) of migrating cells and was not disrupted by TNP-470 (Supplemental Figure S1). To test the association between β-catenin-independent Wnt signaling and caveolin-1 localization (cell polarity), we assayed the effects of downstream PCP activation on TNP-470-mediated loss of cell polarity. Expression of a Dishevelled 2 deletion mutant lacking the aminoterminal DIX domain (ΔDIX-Dvl2) is known to activate Wnt/Ca2+ [29] and PCP signaling independent of Wnt/Frizzled activation [4, 30, 31]. TNP-470 treatment depolarized caveolin-1 subcellular localization even in the presence of a Wnt5a-gradient (Fig. 1c). Activation of β-catenin-independent Wnt signaling through expression of ΔDIX-Dvl2 completely rescued the loss of cell polarity induced by TNP-470 (Fig. 1d). As TNP-470’s cytostatic activity was reported to be relatively selective for endothelial cells over other cell types such as fibroblasts [19, 32], we therefore tested this PCP inhibitor on a cell type resistant to its antiproliferative effects and that expressed caveolin-1 asymmetrically, normal human dermal fibroblasts (NHDFs). Interestingly, the polar localization of caveolin-1 at the trailing end of migrating NHDFs was not altered by TNP-470 (Fig. 1e). Therefore, the activity of TNP-470 to mislocalize caveolin-1 appears to be relatively selective, correlating with its ability to induce cell cycle arrest at nanomolar concentrations in endothelial cells but not fibroblasts.
Fig. 1.

TNP-470, a β-catenin-independent Wnt signaling inhibitor, selectively disrupts endothelial cell polarity. a Typical caveolin-1 (green) polar localization to the trailing edge (white arrowheads) of migrating MPE endothelial cells (DMSO control). b Upon the addition of 10 nM TNP-470, caveolin-1 localizes throughout the cytosol (and is no longer polarized to the trailing edge of the cell). c Proportion of MPE cells displaying a polar localization of caveolin-1 (% Polar) in the presence or absence of a Wnt5a-gradient and/or 10 nM TNP-470 (N = 3–5 Dunn chemotaxis chambers, each N = 12 fields of view (FOV)). d Depolarizing activity of TNP-470 is rescued upon activation of β-catenin-independent signaling by ΔDIX-Dvl2 (N = 3–5 Dunn chemotaxis chambers, each N = 12 FOV) (P >0.05). e Polarization of caveolin-1 in NHDF cells in Dunn chemotaxis chambers. The addition of TNP-470 did not disrupt caveolin-1 localization in NHDF cells (P >0.05). Dashed lines encircle cells to facilitate their visualization
Mutations in Dvl2 affect endothelial cell polarity
Since Dishevelled is known to be a branch point for Wnt/Frizzled signaling, we examined several Dvl2 mutants (Fig. 2a) for their effect on caveolin-1 localization. The aminoterminal DIX domain of Dvl2 is important for its localization to canonical components such as axin and the β-catenin destruction complex [33, 34]. While in Fig. 1d we showed that ΔDIX-Dvl2 expression rescued the TNP-470-induced loss of caveolin-1 polarity, immunohistochemical analyses revealed that Dvl2 and its mutants did not colocalize with caveolin-1 (Fig. 2b–f). Moreover, expression of full-length Dvl2 or ΔDIX-Dvl2 did not disrupt endothelial cell polarity as indicated by caveolin-1 localization (Fig. 2b, c). This indicates that activation of β-catenin-independent Wnt signaling promotes (or in the least, does not disrupt) cell polarity within endothelial cells.
Fig. 2.
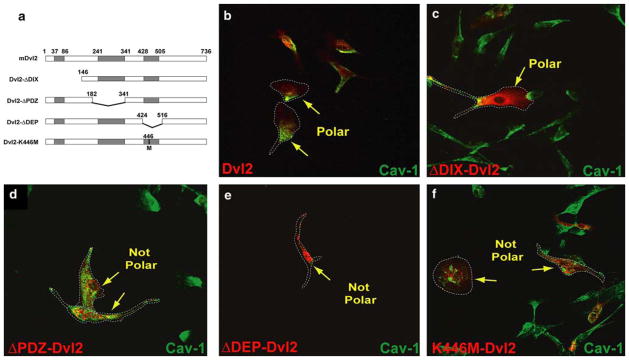
Dvl2 mutants disrupt endothelial cell polarity. a Diagram of HA-Dvl2 constructs in pCMV5 (HA-tags on the N-terminal of each construct are not depicted). Each of the functional domains have been individually deleted. In addition, one construct was generated through the introduction of a point mutation to yield the PCP-inhibitory K446M-Dvl2 construct. Caveolin-1 localization (b–f, green) in MPE cells expressing various HA-Dvl2 (red) constructs: b Dvl2, c ΔDIX-Dvl2, d ΔPDZ-Dvl2, e ΔDEP-Dvl2 and f K446M-Dvl2. Normal asymmetrical caveolin-1 localization was observed in Dvl2 and ΔDIX-Dvl2-expressing cells but was either diminished or mis-localized in cells expressing the others constructs. The minimal K446M point-mutation in Dvl2 was sufficient to bring about this phenotype. Dashed lines encircle cells to facilitate their visualization
In contrast, expression of PDZ or DEP domain-deleted mutants of Dvl2, which suppress β-catenin-independent signaling, resulted in either mislocalization (ΔPDZ-Dvl2) or reduced expression (ΔDEP-Dvl2) of caveolin-1 (Fig. 2d, e). Although we were able to visualize endothelial cells with the ΔPDZ and ΔDEP mutations, the expression of these Dvl2 mutants drastically compromised endothelial cell viability, and we were not able to measure either 3H-thymidine or BrdU incorporation in these cells (data not shown). We therefore hypothesized that the ability of TNP-470 to mislocalize caveolin-1 was linked to β-catenin-independent Wnt-signaling inhibition. We next tested whether a Dvl2 mutant specifically deficient in PCP signaling could also disrupt caveolin-1 localization. Specific inhibition of planar cell polarity signaling was established through overexpression of a DEP domain mutant of Dvl2 (K446M-Dvl2). The DEP domain of Dvl2 is required for its membrane localization and function in PCP signaling [35, 36]. A DEP domain point mutant, K446M-Dvl2 (Drosophila Lys-417; Lys-446 in mouse Dvl2), has shown to act as a selective dominant negative inhibitor of PCP signaling [36, 37] since it specifically abolishes Wnt-induced activation of JNK/SAPK while maintaining Dishevelled localization and canonical signaling [35]. Immunohistochemical data indicated that expression of the K446M-Dvl2 mutation drastically disrupted the polarized distribution of caveolin-1 (Fig. 2f). Given the specificity of the K446M mutation for inhibition of PCP signaling, these results support a model in which PCP signaling coordinates endothelial cell polarity as defined by caveolin-1 subcellular localization.
