

Abstract
In non-excitable cells, thiol-oxidizing agents have been shown to evoke oscillations in cytosolic free Ca2+ concentration ([Ca2+]i) by increasing the sensitivity of the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) to IP3. Although thiol modification of the IP3R is implicated in this response, the molecular nature of the modification(s) responsible for changes in channel activity is still not well understood. Diamide is a chemical oxidant that selectively converts reduced glutathione (GSH) to its disulfide (GSSG) and promotes the formation of protein–glutathione (P-SSG) mixed disulfide, i.e. glutathionylation. In the present study, we examined the effect of diamide, and the model oxidant hydrogen peroxide (H2O2), on oscillations in [Ca2+]i in fura-2-loaded bovine (BAECs) and human (HAECs) aortic endo-thelial cells using time-lapse fluorescence video microscopy. In the absence of extracellular Ca2+, acute treatment with either diamide or H2O2 increased the number of BAECs exhibiting asynchronous Ca2+ oscillations, whereas HAECs were unexpectedly resistant. Diamide pretreatment increased the sensitivity of HAECs to histamine-stimulated Ca2+ oscillations and BAECs to bradykinin-stimulated Ca2+ oscillations. Moreover, in both HAECs and BAECs, diamide dramatically increased both the rate and magnitude of the thapsigargin-induced Ca2+ transient suggesting that Ca2+-induced Ca2+ release (CICR) via the IP3R is enhanced by glutathionylation. Similar to diamide, H2O2 increased the sensitivity of HAECs to both histamine and thapsigargin. Lastly, biochemical studies showed that glutathionylation of native IP3R1 is increased in cells challenged with H2O2. Collectively our results reveal that thiol-oxidizing agents primarily increase the sensitivity of the IP3R to Ca2+, i.e. enhanced CICR, and suggest that glutathionylation may represent a fundamental mechanism for regulating IP3R activity during physiological redox signalling and during pathologicalical oxidative stress.
Key points
In non-excitable cells, oxidative stress increases inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) activity, which can cause Ca2+ oscillations under basal conditions and enhance agonist-stimulated changes in cytosolic free Ca2+ concentration.
Protein S-glutathionylation, the reversible modification of cysteine thiols by glutathione, is elevated in response to oxidative stress, but the consequence of glutathionylation for IP3R function is not known.
In this study we provide evidence that Ca2+-induced Ca2+-release (CICR) via the IP3R is enhanced by oxidant-induced glutathionylation in cultured aortic endothelial cells.
Our results suggest glutathionylation may represent a fundamental mechanism for regulating IP3R activity during physiological redox signalling and during pathological oxidative stress.
Introduction
In most non-excitable cells, including vascular endo-thelial cells, the generation and propagation of oscillations in cytosolic free Ca2+ concentration ([Ca2+]i) are predominantly driven by the release of Ca2+ from the endoplasmic reticulum (ER) via the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R; for reviews see Bootman & Berridge, 1995; Berridge et al. 2000; Foskett et al. 2007). At the cellular level, signal transduction commonly involves not just the generation of Ca2+ oscillations, but rather changes in both the frequency and the amplitude of the oscillatory response (Parekh, 2011), both of which are controlled to a great extent by the integrated regulation of the IP3R by IP3 and Ca2+. Although the underlying molecular mechanism is quite complex (Marchant & Taylor, 1997; Mak et al. 1998), both IP3 and Ca2+ are required for channel activation (Iino, 1990; Bezprozvanny et al. 1991). Furthermore, the interplay between IP3 and Ca2+ is critical to the ‘all-or-nothing’ generation of a Ca2+ oscillation. At a fixed low concentration of Ca2+, IP3 increases IP3R channel open probability which initiates IP3-induced Ca2+ release (IICR). However, in the presence of a fixed low concentration of IP3, Ca2+ activates the IP3R through a process called Ca2+-induced Ca2+ release (CICR). This feed-forward activation of the IP3R by Ca2+ plays a critical role in the rising phase of a global Ca2+ oscillation. The effect of cytosolic Ca2+, however, is biphasic as higher concentrations of Ca2+ inhibit channel activity. This feedback inhibition helps terminate Ca2+ release and contributes to the falling phase of a Ca2+ oscillation. Precise control of Ca2+ release via the IP3R is further complicated at the cellular and tissue level by a differential distribution of IP3R isoforms (Types 1–3) which exhibit different sensitivities to IP3 and Ca2+ (Tu et al. 2005a,b).
Although the IP3R is typically activated by a rise in IP3 following receptor-initiated hydrolysis of phosphoinositol-4,5-bisphosphate (PIP2) by phospholipase C (PLC; Berridge, 1993), Ca2+ release from IP3-sensitive stores is reported to occur in the absence of an increase in IP3 formation during periods of oxidative stress (Rooney et al. 1991; Bird et al. 1993). In non-excitable cells, including endothelial cells, reactive oxygen species and reactive nitrogen species (ROS/RNS), as well as a variety of pharmacological oxidants, cause an increase in Ca2+ oscillations under basal conditions and enhance agonist-stimulated changes in [Ca2+]i. Oxidant-induced changes in IP3R activity can be reversed by reducing agents such as dithiothreitol (DTT), suggesting that alterations in thiol chemistry are responsible for this enhanced Ca2+ response (Bootman et al. 1992; Bird et al. 1993). Furthermore, studies examining the effects of oxidizing reagents on purified IP3Rs indicate that this enhanced activation by IP3 reflects direct modification of the channel protein (Kaplin et al. 1994; Thrower et al. 1996; Poirier et al. 2001). In this regard, a number of residues have been identified on the IP3R as potential sites of redox modification (Joseph et al. 2006; Kang et al. 2008). However, despite the general consensus that oxidative stress sensitizes the IP3R to activation by IP3, the molecular basis for this response remains unknown.
Protein S-glutathionylation, the reversible modification of cysteine (Cys) thiol groups by glutathione (Dalle-Donne et al. 2008; Mieyal et al. 2008), represents a potential mechanism for regulating IP3R activity during physiological redox signalling and during periods of oxidative stress. Glutathione is the most abundant reducing equivalent in mammalian cells and, along with its associative enzyme networks, serves as the primary cellular antioxidant defence system (Meister & Anderson, 1983). In endothelial cells, oxidative insults have been shown to increase the oxidation of reduced glutathione (GSH) to its disulfide (GSSG), and to promote the formation of protein–glutathione (P-SSG) mixed disulfides, i.e. glutathionylation (Schuppe et al. 1992). Redox regulated changes in Ca2+ signalling are attributed to perturbations in the cellular GSH:GSSG ratio (Elliott & Schilling, 1990; Bootman et al. 1992). Furthermore, changes in IP3R activity have been observed following exogenous application of GSSG (Renard et al. 1992; Hilly et al. 1993; Renard-Rooney et al. 1995). However, in most studies investigating oxidant-induced changes in Ca2+ signalling, oxidative insults were initiated by either endogenous generation or exogenous application of ROS/RNS. These reactive species have a variety of cellular targets including proteins, lipids and nucleic acids. Moreover, ROS/RNS can cause thiol modifications other than glutathionylation, such as sulfenation or nitrosylation. Thus, it is difficult to identify the changes in Ca2+ signalling in response to ROS/RNS that are solely attributable to protein glutathionylation.
Diamide, a cell-permeant thiol-oxidizing agent, preferentially and rapidly oxidizes intracellular GSH to GSSG and promotes the formation of P-SSG mixed disulfides without the added complexities associated with ROS/RNS (Kosower et al. 1969, 1972). Diamide is frequently used to identify protein targets of glutathionylation and to examine the subsequent effect of P-SSG formation on protein and/or cellular function (for examples see Barrett et al. 1999; Fratelli et al. 2002). Previously, we showed that low concentrations of diamide cause a dramatic increase in asynchronous single-cell Ca2+ oscillations in cultured aortic endothelial cells (Lock et al. 2011). Diamide, even at high concentrations, did not increase PIP2 hydrolysis indicating that diamide does not activate PLC. However, the diamide response was observed even in the absence of extracellular Ca2+, and was attenuated by inhibition of the IP3R with 2-APB or by inhibition of PLC by U73122 suggesting that diamide sensitizes the IP3R to basal levels of IP3. Consistent with an important role for protein S-glutathionylation, the effect of diamide on Ca2+ dynamics was reversed by DTT, and was dependent upon intracellular GSH and the capacity of the cell to regenerate GSH from GSSG. Finally, biochemical studies showed that glutathionylation of the IP3R was significantly increased following exposure of the cells to diamide. Together these results demonstrate that diamide mobilizes Ca2+ from IP3-sensitive internal Ca2+ stores and suggest that oxidant-induced glutathionylation sensitizes the IP3R. To test this hypothesis, the effect of diamide and H2O2 on IICR and CICR via the IP3R was examined in the present study in two cultured aortic endothelial cell lines of bovine and human origin (BAECs and HAECs) using single-cell Ca2+ imaging. Our results show that both diamide and H2O2 primarily increase sensitivity of the IP3R to Ca2+, i.e. enhanced CICR.
