

Abstract
Identification of mutations in the HOGA1 gene as the cause of autosomal recessive primary hyperoxaluria (PH) type III has revitalized research in the field of PH and related stone disease. In contrast to the well-characterized entities of PH type I and type II, the pathophysiology and prevalence of type III is largely unknown. In this study, we analyzed a large cohort of subjects previously tested negative for type I/II by complete HOGA1 sequencing. Seven distinct mutations, among them four novel, were found in 15 patients. In patients of non-consanguineous European descent the previously reported c.700+5G>T splice-site mutation was predominant and represents a potential founder mutation, while in consanguineous families private homozygous mutations were identified throughout the gene. Furthermore, we identified a family where a homozygous mutation in HOGA1 (p.P190L) segregated in two siblings with an additional AGXT mutation (p.D201E). The two girls exhibiting triallelic inheritance presented a more severe phenotype than their only mildly affected p.P190L homozygous father. In silico analysis of five mutations reveals that HOGA1 deficiency is causing type III, yet reduced HOGA1 expression or aberrant subcellular protein targeting is unlikely to be the responsible pathomechanism. Our results strongly suggest HOGA1 as a major cause of PH, indicate a greater genetic heterogeneity of hyperoxaluria, and point to a favorable outcome of type III in the context of PH despite incomplete or absent biochemical remission. Multiallelic inheritance could have implications for genetic testing strategies and might represent an unrecognized mechanism for phenotype variability in PH.
Keywords: primary hyperoxularia, HOGA1, calcium oxalate, stone disease
Introduction
At the moment three types of primary hyperoxaluria (PH; type I–III) can be accurately defined. In comparison with widespread idiopathic stone disease, they constitute rare autosomal recessive inborn errors of hepatic glyoxylate metabolism with excessive endogenous oxalate synthesis. High urinary oxalate excretion results in calcium-oxalate deposition within the renal parenchyma (nephrocalcinosis) and/or recurrent stone formation (urolithiasis), the clinical hallmarks of PH.1
Type I PH (PHI, MIM# 259900; gene AGXT, MIM# 604285) is caused by deficient or absent activity of liver-specific alanine-glyoxylate-aminotransferase (AGT).2, 3 It represents the most frequent and most severe PH phenotype with end-stage renal disease (ESRD) being the predictable outcome for the majority of adults. Moreover, ESRD within the first years of life, a condition termed infantile oxalosis, occurs in a substantial subgroup of children representing 10–18% of the total cohort. In Europe and North America, PH I accounts for about 1% of childhood ESRD, whereas prevalence rates of up to 10% are reported from some countries in the Middle East and North Africa.4, 5 With advanced renal insufficiency and failure to excrete the metabolic end product oxalic acid, the disease turns into a lethal multisystemic condition making renal replacement therapy and subsequent liver–kidney transplantation mandatory.6, 7, 8, 9, 10
Type II PH (PHII, MIM# 260000; gene GRHPR, MIM# 604296) is a result of deficient glyoxylate reductase/hydroxypyruvate reductase (GRHPR) enzyme activity.11 In general, PHII shows a milder course with the absence of infantile oxalosis and ESRD occurring in about 20% of patients. The subtype seems to be less frequent, with only 10 documented cases compared with 130 cases of PHI in Germany. Until recently a group of patients displaying significant hyperoxaluria (typically ≥1.0 mmol/1.73 m2 per day) with negative AGXT plus GRHPR mutational analysis, termed atypical PH, remained ill-defined.1, 12
In 2010, Belostotsky et al13 identified different mutations in the HOGA1 gene (4-hydroxy-2-oxoglutarate aldolase, formerly known as DHDPSL; MIM# 613597) in some of their non-PHI/II patients causing PH type III (PHIII, MIM# 613616). HOGA1 encodes a mitochondrial protein of 327 amino acids (35 kDa). The enzyme, expressed in liver and kidney, catalyzes the final step of mitochondrial hydroxyproline metabolism from 4-hydroxy-2-oxoglutarate to glyoxylate and pyruvate. Thus, an accumulation of the oxalate precursor glyoxylate could result in subsequent increased oxalate generation and therefore a gain-of-function mechanism was initially proposed by the authors, although activating mutations would be unusual in an autosomal recessive disorder.
Here, we present the results of complete HOGA1 sequencing in a large cohort of non-PHI/II patients referred to us for suspected PH. These findings expand the spectrum of HOGA1 mutations, revealing also a potential European founder mutation and the occurrence of triallelic PHIII in two individuals from a consanguineous family who were initially misclassified as having PHI. In addition, we provide further evidence that loss of HOGA1 enzymatic function is the underlying pathology in PHIII. Furthermore, our clinical data points to characteristic features and the unique outcome associated with this subtype.
Materials and methods
DNA samples from 49 probands of 38 families including 5 consanguineous families with suspected PH referred to our institution were subjected to complete HOGA1 sequencing. Genomic DNA was extracted from isolated peripheral blood lymphocytes by standard procedures. Only probands without identifiable causes of secondary (hyperabsorptive) hyperoxaluria were included in this study. Forty-two patients were previously tested negative for PHI/II by complete sequencing. Additionally we analyzed seven members from consanguineous family 12 with an established diagnosis of PHI in one individual but with two affected siblings, in whom only a single causative AGXT mutation could be identified on the maternal allele. Large deletions/insertions in the AGXT/GRHPR genes on the paternal allele were excluded using multiplex ligation-dependent probe amplification (MLPA kit P305-B1, MRC Holland, The Netherlands).