PCP signaling inhibition suppresses endothelial cell proliferation
Since TNP-470 induces endothelial cell cycle arrest and loss of cell polarity, we wanted to test whether loss of PCP signaling would also induce endothelial cell cycle arrest. Activation of β-catenin-independent signaling via expression of ΔDIX-Dvl2 (Fig. 3a) neither arrested nor stimulated endothelial cell proliferation in comparison to the vector or full-length Dvl2 controls (Fig. 3b). In contrast, expression of the inhibitory K446M-Dvl2 construct significantly blocked proliferation of MPE cells (Fig. 3b). Downregulation of Dvl2 and the PCP component Prickle using shRNA also resulted in decreased endothelial cell proliferation (Supplemental Figure S2). These data suggest that PCP signaling plays an important role in mediating endothelial cell cycle progression.
Fig. 3.

A PCP-deficient point-mutation form of Dvl2 (K446M-Dvl2) disrupts endothelial cell proliferation. a Western blot of Dvl2 constructs in MPE whole cell lysates. b Expression of WT or DIX-deleted forms of Dvl2 did not affect MPE cell proliferation. MPE cells that expressed either ΔPDZ or ΔDEP mutants were not viable and were therefore not included in this assay. However, endothelial cells expressing K446M-Dvl2 were predominantly cytostatic (***; P <0.001, N = 4/group, triplicate experiments)
PCP signaling affects endothelial cell migration independently of proliferation
Since TNP-470 has been shown to inhibit endothelial cell migration [38], we next tested whether endothelial cell migration required PCP signaling. As shown in Fig. 4a, neither Dvl2 nor ΔDIX-Dvl2-expression impeded endothelial cell migration towards a potent angiogenic mitogen, namely, vascular endothelial growth factor (VEGF). Moreover, ΔDIX-Dvl2 overexpression resulted in resistance to the anti-migratory affect of TNP-470. While activation of PCP signaling did not block migration, loss of PCP signaling via expression of the dominant negative K446M-Dvl2 inhibited endothelial cell migration.
Fig. 4.

PCP signaling inhibition disrupts endothelial cell migration in a p53-indpendant manner. a Migration of MPE cells transiently co-transfected with various Dvl2/GFP constructs (**, P <0.01). b TNP-470 disrupts endothelial cell migration independently of p53 status. The difference between migration indices (MI) was not statistically significant (P >0.05). c Expression of various PCP mediators, including Diversin, DAAM-1 and Inversin rescue from TNP-470-Induced Non-canonical signaling inhibition. TNP-470 treatment significantly inhibited migration of MPE cells (*; P <0.05). Migration indices were determined for different positive PCP mediators for protection against TNP-470 and were statistically not distinguishable from the DMSO control (P >0.05) but meaningfully greater than TNP-470 treatment (*<; P <0.05)
Since the cytostatic activity of TNP-470 is rescued upon deletion of p53 [16, 17], we next tested whether the anti-migratory effect of TNP-470 was due to a p53-mediated cell cycle arrest. Interestingly, the migration of endothelial cells derived from p53 null (Δp53) and wild-type mice was comparably sensitive to TNP-470 (Fig. 4b). Thus, the ability of TNP-470 to inhibit migration is independent or upstream of p53-mediated growth arrest.
We next screened several downstream mediators of PCP signaling for their ability to rescue the anti-migratory activity of TNP-470. We focused on PCP components that have been previously shown to activate PCP signaling, namely, Diversin, DAAM-1 and Inversin. Diversin, the mammalian homologue of the Drosophila PCP signaling component, Diego [39, 40], rescues in vivo PCP defects when overexpressed [41]. More recently, Diversin has been shown to activate PCP signaling through Dishevelled association, and this association is disrupted by the PCP-inactivating K446M-Dvl mutation [42]. As shown in Fig. 4c, overexpression of Diversin partially rescues the TNP-470-induced inhibition of endothelial cell migration in response to VEGF. DAAM-1, which is required for the downstream activation of Rho by Wnt/frizzled PCP signaling [10], likewise rescues endothelial cells from drug-induced migration inhibition (Fig. 4c). Lastly, we tested Inversin in this system. Inversin has been shown to mediate PCP signaling in gastrulating Xenopus laevis embryos [41]. Inversin, like the related PCP component Diversin, rescues the anti-migratory effects of the drug (Fig. 4c). Indeed, although the migration indices from Diversin, DAAM-1 and Inversin appeared diminished relative to the normalized DMSO controls, these values were not statistically different. Taken together, the data demonstrate that disruption of the PCP pathway in endothelial cells results in suppressed cellular migration, which is rescued through downstream activation of PCP signaling (such as through ΔDIX-Dvl2, Fig. 4a) or by overexpression of positive PCP mediators (such as Diversin, DAAM-1 or Inversin; Fig. 4c).