Methods
Cell culture
The isolation, culture and characterization of BAECs have been extensively described in previous reports (Colden-Stanfield et al. 1987; Schilling et al. 1988, 1989). Briefly, BAECs were cultured as monolayers on 100 mm plastic cell culture dishes in low glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing l-glutamine and supplemented with 10% heat inactivated fetal bovine serum (FBS), 15 mm Hepes, 28.6 mm sodium bicarbonate, and 1% penicillin–streptomycin–neomycin solution (PSN; Gibco). BAECs, cultured as confluent monolayers which exhibited a cobblestone appearance typical of contact-inhibited endothelial cell cultures, were used for experimentation between passages 12 and 17. HAECs were purchased from Lonza, and cultured on 100 mm plastic dishes in endothelial cell basal medium-2 (EMB-2; Lonza, cat. no. CC-3156) supplemented with growth factors, cytokines, and chemicals (2% FBS, hydrocortisone, human recombinant fibroblast growth factor-B, human recombinant vascular endothelial growth factor, recombinant Long R insulin like growth factor-1, ascorbic acid, human recombinant epidermal growth factor, gentamicin sulfate/amphotericin-B, and heparin) from Lonza (SingleQuots; cat. no. CC-4176). HAECs were cultured as sub-confluent monolayers according to the supplier's instructions, and used for experimentation between passages 2 and 5.
Ca2+ imaging solutions and reagents
Normal Ca2+ buffer (Ca2+-extracellular solution (ECS)) contains (in mm): 10 Hepes, 140 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, and 10 glucose, pH 7.4 at 37°C. Zero Ca2+ buffer (zero Ca2+-ECS) contains (in mm): 10 Hepes, 140 NaCl, 5.4 KCl, 1 MgCl2, 0.3 EGTA, and 10 glucose, pH 7.4 at 37°C. Diamide (diazene dicarboxylic acid bis(N,N-dimethylamide)), 30% hydrogen peroxide (H2O2), histamine (HIST), bradykinin (BK), poly-d-lysine (PDL), thapsigargin (TG), ionomycin, DTT, and DMSO were purchased from Sigma-Aldrich. Ryanodine (Ryn) and bafilomycin were from Calbiochem, and xestospongin C (XeC) was from Cayman Chemicals. Fura-2 acetoxymethyl ester (fura-2/AM), and pluronic F-127 were obtained from Invitrogen. Fura-2/AM was reconstituted using DMSO and 10% pluronic F-127 at a 1:1 ratio to yield a 1 mm stock solution. Diamide, H2O2, and ryanodine stock solutions were prepared in aqueous solution, whereas TG, ionomycin, bafilomycin, and XeC were reconstituted in 100% ethanol. All stock solutions were prepared at a final concentration of 10–100 mm.
Ca2+ imaging
Time-dependent changes in [Ca2+]i were measured in both HAEC and BAEC monolayers, as previously described (Goel & Schilling, 2010). Briefly, cells were cultured on 12 mm glass coverslips for 2–5 days prior to experimentation. For HAECs, coverslips were charged with 1 mg ml−1 PDL to improve cell attachment. Oxidant and agonist responses did not differ in HAECs cultured on PDL-coated coverslips in comparison to cells grown on untreated coverslips. BAECs and HAECs were loaded for 30 min at 37°C in normal Ca2+ containing solution (Ca2+-ECS) with 6 μm and 3 μm fura-2/AM, respectively. Cells were then washed for 30 min at room temperature in Ca2+-ECS, and mounted in a temperature-controlled perfusion chamber on the stage of a Leica DMIRE2 inverted microscope. All recordings were made in the absence of extracellular Ca2+ (zero Ca2+-ECS) to specifically monitor changes in [Ca2+]i related to the release of Ca2+ from internal stores. Solutions were perfused into the recording chamber via an in-line heater, and all fura-2 imaging experiments were performed at 37°C. At 6 s intervals, excitation wavelength alternated between 340 and 380 nm and emission was recorded at 510 nm using filters appropriate for fura-2. Epifluorescence was recorded using a SPOT-RT camera (Diagnostic Instruments, Sterling Heights, MI, USA) and images were acquired and analysed using SimplePCI imaging software (Compix Inc., Cranberry Township, PA, USA).
Data analysis
Over the course of these experiments, slight variation in the sensitivity of HAECs and BAECs to chemical oxidants and receptor agonists were noted. For this reason, controls were always performed in parallel for each experimental protocol. For example, the dose-dependent effect of histamine on Ca2+ oscillations in untreated HAECs is reported in Figs 2 and 9; these represent independent data sets. Traces show [Ca2+]i responses from individual cells as greyscale and black lines. A range of 24–53 HAECs and 46–93 BAECs were monitored per experiment. The difference in the range of cells monitored per experiment in HAECs versus BAECs reflects the different density in which the cell lines are maintained in culture, i.e. ∼70% confluent versus fully confluent, respectively. The number of cells oscillating in response to oxidant or agonist challenge was quantified for each individual experiment as the percentage of the total cells monitored exhibiting at least one [Ca2+]i oscillation during the indicated treatment period. Histograms and individual data points generating agonist dose–response curves represent the average values from multiple monolayers under each condition reported as means ± SEM with n equal to the number of monolayers examined under each condition. The total number of cells and the total number of monolayers tested for each condition is given in the figure legend. Statistical comparison of means was performed using Student's paired t test or ANOVA with post hoc Tukey's test for pairwise comparisons; a P value <0.05 was considered to be statistically significant. The phenotypic shift in the TG response depicted in Figs 5–7, and 10, and Figs S2 and S3 was quantified by cumulative frequency analysis of the peak change in fura-2 fluorescence ratio (peak ratio), and the length of time from TG exposure to the peak change (latency to peak). Differences were identified using the Kruskal–Wallis test and post hoc pairwise comparisons were made using the Mann–Whitney U test with Bonferonni's correction for multiple comparisons; P < 0.01 was considered significant.
Figure 2. Diamide sensitizes HAECs to histamine-stimulated Ca2+ oscillations.
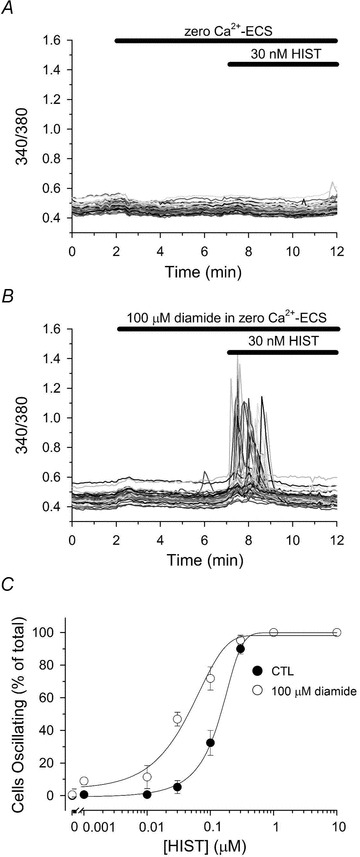
Fura-2-loaded HAECs in zero Ca2+-ECS were left untreated (A) or treated for 5 min with 100 μm diamide (B) prior to challenging with 30 nm histamine (HIST), as indicated by the bars at the top of each panel. C, HAECs were left untreated (filled circles) or treated with 100 μm diamide (open circles) for 5 min prior to stimulation with the indicated [HIST] for an additional 5 min in the continued absence or presence of 100 μm diamide. The number of cells oscillating was quantified as the percentage of the total cells monitored exhibiting at least one oscillation in [Ca2+]i during the 5 min exposure to HIST. Values represents the means ± SEM of 3–5 experiments for each experimental condition. In untreated controls, and total of 156, 129, 153, 177, 218, 165, 137 and 115 cells were analysed for each [HIST] (0, 0.001, 0.01, 0.03, 0.1, 0.3, 1.0 and 10 μm, respectively), whereas 155, 133, 165, 172, 206, 143, 124 and 109 cells were analysed for each [HIST] in diamide-treated monolayers.
