

Background: Surface expression of cardiac ATP-sensitive potassium (KATP) channels impacts cellular energy homeostasis.
Results: Activation of calcium/calmodulin-dependent protein kinase II (CaMKII) results in KATP channel internalization, requiring specific motifs on the Kir6.2 channel subunit.
Conclusion: CaMKII phosphorylation of Kir6.2 promotes endocytosis of cardiac KATP channels.
Significance: This mechanism reveals new targets to improve cardiac energy efficiency and stress resistance.
Keywords: ABC Transporter, Bioenergetics, Endocytosis, Heart, Homology Modeling, Ion Channels, K-ATP Channel, Immunofluorescence, Internalization
Abstract
Cardiac ATP-sensitive potassium (KATP) channels are key sensors and effectors of the metabolic status of cardiomyocytes. Alteration in their expression impacts their effectiveness in maintaining cellular energy homeostasis and resistance to injury. We sought to determine how activation of calcium/calmodulin-dependent protein kinase II (CaMKII), a central regulator of calcium signaling, translates into reduced membrane expression and current capacity of cardiac KATP channels. We used real-time monitoring of KATP channel current density, immunohistochemistry, and biotinylation studies in isolated hearts and cardiomyocytes from wild-type and transgenic mice as well as HEK cells expressing wild-type and mutant KATP channel subunits to track the dynamics of KATP channel surface expression. Results showed that activation of CaMKII triggered dynamin-dependent internalization of KATP channels. This process required phosphorylation of threonine at 180 and 224 and an intact 330YSKF333 endocytosis motif of the KATP channel Kir6.2 pore-forming subunit. A molecular model of the μ2 subunit of the endocytosis adaptor protein, AP2, complexed with Kir6.2 predicted that μ2 docks by interaction with 330YSKF333 and Thr-180 on one and Thr-224 on the adjacent Kir6.2 subunit. Phosphorylation of Thr-180 and Thr-224 would favor interactions with the corresponding arginine- and lysine-rich loops on μ2. We concluded that calcium-dependent activation of CaMKII results in phosphorylation of Kir6.2, which promotes endocytosis of cardiac KATP channel subunits. This mechanism couples the surface expression of cardiac KATP channels with calcium signaling and reveals new targets to improve cardiac energy efficiency and stress resistance.
Introduction
ATP-sensitive potassium (KATP) channels have the unique ability to adjust membrane excitability in response to changes in the energetic status of the cell (1–8). This function serves to reduce myocardial energy consumption and vulnerability to stress (6, 7, 9, 10). The channel complex is formed through physical association of the pore-forming inwardly rectifying potassium channel, Kir6.x, with the regulatory sulfonylurea receptor, SUR4 (3, 6, 11–15). In ventricular myocytes, the primary site of cardiac energy consumption, KATP channels are composed primarily of Kir6.2 and SUR2A subunits (6, 14, 16). The metabolic sensing of these channels occurs through modulation of the ATP sensitivity of Kir6.2 by SUR (5, 11–17). The tight integration of SUR with cellular energetic networks (18–21) as well as the proximity of the channel to sites of ATP consumption in the compartmentalized cellular environment (17, 22) allows KATP channel opening to be regulated not only by severe metabolic stress, such as during ischemia, but also by heart rate acceleration within the range induced by normal physical activity (23, 24). When activated by a reduced ATP/ADP ratio, reflecting either increased cellular metabolic demand or reduced cellular ATP generation, KATP channel-dependent potassium efflux shortens cardiac action potential duration, allowing for a longer diastolic interval that supports myocardial relaxation and restoration of ion gradients and energetic resources as well as limits sodium and calcium entry into the cell and thus reduces energy requirements for ion transport/exchange and contraction (1, 9, 10, 23–28).
There is a growing body of evidence that the ability of cardiac KATP channels to affect cellular excitability and function depends on their abundance at the membrane surface (24, 25, 27, 29–31). For instance, in the heart an increase in functional sarcolemmal KATP channel presence enhances the speed and degree of shortening of action potentials and limits cardiac energy consumption in response to escalating workloads, whereas a decrease in their presence has the opposite effect (24). Additionally, although even a small fraction of the normal population of KATP channels can dramatically shorten the action potential duration in response to severe metabolic stress, the speed at which this myocardially protective action occurs is KATP channel expression-dependent (25). Defining the mechanisms that control cardiac membrane KATP channel expression and associated metabolic signaling is an important step toward understanding how to promote myocardial energy efficiency and stress resistance to prevent or treat cardiac disease.
Myocardial effects of KATP channel current up-regulation are mirrored in many ways by inhibition of calcium/calmodulin-dependent protein kinase II (CaMKII), including shortened action potentials, improved resistance to cell death under metabolic stress, and normalized intracellular calcium homeostasis (32–34), suggesting a potential interaction between KATP channels and CaMKII in a common calcium-related regulatory pathway. CaMKII is a multifunctional kinase, densely expressed in cardiomyocytes, that targets numerous proteins involved in excitation-contraction coupling and excitability to support short-term enhancement of cardiac performance, whereas persistent activation results in adverse cardiac remodeling and dysfunction (35–38). Indeed, inhibition of CaMKII has been shown to increase cardioprotective KATP channel surface presence and current density (29). However, the molecular mechanism underlying this regulation is unknown.
Here we identify CaMKII as a critical regulator of the surface expression and consequent whole cell current capacity of cardiac KATP channels. We find that CaMKII activation triggers rapid internalization of cardiac KATP channels by endocytosis. This process requires phosphorylation of threonine at positions 180 and 224 and an intact YXXØ endocytosis motif, 330YSKF333of Kir6.2. A molecular model of Kir6.2 predicts that the μ2 subunit of the endocytosis adaptor protein, AP2, docks with the KATP channel by interacting with 330YSKF333 and Thr-180 on one and Thr-224 on an adjacent Kir6.2 subunit. These findings provide new insight into the regulation of cardiac KATP channels and provide valuable new targets for the promotion of cardiac energy efficiency and stress resistance.
EXPERIMENTAL PROCEDURES
Mouse Models
Transgenic mice expressing a specific peptide inhibitor of CaMKII (AC3-I, which targets a conserved region of the CaMKII regulatory domain) or a scrambled non-inhibiting peptide (AC3-C), both under control of the cardiac-specific Myh6 promoter (32), were compared with wild-type littermates. The AC3-I mice have been shown to exhibit increased membrane KATP channels not caused by increased transcription or elevated total protein expression (29).
Cardiomyocyte Isolation
Single ventricular cardiomyocytes from anesthetized (Avertin, 2.5% solution of tribromoethanol in 2-methyl-2-butanol, Sigma, 240 mg/kg intraperitoneally) male, heterozygous AC3-I and AC3-C transgenic mice (32) and their wild-type littermates aged 10–12 weeks were enzymatically isolated (24). Hearts were cannulated in situ then rapidly excised and retrogradely perfused at 90 mm Hg at 37 °C, pH 7.4, for 5 min with Hepes buffer (Medium 199, Sigma), 1 min with a “low calcium” medium (100 mmol/liter NaCl, 10 mmol/liter KCl, 1.2 mmol/liter KH2PO4, 5 mmol/liter MgSO4, 20 mmol/liter glucose, 50 mmol/liter taurine, 10 mmol/liter HEPES supplemented with 0.13 mmol/liter CaCl2, 2.1 mmol/liter EGTA), then 13 min with low calcium medium supplemented with 1% bovine serum albumin, 0.2 mmol/liter CaCl2, collagenase (type IV, 22 units/ml, Worthington) and Pronase (100 μg/ml, Serva). Ventricles were dissected away, cut into pieces (∼3 × 3 mm), and incubated at 37 °C for 15 min in the enzyme solution with gentle stirring until dissociated cardiomyocytes could be collected. Protocols conform to the Guide for the Care and Use of Laboratory Animals and were approved by the University of Iowa Institutional Animal Care and Use Committee.