Data collection
Clinical information on each patient including age, gender, age at onset of symptoms, chief complaint, persistence of clinical symptoms, range and persistence of hyperoxaluria and hypercalciuria, renal function, type and number of surgical interventions, and imaging data were reviewed. All families except family 5 and family 24 reside in Germany. The ethnic background of the families is as follows: families 1–4, 6–8, 13–21, 23, 25–36 (German); 9, 24 (Turkish); 12, 31 (Lebanese); 22 (Austrian); 10 (Italian); 5 (Polish); 11, 37, 38 (Syrian). Informed written consent was obtained from all the patients and the participating family members. The study was approved by the institutional review board of the University of Cologne, Medical School.
Mutational analysis of the HOGA1 gene
Mutational analysis was performed by complete exon (1–7) PCR of the HOGA1 gene. Primer sequences for the exons and adjacent intron–exon borders were designed with the Exon Primer software (http://ihg.helmholtzmunechen.de/). Primer sequences and PCR conditions are available on request. Purified PCR products were sequenced by BigDye terminator ready reaction kit v.1.1 (Life Technologies, Bleiswijk, The Netherlands), using a 3500 Genetic Analyzer (Applied Biosystems, Darmstadt, Germany). Resulting sequences were evaluated with the Sequence Pilot software (JSI Medical Systems GmbH, Kippenheim, Germany). All identified mutations were resequenced and segregation analysis was performed in all the available parents. The novel HOGA1 mutations were checked in 100 healthy ethnically matched controls. The following databases were used to obtain genetic information: National Center for Biotechnology Information (NCBI; http://ncbi.nlm.nih.gov/), UCSC Genome Bioinformatics (http://genome.ucsc.edu/), and 1000 Genomes (http://1000genomes.org/). Data on a freeze of about 5000 exomes made available by the NHLBI Exome Sequencing Project (ESP) were checked for the recurrence of any sequence variant identified by us utilizing the Exome Variant Server (http://snp.gs.washington.edu/EVS/). Missense variants were subjected to in silico analysis using the following tools: Mutation Taster (http://neurocore.charite.de/MutationTaster/index.html), PolyPhen-2 (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/), and SIFT (http://sift.jcvi.org/).14, 15, 16, 17
Automated genotyping and copy number analysis
The Affymetrix (Santa Clara, CA, USA) genome-wide Human SNP Array 6.0 utilizing more than 906 600 SNPs and more than 946 000 probes was used for the detection of copy number variations in patients 13, 14, and 15 (family 12). Quantitative data analysis was performed with GTC 3.0.1 (Affymetrix Genotyping Console; Affymetrix) using a reference file of 100 samples (ATLAS Biolabs GmbH, Berlin, Germany) at the following relevant positions: AGXT: chr2: 241 456 835–241 467 210; GRHPR: chr9: 37 412 707–37 426 986; HOGA1: chr10: 99 334 158–99 362 445 (all positions referring to NCBI Build 36.3).
Minigene assay/pSPL3 splicing assay
In vitro analysis of the frequent potential splice-site mutation c.700+5G>T was performed using the pSPL3 splicing assay. Fragments of the human HOGA1 gene containing exon 5, flanked by 500 bp of upstream intronic sequence and 900 bp of downstream intronic sequence, were cloned into the splicing vector pSPL3 generating the plasmids pSPL3-DHDPSL (dihydrodipicolinate synthases)-Exon5-WT and pSPL3-DHDPSL-Exon5+5G>T. Plasmids were transfected into HEK 293T cells and mRNA was reverse transcribed.
Site-directed mutagenesis
The coding sequence of HOGA1 cDNA was amplified from human liver cDNA, cloned into pCIneo vector (Promega, Madison, WI, USA), and its sequence was confirmed. Point mutations were introduced in the plasmid by PCR. The plasmids were sequenced to confirm the distinct mutations and the absence of off-target mutations in the constructs.
Expression of HOGA1 in Cos and CHO cells
HOGA1 wild type and mutants were transfected in Cos and CHO cells using cationic lipids (Transfast, Promega), following the manufacturers guidelines. Expression was analyzed after 48 h, using an anti-human HOGA1 antibody raised in rabbits immunized with recombinant human HOGA1 protein. Mitotracker (Life technologies) was used to label mitochondria. For HOGA1 immunolocalization, cells were fixed and permeabilized by standard methods, and incubated with 1:5000 dilution of rabbit anti-HOGA1 serum in 3% BSA. Alexa-488 anti-rabbit IgG (Life technologies), diluted 1:600 in BSA–PBS, was used as secondary antibody and coverslips were analyzed by confocal microscopy. HOGA1 expression was assessed in cell lysates from the same culture dish. In brief, cells were harvested in RIPA buffer (50 mM TrisHCL pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS), protein concentration of supernatants was measured by bicinchoninic assay (Sigma, St Louis, MO, USA), and 25 μg protein was separated on 10% acrylamide gels in tris-glycine buffer (SDS-PAGE). Anti-HOGA1 antibody, diluted 1:10 000 to probe membranes for 2 h, and HRP-conjugated anti-rabbit IgG secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA), diluted 1:20 000 were used.