Figure 9. H2O2 sensitizes HAECs to histamine-stimulated Ca2+ oscillations.
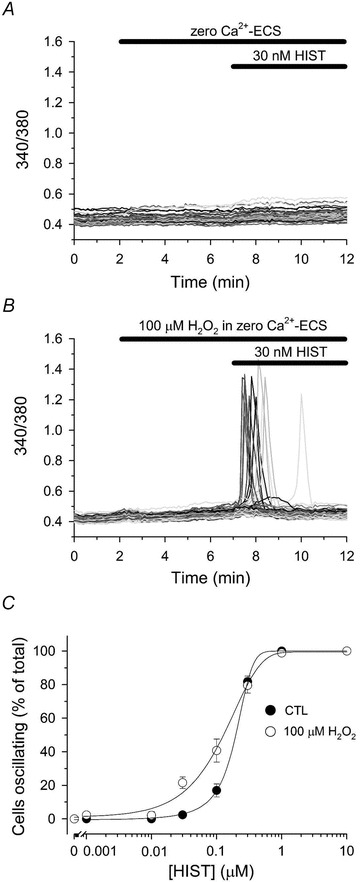
Fura-2-loaded HAECs in zero Ca2+-ECS were left untreated (A) or treated for 5 min with 100 μm H2O2 (B) prior to challenging with 30 nm histamine (HIST) for an additional 5 min, as indicated by the bars at the top of each panel. C, HAECs were left untreated (filled circles) or treated with 100 μm H2O2 (open circles) for 5 min prior to stimulation with the indicated [HIST] for an additional 5 min in the continued absence or presence of H2O2. The number of cells oscillating in response to HIST stimulation was quantified as described in the legend of Fig. 2. Values represent the mean ± SEM of 3–6 experiments for each experimental condition. In untreated controls, a total of 117, 157, 155, 133, 208, 225, 94 and 104 cells were analysed for each [HIST] (0, 0.001, 0.01, 0.03, 0.1, 0.3, 1.0 and 10 μm, respectively), whereas 116, 95, 141, 135, 225, 171, 97 and 104 cells were analysed for each [HIST] in H2O2-treated monolayers.
Figure 5. Diamide enhances thapsigargin-mediated changes in [Ca2+]i of HAECs.
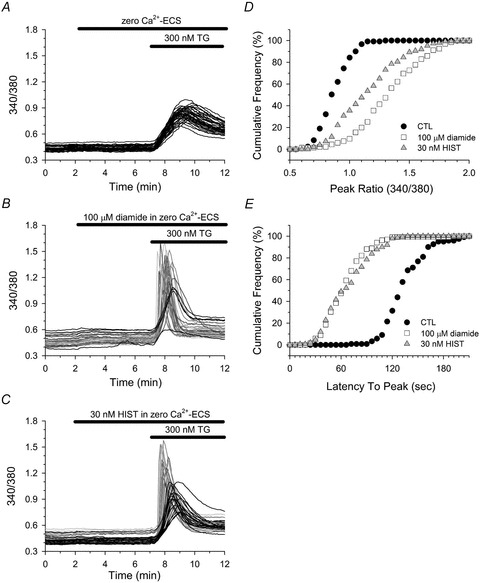
Fura-2-loaded HAECs in zero Ca2+-ECS were left untreated (A), or treated with either 30 nm HIST (C) or with 100 μm diamide (B) for 5 min immediately prior to challenging with 300 nm thapsigargin (TG). A–C, cells were segregated into two phenotypic profiles based on both the rate and the magnitude of the TG-mediated change in [Ca2+]i. Cells represented by black lines displayed a slow rate of rise and a small peak change in [Ca2+]i, whereas cells represented by greyscale lines exhibited an enhanced response characterized by a rapid rate of rise and a large change in [Ca2+]i. D, cumulative frequency of the maximal change in fura-2 fluorescence (peak ratio) observed in response to TG in control (filled circles), diamide (open squares) and HIST (grey triangles) treated cells. E, cumulative frequency of the time from TG exposure to the maximal change in fura-2 fluorescence (latency to peak) for control (filled circles), diamide (open squares), and HIST (grey triangles) treated cells. A total of 121 (control), 135 (diamide) and 149 (HIST) cells were analysed from 4 experiments for each experimental condition. D and E, the TG-induced peak ratio and latency to peak for both diamide- and HIST-treated cells were significantly different from untreated controls; P < 0.001.
Figure 7. Diamide stimulates Ca2+ oscillations in response to 3 nm TG in HAECs.
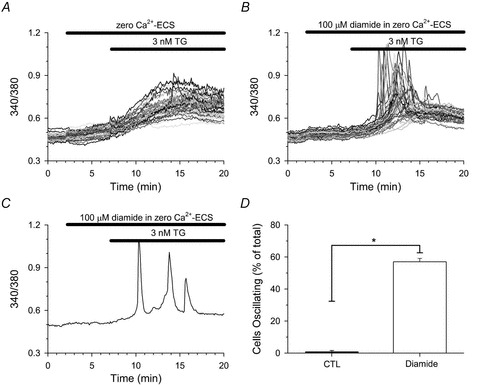
Fura-2 loaded HAECs in zero Ca2+-ECS were left untreated (A) or treated for 5 min with 100 μm diamide (B) prior to stimulation with 3 nm TG in the continued absence or presence of diamide, as indicated by the bars at the top of each panel. C, a representative single-cell trace demonstrating multiple Ca2+ oscillations in response to 3 nm TG in the presence of diamide. D, the number of cells oscillating in response to TG was quantified for each individual experiment as the percentage of the total cells monitored exhibiting at least one oscillation in [Ca2+]i during the 13 min treatment period. A total of 101 (control) and 112 (diamide) cells were analysed for each experimental condition. Values represent the mean ± SEM of 3 experiments per condition. *P < 0.001 compared to paired controls.
Figure 10. H2O2 enhanced TG-mediated changes in [Ca2+]i of HAECs.
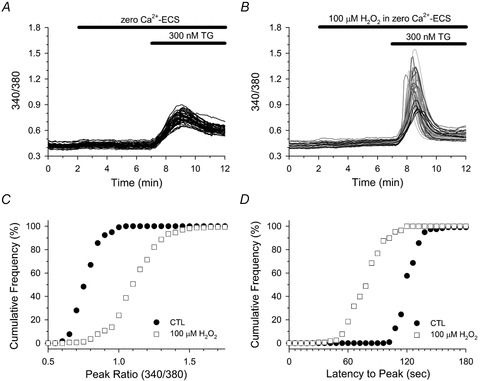
Fura-2-loaded HAECs in zero Ca2+-ECS were left untreated (A), or treated with 100 μm H2O2 (B) for 5 min immediately prior to challenging with 300 nm TG. A and B, individual cell traces are segregated into two phenotypic profiles as described in the legend of Fig. 5. C and D, cumulative frequency analysis of the peak ratio (C) and the latency to peak (D) in response to TG in control (filled circles) and H2O2 (open squares) treated cells. A total of 118 (control) and 168 (H2O2) cells were analysed from 3–4 experiments for each experimental condition. C and D, the TG-induced peak ratio and latency to peak of H2O2-treated cells was significantly different from untreated controls; P < 0.001.
Synthesis of BioGEE
GSH ethyl ester was labelled with sulfo-link- NHS-LC-biotin (Pierce) as previously described (Figtree et al. 2009). Essentially, the reaction was performed by combining 10 mm GSH ethyl ester with 10 mm of biotin reagent in phosphate buffered saline (PBS, pH 8.0). After 1 h at room temperature, 50 mm Tris was added to remove any excess sulfo-link reagent.
Isolation of membranes from BAECs
Membranes were isolated as previously described (Sinkins et al. 2009). Briefly, confluent BAEC monolayers were harvested from the culture dishes by scrapping, centrifuged at 500 g for 5 min, and resuspended in lysis buffer containing 20 mm Tris-Cl, 5 mm EDTA, 1 mm EGTA, and a protease inhibitor mixture. The cell suspension was sonicated on ice, three times for 10 s with a 10 s rest between pulses, using a sonic dismembranator (Fisher) on a power setting of 2.5. The cell lysate was centrifuged at 6000 g for 10 min at 4 °C, and the pellet was discarded. The remaining supernatant was subjected to additional centrifugation at 50,000 g for 30 min, and the resulting microsomal pellets were resuspended in lysis buffer at a protein concentration of 5–10 mg ml−1.