Patch Clamp
Studies were performed using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) integrated with a Nikon TE2000-U microscope. Experiments were performed at 33–35 °C using a temperature controller TC2r (Cell MicroControls, Norfolk, VA).
For whole-cell recording, borosilicate glass pipettes (3–4 megaohms) were filled with internal solution: 140 mmol/liter KCl, 1 mmol/liter MgCl2, 5 mmol/liter EGTA, 5 mmol/liter ATP, 5 mmol/liter HEPES-KOH, pH 7.3. Cardiomyocytes were superfused with Tyrode solution: 136.5 mmol/liter NaCl, 5.4 mmol/liter KCl, 1 mmol/liter CaCl2, 0.53 mmol/liter MgCl2, 5.5 mmol/liter glucose, 5.5 mmol/liter HEPES-NaOH, pH 7.4. Whole-cell current traces were obtained in response to 1-s rectangular pulses from a holding potential of −50 mV to test potentials from −100 to +40 mV. For quantification, whole cell KATP channel current was measured as the difference between base line and pinacidil- and/or 2,3-dinitrophenol (DNP)-stimulated current recorded just before the end of a 1-s applied voltage step from −50 to +40 mV. For analysis, only whole cell recordings in which beginning and ending capacitance were within 10% were used.
For inside-out single channel recording, myocytes were bathed in internal solution 140 mmol/liter KCl, 1 mmol/liter MgCl2, 5 mmol/liter EGTA, 5 mmol/liter HEPES-KOH, pH 7.3, supplemented with glucose (1 g/liter) and various concentrations of ATP. Pipettes (4–5 megaohms) were filled with “Electrode Solution”: 140 mmol/liter KCl, 1 mmol/liter CaCl2, 1 mmol/liter MgCl2, 5 mmol/liter HEPES-KOH, pH 7.3. Pipette holding potential was +60 mV. Patches were excised in 25 μmol/liter ATP to prevent run-down.
All recordings were monitored, sampled, and analyzed using pCLAMP software (Molecular Devices). KATP channel activity in membrane patches (inside-out configuration) was calculated relative to activity in the absence of ATP and fitted by the Hill equation (y = [IC50]/([IC50]h + [ATP]h), where h is the Hill coefficient, [ATP] is the ATP concentration, and [IC50] is the half-maximal inhibitory ATP concentration. Single channel activity was analyzed as previously described (39).
Whole Heart Biotinylation
Hearts were excised from anesthetized mice and retrogradely perfused at 90 mm Hg with Krebs-Henseleit buffer bubbled with 95% O2, 5% CO2 at 37 °C and pH 7.4. The AV node was mechanically ablated, and hearts were paced at 150 or 100 ms cycle length (i.e. 400 or 600 beats/min, Bloom Electrophysiology, Fischer Imaging Corp., Denver, CO) using a platinum pacing catheter positioned in the right ventricle (NuMed; Hopkinton, NY). Coronary flow was measured in series with the aortic cannula (T402, Transonic Systems, Ithica, NY). After 25 min the hearts and perfusion system were cooled to 4 °C (Isotemp 3006D, Fisher Scientific, Inc., Pittsburgh, PA) and perfusion buffer was switched from Krebs-Henseleit buffer to PBS containing 1.0 mg/ml Sulfo-NHS-biotin (ThermoScientific) and supplemented with glucose 5 mmol/liter. During the cooled stage, hearts were asystolic, pacing was discontinued, and perfusion was changed from constant pressure to constant flow to maintain coronary flow at the level measured immediately before cooling. After 25 min of perfusion with biotin, hearts were perfused with PBS containing 15 mmol/liter glycine for 5 min at 4 °C to quench unbound biotin. Immediately after this ventricles were freeze-clamped in liquid nitrogen. The tissue was lysed in buffer containing 10 mmol/liter Tris-Base, 50 mmol/liter NaCl, 5 mmol/liter EDTA, 50 mmol/liter NaF, 30 mmol/liter Na4P2O7, 1.25% Triton X-100, and protease inhibitor mixture (Roche Applied Science) at pH 7.4. Biotinylated (cell surface) proteins were isolated by incubating the tissue lysate with immobilized NeutrAvidin beads (ThermoScientific) for 12 h at 4 °C. After separation by 3–8% gradient Nu-Page Tris acetate gel (Invitrogen), biotinylated Kir6.2 was detected by immunoblot with anti-Kir6.2 antibody (Novus) and HRP-conjugated secondary antibody (Santa Cruz Biotechnology).
Cell Culture and cDNA Expression
HEK293T/17 cells (ATCC, CRL-11248) were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 2 mmol/liter glutamine, and penicillin-streptomycin. Cells were grown on coverslips for immunofluorescence in a 24-well plate and in 100 ml of tissue culture dishes for immunoprecipitation and phosphorylation assays. Cells were transfected with pcDNA constructs for WT or mutant KATP channel subunits and CaMKII (see below) using Lipofectamine 2000 (Invitrogen).
Immunofluorescence
HEK293T/17 cells were grown on 13 mm fibronectin-coated glass coverslips and transfected (Lipofectamine 2000; Invitrogen) with cDNA for 1) Ki6.2-HA or its mutants, 2) SUR2A and 3) CaMKII-GFP, 1:5:1 ratio for 48 h at 37 °C. 48 h after transfection cells were blocked with 5% BSA in PBS (30 min at 4 °C) labeled with primary anti-HA monoclonal mouse antibody (1:1000, Clone H-11, Covance) for 1 h at 4 °C. Cells were then washed with phosphate-buffered saline (PBS+/+), then incubated at 37 °C for 30 min in the presence of A23187, fixed with 4% paraformaldehyde in PBS (15 min), permeabilized with 0.1% Triton X-100 (10 min), and blocked with 1% BSA in PBS for 1 h at room temperature. Cells were labeled with Alexa 568-conjugated goat anti-mouse secondary antibody (1:500, Invitrogen) for 1 h at room temperature in a dark chamber. Cells were washed and mounted with vectashield on glass slides for confocal microscopic analysis.
Confocal Fluorescence Imaging
Cells were imaged on a Zeiss LSM 510META laser scanning confocal microscope under a 63× oil immersion lens. Middle cuts through cells were analyzed using ImageJ Software (National Institutes of Health, public domain). The ratio of intracellular:total intensity was calculated.
Constructs
The Kir6.2 sequence containing an HA tag and an 11-amino acid linker was used as “WT” Kir6.2 (30) and served as a template to generate the following mutant constructs using QuikChange II Site-directed Mutagenesis kit (Stratagene): Kir6.2Δ26-HA, Kir6.2Δ36-HA, Kir6.2T180A-HA, Kir6.2T224A-HA, Kir6.2T224E-HA, and Kir6.2Y330C-HA. The Kir6.2T180A-HA construct was used as a template to generate Kir6.2A180T-HA. We also used previously described constructs for SUR2A (40), constitutively active CaMKII-GFP (41), and CaMKII-GFP (41).