Aberrant PCP signaling disrupts endothelial cell network formation in vitro
To investigate whether activation or disruption of β-catenin-independent Wnt signaling further disrupts endothelial cell network formation, we next cultured MPE cells transfected with various Dishevelled mutant constructs in Matrigel™. Endothelial cells are capable of spontaneously forming cellular extensions and coordinated networks on a Matrigel™ basement membrane. The ability of endothelial cells to form these structures is an established quantifiable in vitro assay to assess angiogenic potential [24]. Prior studies have shown that both activation and disruption of PCP perturb cell movements [6, 43, 44] and can cause gastrulation defects [4, 31, 45–47]. Other evidence suggests that spatial and temporal timing of PCP signaling is important for higher order coordination of several tissues [5, 48]. Therefore, we next determined whether alteration of PCP signaling could affect the intercellular organization typically observed in endothelial cell network formation.
TNP-470 has been previously been reported to inhibit endothelial cell network formation on a Matrigel™ matrix [38, 49]. In reproducing these experiments, we noted that drug-induced PCP inhibition predominantly disrupted the number of extensions formed in this assay more so than their length (Fig. 5a–b). This suggested that TNP-470 inhibits the very early stages of angiogenesis, likely during the initial branching from pre-existing vasculature. This is consistent with the less drastic effects on the extension length where the branch point would have already been established. Therefore, it was expected that PCP signaling inhibition by genetic means would disrupt network formation in a similar manner. Interestingly, whereas Dvl2 expression significantly increased the number of cell extensions formed, expression of either activating ΔDIX-Dvl2 or inactivating K446M-Dvl2 constructs drastically suppressed this number (Fig. 5c–g). The over-activation of β-catenin-independent signaling through ΔDIX-Dvl2 expression prevented the formation of a coordinated network not directly through defects in migration but rather via extensive clumping of cells (Fig. 5d). This observation is different from the proliferation and migration data (Figs. 3, 4) where ΔDIX-Dvl2 expression had little effect. However, the pervasive cell adhesions indicate a clear role for cell–cell interactions mediated by PCP signaling independent of cell growth and movement. This is consistent with other reports that β-catenin-independent signaling can modulate extracellular matrix deposition and cell adhesion [50–52]. Thus, coordination of several cell types, including endothelial cells described herein, require PCP signaling in a temporally and spatially regulated manner. Interestingly, endothelial cells with disrupted PCP signaling (Fig. 5f–g) do not significantly form coordinated networks. Hence, ectopic activation as well as disruption of PCP signaling both resulted in deleterious (although not identical) effects on vascular network formation in vitro.
Fig. 5.

Both over-activation and inhibition of PCP signaling disrupt endothelial cell network formation. (a–b) TNP-470 inhibits the initiation of endothelial network formation in vitro. MPE cells were plated onto Matrigel™ with or without TNP-470. a The number of extensions formed was drastically reduced in the presence of 10-nM TNP-470. b Cell-extension length was reduced by approximately 25% with 10-nM TNP-470. c–f Networks formed by MPE cells expressing various Dvl2 constructs (as labeled). g Quantification of cellular extensions formed depicted in (c–f) (N = 8/group). h–j MPE cells co-expressing Dvl2 mutations and GFP were mixed with non-transfected MPE (mosaic analysis) and incubated on Matrigel™. h Phase contrast and i green fluorescence images of MPE cells within the mosaic analysis. Cell extensions were thereafter scored as GFP-positive (green arrows) or GFP-negative (white arrows) if cell extensions were devoid of GFP-positive cells. j Quantification of extensions formed in the mosaic analysis (N = 8/group). Values within each of the groups (GFP+ vs. GFP−) were statistically different (P <0.01). The non-mutant Dvl2 group displayed more extensive networks over other test conditions (P <0.05). The quantities were significantly different for the Dvl2-autonomous (increase, P <0.05) and K446M-Dvl2-autonomous (decrease, P <0.01) groups. All scale bars 200 μm
Given the need for cell–cell communication during angiogenesis, we hypothesized that loss of PCP signaling in an individual endothelial cell is likely to affect the coordinated growth and migration of neighboring cells during neovascularization. To examine the ability of endothelial cells with misregulated PCP signaling to coordinate intercellular communication, GFP-positive MPE cells expressing various Dvl2 mutants were co-cultured with wild-type (GFP-negative) endothelial cells in Matrigel™ assays (Fig. 5h–j). The Dvl2 and ΔDIX-Dvl2 cells in these mosaic analyses coordinated with wild-type endothelial cells to form cellular extensions in a similar manner as the vector control (Fig. 5j, note the GFP-positive cells). In fact, many of the networks formed (all but the K446M-Dvl2 expressing cells) contained GFP-positive cells bearing the genetic modifications. Clearly, these cells are still capable of coordinating with neighboring cells to form complex networks. However, cells expressing the PCP-inactivating K446M-Dvl2 were incapable of contributing significantly to the networks formed (Fig. 5j). Therefore, PCP signaling coordinates non-autonomous signaling with other endothelial cells, consistent with cell non-autonomous signaling behavior observed in vertebrate convergent–extension movements [4, 11, 53].
Interestingly, while proliferation is compromised in endothelial cells with suppressed Dvl2 expression (Supplemental Figure S2), these cells maintained their ability to migrate and coordinate to form networks on Matrigel (Supplemental Figure S3). In both cases the lack of Dvl2 served to rescue from TNP-470 treatment, suggesting that Dvl2 is required for the biological activities of TNP-470. However, as endothelial cells were still capable of functioning in these assays despite Dvl2-silencing, this suggests that other Dvls can replace function of Dvl2 in endothelial cells. A redundant role for Dvls is supported through mouse knockout studies as viable adult Dvl2 knockout mice have been generated (despite high prenatal mortality in addition to cardiac and neuronal and other developmental deficiencies) [54].