Glutathionylation of IP3R1 in vivo
BAECs, harvested by scraping, were washed and re-suspended in 1.5 ml Ca2+-ECS and incubated with 500 μm BioGEE for 180 min at 37°C. BioGEE-loaded cells were washed with Ca2+-ECS and either left untreated or subjected to oxidative challenge with either 0.1 mm H2O2, or 1 mm H2O2 for 10 min at room temperature. Following treatment, the cells were washed with Ca2+-ECS and membrane preparations were generated as described above. For each sample, membrane preparations were divided into two aliquots; one aliquot from each sample was incubated with 20 mm DTT. Membrane aliquots were then solubilized in PBS containing 1% Triton X-100 for 30 min on ice. Following solubilization membrane lysates were cleared by centrifugation, and biotinylated proteins were extracted with streptavidin-agarose beads by overnight incubation at 4°C.
Immunoblots
Following in vivo glutathionylation reactions, proteins captured on streptavidin–agarose beads were fractionated by SDS-PAGE and electrotransferred to PVDF membrane (100 V for 1 h) in CAPS/methanol buffer. Blots were probed with anti-IP3 receptor type 1 antibody (Millipore; cat. no. 07-514) and detected, following incubation with HRP-conjugated IgG, by SuperSignal West Pico chemiluminescent substrate (Pierce).
Results
Effect of diamide on [Ca2+]i in cultured aortic endothelial cells
The acute effect of diamide on [Ca2+]i of two independent cultured endothelial cell lines – bovine (BAECs) and human (HAECs) aortic endothelial cells – was investigated by single-cell imaging of fura-2-loaded monolayers. Previously, we reported that diamide produced a concentration-dependent increase in asynchronous Ca2+ oscillations in BAECs by stimulating the release of Ca2+ from IP3-sensitive stores (Lock et al. 2011). HAECs, however, were unexpectedly resistant to diamide-induced oscillations in [Ca2+]i. As shown in Fig. 1A and quantified in Fig. 1C, HAECs challenged with 100 μm diamide exhibited Ca2+ oscillations in only 1.5 ± 0.9% (mean ± SEM) of the total cells monitored, whereas oscillations were not observed in paired controls. Challenge with increasing concentrations of diamide (up to 1.0 mm) had no effect on the number of HAECs responding (data not shown). In comparison, BAECs challenged with 100 μm diamide displayed a significant increase in the number of cells exhibiting asynchronous Ca2+ oscillations when compared to paired untreated controls (Fig. 1B and quantified in Fig. 1C).
Figure 1. Effect of diamide on [Ca2+]i in HAECs and BAECs.
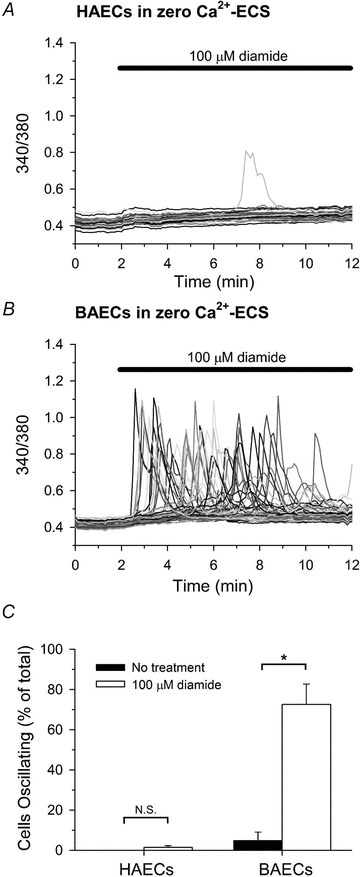
Fluorescence ratio (340/380) was recorded from single fura-2-loaded HAECs (A) and BAECs (B) bathed in zero Ca2+-ECS during treatment with 100 μm diamide, as indicated at the top of each panel. In this and all subsequent figures, a representative experiment (i.e. a single coverslip) is shown with individual cells depicted as different grey scale traces. C, The number of cells oscillating in response to diamide was quantified for each individual experiment as the percent of the total cells monitored exhibiting at least one oscillation in [Ca2+]i during the 10 min treatment period. A total of 118 (untreated) and 133 (100 μm diamide) HAECs, and 231 (untreated) and 202 (100 μm diamide) BAECs were analysed per experimental condition. Values represent the mean ± SEM of 3–4 experiments (coverslips) for each experimental condition. *P < 0.05 compared to paired-untreated controls.
Diamide increases the sensitivity of cultured aortic endothelial cells to receptor-stimulated Ca2+ oscillations
Our previous studies showed that the diamide-induced change in [Ca2+]i in BAECs is blocked by inhibition of PLC by U73122 or by inhibition of the IP3R by 2-APB suggesting that diamide sensitizes the IP3R to basal levels of IP3. Thus, the lack of sensitivity of HAECs to diamide may reflect the lack of sufficient IP3 to initiate an oscillation under resting conditions. If this is true, we reasoned that low concentrations of diamide that have little or no effect on Ca2+ oscillations, should enhance the response to receptor agonists that generate IP3. To test this hypothesis, HAECs bathed in Ca2+ free medium, were left untreated or treated with diamide for 5 min immediately prior to stimulation with HIST in the continued absence or presence of diamide (Fig. 2). Under control conditions, 30 nm HIST had no effect on [Ca2+]i (Fig. 2A). However, pretreatment with 100 μm diamide dramatically increased in the number cells oscillating in response to 30 nm HIST (Fig. 2B). Diamide pretreatment significantly increased the number of HAECs responding to every submaximal concentration of HIST tested, whereas neither untreated cells, nor cells treated with diamide alone exhibited Ca2+ oscillations in the absence of receptor stimulation. This increased sensitivity to HIST-stimulated Ca2+ oscillations is reflected in a 3- to 5-fold leftward shift in the HIST dose–response curve in HAECs pretreated with diamide relative to paired-untreated controls (Fig. 2C). In a parallel set of experiments (Fig. 3), we confirmed that this effect of diamide is related to a change in IP3R function since the response was unaffected by Ryn (Fig. 3C and D) and significantly attenuated by XeC (Fig. 3B and D).
Figure 3. Effects of diamide are attenuated by xestospongin C but not ryanodine.
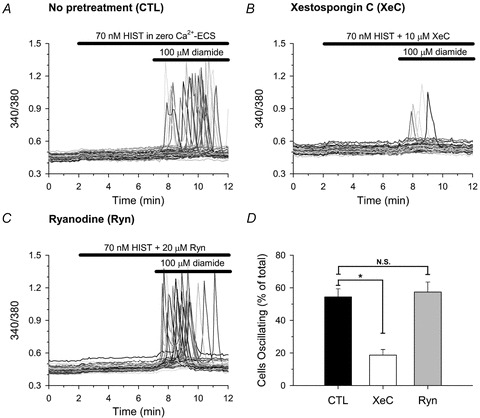
Fura-2-loaded HAECs were left untreated (A), or pretreated with either 10 μm xestospongin C (XeC) for 30 min (B), or 20 μm ryanodine (Ryn) for 10 min (C) immediately prior to sequential exposure, in zero Ca2+-ECS, to 70 nm HIST for 5 min followed by 100 μm diamide for an additional 5 min in the continued presence of HIST, as indicated by the bars at the top of each panel. All recordings in panels B and C were made in the continued presence of the indicated inhibitor. D, the number of cells oscillating in response to diamide was quantified as described in the legend to Fig. 1. A total of 139 (untreated), 132 (XeC pretreated), and 118 (Ryn pretreated) cells were analysed per experimental condition. Values represents the mean ± SEM of 3 experiments for each condition. *P < 0.01 compared to paired-untreated controls.
A similar effect of diamide on BK-mediated changes in [Ca2+]i was observed in BAECs (Fig. 4). Cells, which did not respond to 100 pm BK under control conditions (Fig. 4A), exhibited a robust Ca2+ response to 100 pm BK when briefly pretreated with 20 μm diamide, a threshold concentration that has only modest affects on Ca2+ oscillations (Fig. 4B). Once again, diamide pretreatment significantly increased the number of cells oscillating in response to every submaximal agonist concentration tested, and altogether produced a 2- to 3-fold leftward shift in the BK dose–response relationship when compared to paired-untreated controls (Fig. 4C). Since BAECs do not express Ryn receptors (Schilling & Elliott, 1992), collectively these results demonstrate that diamide increases the sensitivity of cultured aortic endothelial cells to receptor-stimulated Ca2+ oscillations, and suggests that P-SSG formation enhances the sensitivity of the IP3R to IICR.
Figure 4. Diamide sensitizes BAECs to bradykinin-stimulated Ca2+ oscillations.