Western Blot
Whole cell protein extracts (24) were used for immunoblotting with CaMKII (pan), phospho-CaMKII (Thr-286, Cell Signaling), and anti-GAPDH (Santa Cruz Biotechnologies) antibodies.
Drugs
The following drugs were purchased from Sigma: isoproterenol, A23187, pinacidil, DNP, chelerythrine, and dynasore. Dominant-negative dynamin and scrambled control were synthesized (bioSYNTHESIS) according to the published sequence (30).
Biotinylation
HEK293T/17 cells were transfected (Lipofectamine 2000; Invitrogen) with cDNA for Kir6.2-HA, SUR2A, and CaMKII-GFP. The cells were cultured in Dulbecco's modified Eagle's medium. 48 h after transfection the cells were washed with phosphate-buffered saline (PBS+/+). The cells were incubated at 37 °C for 0, 10, or 30 min in the presence of A23187 and then placed on ice. Channels remaining at the cell surface were incubated with 0.5 mg/ml Sulfo-NHS-biotin (ThermoScientific) in PBS+/+ for 30 min on ice and then quenched with 100 mmol/liter glycine in PBS+/+ for 10 min on ice. The cells were lysed in Nonidet P-40 lysis buffer (0.4% sodium deoxycholate, 1% Nonidet P-40, 63 mmol/liter EDTA, 50 mm Tris-HCl, pH 8, and protease inhibitor mixture). Biotinylated (cell surface) proteins were isolated by incubating the cell lysate with immobilized NeutrAvidin beads (ThermoScientific) for 12 h at 4 °C. After separation by a 3–8% gradient Nu-Page Tris acetate gel (Invitrogen), biotinylated Kir6.2-HA was detected by immunoblot with anti-HA monoclonal antibody (Covance) and HRP-conjugated secondary antibody (Cell Signaling).
Immunoprecipitation
The left ventricle was isolated from WT mice and freeze-clamped. The tissue was ground in liquid N2 until powder. Tissue was added to lysis buffer containing protease and phosphatase inhibitors (Roche Applied Science), vortexed, and placed on ice for 1 h with sonication for 15 s every 15 min. After centrifugation at 14,000 rpm for 1 h, protein was quantified (Bio-Rad). The lysed ventricle tissue sample (100 μg/100 μl of total protein), and lysis buffer with protease inhibitors (100 μl) was added to Dynabeads (Bio-Rad) conjugated with anti-Kir6.2 (Santa Cruz Biotechnology) or anti-CaMKII (Cell Signaling) antibodies. After elution, Western blot with anti-CaMKII or anti-Kir6.2 antibodies was performed.
Phosphorylation
HEK cells transfected with WT and mutant Kir6.2-HA and SUR were lysed (see method under whole heart biotinylation), and the HA-tagged proteins were isolated using immunoprecipitation with agarose complexed with anti-HA antibody (ThermoScientific). Agarose beads complexed with channel proteins were resuspended in Hepes/magnesium acetate buffer: 50 mm Hepes, 10 mm magnesium acetate, 0.5 mm CaCl2, 1 mg/ml BSA, 1 μm calmodulin, and incubated with [γ-32]ATP (1–1.5 μCi, PerkinElmer Life Sciences) with or without activated CaMKII (250 ng). The CaMKII, purified from Sf9 insect cells (see below), was activated by 1 μm calmodulin and 0.5 mm CaCl2 in the presence of 150 μm ATP (42). Reactions were quenched, and proteins were eluted from the beads by LDS-sample buffer (Invitrogen). Eluted proteins were separated on a 3–8% gradient Nu-Page Tris acetate gel (Invitrogen) by electrophoresis at 200 V for 30 min. Phosphorylation was assessed by 32P incorporation.
Mouse Kir6.2 fragments containing mutations and their WT control fragments were synthetized (bioSYNTHESIS) as follows (mutations are in bold): Thr-224 (3.32 kDa)/T224A (3.29 kDa), MQVVRKTT(A)SPEGEVVPLHQVDIPMENGVGGN; Thr-180 (4.56 kDa)/T180A (4.53 kDa), MKTAQAHRRAET(A)LIFSKHAVITLRHGRLCFMLRVGDLRK.
The peptides (10 μg each) were incubated with [γ-32]ATP and activated CaMKII. Phosphorylation was assessed by quantifying 32P incorporation using densitometric analysis of Western blots after separation on a 4–12% Bis-tris gel (Invitrogen) by electrophoresis at 200 V for 30 min.
CaMKII Purification
Sf9 cells infected with recombinant CaMKII baculovirus were harvested 48 h post-infection by centrifugation. Cells were resuspended in homogenization buffer (50 mmol/liter Tris-HCl, pH 7.5, 300 mmol/liter NaCl, 5% betaine, 1 mmol/liter EDTA, 1 mmol/liter EGTA, 1mmol/liter DTT, protease inhibitors), and sonicated to release cell contents. Cell lysate was then centrifuged at 15,000 × g for 20 min at 4 °C. The supernatant was removed and added to 3 volumes of equilibration buffer (50 mmol/liter Tris-HCl, pH 7.5, 100 mmol/liter NaCl, 2 mmol/liter CaCl2, 4 mmol/liter magnesium acetate, 1 mmol/liter DTT, protease inhibitors) and CaM-agarose and incubated for 1 h at 4 °C. Resin was washed 3 times with wash buffer (50 mmol/liter Tris-HCl, pH 7.5, 1 mmol/liter CaCl2, 1 mmol/liter magnesium acetate, 1 mmol/liter DTT, protease inhibitors) followed by 2 times with 1 mol/liter NaCl wash buffer (50 mmol/liter HEPES, pH 7.5, 1 mol/liter NaCl, 1 mmol/liter CaCl2, 1 mmol/liter magnesium acetate, 1 mmol/liter DTT, protease inhibitors) and 2 times with 1 mol/liter NaCl wash buffer without protease inhibitors. CaMKII was eluted from the resin in 5 fractions of elution buffer (50 mmol/liter HEPES, pH 7.5, 100 mmol/liter NaCl, 2.5 mmol/liter EDTA, 1 mmol/liter DTT). Protein fractions were dialyzed overnight at 4 °C into storage buffer (50 mmol/liter HEPES pH 7.5, 1 mmol/liter EDTA, 50% glycerol, 10% ethylene glycol). The fractions containing CaMKII protein were determined by SDS-PAGE.
CaMKII Activity Assay
The protein lysate/purified protein was mixed 1:1 with 50 mmol/liter HEPES, pH 7.5, 10 mmol/liter magnesium acetate, 0.5 mmol/liter CaCl2, 0.001 mmol/liter calmodulin, 1 mg/ml BSA, 1 mmol/liter DTT, 0.4 mmol/liter [γ-32]ATP (200–500 cpm/pmol), 0.02 mmol/liter syntide. The mixture was incubated for 10 min at 30 °C. The reaction was stopped with SDS-PAGE loading buffer or by spotting on Whatman P81 paper. 5 washes of 10 min each were performed before counts obtained by scintillator.