Urinary oxalate and calcium excretion, plasma oxalate (Pox) monitoring
Urinary oxalate and calcium excretion was measured by standard procedures. A daily urinary oxalate excretion of <0.50 mmol (<45 mg)/1.73 m2 and a daily calcium excretion <0.1 mmol/kg (<4 mg/kg) body weight in children, respectively, <8 mmol/d in adults were considered normal for 24 h collected urine samples. In spot samples the age-related reference values for urinary oxalate and calcium:creatinine ratios were used.18 Pox concentration was determined as previously described by ion chromatography (normal range: 6.3±1.3 μmol/l).19
Results
HOGA1 analysis
Complete Sanger sequencing of HOGA1 in 48 probands tested negative for PHI/II identified recessive mutations in 15 patients (31%) from 12 different families (Figures 1a and b). A synopsis of relevant clinical, biochemical and genetic data is given in Table 1 (positive cases) and Table 2 (negative cases).
Figure 1.

(a) HOGA1 mutations identified in patients with PHIII. Electropherograms of the predominant splice-site mutation c.700+5G>T and the four novel HOGA1 point mutations compared with the parental carriers (if available) or wild-type sequence. (b) Pedigree and corresponding electropherograms of kindred 12. Segregation of the c.569C>T (p.P190L) HOGA1 mutation and the c.603C>A (p.D201E) AGXT mutation in a consanguineous kindred from Lebanon.
Table 1. Synopsis of genetic, clinical, and biochemical findings of patients with recessive PHIII mutations (white cells) and additional probands from family 12 (shaded cells, see Figure 1b for details).
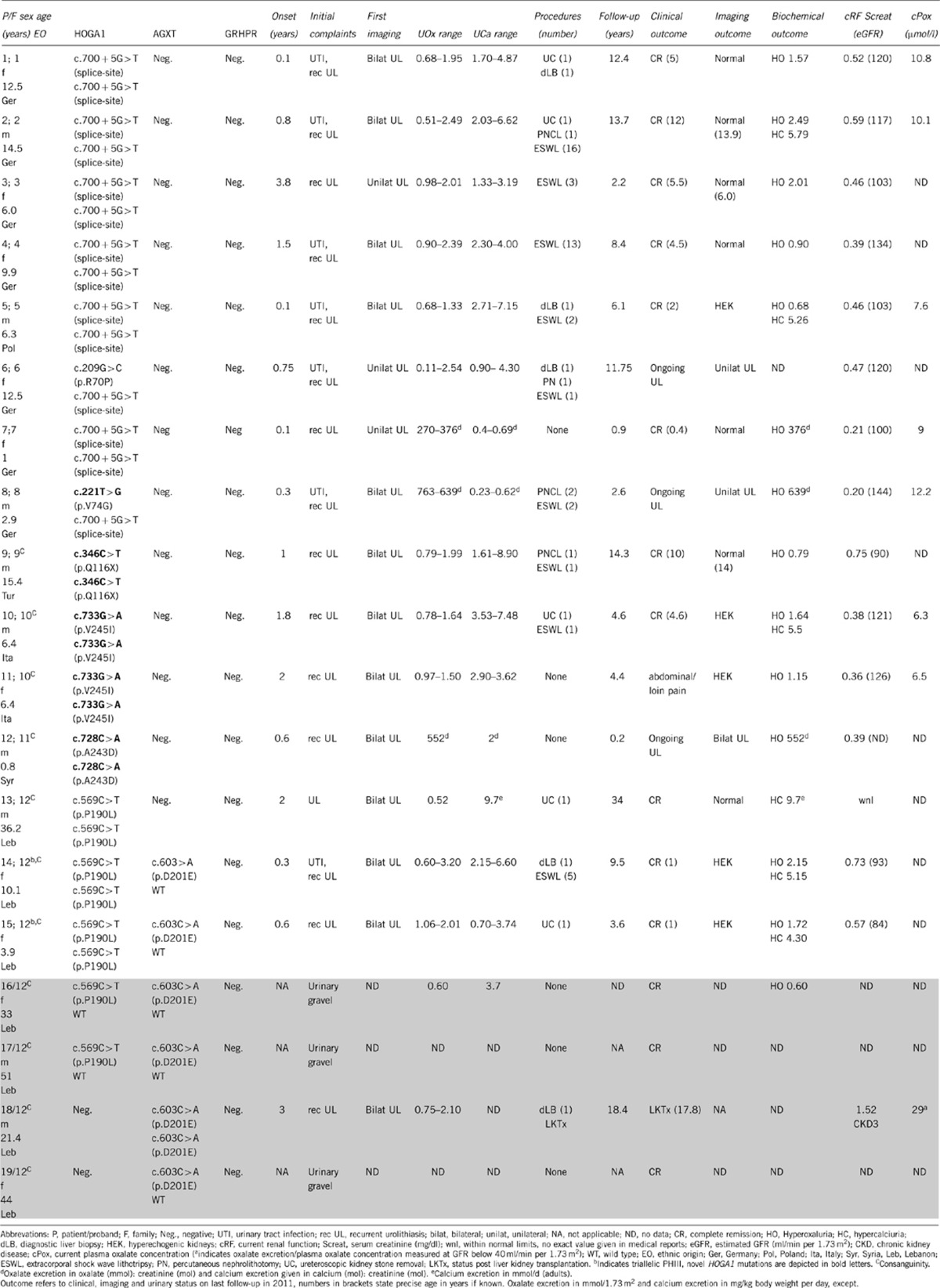
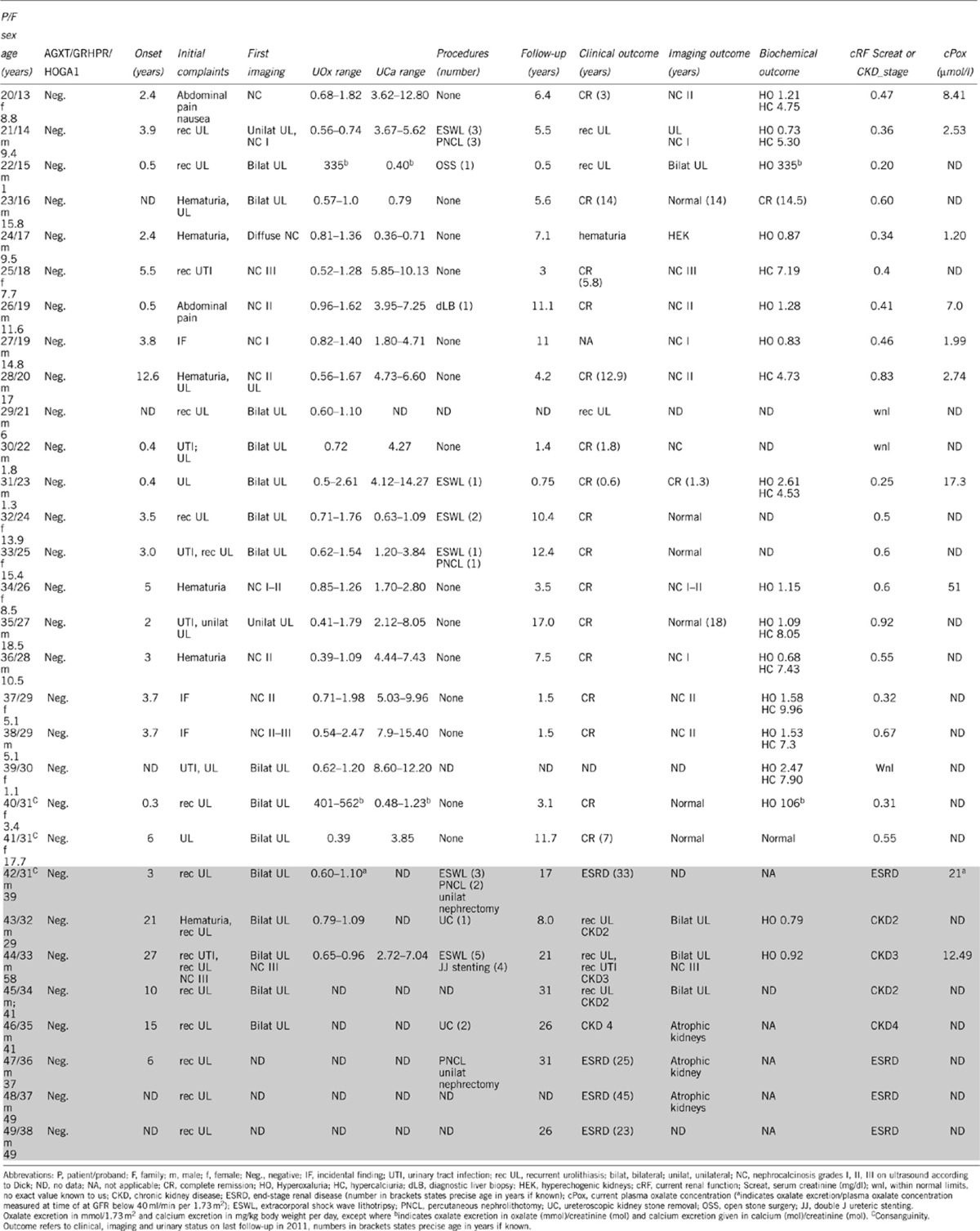
Table 2. Synopsis of clinical and biochemical findings of patients tested negative for PHI–III with normal renal function (white cells) and with CKD/ESRD (shaded cells).