Diminished β-catenin-independent Wnt signaling in a vertebrate model results in vascular defects
Given our findings that perturbation of PCP signaling disrupts endothelial polarity, growth, migration and network formation in cell culture assays, we next investigated the effect of PCP inhibition on in vivo angiogenesis. A transgenic zebrafish line expressing the Fli1-promoter (endothelial cell specific)—driven GFP strain (Tg(fli:EGFP)) [55] was crossed into the Wnt5 mutant, pipetail (ppt) genetic background. Homozygous pipetail (ppt−/−) embryos display shortened body length with head cartilage and tail defects [56]. Wildtype-like (ppt+/+, or ppt+/−) Tg(fli:EGFP) animals displayed ordered angiogenesis throughout the animal including formation of well-defined intersegmental (IS) vessels (Fig. 6a, e; arrows), and a robust meshwork of the dorsal aorta (DA) and posterior cardinal vein (PCV) (Fig. 6a, e) in the trunk [57]. Ppt mutants displayed loss or disorganization of IS vessels (Fig. 6b, f; arrows), reduced DA and PCV (Fig. 6b, arrowhead) as well as a shortened yolk extension. In addition, ppt animals showed a disruption of the dorsal vasculature including the Duct of Cuvier (DC), which will become the future common cardinal vein (CCV) (Fig. 6c vs. d). The vascular defects are not due to developmental delay as the defect persists through 48 hpf (Fig. 6e vs. f) and is consistent with the endothelial migration defects caused by diminished PCP regulation. We therefore conclude that the coordination of vascular networks has been compromised in this mutant model.
Fig. 6.

Wnt5-deficient (ppt) zebrafish display aberrant angiogenesis in vivo. a–b Tg(fli:EGFP) expression (28–30 hpf) as detected by whole mount. An in situ Fli probe shows ordered intersegmental vessel (IS) development in wt-like embryos (a, arrow) as well as proper development of the dorsal aorta (DA), and posterior cardinal vein (PCV) (arrowhead). b Similarly staged ppt mutants display disrupted angiogenesis with defective IS vessel formation (arrow) and reduced DA/PCV expression (arrowhead). c–d Fli-GFP expression in vivo. WT (c) and ppt (d) expression of Fli-GFP. Loss of Wnt5 results in disruption of the Duct of Cuvier/CCV. (e–f) Live in vivo Fli-GFP images of 48 hpf embryos. Irregular vessel formation is seen in ppt relative to WT control. g–h Whole mount staining of WT and ppt embryo head regions. g Robust vasculature can be observed in the anterior and middle cerebral veins within WT embryos. These vessels are diminished and aberrantly patterned within ppt embryos (h)
The disruption in ppt mutants predominantly impacts the tail and trunk and it has been shown that somite mutants can have secondary vascular defects [58]. Therefore, we characterized vasculature defects in regions independent of the tail, such as over the yolk (Duct of Cuvier) and reduced or absent anterior cerebral vein (ACeV) and middle cerebral vein (MCeV) within the head (Fig. 6g vs. h). These phenotypes suggest that the defects are less likely due to improper somite formation. Moreover, pharmacological treatment with TNP-470 after gastrulation has initiated does not disrupt somite patterning but results in vascular defects (Fig. 7a vs. b). DMSO treated (n = 21) displayed normal vasculature while greater than 95% of TNP-470 treated (n = 37) had defects.
Fig. 7.
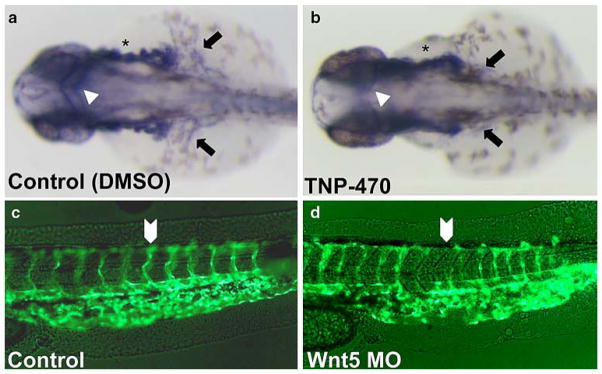
Vascular defects due to chemical inhibition of PCP signaling mimic those of ppt zebrafish. a–b Tg(fli:EGFP) expression (28–30 hpf) as detected by whole mount. Relative to WT (a) vasculature, TNP-470-treated animals (b) displayed disrupted vessel growth (asterisks), disrupted common cardinal vein formation (arrows) and markedly diminished middle cerebral vein (arrowheads). c–d Wnt5 MO-injected embryos evaluated for vasculature defects. Fluorescent imaging of Control (c) and Wnt MO-injected embryos (d), lateral view of the tail. Wnt5-MO injection (d) disrupts intersegmental angiogenesis in Tg(fli:EGFP)
Complementing our ppt mutant genetic data, Wnt5 antisense morpholino (MO) microinjection phenocopied the vascular defects observed with the Wnt5-deficient pipetail mutants (Fig. 7c vs. d). The vascular phenotypes in vivo closely mimicked the migration and coordination defects observed in vitro. In particular, we have noted coordination defects in IS vessels with respect to aberrant patterning in ppt mutants. Suppressed migration of endothelial cells was also found in mutant animals. Vascular defects were consistent between all of these groups including the defects in the cerebral veins, the IS vascularization, duct of Cuvier and common cardinal vein. In addition, transplantation of Wnt5-MO injected cells into wild-type host demonstrate aberrant vessel outgrowth in the region of Wnt-depleted cells (Supplemental Figure S4). Taken together, our model suggests that inhibition of β-catenin-independent Wnt signaling is disruptive to endothelial cell migration and angiogenesis in vivo.
Discussion
Wnt signaling plays an important role in mediating cell–cell communication and pattern formation during development. Previous studies have demonstrated a role for canonical Wnt/β-catenin signaling in the complex series of events that lead to angiogenesis or neovascularization [59–63]. Here, we report that a β-catenin-independent Wnt signaling cascade, the planar cell polarity (PCP) pathway also plays a critical role in endothelial proliferation, migration and coordination. Furthermore, we confirmed the in vivo significance of these cell culture results by demonstrating that Wnt5 mutant zebrafish display impaired angiogenesis. Below, we discuss what our results reveal about the requirement for PCP signaling in the developing endothelium and propose a model that potentially links this signaling pathway with a critical p53-dependent cell cycle checkpoint.