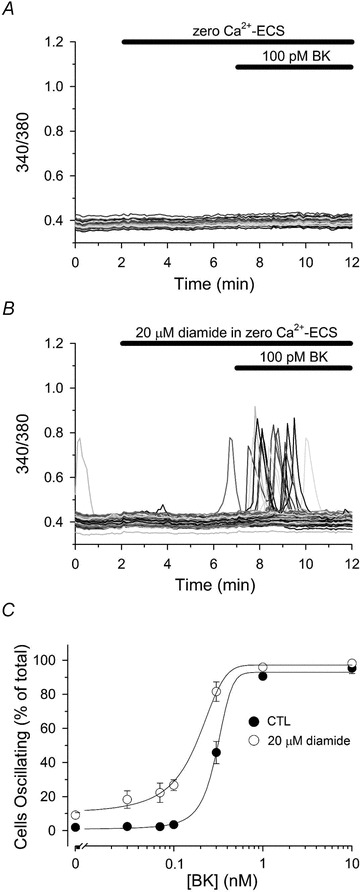
Fura-2-loaded BAECs in zero Ca2+-ECS were left untreated (A) or treated for 5 min with 20 μm diamide (B) prior to challenging with 100 pm bradykinin (BK), as indicated by the bars at the top of each panel. C, BAECs were left untreated (filled circles) or treated with 20 μm diamide (open circles) for 5 min prior to stimulation with the indicated [BK] for an additional 5 min in the continued absence or presence of diamide. Values represent the mean ± SEM of 3–8 experiments for each experimental condition. In untreated controls, a total of 401, 371, 530, 468, 345, 193 and 196 cells were analysed for each [BK] (0, 0.03, 0.07, 0.1, 0.3, 1.0 and 10 nm, respectively), whereas 340, 329, 436, 471, 316, 225, 180, cells were analysed for each [BK] in diamide-treated monolayers.
Diamide enhances thapsigargin-mediated changes in the [Ca2+]i of cultured aortic endothelial cells
Inhibition of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) pump by thapsigargin (TG) is a commonly used method for increasing [Ca2+]i in the absence of receptor stimulation and in the absence of phosphoinositide hydrolysis (Lytton et al. 1991). It is thought that TG-induced rise in [Ca2+]i reflects the passive leak of Ca2+ from the ER following SERCA inhibition. However, the results of our next experiments revealed a surprising finding. As previously reported in most non-excitable cell types, in the absence of extracellular Ca2+, TG elicited a near homogeneous Ca2+ response in HAECs under control conditions (Fig. 5A, black traces). The response in each cell was characterized by a relatively slow rate of rise and small peak change in [Ca2+]i. In contrast, the TG response of HAECs briefly pretreated with a low concentration of diamide could be segregated into two phenotypic profiles. The first profile was that seen in controls, i.e. slow rate of rise and low amplitude, whereas the second profile was characterized by a rapid rate to rise and large peak change in [Ca2+]i (Fig. 5B; grey-scale traces), suggesting that diamide may sensitize the IP3 receptor to CICR. If this is true we reasoned that a low concentration of HIST, which would increase IP3 but not produce oscillations, would also sensitize HAECs to TG. As seen in Fig. 5C, the TG-induced Ca2+ release in HIST pre-treated cells was phenotypically similar both in magnitude and time course to that observed in the diamide-treated group, consistent with enhanced CICR. In HAECs, TG evoked a CICR response in 87.1 ± 3.5% of the total cells pretreated with 100 μm diamide and 58.8%± 9.3% of cells pretreated with 30 nm HIST, whereas a CICR profile in response to TG was observed in only 1 out of 121 cells analysed under control conditions. The phenotypic change in the TG response (i.e. enhanced CICR) was quantified in an unbiased manner using cumulative frequency analysis of the peak ratio and the latency to peak. As seen in Fig. 5D and E, both diamide and HIST produced a significant increase in the peak ratio and a significant decrease in the latency to peak following TG exposure in HAECs.
Similar to the Ca2+ response to TG observed in HAECs, BAECs briefly pretreated with a subthreshold concentration of either diamide (Fig. 6B) or BK (Fig. 6C) also exhibited an enhanced Ca2+ response to TG. However, unlike HAECs, BAECs exhibited a heterogeneous response, consisting of both slow (black traces) and fast (grey traces) Ca2+ profiles, even under control conditions (Fig. 6A). In some cells, a rapid release of Ca2+ was observed after a slow rise in [Ca2+]i, providing further evidence that the fast response reflects all-or-nothing CICR from stores (inset Fig. 6A). Altogether, a CICR profile in response to TG was observed in 74.4 ± 4.9% of diamide-treated and 52.4%± 9.3% of BK-treated cells, whereas only 32.3%± 4.5% of untreated BAECs exhibited a CICR response to TG. As seen in Fig. 6D and E, both diamide and BK significantly increased the peak ratio and significantly decrease the latency to peak in response to TG in BAECs.
Figure 6. Diamide enhances TG-mediated changes in [Ca2+]i of BAECs.
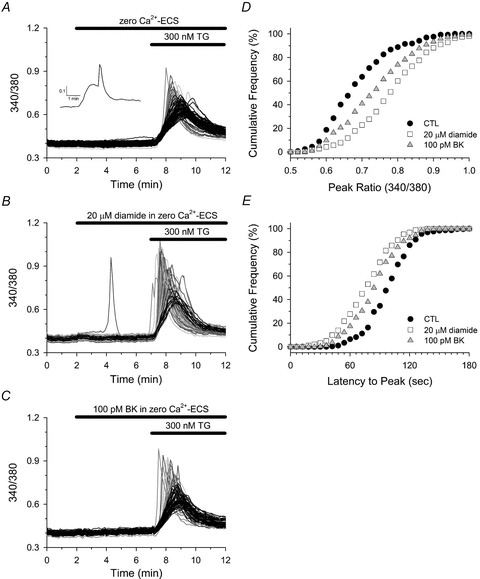
Fura-2-loaded BAECs in zero Ca2+-ECS were left untreated (A), or treated with either 100 pm BK (C) or 20 μm diamide (B) for 5 min immediately prior to challenging with 300 nm TG. A–C, individual cell traces are segregated into two phenotypic profiles as described in the legend of Fig. 5. Inset in A shows a single-cell trace exhibiting a delayed CICR response to TG that was sometimes observed. D, cumulative frequency of the peak ratio (D) and the latency to peak (E) observed in response to TG in control (filled circles), diamide- (open squares) and BK-treated (grey triangles) cells. A total of 294 (control), 281 (diamide) and 276 (BK) cells were analysed from 4 experiments for each experimental condition. D and E, the TG-induced peak ratio and latency to peak of both diamide- and BK-treated cells were significantly different from untreated controls; P < 0.001.
Diamide is expected to increase glutathionylation of a number of cellular proteins, some of which may impact Ca2+ homeostasis. For example, diamide may affect the Ca2+ buffer capacity of the cytoplasm and/or alter the Ca2+ load of the internal stores by stimulation of SERCA, both of which may contribute to the enhanced response to TG. However, at the concentrations used, diamide had no significant effect on basal [Ca2+]i or on the peak response to maximum concentrations of HIST, BK, or ionomycin (Fig. S1). Furthermore, diamide had no significant effect on the kinetics of these responses. Thus, alterations in stored Ca2+ or in the buffer capacity of the cytoplasm seem unlikely. Additionally, the diamide-induced change in TG response was unaffected by blockade of the Ryn receptor, or by inhibition of lysosmes by bafilomycin, but could be partially (but significantly) attenuated by XeC (Figs S2 and S3). To further demonstrate the impact of diamide on CICR via the IP3R, we examined a near threshold concentration of TG. As seen in Fig. 7, 3 nm TG produced a slow gradual rise in [Ca2+]i in HAECs when measured in the absence of extracellular Ca2+ under control conditions. However, in the presence of diamide, CICR was seen in 57.0 ± 2.1% of cells examined (Fig. 7B and D) and 14.1 ± 1.3% exhibited repetitive Ca2+ oscillations over the recording period (Fig. 7C). Collectively these results demonstrate IP3R activation can shape the change in [Ca2+]i following depletion of the ER Ca2+ store by a process of CICR, and suggest that diamide increases the sensitivity of the IP3R to cytosolic Ca2+. Moreover, these results indicate that the stimulatory effect of diamide on IP3R activity is down-stream of receptor activation.