The c.700+5G>T splice-site mutation was by far the most frequent pathogenic variant found in 14 of 30 disease alleles (allelic frequency 47%). The mutation was exclusively detected in unrelated, non-consanguineous families from different parts of Germany (n=7) and Poland (n=1), with six patients bearing the mutation in homozygous state and only two patients showing compound heterozygosity for the splice site and a missense mutation, p.V74G (patient 8, novel) or p.R70P (patient 6, reported). Segregation analysis, proved all parents to be heterozygous carriers. Analysis of the c.700+5G>T mutation using a minigene assay to assess splicing in vitro showed activation of a new splice site 52 bases downstream from the wild-type donor splice site. The insertion of 51 nucleotides of intron five would lead to an in-frame insertion of 17 amino acids to the native protein (Figure 2).
Figure 2.

pSPL3 splicing assay and sequencing result for the frequent splice-site mutation c.700+5G>T compared with wild-type cDNA. Fragments of the human HOGA1 gene containing exon 5 were cloned into the splicing vector pSPL3 generating plasmids pSPL3-HOGA1-Exon5-WT and pSPL3-HOGA1-Exon5+5G>T. After transfection mRNA was extracted from cells and reverse transcribed cDNA was amplified using specific primers within flanking pSPL3 exons. The G>T sequence alteration on position +5 cripples the wild-type donor site and activates an aberrant donor site on position +52, which leads to the in-frame insertion of 51 nucleotides of intron 5 (17 amino acids) into the native protein. The interrupted black lines mark the spliced out intronic sequences. The blue box indicates HOGA1 exon 5 while the blue lines represent the flanking 500 bp of upstream and 900 bp downstream intronic sequence of HOGA1 exon 5. The gray boxes mark pSPL3 exons A and B with the adjacent black lines depicting the flanking intronic sequence of the pSPL3 exons. The red box represents the 51 bp insertion resulting from the c.700+5G>T splice-site mutation.