Wnt signaling in endothelial cell proliferation and angiogenesis
A role for canonical Wnt/β-catenin signaling in endothelial cell biology is well established [60]. Both Wnt1 addition as well as expression of constitutively active β-catenin (S37A) induced proliferation and survival of cultured endothelial cells [63]. In addition, β-catenin stabilization has been observed in proliferating endothelium during neovascularization following myocardial infarction [59] and in endothelial cells within glioblastomas [64]. This correlation is supported by murine knockout studies in which a targeted deletion of a canonical Wnt receptor gene, frizzled-5, resulted in lethal defects in yolk sac angiogenesis with reduced endothelial cell proliferation [65]. Since Dvl2 overexpression has been shown to induce Wnt canonical signaling, our results showing improved network formation in Matrigel™ upon Dvl2 overexpression (Fig. 5e, g) also support the idea that canonical Wnt/β-catenin signaling plays a critical role in endothelial cell biology.
In comparison to the established significance of β-catenin/Wnt signaling in angiogenesis, the role of β-catenin-independent Wnt signaling in endothelial cell biology and neovascularization is less well understood. Recent studies have shown that the non-canonical Wnt5a induces endothelial cell proliferation and enhances cell survival [14, 15]. However, we found that activation of the PCP pathway via expression of ΔDIX-Dvl2 did not induce endothelial cell proliferation (Fig. 3). Our data, instead, support a model in which PCP signaling provides a permissive signal for endothelial cell proliferation and migration to occur.
By analogy with the polarized plane of wing epithelial cells on the adult Drosophila, we propose that during angiogenesis actively dividing endothelial cells migrating along a gradient of angiogenic factors behave as a cylindrical sheet of cells, coordinating their polarization, migration and formation into a nascent tubular structure through cell–cell communication. Unlike other cell types, such as fibroblasts, which lack a need to coordinate their polarity with neighboring cells, we posit that endothelial cells fail to divide if they cannot process the necessary PCP signaling derived from their neighbors to determine the direction in which they should divide.
In support of this hypothesis, we have shown that disruption of PCP signaling either by using a small molecule PCP inhibitor or by expression of a dominant negative Dishevelled DEP domain mutant (K446M-Dvl2) leads to loss of endothelial cell proliferation, polarization, migration and ability to incorporate into a nascent network in cell culture. In addition, overexpression of the downstream PCP effector proteins Diversin, DAAM-1 or Inversin each rescued the migrational suppression induced by chemical inhibition of PCP (Fig. 4). Similar in vivo rescue experiments, could have effects on gastrulation and, therefore, more global effects outside angiogenesis. However, the selectivity of TNP-470 for endothelial cells and the relatively mild effects of the Wnt5 MO allowed us to examine a mild phenotype that showed minimal perturbations globally in these fish.
These cell culture results are consistent with our observed in vivo link between angiogenesis and PCP signaling. Wnt5 mutant (ppt) zebrafish, which have decreased PCP function, display previously overlooked angiogenesis defects (Figs. 6, 7). While somite defects could contribute to vascular defects, the gross morphology of the anterior regions of ppt and wild-type fish are very similar, yet, distinguishable differences in the head vasculature of ppt fish can be observed, specifically in the ACeV and MCeV (Fig. 6g, h). In addition, late stage treatment with TNP-470 as well as transplantation of Wnt5-MO depleted cells in a wild-type host both lead to vascular defects. Thus, it is less likely that secondary defects could explain these observations in the ppt mutant. These data, therefore, demonstrate a connection between PCP signaling and endothelial cell coordination and, moreover, were corroborated in Prickle antisense morpholino injected embryos (H. Griesbach and D. Slusarski, unpublished observations). However, Wnt5a has recently been published to affect planar polarity signaling in mouse cochlea [66] but has not been reported to have a vascular phenotype [67]. Since the ppt zebrafish clearly show vascular defects in development, this may suggest a greater redundancy for Wnt signaling in mammals.
A potential link between PCP signaling and the cell cycle
The mechanism by which inhibition of PCP signaling is interpreted by the cell cycle machinery remains an outstanding question. Previous studies showed that loss of RhoA-mediated actin cytoskeletal integrity can lead to a p27KIP1-induced G1/S cell cycle arrest [68, 69]. Given that RhoA activation is downstream of Wnt5/Wnt11 signaling, it is thus conceivable that disruption of PCP signaling could lead to abnormal actin cytoskeleton organization and subsequent cell cycle arrest.
Interestingly, TNP-470-mediated inhibition of PCP signaling also leads to a G1/S endothelial cell cycle arrest albeit via a different cyclin dependent kinase inhibitor, p21CIP/WAF [19, 70]. Induction of p21CIP/WAF levels in TNP-470-treated cells is most likely at the transcriptional level since inactivation of the p21CIP/WAF upstream trans-activator p53 renders them insensitive to the drug [19, 70]. The possibility that the endothelial cell depolarization observed here is a simple consequence of TNP-470-mediated growth arrest was refuted by our finding that TNP-470 still blocks the Δp53 MPE cell migration despite the inability to elicit growth arrest in these cells.
The PCP pathway as a potential therapeutic target
Taken together, our results suggest a model in which endothelial cells respond to pro-angiogenic stimuli in a manner requiring the PCP pathway. Thus, misregulation of PCP signaling in endothelial cells leads to a p53-dependent cell cycle arrest to prevent uncoordinated and non-directed growth. This model also explains the exquisite sensitivity of endothelial cells to the PCP inhibitor TNP-470 (pi-comolar IC50 value) whereas other cell types are resistant; cells such as fibroblasts do not need to grow and migrate as a polarized collection of cells and thus do not possess this PCP-dependent p53 endothelial cell cycle checkpoint. Given that pathological angiogenesis occurs in several diseases including diabetic retinopathy, age-related macular degeneration [71] and most notably in cancer [72, 73], anti-angiogenic therapeutic strategies are very attractive [74]. Whereas the potent angiogenic inhibitor endostatin has been shown to inhibit β-catenin/canonical Wnt signaling [62], our findings suggest that targeting the PCP pathway could yield additional anti-angiogenic agents.