Homology Modeling of Kir6.2
The amino acid sequence of mouse Kir6.2 (UniProt ID Q61743) was employed as a query for homology modeling. The N-terminal segment Met-1—Arg-25 and the C-terminal segment Ler-355–Ser390 were deleted before any calculation. The mouse Kir3.2 channel in closed state co-crystallized with K+, Na+, and a phosphatidylinositol diphosphate analog (PDB coda 3SYA) was used as a template for the homology modeling. The sequence of Kir6.2 was aligned to that of Kir3.2 using T-Coffee (43). The alignment was manually refined with VMD1.9.1 and the Multiseq plugin. Homology modeling was performed with Modeler 9.10. One hundred models of the Kir6.2 homotetramer were generated and ranked according to their DOPE score. The model with the lowest score was selected. The best model was minimized in five successive stages using the CHARMM27 force field and NAMD2.9: 1) minimization of the hydrogen atoms (1,000 steps); the position of non-hydrogen atoms was restrained by a force constant of 2 kcal/mol/Å2; 2) minimization of the amino acid side chains (2,000 steps) with position restraints applied on carbons Cβ plus the backbone; 3) minimization of the whole amino acid side chains (4,000 steps) with position restraints applied on the backbone; 4) minimization (10,000 steps) with position restraints applied on carbons Cα; 5) minimization of the whole Kir6.2 homotetramer (15,000 steps). The phosphorylation (PO42−) of Thr-180 and/or Thr-224 was introduced before stage 1. The final models were checked with the What If and the JCSG protein structure validation servers. One disulfide bridge between Cys110 and Cys142 was defined during the calculation. The coordinates of the four K+ ions and the four phosphatidylinositol diphosphate analogues initially present in the x-ray structures of Kir3.2 were preserved in the Kir6.2 model. The position of all their non-hydrogen heteroatoms was restrained over all the minimization process. The phosphatidylinositol diphosphate analog was modeled using the CHARMM36 parameters. Analysis of the results was performed with VMD 1.9.1.
The model of μ2 is based on the published structure of μ2 co-crystallized with the TGN38 internalization signal peptide DYQRN (PDB code 1BXX). The peptide YQ was superimposed with the YS of YSQKF and μ2 positioned by trial and error. The position with the fewest steric conflicts was selected.
Statistical Analysis
Results are expressed as the mean ± S.E. Comparisons between two groups were made using the 2-sided Student's t test and appropriate post hoc tests. A p value < 0.05 was considered statistically significant.
RESULTS
CaMKII Activation Reduces Whole Cell KATP Channel Current and Cell Membrane Expression
Transgenic mice expressing a specific peptide inhibitor of CaMKII (AC3-I) exhibit an increased presence of membrane KATP channels not caused by increased transcription or elevated total protein expression (29). CaMKII is activated by signals that increase intracellular calcium, including catecholamine stimulation (32). Using real-time patch clamp monitoring of the KATP channel, whole cell current induced by the opener pinacidil, and metabolic inhibition by DNP, we found that application of isoproterenol significantly reduces KATP current in isolated ventricular cardiomyocytes within 2–3 min (Fig. 1, A and D, fractional reduction = 0.66 ± 0.1, p < 0.05). A similar significant and prompt reduction was found in ventricular cardiomyocytes from transgenic hearts expressing a non-CaMKII-inhibiting control peptide, AC3-C (Fig. 1, B and D, fractional reduction = 0.58 ± 0.08, p < 0.05, p = NS versus WT). In contrast, very little KATP channel current reduction was observed in cardiomyocytes isolated from transgenic mice expressing the CaMKII inhibitory peptide, AC3-I (Fig. 1, C and D, fractional reduction = 0.14 ± 0.06, p < 0.05 versus WT), supporting the hypothesis that the isoproterenol effect on reducing whole cell KATP channel current in the WT cardiomyocytes occurred through CaMKII activation. In WT and AC3-I cardiomyocytes, the reduction of stimulated KATP channel whole cell current in response to isoproterenol was reversed within ∼5 min upon washout of isoproterenol, indicating that the phenomenon is not related to channel rundown (Fig. 1, A and B). KATP channel current may also be regulated by calcium activated forms of PKC (30, 44–49). To test for a potential role of PKC in the witnessed reduction of KATP channel current, we examined the isoproterenol effect in the presence of the PKC inhibitor, chelerythrine. Despite pretreatment with chelerythrine, isoproterenol induced a significant decrease in whole cell KATP channel current in isolated cardiomyocytes (Fig. 1, E–G, additional fractional reduction with isoproterenol = 51.2 ± 7.6, p < 0.05 versus reduction by chelerythrine alone).
FIGURE 1.

CaMKII effect on native KATP current in isolated ventricular cardiomyocytes. Shown is representative whole cell KATP channel current, stimulated by 100 μmol/liter pinacidil (pin) and 50 μmol/liter DNP, measured during application and wash-out of the CaMKII activator 500 nmol/liter isoproterenol (iso) in isolated cardiomyocytes from WT mice (A), trangenic mice expressing a scrambled, control peptide AC3-C (B), and transgenic mice expressing the CaMKII inhibiting peptide, AC3-I (C). pF, picofarads. D, shown are summary data: AC3-I (n = 12) versus WT (n = 4) and AC3-C (n = 3); *, p < 0.05. E, representative whole cell KATP channel current, stimulated by 100 μmol/liter pinacidil and 50 μmol/liter DNP, was measured during application of the PKC inhibitor, 5 μmol/liter chelerythrine (che), and 500 nmol/liter isoproterenol in an isolated WT murine cardiomyocyte. F, shown are summary data for chelerythrine and isoproterenol effects on pinacidil- and DNP-stimulated KATP channel current in isolated WT murine cardiomyocytes (n = 5; *, p < 0.05 versus base line, the isoproterenol bar represents additional reduction beyond that attributed to chelerythrine application). G, shown is an example of whole cell current tracings measured at points marked from the graph in E. H, On left ventricular lysates from WT mice, immunoprecipitation was performed with the anti-Kir6.2 antibody and probed for CaMKII by Western blot (IB) with the anti-CaMKII Ab. I, immunoprecipitation was performed with the anti-CaMKII Ab and probed for Kir6.2 by Western blot with the anti-Kir6.2 Ab. J, shown are Western blots of biotin-labeled membrane and whole cell fractions of ventricular lysates from isolated hearts paced at 150 or 100 ms cycle length (C.L.). Membrane and total protein fractions are probed with anti-Kir6.2 antibody and with antibodies for Na+/K+ ATPase and GAPDH as controls. K, shown are summary data for the fractional change in Kir6.2 expression with pacing cycle length of 100 versus 150 ms (*, p < 0.05, n = 2 hearts under each pacing condition for WT and AC3-I and n = 3 each for AC3-C).
The interaction of CaMKII and KATP channels in cardiomyocytes was further confirmed by co-immunoprecipitation. Anti-CaMKII Ig co-immunoprecipitated Kir6.2 and anti-Kir6.2 antibodies co-immunoprecipitated CaMKII from detergent-soluble lysates generated from left ventricles of WT mice (Fig. 1, H and I).