Effect of H2O2 on [Ca2+]i of cultured aortic endothelial cells
To determine if a physiologically relevant oxidant produces similar changes in IP3R activity, the effect of hydrogen peroxide (H2O2) on the [Ca2+]i of HAECs and BAECs was investigated under identical conditions to those described above for diamide. In the absence of extracellular Ca2+, HAECs challenged with 100 μm H2O2 did not exhibit oscillations in [Ca2+]i, nor were Ca2+ oscillations observed in paired-untreated controls (0 ± 0%vs. 0 ± 0%, respectively; Fig. 8A and quantified in Fig. 8C). BAECs, on the other hand, exhibited a significant increase in the number of cells oscillating in response to 100 μm H2O2 when compared to paired-untreated controls (10.3 ± 0.8%vs. 1.2 ± 0.7%, respectively; Fig. 8B and quantified in Fig. 8C).
Figure 8. Effect of H2O2 on [Ca2+]i in HAECs and BAECs.
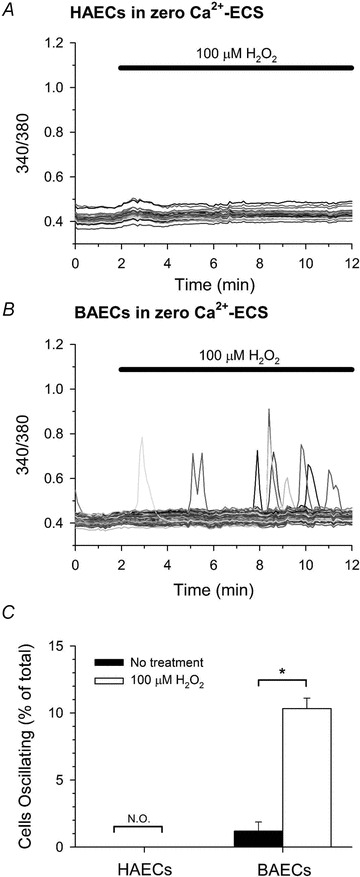
Fura-2 fluorescence ratio was recorded from single HAECs (A) and BAECs (B) in zero Ca2+-ECS during treatment with 100 μm H2O2, as indicated at the top of each panel. C, the number of cells oscillating in response to H2O2 was quantified as described in the legend of Fig. 1. A total of 111 (untreated) and 151 (H2O2) HAECs, and 217 (untreated) and 248 (H2O2) BAECs were analysed. Values represent the mean ± SEM of 3–4 experiments for each experimental condition. *P < 0.001 compared to paired-untreated controls; N.O., not observed.
H2O2 increases the sensitivity of HAECs to HIST- and TG- stimulated changes in [Ca2+]i
To determine if H2O2-induced Ca2+ oscillations reflect an increased sensitivity of the IP3R, the effect of H2O2 on HIST- and TG-mediated changes in [Ca2+]i was examined in HAECs. Similar to the effects seen with diamide, brief pretreatment of HAECs with 100 μm H2O2 significantly increased the number of cells oscillating in response to a submaximal concentration of HIST, and altogether resulted in a 2- to 3-fold shift in the HIST dose–response relationship when compared to paired-untreated controls (Fig. 9). Again, analogous to effect of diamide, HAECs pretreated with H2O2 exhibited an enhanced response to TG, indicative of IP3R activation by CICR, when compared to paired-untreated controls (Fig. 10). Collectively, these results demonstrate that H2O2, like diamide, can increase the sensitivity of the IP3R to activation by IICR and CICR.
Oxidative stress promotes glutathionylation of the IP3R1 in vivo
Despite substantial evidence that thiol-oxidizing agents directly modify the IP3R, the molecular nature of the modification(s) responsible for changes in IP3R activity are still not well understood. The type 1 IP3R (IP3R1) is expressed in vascular endothelial cells (Grayson et al. 2004) and we previously reported that glutathionylation of native type 1 IP3R (IP3R1) is increased in BAECs treated with diamide (Lock et al. 2011). To determine if glutathionylation occurs in response to a physiological oxidant, the effect of H2O2 on glutathionylation of native IP3R1 was investigated in BAECs loaded with biotin-GSH ethyl ester (BioGEE). BioGEE is a membrane-permeant form of biotin-labelled GSH that is trapped within the cell by the action of cellular esterases. Following 10 min treatment with H2O2, glutathionylated proteins from BioGEE-loaded cells were captured using streptavidin–agarose beads and probed for IP3R1 by Western blot as described in Methods. As seen in Fig. 11, IP3R1 glutathionylation was increased in response to H2O2. Consistent with thiol modification, oxidant-induced glutathionylation of the IP3R was reversed by addition of excess DTT prior to avidin pull-down. These results suggest that glutathionylation of IP3R1 may be a common response to oxidative stress.
Figure 11. Glutathionylation of native IP3R1 in vivo.
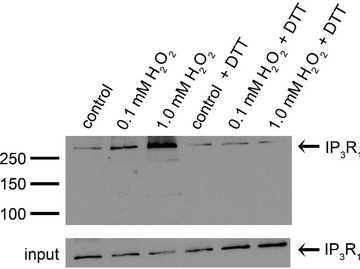
BioGEE-loaded BAECs were suspended in Ca2+-ECS and cells were divided equally into aliquots as indicated above each lane. Cells were left untreated (control) or incubated for 10 min with 0.1 mm or 1.0 mm H2O2. Following treatment, the membrane lysates from one sample of each of the indicated conditions tested was incubated with 20 mm DTT prior to pull-down. Biotinylated proteins were extracted from cleared lysates with avidin–agarose beads and probed for the IP3R1 by Western Blot. Aliquots of cell lysates prior to pull-down are shown as input controls. Result shown is representative of 3 independent experiments.
Discussion
In the present study, we utilized the thiol-oxidant diamide to investigate the consequences of protein S-glutathionylation on IP3R function in intact cultured aortic ECs. Diamide reacts with GSH and promotes P-SSG formation in a well-established two step reaction (Kosower et al. 1969; Kosower & Kosower, 1995). Intracellularly, diamide rapidly and preferentially reacts with GSH producing a diamide-GS intermediate which can either react with another molecule of GSH (producing GSSG) or react with a protein thiol (P-SH) giving rise to P-SSG. Protein de-glutathionylation is achieved primarily through the enzymatic action of glutaredoxin, which uses GSH to produce P-SH and GSSG (Shelton et al. 2005). Reduced GSH is regenerated from GSSG by glutathione reductase at the expense of NADPH. Since the concentration of GSH in the cytosol is generally 1–10 mm (Meister & Anderson, 1983), low concentrations of diamide (e.g. 20–100 μm) are thought to shift the steady-state equilibrium in favour of P-SSG. Previously we found that diamide produced asynchronous Ca2+ oscillations in BAECs (Lock et al. 2011). This effect of diamide was prevented by inhibition of PLC or by blockade of the IP3R, but diamide did not increase hydrolysis of PIP2. Together these results suggested that diamide sensitizes the IP3R to basal levels of IP3. However, the extent to which this reflects enhanced sensitivity to IP3 or to cytosolic Ca2+ is difficult to distinguish at the cellular level. Since IP3 is not changing during challenge with diamide, an increase in Ca2+ oscillations likely reflects an increased sensitivity of the IP3R to Ca2+, i.e. enhanced CICR. The results of the present study demonstrate that CICR is indeed enhanced following diamide treatment. Furthermore, we also found that exogenous application of H2O2 produced similar changes in IP3R activity and, like diamide, H2O2 increased glutathionylation of native IP3R1 in BAECs. Although an increased sensitivity of the IP3R to IP3 in response to oxidative stress has been reported previously (Rooney et al. 1991; Bird et al. 1993), to our knowledge this is the first demonstration that thiol-oxidizing agents enhance the sensitivity of the IP3R to CICR. This may reflect a direct modification of the IP3R by glutathionylation.