Supplementary Material
Acknowledgments
We would like to thank John Hines for his critical review of the manuscript. This work was supported by grants from the NIH (CA083049 to CMC and CA112369 to DCS). PC is the recipient of the Leukemia and Lymphoma Society Fellowship.
Abbreviations
- Dvl
Dishevelled
- PCP
Planar cell polarity
- NHDF
Normal human dermal fibroblasts
- MPE
Murine pulmonary endothelial
- GFP
Green fluorescent protein
- MetAP-2
Methionine aminopeptidase 2
- IS
Intersegmental
- MO
Morpholino
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10456-008-9116-2) contains supplementary material, which is available to authorized users.
The authors have declared no competing interests.
Contributor Information
Pasquale Cirone, Department of Molecular, Cellular and Developmental Biology, Yale University, Kline Biology Tower Room 400, P.O. Box 208103, New Haven, CT 06520-8103, USA.
Shengda Lin, Department of Biology, University of Iowa, Iowa City, IA 52242, USA.
Hilary L. Griesbach, Department of Biology, University of Iowa, Iowa City, IA 52242, USA
Yi Zhang, Department of Molecular, Cellular and Developmental Biology, Yale University, Kline Biology Tower Room 400, P.O. Box 208103, New Haven, CT 06520-8103, USA.
Diane C. Slusarski, Department of Biology, University of Iowa, Iowa City, IA 52242, USA
Craig M. Crews, Email: craig.crews@yale.edu, Department of Molecular, Cellular and Developmental Biology, Yale University, Kline Biology Tower Room 400, P.O. Box 208103, New Haven, CT 06520-8103, USA. Department of Chemistry, Yale University, New Haven, CT 06520-8103, USA. Department of Pharmacology, Yale University, New Haven, CT 06520-8103, USA
References
- 1.Liebner S, Cavallaro U, Dejana E. The multiple languages of endothelial cell-to-cell communication. Arterioscler Thromb Vasc Biol. 2006;26:1431–1438. doi: 10.1161/01.ATV.0000218510. 04541.5e. [DOI] [PubMed] [Google Scholar]
- 2.Fanto M, McNeill H. Planar polarity from flies to vertebrates. J Cell Sci. 2004;117:527–533. doi: 10.1242/jcs.00973. [DOI] [PubMed] [Google Scholar]
- 3.Gong Y, Mo C, Fraser SE. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature. 2004;430:689–693. doi: 10.1038/nature02796. [DOI] [PubMed] [Google Scholar]
- 4.Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, et al. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000;405:76–81. doi: 10.1038/35011068. [DOI] [PubMed] [Google Scholar]
- 5.Myers DC, Sepich DS, Solnica-Krezel L. Convergence and extension in vertebrate gastrulae: cell movements according to or in search of identity? Trends Genet. 2002;18:447–455. doi: 10.1016/S0168-9525(02)02725-7. [DOI] [PubMed] [Google Scholar]
- 6.Wallingford JB, Vogeli KM, Harland RM. Regulation of convergent extension in Xenopus by Wnt5a and Frizzled-8 is independent of the canonical Wnt pathway. Int J Dev Biol. 2001;45:225–227. [PubMed] [Google Scholar]
- 7.Arevalo JC, Chao MV. Axonal growth: where neurotrophins meet Wnts. Curr Opin Cell Biol. 2005;17:112–115. doi: 10.1016/j.ceb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005;8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- 9.Malbon CC, Wang HY. Dishevelled: a mobile scaffold catalyzing development. Curr Top Dev Biol. 2006;72:153–166. doi: 10.1016/S0070-2153(05)72002-0. [DOI] [PubMed] [Google Scholar]
- 10.Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 2001;107:843–854. doi: 10.1016/S0092-8674(01)00614-6. [DOI] [PubMed] [Google Scholar]
- 11.Marlow F, Topczewski J, Sepich D, Solnica-Krezel L. Zebrafish Rho kinase 2 acts downstream of Wnt11 to mediate cell polarity and effective convergence and extension movements. Curr Biol. 2002;12:876–884. doi: 10.1016/S0960-9822(02)00864-3. [DOI] [PubMed] [Google Scholar]
- 12.Yamanaka H, Moriguchi T, Masuyama N, Kusakabe M, Hanafusa H, Takada R, et al. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002;3:69–75. doi: 10.1093/embo-reports/kvf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright M, Aikawa M, Szeto W, Papkoff J. Identification of a Wnt-responsive signal transduction pathway in primary endothelial cells. Biochem Biophys Res Commun. 1999;263:384–388. doi: 10.1006/bbrc.1999.1344. [DOI] [PubMed] [Google Scholar]
- 14.Masckauchan TN, Agalliu D, Vorontchikhina M, Ahn A, Parmalee NL, Li CM, et al. Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol Biol Cell. 2006;17:5163–5172. doi: 10.1091/mbc.E06-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CW, Yeh JC, Fan TP, Smith SK, Charnock-Jones DS. Wnt5a-mediated non-canonical Wnt signalling regulates human endothelial cell proliferation and migration. Biochem Biophys Res Commun. 2008;365:285–290. doi: 10.1016/j.bbrc.2007.10.166. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Yeh JR, Mara A, Ju R, Hines JF, Cirone P, et al. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001–1009. doi: 10.1016/j.chembiol.2006. 07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeh JR, Ju R, Brdlik CM, Zhang W, Zhang Y, Matyskiela ME, et al. Targeted gene disruption of methionine aminopeptidase 2 results in an embryonic gastrulation defect and endothelial cell growth arrest. Proc Natl Acad Sci USA. 2006;103:10379–10384. doi: 10.1073/pnas.0511313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Neo SY, Han J, Lin SC. Dimerization choices control the ability of axin and dishevelled to activate c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 2000;275:25008–25014. doi: 10.1074/jbc.M002491200. [DOI] [PubMed] [Google Scholar]