All other mutations identified were homozygous point mutations (p.P190L, p.A243D, and p.V245I) occurring in single consanguineous families, including a homozygous stop mutation in exon 3 (p.Q116X). Again, segregation analysis confirmed all parents to be heterozygous carriers (except for family 10 due to missing parental material), ruling out de novo mutations or large deletions on one paternal allele.
None of the novel point mutations were listed as SNPs in databases (dbSNP, 1000 Genome Project) nor could they be detected in 100 ethnically matched controls, and all were predicted to be disease causing by the Mutation Taster, Polyphen-2, and SIFT software. Multisequence alignment indicated strong evolutionary conservation of these positions and, most importantly, data on 5000 exomes available from the NHLBI ESP did not contain any distinct HOGA1 mutation identified by us. To further categorize their functional consequences, constructs were introduced into Cos and CHO cells, and protein expression was analyzed by western blot and confocal miscroscopy. All missense mutants showed similiar expression levels and regular mitochondrial localization while the p.Q116X was not expressed, as expected (Figures 3a and b).
Figure 3.

(a) Subcellular localization of HOGA1 protein in transfected Cos cells. Top panel: confocal microscopy of Cos cells transfected with wild-type human HOGA1 cDNA in pCIneo vector showed the typical filamentous network of mitochondria after incubation with rabbit anti-human HOGA1 antibody and secondary Alexa-488-conjugated anti-rabbit antibody (green fluorescence, left image), which largely coincide with red signal from the mitochondria, visualized using MitoTracker Red (red fluorescence, central image), as can be demonstrated by merging both confocal images (yellow signal, right image). Bottom panel: confocal microscopy of Cos cells transfected with human HOGA1 variants showed mitochondrial subcellular localization after incubation with rabbit anti-human HOGA1 antibody, like above (green fluorescence). The pattern coincided in all cases with MitoTracker labeled mitochondria (data not shown). (b) Western blot of HOGA1 protein in transfected Cos cells. Top panel: 25 μg protein from Cos cells transfected with either wild-type (wt) or mutant HOGA1 cDNAs were electrophoresed in denaturing acrylamide gels, transferred to nitrocellulose and probed with rabbit antibody raised against recombinant human HOGA1. A band slightly above the expected 35.2 kDa of the HOGA1 monomer is observed in all lanes except mutant p.Q116X and untransfected Cos cells (cos). Molecular weight markers of 35 and 42 kDa are shown to the left (arrows). Ten μg mouse liver protein (Lv) were loaded on the right-most lane as a positive control. Bottom panel: loading control showing similar amounts of protein per lane when the filter was re-probed with mouse anti-actin antibody (except for the liver control; last lane to the right, Lv; where the signal could only be seen upon longer exposures of the film (data not shown)).
In none of the eight PHI/II-negative patients with chronic kidney disease (CKD) or ESRD suffering from recurrent calcium oxalate stone disease we were able to confirm a diagnosis of PHIII (Table 2). Only a single missense HOGA1 variant p.A148V (rs149896877) was identified in heterozygous state in patient 42 and his two offspring (patients 40, 41). This sequence change could also be found at low frequency (3/10758) on the exome variant server, it was predicted as benign, and the expression and subcellular mitochondrial localization in vitro were unremarkable (data not shown).
In a consanguineous Lebanese kindred (family 12; Figure 1b and Table 1, patients 13–19) we failed to establish a molecular diagnosis of PHI in two girls (patients 14, 15) presenting with typical features of infantile onset PH. A definite diagnosis of PHI had previously been made in another family member (patient 18) by liver biopsy (AGT activity 4.4 μmol/h/mg protein, reference range 19.1–47.9) and had been reconfirmed by molecular testing revealing homozygosity for the AGXT p.D201E mutation.20 Complete AGXT/GRHPR sequencing and MLPA performed in patients 14 and 15, however, identified the familial p.D201E mutation only in heterozygous state, segregating from the mother. Both girls were initially misclassified as having PHI, based on their severe phenotype in conjunction with results obtained on a liver biopsy from the older girl showing reduced liver AGT activity (15.8 μmol/h/mg protein; a level also compatible with carrier status in an asymptomatic proband).
Subsequent HOGA1 sequencing of the father (patient 13) and his two daughters, proved all three to be homozygous for the p.P190L mutation in exon 4, demonstrating triallelic inheritance, defined as the presence of a disease-modulating heterozygous AGXT mutation, in both offspring affected by PHIII. Onset of recurrent urolithiasis in the girls was 0.3 and 0.6 years compared with a single stone episode requiring surgery in their father at the age of two years. Now, at the age of 36 the father is clinically unremarkable apart from marked hypercalciuria (9.7 mmol/d), while his daughters demonstrate persistent substantial hyperoxaluria accompanied by intermittent hypercalciuria. Reanalysis of the HOGA1 position with the Affymetrix SNP array 6.0 could not detect any copy number variations indicating a deletion of exon 4 on one paternal allele (data not shown). The mother (proband 16) carried heterozygous alterations in both genes (AGXT, HOGA1), and her past medical history stated urinary gravel, but she denied actual urolithiasis. Only a single collected 24 h urine sample from her was available and displayed normal excretion parameters apart from a moderately elevated urinary oxalate of 0.6 mmol/1.73 m2 per day. Rechecking segregation of the p.P190L mutation in additional family members yielded that the father (proband 19) of the PHI index case also carried the HOGA1 and the AGXT mutation in heterozygous state. He denied urolithiasis and objected repeat urinary analysis and ultrasound examination, but his medical records repeatedly mentioned the spontaneous passage of gravel-like material.