In the presence of increased pacing rates, previously linked to CaMKII activation (50), the membrane fraction of ventricular lysates from biotinylated isolated hearts (WT and AC3-C) exhibited lower Kir6.2 membrane expression (fractional difference in ratio of Kir6.2 expression normalized to Na+/K+ ATPase expression at 100 compared with 150-ms pacing cycle length = −0.76 ± 0.09 for WT and −0.66 ± 0.18 for AC3-C, p < 0.05). This difference is similar in magnitude to the whole cell current change observed with isoproterenol treatment of isolated WT and AC3-C cardiomyocytes (Fig. 1D), indicating that the current change can be mostly accounted for by a reduction in sarcolemmal expression of KATP channels. A reduction in surface channel expression at higher pacing rates as assessed by whole heart biotinylation was not seen in hearts expressing a peptide inhibitor of CaMKII (AC3-I, Fig. 1, J and K, fractional difference .01±.13, p = NS), similar to the absence of whole cell current reduction observed in isolated cardiomyocytes from AC3-I mice (Fig. 1D). The expression of Na+/K+ ATPase and GAPDH in membrane and total fractions, respectively, did not show significant changes with faster pacing (p = NS).
We next developed a heterologous cell expression system to dissect the components of the CaMKII-KATP channel pathway. We examined HEK cells with and without exogenous overexpression of CaMKII and found that HEK cells had very little native CaMKII expression (Fig. 2A). We then performed a densitometric analysis of immunoblots from HEK cells with exogenous CaMKII overexpression and found that their exposure to the calcium ionophore A23187 resulted in a significant increase in phosphorylated (activated) CaMKII (0.75 ± 0.09 versus 1.29 ± 0.07, n = 3 each, p < 0.05).
FIGURE 2.

CaMKII effect on recombinant KATP channels in HEK cells. A, HEK cells with/without exogenous overexpression of CaMKII and with/without exposure to A23187 5 μmol/liter were lysed and probed by immunoblot. GAPDH expression served as a control. B, HEK cells expressing Kir6.2-HA, SUR2A, and CaMKII-GFP underwent exposure to 10 μmol/liter A23187 for 0, 10, or 30 min 37 °C. At each time point, groups of cells were cooled to 4 °C and biotinylated. Surface and total protein from lysed cells were assayed for Kir6.2 by anti-HA immunoblot. Cells expressing only CaMKII-GFP without KATP channel subunits served as the negative control. C, shown are summary data of biotinylation experiments in HEK cells, indicating a time-dependent reduction in Kir6.2 surface expression with A23187 exposure (n = 3 each; *, p < 0.05). D, shown is representative whole cell KATP channel current, stimulated by 50 μmol/liter pinacidil (pin) and 50 μmol/liter DNP, measured during application and wash-out of the 5 μmol/liter calcium ionophore A23187 in HEK cells engineered to express Kir6.2, SUR2A, and CaMKII. pF, picofarads. E, shown are summary data from these experiments performed in the presence and absence of CaMKII overexpression (*, p < 0.05). F, the same experiment was performed as in D but with dialysis of the endocytosis inhibitor (DN) dynamin 100μmol/liter through the patch pipette. G, shown are summary data from the experiments performed the presence of dominant negative dynamin or a non-inhibiting, scrambled peptide (100 μmol/liter each, n = 5 each; *, p < 0.05).
To confirm the effect of CaMKII activation on the surface expression of KATP channels in this model, we used a cell surface biotinylation assay. Extracted total and biotinylated proteins from HEK cells transfected with CaMKII and the cardiac KATP channel subunits Kir6.2 and SUR2A with and without CaMKII activation by A23187 were assayed by Western blot. This indicated that CaMKII activation reduced the fraction of surface versus total HA-tagged Kir6.2 subunits in a time-dependent manner (Fig. 2, B and C, % of expression at time 0 = 67 ± 8 at 10 min and 17 ± 11 at 20 min, p < 0.05).
These changes in the presence of KATP channels at the cell membrane were further confirmed by measuring whole cell KATP channel current by patch clamp. A23187 dramatically reduced KATP channel current (Fig. 2, D and E, n = 4, fractional reduction = 0.49 ± 0.04, p < 0.05). Similar to the finding in cardiomyocytes, the effect was reversible with washout of the ionophore (Fig. 2D). The dependence of this phenomenon on CaMKII activation was confirmed in HEK cells expressing KATP channel subunits but without exogenous CaMKII overexpression. In these cells very little KATP channel current reduction by A23187 was observed (Fig. 2E, n = 5, fractional reduction = 0.06 ± 0.03, p < 0.05 versus cells with CaMKII). These biochemical and biophysical data indicate that CaMKII directly interacts with cardiac KATP channels and that CaMKII activation promotes a reversible reduction in the presence of KATP channels at the cell membrane.
CaMKII Activation Reduces KATP Channel Membrane Expression through Endocytosis
The above whole cell patch clamp experiments and biotinylation experiments in HEK cells expressing KATP channel subunits and CaMKII indicate a reversible down-regulation of membrane KATP channel subunits and KATP channel current without a change in total cellular Kir6.2, suggesting endocytosis as a possible mechanism. To test this, we tracked whole cell KATP channel current when endocytosis was prevented by the addition of a dominant negative dynamin peptide (30) to the patch pipette solution. We found that KATP current reduction in response to CaMKII activation by A23187 was eliminated in the presence of dominant negative (DN) dynamin (Fig. 2, F and G). In contrast, an inactive scrambled peptide had no effect on the KATP channel current response to A23187 (Fig. 2G, n = 5 each, fractional current reduction = 0.40 ± 0.05 for scrambled, p < 0.05, and 0.03 ± 0.04 for dominant negative dynamin, p = NS versus base line). These data implicate enhanced endocytosis of channel subunits as the dominant mechanism for the witnessed CaMKII activation-dependent decrease in whole cell KATP channel current capacity.
We confirmed these data using immunofluorescence to detect internalization of labeled surface Kir6.2 subunits in response to A23187. HEK cells transfected with Kir6.2-HA, SUR2A, and CaMKII-GFP were incubated with an anti-HA antibody to allow the antibodies to bind the surface channels and undergo internalization. We then treated cells with A23187, permeabilized, and stained them with red fluorescence-conjugated secondary antibody. Thus, only channels that were initially at the cell membrane and either remained there or became internalized were labeled. When CaMKII was not overexpressed (Fig. 3, A and B) or when CaMKII was overexpressed but cells were not treated with A23187 (Fig. 3, C and D), we detected bright red fluorescence only on the cell surface (n = 16 cells from 1 transfection, fluorescence ratio 0.52 ± 0.04, p < 0.05 versus control and n = 16 cells from 1 transfection, fluorescence ratio .52±.03, p < 0.05 versus control, respectively). However, when cells had CaMKII overexpression and were treated with A23187 (Fig. 3, E and F), we detected distinct puncta of red fluorescence in the cytoplasm, consistent with KATP channel subunit internalization (n = 34 cells from 2 transfections, cytosolic/total cellular intensity ratio = 0.71 ± 0.02). This effect was eliminated in CaMKII-overexpressing cells that had treatment with A23187 and the membrane-permeable endocytosis inhibitor dynasore (Fig. 3, G and H, n = 18 cells from 1 transfection, fluorescence ratio 0.48 ± 0.01, p < 0.05 versus control). Taken together, measurement of membrane currents, biotinylation of surface proteins, and immunodetection of KATP channel subunits indicates that CaMKII activation reduces the cardiac KATP channel current capacity of cells by enhancing endocytosis.
FIGURE 3.