- 19.Yeh JR, Mohan R, Crews CM. The antiangiogenic agent TNP-470 requires p53 and p21CIP/WAF for endothelial cell growth arrest. Proc Natl Acad Sci USA. 2000;97:12782–12787. doi: 10.1073/pnas.97.23.12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beardsley A, Fang K, Mertz H, Castranova V, Friend S, Liu J. Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J Biol Chem. 2005;280:3541–3547. doi: 10.1074/jbc.M409040200. [DOI] [PubMed] [Google Scholar]
- 21.Zicha D, Dunn GA, Brown AF. A new direct-viewing chemotaxis chamber. J Cell Sci. 1991;99(Pt 4):769–775. doi: 10.1242/jcs.99.4.769. [DOI] [PubMed] [Google Scholar]
- 22.Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, et al. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem. 2000;275:10661–10672. doi: 10.1074/jbc. 275.14.10661. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi N, Anand-Apte B, Lee M, Sasaki T, Fukai N, Shapiro R, et al. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999;18:4414–4423. doi: 10.1093/emboj/18.16.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicosia RF, Ottinetti A. Modulation of microvascular growth and morphogenesis by reconstituted basement membrane gel in three-dimensional cultures of rat aorta: a comparative study of angiogenesis in matrigel, collagen, fibrin, and plasma clot. In Vitro Cell Dev Biol. 1990;26:119–128. doi: 10.1007/BF02624102. [DOI] [PubMed] [Google Scholar]
- 25.Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/S1074-5521(02)00248-X. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida A, Anand-Apte B, Zetter BR. Differential endothelial migration and proliferation to basic fibroblast growth factor and vascular endothelial growth factor. Growth Factors. 1996;13:57–64. doi: 10.3109/08977199609034566. [DOI] [PubMed] [Google Scholar]
- 27.Grande-Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman-Storer CM, Valdivielso JM, et al. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J Cell Biol. 2007;177:683–694. doi: 10.1083/jcb.200701006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parat MO, Anand-Apte B, Fox PL. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol Biol Cell. 2003;14:3156–3168. doi: 10.1091/mbc.E02-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheldahl LC, Slusarski DC, Pandur P, Miller JR, Kuhl M, Moon RT. Dishevelled activates Ca2+flux, PKC, and CamKII in vertebrate embryos. J Cell Biol. 2003;161:769–777. doi: 10.1083/jcb. 200211094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–118. doi: 10.1016/S0092-8674(00)81226-X. [DOI] [PubMed] [Google Scholar]
- 31.Tada M, Smith JC. Xwnt11 is a target of Xenopus Brachyury: regulation of gastrulation movements via Dishevelled, but not through the canonical Wnt pathway. Development. 2000;127:2227–2238. doi: 10.1242/dev.127.10.2227. [DOI] [PubMed] [Google Scholar]
- 32.Kusaka M, Sudo K, Fujita T, Marui S, Itoh F, Ingber D, et al. Potent anti-angiogenic action of AGM-1470: comparison to the fumagillin parent. Biochem Biophys Res Commun. 1991;174:1070–1076. doi: 10.1016/0006-291X(91)91529-L. [DOI] [PubMed] [Google Scholar]
- 33.Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol Cell Biol. 1999;19:4414–4422. doi: 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, et al. Dishevelled proteins lead to two signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. J Biol Chem. 1999;274:129–134. doi: 10.1074/jbc.274.1.129. [DOI] [PubMed] [Google Scholar]
- 35.Moriguchi T, Kawachi K, Kamakura S, Masuyama N, Yamanaka H, Matsumoto K, et al. Distinct domains of mouse dishevelled are responsible for the c-Jun N-terminal kinase/stress-activated protein kinase activation and the axis formation in vertebrates. J Biol Chem. 1999;274:30957–30962. doi: 10.1074/jbc.274. 43.30957. [DOI] [PubMed] [Google Scholar]
- 36.Park TJ, Gray RS, Sato A, Habas R, Wallingford JB. Subcellular localization and signaling properties of dishevelled in developing vertebrate embryos. Curr Biol. 2005;15:1039–1044. doi: 10.1016/j.cub.2005.04.062. [DOI] [PubMed] [Google Scholar]
- 37.Pan WJ, Pang SZ, Huang T, Guo HY, Wu D, Li L. Characterization of function of three domains in dishevelled-1: DEP domain is responsible for membrane translocation of dishevelled-1. Cell Res. 2004;14:324–330. doi: 10.1038/sj.cr.7290232. [DOI] [PubMed] [Google Scholar]
- 38.Farinelle S, Malonne H, Chaboteaux C, Decaestecker C, Dedecker R, Gras T, et al. Characterization of TNP-470-induced modifications to cell functions in HUVEC and cancer cells. J Pharmacol Toxicol Methods. 2000;43:15–24. doi: 10.1016/S1056-8719(00)00080-0. [DOI] [PubMed] [Google Scholar]
- 39.Schwarz-Romond T, Asbrand C, Bakkers J, Kuhl M, Schaeffer HJ, Huelsken J, et al. The ankyrin repeat protein Diversin recruits Casein kinase Iepsilon to the beta-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 2002;16:2073–2084. doi: 10.1101/gad.230402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tissir F, Bar I, Goffinet AM, Lambert De Rouvroit C. Expression of the ankyrin repeat domain 6 gene (Ankrd6) during mouse brain development. Dev Dyn. 2002;224:465–469. doi: 10.1002/dvdy.10126. [DOI] [PubMed] [Google Scholar]
- 41.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moeller H, Jenny A, Schaeffer HJ, Schwarz-Romond T, Mlodzik M, Hammerschmidt M, et al. Diversin regulates heart formation and gastrulation movements in development. Proc Natl Acad Sci USA. 2006;103:15900–15905. doi: 10.1073/pnas.0603808103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Calisto J, Araya C, Marchant L, Riaz CF, Mayor R. Essential role of non-canonical Wnt signalling in neural crest migration. Development. 2005;132:2587–2597. doi: 10.1242/dev.01857. [DOI] [PubMed] [Google Scholar]
- 44.Park M, Moon RT. The planar cell-polarity gene stbm regulates cell behaviour and cell fate in vertebrate embryos. Nat Cell Biol. 2002;4:20–25. doi: 10.1038/ncb716. [DOI] [PubMed] [Google Scholar]