Out of the 23 parents being potential HOGA1 carriers the mothers from families 2 and 5 reported a single episode of urolithiasis associated with recurrent urinary tract infections during adulthood, while medical history of all other parents was unremarkable.
Outcome
Looking at the phenotype of our PHIII cohort, most patients show an early onset and rather fulminant initial clinical course resembling PHI in some aspects. Median onset was 0.75 years (range 0.1–3.8 years; 1.05±1.02 years mean±SD) compared with 2.0 years in 130 fully genotyped cases of the German PHI cohort (7.34±13.11 mean±SD). Severity of the condition was also reflected by a high rate of interventional urological procedures (12 out of 15 patients) and documented liver biopsy in four children at a time when definite diagnosis relied mostly on determination of enzymatic activity. Recurrent urolithiasis and urinary tract infections were the leading symptoms in all PHIII patients, yet severe medullary or diffuse nephrocalcinosis on initial or follow-up ultrasound examination could only be noted among the non-PHIII group (Table 2). The range of oxalate excretion observed in PHIII was highly variable (from within normal limits to severe hyperoxaluria), but largely overlapping with the range seen in PHI and PHII. Next to persistent hyperoxaluria, temporary hypercalciuria of variable severity (range 4.87–8.90 mg/kg body weight/day) was noted in 9 out of 15 patients (60%), a phenomenon not observed in any of our PHI/II patients. On the other hand, the combination of hyperoxaluria plus intermittent hypercalciuria could also be found in comparable frequency among individuals tested negative for PHIII (Table 2). Of the 15 patients with recessive HOGA1 mutations complete clinical remission was noted in 11 (out of 15) patients (73%) at a median age of 4.5 years (range 0.4–12.0 years). Clinical remission was paralleled by complete normalization of ultrasound findings in seven of them, and only minor residual kidney hyperechogenicity remaining in another five patients. Only three patients exhibited ongoing urolithiasis at the age of 0.8, 2.9, and 12.5 years, respectively, while a fourth patient still complained about loin pains at the age of 6.4 years in the absence of detectable calculi on serial ultrasound examination.
In contrast to overt clinical and radiological remission, persistence of hyperoxaluria was observed in 13 patients, with severe hyperoxaluria above 1 mmol/1.73 m2 per day detectable in seven patients. Hypercalciuria in conjunction with hyperoxaluria occurred in three patients, while isolated severe hypercalciuria (9.7 mmol/d) was noted in the adult patient on last follow-up. After a median follow-up time of 6.1 years (range 0.2–34 years) mean estimated GFR was 112±18 ml/min per 1.73 m2 (mean±SD), and no patient showed apparent impairment of renal function or has progressed to ESRD.
Discussion
Clinical diagnosis of PH is based on the hallmarks of recurrent urolithiasis and/or nephrocalcinosis commonly presenting in childhood in combination with the biochemical finding of massively elevated urinary oxalate excretion in the absence of secondary causes. As clinical features especially in infants may be hard to distinguish between the three PH types and prognosis is largely depending on the subtype, a correct diagnosis is of paramount importance. Although the absolute amount of oxalate excretion allows some discrimination, with PHI patients showing the highest and PHIII the lowest oxalate levels,21 the large overlap between the types and intra-individual variation of oxalate excretion makes biochemical subtyping an impractical and erroneous approach. With PHIII detected in 31% of formerly unclassified PH patients, this entity is making up for about 10% of all proven cases and is currently the second most frequent PH type in Germany.
In this paper, we describe four novel mutations and 15 new PHIII cases from 12 families including first triallelic PHIII cases, where next to a homozygous HOGA1 mutation a potentially disease-modulating AGXT mutation could be identified. With the detection of a novel homozygous stop mutation in exon 3 and identification of a frequent homozygous HOGA1 splice-site mutation c.700+5G>T, initially reported in heterozygous state as c.701+4G>T,13 we provide further proof that loss-of-function is likely the underlying mechanism in PHIII. Employing a minigene assay, we were able to detect activation of a cryptic splice-site 51 bp downstream of the wild-type site leading to the introduction of 17 codons in-frame. While this manuscript was being prepared, Monico et al19 have reported similar findings regarding the c.700+5G>T splice-site mutation by utilizing EBV-transformed patient lymphocytes. The high frequency of the c.700+5G>T splice-site mutation among unrelated families from central Europe suggests a founder mutation in our population- an important information in terms of rational genetic testing. The in-frame deletion c.944_946delAGG frequently observed in individuals from Ashkenazi Jewish descent could not be found in our population.