The mobilization of intracellular Ca2+ stores in response to thiol-oxidizing agents has been reported in a wide range of non-excitable cell types including hepatocytes (Rooney et al. 1991; Bird et al. 1993), Hela cells (Bootman et al. 1992), hamster eggs (Miyazaki et al. 1992), pancreatic acinar cells (Klonowski-Stumpe et al. 1997), platelets (vanGorp et al. 1997), and aortic ECs (Hu et al. 1998). Although H2O2 was shown to stimulate single-cell Ca2+ oscillations in cultured HAECs in a prior study, in our hands neither H2O2, nor diamide evoked an oscillatory response in HAECs under basal conditions. In contrast, both diamide and H2O2 were effective in eliciting Ca2+ oscillations in BAECs. These results are reminiscent of a study by Bird et al., in which they observed Ca2+ oscillations in response to tert-butyl hydroperoxide (t-BOOH) in primary rat hepatocytes, but not in hepatocytes isolated from guinea pig (Bird et al. 1993). In their study, if the intracellular IP3 was elevated by a low concentration of receptor agonist or by microinjection of IP3, t-BOOH-induced Ca2+ oscillations were then observed in guinea pig hepatocytes. Similarly, we found that brief (5 min) pretreatment of either HAECs or BAECs with diamide, at a concentration which did not stimulate Ca2+ oscillations, significantly increased the number of cells oscillating in response to either HIST or BK, respectively. Moreover, H2O2 pretreatment increased the sensitivity of HAECs to HIST-induced oscillations. Since both HIST and BK initiate Ca2+ oscillations by activating PLC and elevating IP3, both diamide and H2O2 may decrease the threshold for activation of the IP3R by IP3. However, since a local rise in [Ca2+]i in response to IP3 binding can stimulate Ca2+ release through a feed-forward process involving CICR, the increased IP3R activity during an oxidative challenge could also reflect changes in the Ca2+ sensitivity of the receptor.
It is well established that IP3R activity is regulated by cytosolic Ca2+ (Taylor, 1998). In both single channel studies (Bezprozvanny et al. 1991; Tu et al. 2005a; Ionescu et al. 2006) and permeabilized cell models (Iino, 1990; Marshall & Taylor, 1993; Bootman et al. 1995; Marchant & Taylor, 1997) low concentrations of cytosolic Ca2+ are required for IP3-mediated activation of the IP3R, and have been shown to stimulate IP3R activity at a fixed concentration of IP3. Moreover, increasing [Ca2+]i has been shown to augment IP3 binding to the IP3R (Hilly et al. 1993; Cardy et al. 1997). However, despite a clear role for Ca2+ in the regulation of IP3R activity, the effect of thiol-oxidants on the Ca2+ sensitivity of the IP3R has not been investigated. We took advantage of the SERCA inhibitor TG to address whether changes in [Ca2+]i can influence IP3R activity during an oxidative challenge in intact cells. TG-mediated inhibition of SERCA leads to a transient rise in [Ca2+]i due to passive leak of Ca2+ from the ER (Lytton et al. 1991). Our results reveal that both diamide and H2O2 increased the sensitivity of the IP3R to a rise in [Ca2+]i induced by TG. This increased sensitivity toward cytosolic Ca2+ resulted in a rapid Ca2+ transient when cells were challenged with TG suggestive of CICR via the IP3R. Moreover, regenerative Ca2+ oscillations, a well-established characteristic of IP3-sensitive stores, were observed in response to near threshold concentrations of TG in diamide-treated cells. Concordant with a role for the IP3R in the Ca2+ response to TG, low concentrations of HIST or BK produced a similar shift in the Ca2+ profile following TG exposure. A role for CICR via the IP3R in shaping the kinetics of the Ca2+ transient induced by TG is consistent with the observations that TG-mediated changes [Ca2+]i are dependent upon the basal level of IP3 (Smith & Gallacher, 1994), and can be attenuated by the IP3R blocker 2-APB (Luo et al. 2001). Taken together, our results suggest thiol-oxidizing agents increase the sensitivity of the IP3R to activation by cytosolic Ca2+. Additionally, our results demonstrate that the stimulatory effects of both diamide and H2O2 on IP3R activation are downstream of receptor stimulation and are independent of PLC activity and IP3 formation.
Oxidant-induced changes in IP3R activity are thought to reflect a direct thiol modification of the receptor/channel. Functional IP3Rs contain over 200 Cys residues (∼60 Cys per monomer), and although a number of Cys have been identified as potential sites of redox modification (Joseph et al. 2006; Kang et al. 2008), a role for glutathionylation has not been defined. Our results demonstrate that, in addition to diamide (Lock et al. 2011), glutathionylation of native IP3R1 in cultured aortic ECs occurs in response to H2O2. The increase in IP3R glutathionylation in response to thiol-oxidizing agents may be indirect, and we cannot unequivocally exclude the possibility that glutathionylation of a tightly bound accessory/regulatory protein is responsible for the increase in IP3R1 detected by Western blot following avidin pull-down. However, given the evidence that thiol-oxidizing agents can directly modify the IP3R (Kaplin et al. 1994; Thrower et al. 1996; Poirier et al. 2001), a direct modification by glutathionylation seems plausible. In preliminary studies using BAECs permabilized with saponin, we found that IP3R1 is not glutathionylated in its cytoplasmic domain (unpublished observation) whereas robust glutathionylation of IP3R1 was observed in isolated BAEC microsomes under similar conditions (Lock et al. 2011). This would suggest that the IP3R1 is glutathionylated in an ER luminal domain. The IP3R monomer has six membrane spanning segment; the NH2- and COOH-termini are cytoplasmic and there are three luminal loops, L1–L3. L3 has four Cys residues that are conserved in type-1, -2, and -3 IP3R. Previous studies by Mikoshiba's group showed that the ER resident protein, ERp44, binds tightly to the type-1 receptor, but not to type-2 or type-3 (Higo et al. 2005). Binding of ERp44 to the IP3R is sensitive to redox potential and is favoured under reducing conditions and inhibited under oxidizing conditions. Binding of ERp44 to the IP3R requires reduced Cys residues, and binding inhibits channel activity. Importantly, oxidized conditions will also favour protein S-glutathionylation. Thus, glutathionylation of one or more of these Cys residues may block interaction of ERp44 with IP3R and hence prevent inhibition of the channel activity by ERp44. Further studies will be necessary to determine the role of these residues in IP3R glutathionylation and the subsequent effect on IP3R function.
Acknowledgments
This work was supported by grant HL097355, and J.T.L. was supported in part by training grant HL007887, both from the National Heart Lung and Blood Institute (USA).
Glossary
- 2-APB
2-aminoethyl diphenyl borate
- BAEC
bovine aortic endothelial cell
- BK
bradykinin
- BioGEE
biotinylated glutathione ethyl ester
- CICR
Ca2+-induce Ca2+ release
- Cys
cysteine
- EC
endothelial cell
- ECS
extracellular solution
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HAEC
human aortic endothelial cell
- HIST
histamine
- H2O2
hydrogen peroxide
- IP3R
inositol 1,4,5-trisphosphate receptor
- IICR
IP3-induced Ca2+ release
- PLC
phospholipase C
- P-SH
protein-thiol
- P-SSG
protein-S-glutathione
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Ryn
ryanodine
- TG
thapsigargin
- XeC
xestospongin C
Author contributions
All experiments were performed in the laboratory of W.P.S. Conception and design of research by J.T.L., W.G.S. and W.P.S. Data were collected by J.T.L. and W.G.S., and analysed by J.T.L., W.G.S. and W.P.S. The manuscript was written by J.T.L and W.P.S with assistance from W.G.S. All authors have read and approved the final version of the manuscript.