- 45.Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 2003;17:295–309. doi: 10.1101/gad.1022203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kilian B, Mansukoski H, Barbosa FC, Ulrich F, Tada M, Heisenberg CP. The role of Ppt/Wnt5 in regulating cell shape and movement during zebrafish gastrulation. Mech Dev. 2003;120:467–476. doi: 10.1016/S0925-4773(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 47.Moon RT, Campbell RM, Christian JL, McGrew LL, Shih J, Fraser S. Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development. 1993;119:97–111. doi: 10.1242/dev.119.1.97. [DOI] [PubMed] [Google Scholar]
- 48.Liao G, Tao Q, Kofron M, Chen JS, Schloemer A, Davis RJ, et al. Jun NH2-terminal kinase (JNK) prevents nuclear {beta}-catenin accumulation and regulates axis formation in Xenopus embryos. Proc Natl Acad Sci USA. 2006;103:16313–16318. doi: 10.1073/pnas.0602557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kragh M, Hjarnaa PJ, Bramm E, Binderup L. A versatile in vivo chamber angiogenesis assay for measuring anti-angiogenic activity in mice. Oncol Rep. 2004;11:303–307. [PubMed] [Google Scholar]
- 50.Goto T, Davidson L, Asashima M, Keller R. Planar cell polarity genes regulate polarized extracellular matrix deposition during frog gastrulation. Curr Biol. 2005;15:787–793. doi: 10.1016/j.cub.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 51.Na J, Lykke-Andersen K, Torres Padilla ME, Zernicka-Goetz M. Dishevelled proteins regulate cell adhesion in mouse blastocyst and serve to monitor changes in Wnt signaling. Dev Biol. 2007;302:40–49. doi: 10.1016/j.ydbio.2006.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witzel S, Zimyanin V, Carreira-Barbosa F, Tada M, Heisenberg CP. Wnt11 controls cell contact persistence by local accumulation of Frizzled 7 at the plasma membrane. J Cell Biol. 2006;175:791–802. doi: 10.1083/jcb.200606017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keller R, Shih J, Sater AK, Moreno C. Planar induction of convergence and extension of the neural plate by the organizer of Xenopus. Dev Dyn. 1992;193:218–234. doi: 10.1002/aja.1001930303. [DOI] [PubMed] [Google Scholar]
- 54.Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, et al. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838. doi: 10.1242/dev.00164. [DOI] [PubMed] [Google Scholar]
- 55.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 56.Rauch GJ, Hammerschmidt M, Blader P, Schauerte HE, Strahle U, Ingham PW, et al. Wnt5 is required for tail formation in the zebrafish embryo. Cold Spring Harb Symp Quant Biol. 1997;62:227–234. [PubMed] [Google Scholar]
- 57.Isogai S, Horiguchi M, Weinstein BM. The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev Biol. 2001;230:278–301. doi: 10.1006/dbio.2000.9995. [DOI] [PubMed] [Google Scholar]
- 58.Shaw KM, Castranova DA, Pham VN, Kamei M, Kidd KR, Lo BD, et al. Fused-somites-like mutants exhibit defects in trunk vessel patterning. Dev Dyn. 2006;235:1753–1760. doi: 10.1002/dvdy. 20814. [DOI] [PubMed] [Google Scholar]
- 59.Blankesteijn WM, van Gijn ME, Essers-Janssen YP, Daemen MJ, Smits JF. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am J Pathol. 2000;157:877–883. doi: 10.1016/s0002-9440(10)64601-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goodwin AM, D’Amore PA. Wnt signaling in the vasculature. Angiogenesis. 2002;5:1–9. doi: 10.1023/A:1021563510866. [DOI] [PubMed] [Google Scholar]
- 61.Goodwin AM, Sullivan KM, D’Amore PA. Cultured endothelial cells display endogenous activation of the canonical Wnt signaling pathway and express multiple ligands, receptors, and secreted modulators of Wnt signaling. Dev Dyn. 2006;235:3110–3120. doi: 10.1002/dvdy.20939. [DOI] [PubMed] [Google Scholar]
- 62.Hanai J, Gloy J, Karumanchi SA, Kale S, Tang J, Hu G, et al. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol. 2002;158:529–539. doi: 10.1083/jcb.200203064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masckauchan TN, Shawber CJ, Funahashi Y, Li CM, Kitajewski J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8:43–51. doi: 10.1007/s10456-005-5612-9. [DOI] [PubMed] [Google Scholar]
- 64.Yano H, Hara A, Takenaka K, Nakatani K, Shinoda J, Shimokawa K, et al. Differential expression of beta-catenin in human glioblastoma multiforme and normal brain tissue. Neurol Res. 2000;22:650–656. doi: 10.1080/01616412.2000.11740735. [DOI] [PubMed] [Google Scholar]
- 65.Ishikawa T, Tamai Y, Zorn AM, Yoshida H, Seldin MF, Nishikawa S, et al. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 2001;128:25–33. doi: 10.1242/dev.128.1.25. [DOI] [PubMed] [Google Scholar]
- 66.Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel KP, et al. Wnt5a functions in planar cell polarity regulation in mice. Dev Biol. 2007;306:121–133. doi: 10.1016/j.ydbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 68.Huang S, Chen CS, Ingber DE. Control of cyclin D1, p27(Kip1), and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol Biol Cell. 1998;9:3179–3193. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2–p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279:26323–26330. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Griffith EC, Sage J, Jacks T, Liu JO. Cell cycle inhibition by the anti-angiogenic agent TNP-470 is mediated by p53 and p21WAF1/CIP1. Proc Natl Acad Sci USA. 2000;97:6427–6432. doi: 10.1073/pnas.97.12.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/S1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 72.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 73.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 74.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.