CaMKII-dependent internalization of KATP channels and Kir6.2 subunit dependence. Immunofluorescence confocal imaging was performed on HEK cells expressing HA-tagged Kir6.2, WT SUR2A, and GFP-tagged CaMKII or GFP alone without CaMKII. Anti-HA antibody was applied at the beginning of the experiment, and cells were washed. After a 30-min exposure to 10 μmol/liter A23187 to activate CaMKII, cells were cooled, fixed, and permeabilized, and secondary antibody was applied (red) to track surface Kir6.2-HA movement. Shown are cells expressing Kir6.2-HA, SUR2A, and GFP without CaMKII (A and B), Kir6.2-HA, SUR2A, and CaMKII-GFP but not stimulated with A23187 (C and D), Kir6.2-HA, SUR2A, and CaMKII-GFP with application of A23187(E and F), and Kir6.2-HA, SUR2A, and CaMKII-GFP stimulated with A23187 in the presence of the endocytosis inhibitor, dynasore 50μmol/liter (G and H). I and J, immunofluorescence confocal microscopy was performed on HEK cells expressing Kir6.2Δ36-HA (red) and CaMKII-GFP following the same protocol. K, representative whole cell KATP channel current was stimulated by 200 μmol/liter DNP in response to 5 μmol/liter A23187 in a HEK cell expressing Kir6.2Δ36 and CaMKII. L, shown are summary data for DNP-stimulated whole cell KATP channel current from HEK cells expressing CaMKII and Kir6.2Δ26 (n = 4) or Kir6.2Δ36 (n = 5) in response to CaMKII activation by A23187 (*, p < 0.05).
CaMKII-dependent Endocytosis of KATP Channels Does Not Require the SUR Subunit
Because cardiac KATP channels are composed of pore-forming and regulatory subunits, we sought to determine whether both were required for CaMKII-triggered endocytosis. WT Kir6.2 requires SUR for normal assembly and trafficking to the cell membrane. To circumvent the interdependence of Kir6.2 and SUR, we transfected HEK cells with an HA-tagged Kir6.2 truncation mutant (Kir6.2Δ36) that can traffic to the membrane without the SUR subunit (51). Application of A23187 resulted in internalization of the Kir6.2Δ36-HA subunit to a similar degree as HA-tagged WT KATP channels as demonstrated by immunofluorescence (Fig. 3, I and J, n = 36 cells from 3 transfections, cytosolic/total cellular intensity ratio = 0.58 ± 0.02, p = NS versus WT) with a significant reduction in DNP-stimulated KATP channel current as demonstrated by whole cell patch clamp (Fig. 3, K and L, n = 5, fractional reduction = 0.89 ± 0.04, p < 0.05). Similar findings were also seen for Kir6.2Δ26, which can also traffic to the membrane without SUR (51) (Fig. 3L, n = 4, fractional reduction = 0.95 ± 0.02, p < 0.05, immunofluorescence not shown, n = 24 cells from 3 transfections, cytosolic/total cellular intensity ratio = 0.60 ± 0.02, p = NS versus WT channels). These findings support that CaMKII-induced internalization of membrane cardiac KATP channels is mediated through Kir6.2 and does not require SUR.
Phosphorylation of Thr-180 and Thr-224 on Kir6.2 Are Required for KATP Channel Endocytosis
CaMKII regulates the function of proteins by targeting specific serines and threonines embedded within consensus sequences. To further investigate regions on Kir6.2 that may be required for CaMKII-mediated KATP channel endocytosis, we investigated four sites identified through scanning the Kir6.2 sequence for the RXX(S/T) motif and through literature reports of sites with other serine/threonine kinase interactions (44, 45, 47, 52, 53). We identified candidate sites at Ser-37, Thr-180, Thr-224, and Ser-372. In experiments to determine the impact of these sites on endocytosis of cardiac KATP channels, we found no effect of S37A and S372A mutations on KATP channel current or membrane expression (data not shown), and thus further results and discussion focus only on the Thr-180 and Thr-224 sites.
We probed isolated WT and mutant Kir6.2-HA-containing channels for in vitro phosphorylation by CaMKII. Mutation of Thr-180 or Thr-224 to a non-phosphorylatable alanine (T224A and T180A) resulted in only a faint signal for incorporation of the radio-labeled γ-phosphate from ATP in the presence of activated CaMKII, and this signal was further attenuated in the presence of double T180A/T224A mutation in Kir6.2 compared with WT (Fig. 4A).
FIGURE 4.

CaMKII effect in Kir6.2T224A and Kir6.2T180A mutants. A, in isolated recombinant cardiac KATP channels with WT or mutated Kir6.2, an in vitro phosphorylation assay was performed in the presence and absence of CaMKII. Kir6.2-HA subunits were mutated from threonine to alanine at Thr-224 or Thr-180 or both. Kir6.2-HA is identified as a double band at ∼37 kDa. The prominent signal near 50 kDA level represents autophosphorylated CaMKII. B, in WT and mutated peptide fragments of Kir6.2, in vitro phosphorylation assay was performed in the presence and absence of CaMKII. Fragments were mutated from threonine to alanine at Thr-224 or Thr-180. Differences in band migration are attributed to different sizes and charge of the fragments. The WT Thr-180 and mutant T180A fragments are identified at ∼3.3 kDa (best seen for Thr-180 + CaMKII and faintly for T180A + CaMKII), whereas the Thr-224 and T224A fragments are identified at ∼4.5 kDa (best seen for Thr-224 + CaMKII). C–H, representative immunofluorescence confocal imaging was performed in HEK cells following the same protocol as in Fig. 3. C and D, cells expressing CaMKII-GFP, Kir6.2-HA, and SUR2A are shown. E and F, cells expressing CaMKII-GFP, Kir6.2T180A-HA, and SUR2A are shown. G and H, cells expressing CaMKII-GFP, Kir6.2T224A-HA and SUR2A. I, summary data for immunofluorescence imaging is shown. For WT, n = 34 cells were from 2 transfections. For T224A, n = 4 transfections, 48 cells; *, p < 0.05 versus WT. For T180A, n = 4 transfections, 42 cells; *, p < 0.05 versus WT. J, representative whole cell KATP channel current was stimulated by 50 μmol/liter pinacidil (pin) and 50 μmol/liter DNP in a HEK cell expressing CaMKII-GFP, Kir6.2T224A, and SUR2A in response to A23187 5 μmol/liter. pF, picofarads. K, summary data of pinacidil- and DNP-stimulated whole cell KATP channel current in response to A23187 in HEK cells expressing CaMKII-GFP, SUR2A, and WT Kir6.2 versus Kir6.2T224A (*, p < 0.05).
Next, we generated segments of the Kir6.2 subunit surrounding Thr-224 and Thr-180 to further demonstrate phosphorylation of these sites by CaMKII (Fig. 4B). Mutation to T224A and T180A in these fragments eliminated incorporation of the radiolabeled γ-phosphate from ATP in the presence of activated CaMKII (Fig. 4B).
In HEK cells expressing CaMKII, SUR2A, and Kir6.2T224A-HA or Kir6.2T180A-HA, much less internalization of KATP channels in response to treatment with A23187 was appreciated compared with cells expressing HA-tagged WT Kir6.2 (Fig. 4, C–H). The cytosolic/total cellular intensity was 0.61 ± 0.02 for WT versus 0.39 ± 0.01 for T224A and 0.37 ± 0.02 for T180A, p < 0.05 versus WT for both mutants, Fig. 4I).