Supplementary material
FIGURE S1
FIGURE S2
FIGURE S3
References
- Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signaling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3-gated and calcium-gated channels from endoplasmic-reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bird GS, Burgess GM, Putney JW. Sulfhydryl-reagents and cAMP-dependent kinase increase the sensitivity of the inositol 1,4,5-trisphosphate receptor in hepatocytes. J Biol Chem. 1993;268:17917–17923. [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Missiaen L, Parys JB, Desmedt H, Casteels R. Control of inositol 1,4,5-trisphosphate-induced Ca2+ release by cytosolic Ca2+ Biochem J. 1995;306:445–451. doi: 10.1042/bj3060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman MD, Taylor CW, Berridge MJ. The thiol reagent, thimerosal, evokes Ca2+ spikes in Hela-cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1992;267:25113–25119. [PubMed] [Google Scholar]
- Cardy TJA, Traynor D, Taylor CW. Differential regulation of types-1 and -3 inositol trisphosphate receptors by cytosolic Ca2+ Biochem J. 1997;328:785–793. doi: 10.1042/bj3280785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial-cells. Circ Res. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Milzani A, Gagliano N, Colombo R, Giustarini D, Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal. 2008;10:445–473. doi: 10.1089/ars.2007.1716. [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Schilling WP. Carmustine augments the effects of tert-butyl hydroperoxide on calcium signaling in cultured pulmonary-artery endothelial-cells. J Biol Chem. 1990;265:103–107. [PubMed] [Google Scholar]
- Figtree GA, Liu C-C, Bibert S, Hamilton EJ, Garcia A, White CN, Chia KKM, Cornelius F, Geering K, Rasmussen HH. Reversible oxidative modification a key mechanism of Na+-K+ pump regulation. Circ Res. 2009;105:185–U187. doi: 10.1161/CIRCRESAHA.109.199547. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung K-H, Mak D-OD. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel M, Schilling WP. Role of TRPC3 channels in ATP-induced Ca2+ signaling in principal cells of the inner medullary collecting duct. Am J Physiol Renal Physiol. 2010;299:F225–F233. doi: 10.1152/ajprenal.00670.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJH, Hill CE. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium. 2004;36:447–458. doi: 10.1016/j.ceca.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell. 2005;120:85–98. doi: 10.1016/j.cell.2004.11.048. [DOI] [PubMed] [Google Scholar]
- Hilly M, Pietrirouxel F, Coquil JF, Guy M, Mauger JP. Thiol reagents increase the affinity of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1993;268:16488–16494. [PubMed] [Google Scholar]
- Hu QH, Corda S, Zweier JL, Capogrossi MC, Ziegelstein RC. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation. 1998;97:268–275. doi: 10.1161/01.cir.97.3.268. [DOI] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in smooth-muscle cells of the guinea-pig taenia ceci. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu L, Cheung K-H, Vais H, Mak D-OD, White C, Foskett JK. Graded recruitment and inactivation of single InsP3 receptor Ca2+-release channels: implications for quartal Ca2+ release. J Physiol. 2006;573:645–662. doi: 10.1113/jphysiol.2006.109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SK, Nakao SK, Sukumvanich S. Reactivity of free thiol groups in type-I inositol trisphosphate receptors. Biochem J. 2006;393:575–582. doi: 10.1042/BJ20050889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Kang J, Kwon H, Frueh D, Yoo SH, Wagner G, Park S. Effects of redox potential and Ca2+ on the inositol 1,4,5-trisphosphate receptor L3-1 loop region: implications for receptor regulation. J Biol Chem. 2008;283:25567–25575. doi: 10.1074/jbc.M803321200. [DOI] [PubMed] [Google Scholar]
- Kaplin AI, Ferris CD, Voglmaier SM, Snyder SH. Purified reconstituted inositol 1,4,5-trisphosphate receptors – thiol reagents act directly on receptor protein. J Biol Chem. 1994;269:28972–28978. [PubMed] [Google Scholar]
- Klonowski-Stumpe H, Schreiber R, Grolik M, Schulz HU, Haussinger D, Niederau C. Effect of oxidative stress on cellular functions and cytosolic free calcium of rat pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 1997;272:G1489–G1498. doi: 10.1152/ajpgi.1997.272.6.G1489. [DOI] [PubMed] [Google Scholar]
- Kosower EM, Kosower NS, Kinon BJ, Correa W. Glutathione. VII. Differentiation among substrates by thiol-oxidizing agent, diamide. Biochim Biophys Acta. 1972;264:39–44. doi: 10.1016/0304-4165(72)90114-6. [DOI] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for intracellular oxidation of glutathione to disulfide. Biochem Biophys Res Commun. 1969;37:593–596. doi: 10.1016/0006-291x(69)90850-x. [DOI] [PubMed] [Google Scholar]
- Lock JT, Sinkins WG, Schilling WP. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H493–H506. doi: 10.1152/ajpheart.01073.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Broad LM, Bird GSJ, Putney JW. Signaling pathways underlying muscarinic receptor-induced [Ca2+] i oscillations in HEK293 cells. J Biol Chem. 2001;276:5613–5621. doi: 10.1074/jbc.M007524200. [DOI] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic-reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- Mak DOD, McBride S, Foskett JK. Inositol 1,4,5-tris-phosphate activation of inositol tris-phosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci U S A. 1998;95:15821–15825. doi: 10.1073/pnas.95.26.15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant JS, Taylor CW. Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr Biol. 1997;7:510–518. doi: 10.1016/s0960-9822(06)00222-3. [DOI] [PubMed] [Google Scholar]
- Marshall ICB, Taylor CW. Biphasic effects of cytosolic Ca2+ on Ins(1,4,5)P3-stimulated Ca2+ mobilization in hepatocytes. J Biol Chem. 1993;268:13214–13220. [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S, Shirakawa H, Nakada K, Honda Y, Yuzaki M, Nakade S, Mikoshiba K. Antibody to the inositol trisphosphate receptor blocks thimerosal-enhanced Ca2+-induced Ca2+ release and Ca2+ oscillations in hamster eggs. FEBS Lett. 1992;309:180–184. doi: 10.1016/0014-5793(92)81090-9. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Decoding cytosolic Ca2+ oscillations. Trends Biochem Sci. 2011;36:78–87. doi: 10.1016/j.tibs.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Poirier SN, Poitras M, Laflamme K, Guillemette G. Thiol-reactive agents biphasically regulate inositol 1,4,5-trisphosphate binding and Ca2+ release activities in bovine adrenal cortex microsomes. Endocrinology. 2001;142:2614–2621. doi: 10.1210/endo.142.6.8195. [DOI] [PubMed] [Google Scholar]
- Renard-Rooney DC, Joseph SK, Seitz MB, Thomas AP. Effect of oxidized glutathione and temperature an inositol 1,4,5-trisphosphate binding in permeabilized hepatocytes. Biochem J. 1995;310:185–192. doi: 10.1042/bj3100185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard DC, Seitz MB, Thomas AP. Oxidized glutathione causes sensitization of calcium release to inositol 1,4,5-trisphosphate in permeabilized hepatocytes. Biochem J. 1992;284:507–512. doi: 10.1042/bj2840507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney TA, Renard DC, Sass EJ, Thomas AP. Oscillatory cytosolic calcium waves independent of stimulated inositol 1,4,5-trisphosphate formation in hepatocytes. J Biol Chem. 1991;266:12272–12282. [PubMed] [Google Scholar]
- Schilling WP, Elliott SJ. Ca2+ signaling mechanisms of vascular endothelial cells and their role in oxidant-induced endothelial-cell dysfunction. Am J Physiol Heart Circ Physiol. 1992;262:H1617–H1630. doi: 10.1152/ajpheart.1992.262.6.H1617. [DOI] [PubMed] [Google Scholar]
- Schilling WP, Rajan L, Strobljager E. Characterization of the bradykinin-stimulated calcium influx pathway of cultured vascular endothelial cells. Saturability, selectivity, and kinetics. J Biol Chem. 1989;264:12838–12848. [PubMed] [Google Scholar]
- Schilling WP, Ritchie AK, Navarro LT, Eskin SG. Bradykinin-stimulated calcium influx in cultured bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 1988;255:H219–H227. doi: 10.1152/ajpheart.1988.255.2.H219. [DOI] [PubMed] [Google Scholar]
- Schuppe I, Moldeus P, Cotgreave IA. Protein-specific S-thiolation in human endothelial cells during oxidative stress. Biochem Pharmacol. 1992;44:1757–1764. doi: 10.1016/0006-2952(92)90069-u. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- Sinkins WG, Estacion M, Prasad V, Goel M, Shull GE, Kunze DL, Schilling WP. Maitotoxin converts the plasmalemmal Ca2+ pump into a Ca2+-permeable nonselective cation channel. Am J Physiol Cell Physiol. 2009;297:C1533–C1543. doi: 10.1152/ajpcell.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Gallacher DV. Thapsigargin-induced Ca2+ mobilization in acutely isolated mouse lacrimal acinar cells is dependent on a basal level of Ins(1,4,5)P-3 and is inhibited by heparin. Biochem J. 1994;299:37–40. doi: 10.1042/bj2990037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW. Inositol trisphosphate receptors: Ca2+-modulated intracellular Ca2+ channels. Biochim Biophys Acta. 1998;1436:19–33. doi: 10.1016/s0005-2760(98)00122-2. [DOI] [PubMed] [Google Scholar]
- Thrower EC, Duclohier H, Lea EJA, Molle G, Dawson AP. The inositol 1,4,5-trisphosphate-gated Ca2+ channel: effect of the protein thiol reagent thimerosal on channel activity. Biochem J. 1996;318:61–66. doi: 10.1042/bj3180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HP, Wang ZN, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J. 2005a;88:1056–1069. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HP, Wang ZN, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys J. 2005b;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vanGorp RMA, vanDamMieras MCE, Hornstra G, Heemskerk JWM. Effect of membrane-permeable sulfhydryl reagents and depletion of glutathione on calcium mobilisation in human platelets. Biochem Pharmacol. 1997;53:1533–1542. doi: 10.1016/s0006-2952(97)82444-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.