Western blot analysis of all novel PHIII missense mutations showed stable expression and regular mitochondrial subcellular localization in transfected Cos and CHO cells. These changes, predicted to be pathogenic by algorithms based on sequence conservation and protein structure, could affect the catalytic activity. The structure of human HOGA1 protein has been recently published,22 defining key residues of the β-strand 5, part of the active site such as Lys196, which forms a Schiff base with pyruvate, and Ser198, involved in the positioning of the substrate. Thus, the mutation p.P190L, located in the adjacent turn and involving a proline, is likely to disrupt the active site. Missense mutations p.A243D and p.V245I are located at the last β-strand of the (α/β)8 domain and have the potential to disrupt the hydrophobic core of the barrel and thereby affect protein folding, tetramer formation or alter the distribution of HOGA1 quaternary structure isoforms. The p.V74G mutation is located close to the G76-X-X-G-E80 pyruvate-binding motif and may affect substrate binding. On the other hand, the conservative change p.A148V, located at the beginning of the α-helix 4, does not affect residues located near the active site or involved in the dimer interphase. Its benign nature is further supported by bioinformatics predictions and its presence in the exome databases. There might be subtle differences in expression levels that could not be detected by our in vitro experiments. More information could potentially be obtained from immunohistochemistry or confocal microscopy performed on the kidney and the liver sections in the future, as some patients did undergo biopsy for diagnostic purpose. Differences in the efficiency of HOGA1 mitochondrial import and leader peptide processing constitute a potential third pathomechanism apart from affecting enzymatic activity and oligomeric state. Close examination of western blot banding seems to reveal two bands with a very small difference in size. The lower one possibly resembles the mature form, after removing the mitochondrial targeting sequence, and some mutations seem to produce stronger unprocessed HOGA fractions than others. This finding could be explored by performing in vitro transcription–translation and mitochondrial import experiments.
Catabolism of 4-hydroxyproline derived from endogenous (collagen turnover) and dietary sources has been shown to result in significant glyoxylate and oxalate generation, which makes this pathway an interesting tool for understanding the full metabolic spectrum of PH including the so-called gut and kidney axis in hyperoxaluria.23, 24, 25 However, the precise molecular mechanisms that result in hyperoxaluria as a consequence of defective 4-hydroxyproline catabolism due to HOGA1 mutations remain unknown, as much as the pathogenesis and the significance of hypercalciuria. Riedel and colleagues22 demonstrated that in contrast to related bacterial enzymes termed DHDPS human HOGA1 favours forward cleavage of 4-hydroxy-2-oxo-glutarate to glyoxylate and pyruvate.26 Glyoxylate is a highly reactive molecule that is metabolized, in a compartment-dependent manner by either GR (cytosol, mitochondria) to glycolate, by alanine-glyoxylate-aminotransferase (AGT; peroxisome) to glycine or by cytosolic lactate dehydrogenase to oxalate. Mitochondrial buildup of 4-hydroxy-2-oxoglutarate caused by HOGA1 blockade and subsequent leakage to the cytosol where a non-specific pyruvate aldolase would catalyze the formation of glyoxylate has been hypothesized.21
This study underscores the favorable outcome of PHIII in the context of PH in accordance with previous findings.13, 21 Moreover, we show that clinical remission occurs in PHIII not because of biochemical remission but despite persisting hyperoxaluria (and to a lesser degree hypercalciuria) in the majority of patients. For the lack of a sufficient number of adults with long-term follow-up and potential sequelae arising from recurrent urolithiasis, we should still be cautious to call PHIII a benign condition. However, the absence of HOGA1 mutations in our series of unclassified calcium oxalate urolithiasis with CKD/ESRD is reassuring. A self-limiting clinical course in PHIII might be one reason why the number of adult probands identified is so low. It would be intriguing to speculate on age-dependent changes in metabolism and/or diet accounting for the high rate of clinical remission seen with this type, a hypothesis that is challenged by the persistence of hyperoxaluria. In summary, early onset of calcium oxalate urolithiasis that becomes more and more quiescent with age, the presence of intermittent hypercalciuria, and the absence of marked nephrocalcinosis and renal insufficiency all provide valuable clinical clues to a diagnosis of PHIII.
We here report the occurrence of mutations in two genes HOGA1 and AGXT involved in glyoxylate metabolism in a single consanguineous family with several affected members. The two patients with triallelic mutations presented with the highest and most constant oxalate excretion among our cohort and clearly demonstrated a more severe PHIII phenotype compared with the (p.P190L homozygous) father.
Interestingly, both heterozygous AGXT/HOGA1 carriers (patients 16, 17) had appointments for urinary gravel, but denied any episodes of stone passage. In the absence of confirmed urolithiasis, with sparse or even lacking biochemical and imaging information we classified them as unclear cases. Digenic inheritance of PH remains unknown. The association of heterozygous PHIII mutations to idiopathic calcium oxalate urolithiasis reported by Monico et al21 presents an exciting hypothesis, in particular in conjunction with the observations from this family, although the low rate of urolithiasis observed in our parental cohort may question this conclusion.27
Our findings are of particular relevance as mutation screening is usually halted as soon as causative mutations have been identified in a first gene. In the light of our working hypothesis that in some cases the PH phenotype may be modulated by mutations in other genes affecting glyoxylate metabolism, complete sequencing of all known causative genes should be considered, notably in those patients who lack a second causative mutation or those with an atypical presentation. This would not only prevent misclassification, but furthermore allow the uncovering of multiallelic inheritance, which may constitute one of the long-sought mechanisms for inter- and intra-familial phenotype variability in PH.
Obviously, much can be gained from a thorough investigation; nevertheless, the specific etiology of PH remains to be elucidated in a significant proportion of patients. Other genes involved in glyoxylate metabolism are promising first candidates. Unraveling the precise molecular and biochemical pathogenesis behind PHIII, a disorder where hyperoxaluria is present from birth, might have therapeutic implications even for the treatment of the more severe PH types. So far, we have thought that the time and the severity of hyperoxaluria in the latter types directly correlate with the clinical course, a concept that now might need some rethinking.