Similarly, in HEK cells expressing CaMKII, SUR2A, and Kir6.2T224A-HA, the whole cell KATP channel current reduction after CaMKII activation was diminished compared with cells with HA-tagged WT Kir6.2 subunits (Fig. 4, J and K, n = 6, fractional reduction = 0.05 ± 0.03 for T224A versus n = 4, fractional reduction = 0.49 ± 0.04 for WT, p < 0.05). However, although surface presence of KATP channels in HEK cells expressing CaMKII, SUR2A, and Kir6.2T180A-HA was clearly seen by immunofluorescence (Fig. 4F), we were unable to identify significant pinacidil and DNP-stimulated whole cell KATP channel current (n = 20 cells). In rare cases (n = 4 of 30 patches), a single KATP channel was identified by inside-out patch clamp in the presence of low (0–25 μm) ATP in cells expressing Kir6.2T180A-HA (Fig. 5C). Similar electrophysiologic findings were obtained in cells transfected instead with Kir6.2T180A without the HA tag (data not shown).
FIGURE 5.
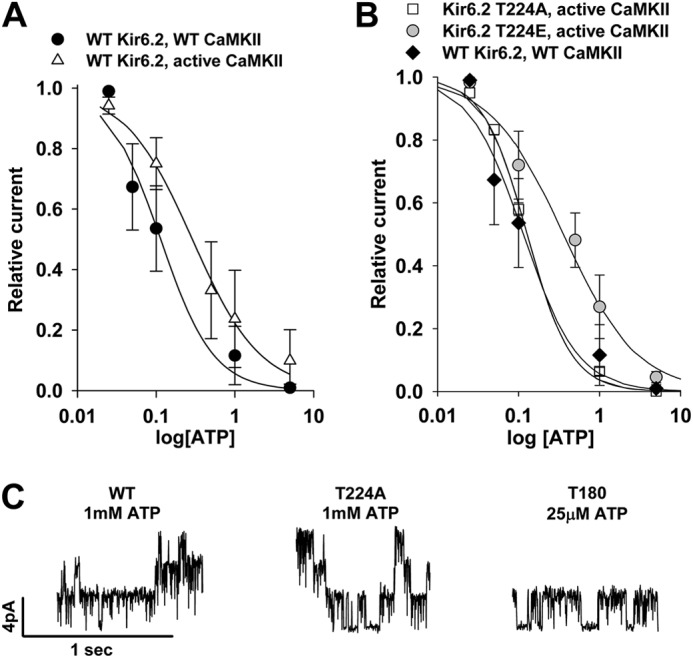
CaMKII and mutation effects on biophysical properties of KATP channels. A, shown is KATP channel ATP sensitivity in patches from HEK cells expressing WT Kir6.2, SUR2A, and WT versus constitutively active CaMKII. Symbols indicate raw data. Lines indicate fitted Hill equation. B, KATP channel ATP sensitivity in patches from cells expressing constitutively active CaMKII, SUR2A, and Kir6.2T224A or Kir6.2T224E versus cells expressing WT Kir6.2, SUR2A, and WT CaMKII is shown. Symbols indicate raw data. Lines indicate the fitted Hill equation. C, shown are examples of single-channel inside-out recordings from HEK cells expressing WT CaMKII, SUR2A, and WT Kir6.2 in 1 mmol/liter ATP (left), Kir6.2T224A in 1 mmol/liter ATP (middle), or Kir6.2T180A in 25 μmol/liter ATP (right). The last panel is not entirely representative as the majority of patches from cells expressing Kir6.2T180A had no KATP channels even in the absence of ATP.
We also examined whether there could be a contribution to whole cell KATP channel current reduction by a CaMKII activation-dependent change in KATP channel biophysical properties. We observed a rightward shift in the ATP sensitivity (decreased ATP sensitivity) of KATP channels in inside-out patches from cells expressing WT KATP channel subunits and constitutively active CaMKII (n = 7, Hill = 1, IC50 = 290 μm) versus WT CaMKII (n = 3, Hill = 1.3, IC50 = 110 μm, Fig. 5A). Expression of Kir6.2T224A with SUR2A and constitutively active CaMKII eliminated the rightward shift in ATP sensitivity (n = 4, Hill = 1.6, IC50 = 128 μm, Fig. 5B), whereas expression of Kir6.2T224E (to mimic phosphorylation of the Kir6.2 subunit) with SUR2A and constitutively active CaMKII restored the rightward shift in ATP sensitivity (n = 7, Hill = 0.96, IC50 = 360 μm, Fig. 5B). These data indicate that CaMKII-dependent phosphorylation of Kir6.2 at Thr-224 results in a reduction in KATP channel ATP sensitivity. However, this decrease in ATP sensitivity of the channel would oppose, not contribute to or account for, the reduction in whole cell KATP channel current in the presence of CaMKII activation.
Expression of constitutively active CaMKII caused no differences in KATP single channel conductance (70.6 ± 0.5 versus 71.1 ± 0.8 picosiemens with WT CaMKII) or open probability (0.81 ± 0.05 versus 0.84 ± 0.03 with WT CaMKII).
Mutations of the Kir6.2 Thr-180 residue resulted in loss of normal channel gating. Overall activity of channels with the Kir6.2T180A mutation was rare, and these channels appeared to have dramatically increased ATP sensitivity (n = 3, Hill = 1.8, IC50 = 40 μm). No KATP channel current was detected in the presence of the Kir6.2T180E mutant. These data indicate that the Thr-180 site is essential for normal regulation of channel gating.
Single channel conductance was unchanged in channels comprised of WT Kir6.2 versus Kir6.2T224A or Kir6.2T180A (71.4 ± 0.8 versus 70.9 ± 0.6 versus 70.1 ± 0.8 picosiemens, respectively, n = 3 each, Fig. 5C). Overall, these data show that CaMKII-dependent phosphorylation of the Thr-224 and Thr-180 residues of Kir6.2 has a role in regulating channel gating and is critical to promote endocytosis of the KATP channel subunits.
The Kir6.2 Internalization Motif 330YSKF333 Is Required for CaMKII-induced KATP Channel Down-regulation
Patients with a genetic form of neonatal diabetes have been found to harbor Y330C and F333I mutations of Kir6.2 that inhibit spontaneous endocytosis of KATP channels at 37 °C and result in a ∼2-fold increase in surface channels (54). Here, we examined whether the 330YSKF333 sequence is required for internalization of cardiac KATP channels driven by CaMKII activation. In HEK cells expressing Kir6.2Y330C-HA, SUR2A, and CaMKII, pinacidil- and DNP-stimulated KATP channel current was reduced very little by application of A23187 (fractional reduction 0.05 ± 0.02, n = 6, p < 0.05 versus 0.49 ± 0.04, n = 4 for WT Kir6.2, Fig. 6, A and B). Similarly, there was little cytosolic appearance of labeled Kir6.2Y330C-HA after stimulation of HEK cells with A23187 (Fig. 6 C and D, n = 44 cells from 4 transfections, cytosolic/total cellular intensity = .20±.01, p < 0.05 versus WT). These findings indicate that an intact 330YSKF333 motif is required for CaMKII-induced endocytosis of cardiac KATP channels.
FIGURE 6.

Mutation of YXXØ internalization motif interferes with CaMKII-induced KATP channel down-regulation. A, shown is representative whole cell KATP channel current, stimulated by 50 μmol/liter pinacidil (pin) and 50 μmol/liter DNP, measured during application and wash-out of 5 μmol/liter A23187 in HEK cells engineered to express Kir6.2Y330C-HA, SUR2A, and CaMKII-GFP. pF, picofarads. B, shown are summary data from whole cell patch clamp experiments performed with Kir6.2-HA (n = 4) versus Kir6.2Y330C-HA (n = 6; *, p < 0.05). C and D, shown are representative confocal fluorescence images of HEK cells expressing Kir6.2Y330C-HA, SUR2A, and CaMKII-GFP after treatment with A23187.