Acknowledgments
We are grateful to all patients and family members who participated in this study, and to the following referring physicians: Magdalena Riedl (Innsbruck), Jens König (Münster), Kay Latta (Frankfurt), Sabine Ponsel (Munich) and Katharina Hohenfellner (Traunstein) for providing clinical information on PHIII negative cases. We thank Karin Boss for critically reading the manuscript. The authors would like to thank the NHLBI GO ESP and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). This work was supported in part by a Koeln Fortune Program grant (25/2008)/Faculty of Medicine, University of Cologne (to BB Beck).
The authors declare no conflict of interest.
References
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer HE, Dormer AE, Scowen EF, Watts RW. Primary hyperoxaluria. Lancet. 1957;273:320–322. doi: 10.1016/s0140-6736(57)92210-9. [DOI] [PubMed] [Google Scholar]
- Danpure C, Jennings P. Peroxisosomal alanine glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS. 1986;201:20–24. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- Kamoun A, Lakhoua R. Endstage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10:479–482. doi: 10.1007/s004670050143. [DOI] [PubMed] [Google Scholar]
- Al-Eisa AA, Samham M, Naseef M. End-stage renal disease in Kuwaiti children: an 8 year experience. Transplant Proc. 2004;36:1788–1791. doi: 10.1016/j.transproceed.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Hoppe B, Graf D, Offner G, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol. 1996;110:488–492. doi: 10.1007/s004670050145. [DOI] [PubMed] [Google Scholar]
- Hoppe B, Kemper MJ, Bokenkamp A, Portale A, Cohn R, Langman CB. Plasma calcium-oxalate supersaturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int. 1999;56:268–274. doi: 10.1046/j.1523-1755.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. 2006;70:1642–1648. doi: 10.1038/sj.ki.5001806. [DOI] [PubMed] [Google Scholar]
- Jamieson NV, European PH I Transplantation Study Group A 20-year experience of combined liver-kidney transplantation for primary hyperoxaluria (PH1): the European transplant registry experience 1084–2004. Am J Nephrol. 2005;25:282–289. doi: 10.1159/000086359. [DOI] [PubMed] [Google Scholar]
- Brinkert F, Ganschow R, Helmke K, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87:1415–1421. doi: 10.1097/TP.0b013e3181a27939. [DOI] [PubMed] [Google Scholar]
- Cramer S, Ferree P, Lin K, Milliner D, Holmes R. The gene encoding hydroxypyruvate reductase is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999;8:2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- Monico CG, Persson M, Ford GC, Rumsby G, Milliner DS. Potential mechanisms of marked hyperoxaluria not due to primary hyperoxaluria I or II. Kidney Int. 2002;62:392–400. doi: 10.1046/j.1523-1755.2002.00468.x. [DOI] [PubMed] [Google Scholar]
- Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Gen. 2010;87:392–399. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Variant Server, NHLBI Exome Sequencing Project (ESP) Seattle, WA. (URL: http://snp.gs.washington.edu/EVS/ (accessed September 2011).
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein functions using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:2489. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Hesse A, Tiselius HG, Siener R, Hoppe B.Urinary Stones3rd ednBasel: Karger; 2009 [Google Scholar]
- Hoppe B, Kemper MJ, Hvizd MG, Sailer DE, Langman CB. Simultaneous determination of oxalate, citrate and sulfate in children's plasma with ion chromatography. Kidney Int. 1998;53:1348–1352. doi: 10.1046/j.1523-1755.1998.00891.x. [DOI] [PubMed] [Google Scholar]
- Williams EL, Acquaviva C, Amoroso A, et al. Primary Hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mut. 2009;30:910–917. doi: 10.1002/humu.21021. [DOI] [PubMed] [Google Scholar]
- Monico CG, Rossetti S, Belostotsky R, et al. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol. 2011;6:2289–2295. doi: 10.2215/CJN.02760311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel TJ, Johnson LC, Knight J, Hantgan RR, Holmes RP, Lowther TW. Structural and biochemical studies of human 4-hydroxy-2-oxo-glutarate aldolase: implications for hydroxyproline metabolism in primary hyperoxaluria. PLoS ONE. 2011;6:e26021. doi: 10.1371/journal.pone.0026021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama T, Fujita K, Suzuki M, et al. Control of oxalate formation from L-hydroxyproline in liver mitochondria. J Am Soc Nephrol. 2003;14:939–946. doi: 10.1097/01.asn.0000059310.67812.4f. [DOI] [PubMed] [Google Scholar]
- Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney Int. 2006;70:1929–1934. doi: 10.1038/sj.ki.5001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robijn S, Hoppe B, Vervaet BA, D'Hase PC, Verhulst A. Hyperoxaluria: a gut-kidney axis. Kidney Int. 2011;80:1146–1158. doi: 10.1038/ki.2011.287. [DOI] [PubMed] [Google Scholar]
- Mirwaldt C, Koerndorfer I, Huber R. The crystal structure of Dihydropicolinate Synthase from Escherichia coli at 2.5 A resolution. J Mol Biol. 1995;246:227–239. doi: 10.1006/jmbi.1994.0078. [DOI] [PubMed] [Google Scholar]
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest. 2005;115:2598–2608. doi: 10.1172/JCI26662. [DOI] [PMC free article] [PubMed] [Google Scholar]