A Molecular Model Suggests Docking of the Endocytosis Adaptor Protein μ2 Subunit Requires CaMKII Phosphorylation Sites on Neighboring Kir6.2 Subunits
The Kir6.2 C terminus including residues 178–364 (which would include the Thr-180 and Thr-224 sites and the 330YSKF333motif) has been shown to interact directly with the μ2 subunit of the endocytosis adaptor protein, AP2 (54). Here, to investigate the effect of phosphorylation of Thr-180 and Thr-224 on this relationship, we formulated a molecular homology model of Kir6.2 based on the crystal structure of mouse Kir3.2. The model predicts that Thr-224 and Thr-180 are too distant from the 330YSKF333 sequence to directly interact and that phosphorylation of Thr-224 and Thr-180 is unlikely to induce large enough conformational changes to alter accessibility of the 330YSKF333 sequence (Fig. 7, A and B).
FIGURE 7.

Homology model of Kir6.2. A, a three-dimensional model of Kir6.2 presented as a solvent-accessible surface (except the phosphatidylinositol diphosphate analog) focused on the ATP binding site indicates the position of Thr-180 (red) within the ATP binding pocket (blue) adjacent to the YSKF internalization motif (green). Thr-224 (also red) is seen in the bottom right corner of the image. B, the same model shows little change in the relationship of Thr-180 or Thr-224 to YSKF with phosphorylation of both Thr-180 and Thr-224. The calculated residue displacements (root mean square deviation) induced by phosphorylation of Kir6.2 are: Kir6.2 + Thr-180(P) = 0.39 Å, Kir6.2 + Thr-224(P) = 0.17 Å, and Kir6.2 + Thr-180(P) + Thr-224(P) = 0.43 Å. The root mean square deviation is slightly larger when Thr-180 is phosphorylated as Thr-180 is embedded in the Kir6.2 homotetramer, poorly accessible to the solvent. As a consequence, the neighbor residues as well as the backbone of Thr-180 adapt their conformation due to introduction of the bulky phosphate group. In contrast, Thr-224 is well exposed on the surface of Kir6.2. The phosphorylation of this amino acid has little consequence on neighbor residues. Both Thr-180 and Thr-224 are remote from the YSKF signal. The closest residue of the YSKF motif to Thr-180 is Tyr-330. The distance between the phosphate/hydroxyl of Thr-180 and the hydroxyl of Tyr-330 is, respectively, 9.3, 9.6, 9.3, and 9.7 Å in Kir6.2 and Kir6.2 phosphorylated at Thr-180, Kir6.2 phosphorylated at Thr-224, and Kir6.2 phosphorylated at both Thr-180 and Thr-224. The closest YSKF residue to Thr-224 is Lys-332. The distance between the phosphate/hydroxyl of Thr-224 and the ammonium of Lys-332 is, respectively, 18.8, 19.0, 19.8, and 19.8 Å in Kir6.2, Kir6.2 phosphorylated at Thr-180, Kir6.2 phosphorylated at Thr-224, and Kir6.2 phosphorylated at both Thr-180 and Thr-224. C, a side view model of Kir6.2, with two subunits removed for clarity, indicates proposed docking with μ2 (large, green). The ATP binding pocket (blue), YSKF (green), Thr-180 (upper red), and Thr-224 (lower red) are also shown. D, an end-on view of the Kir6.2 model shows the relationship of docked μ2 (large, green) with adjacent subunits of Kir6.2. Thr-224 (red) is largely obscured by μ2. Kir6.2 subunits A and C are white, and subunits B and D are gray.
However, docking of the known structure of the μ2 subunit of the endocytosis adaptor protein, AP2, to the modeled Kir6.2 suggests that μ2 could bind 330YSKF333 and interact with the nearby Thr-180 via a long loop (possibly the lysine-rich Asn-217—Asp-244, Fig. 7, C and D), assuming the ATP binding pocket was unoccupied. The μ2 subunit could simultaneously interact with Thr-224 of the neighboring Kir6.2 subunit via a loop (possibly Ile-159—Asn-171, Asn-217—Asp-244, or Val-406—Ser-414, Fig. 7, C and D). The arginine-rich N terminus could be electrostatically compatible with a Thr-224 phosphate. Thus, the model suggests a possible role for the Thr-180 and Thr-224 residues in coordinating the docking of μ2 with Kir6.2 at 330YSKF333.
DISCUSSION
Cardiac KATP channels are critical for cellular energetic homeostasis and protection from injury and arrhythmia (1, 6, 26). As such, deciphering their regulation is key to understanding both the normal physiology of cardiac adaptation to varying workloads and how functional/metabolic diseases such as heart failure occur and progress. This study establishes a previously unrecognized mechanism for regulation of the surface expression and whole cell current capacity of sarcolemmal KATP channels by CaMKII.
CaMKII can be viewed as an ideal candidate for cardiac workload-dependent regulation of KATP channels by endocytosis. First, CaMKII is a vital regulator of cardiac excitation-contraction coupling and, thus, cardiac energy consumption (35–37). Second, CaMKII activation underlies increased cardiac performance during heart rate acceleration (38, 50), an established trigger of KATP channel opening (23, 24). Finally, CaMKII has been linked to endocytosis of membrane proteins in other cell types (55, 56). Here, we reveal that activation of CaMKII induces endocytosis of cardiac KATP channels. We propose that this phenomenon is critical for augmented myocardial performance in response to increased bodily demand. We previously demonstrated that a higher density of surface sarcolemmal KATP channels results in a more rapid adjustment of ventricular action potential duration and thus higher fidelity matching of energy consumption to available metabolic resources under a sudden increase in cardiac workload or under metabolic insult (24, 25). However, during persistent, intense cardiac performance and augmented energy consumption, a high surface density of KATP channels could be a liability for the cell as the continued recruitment of KATP channel current could render the membrane less excitable (1, 5, 6, 57). A reduction in the overall surface abundance of KATP channels through endocytosis promoted by CaMKII activation would effectively reduce the gain in the energetic feedback circuit, thereby supporting enhanced myocardial performance, whereas the rapid restoration of KATP channel surface density when CaMKII activation is abated would permit the cell to “reset” to its base-line level of action potential responsiveness. This hypothesis is supported by the blunted contractility at high pacing rates of hearts expressing a CaMKII inhibiting peptide (50).
On the other hand, chronic CaMKII activation, as occurs in disease states such as heart failure (35, 37, 38), would prohibit resetting of the normal baseline KATP channel surface density. This would blunt the protective shortening of action potentials under repeated acute stress, thereby predisposing myocytes to worsened energetic insult and injury that would promote disease progression. In this way the plasticity of KATP channel surface presence can be viewed as vital for optimal cardiac function.
This series of experiments indicates that mutation of any one of Thr-180, Thr-224, or Tyr-330 is sufficient to interfere with CaMKII-mediated endocytosis. Based on our homology model of the Kir6.2 subunit and consistent with that of others (58, 59), Thr-180 is likely located in or near the channel pore at the site of ATP binding and adjacent to the internalization motif 330YSKF333. Our model suggests that Thr-180 is unlikely to directly interact with 330YSKF333 and that phosphorylation of Thr-180 is unlikely to alter accessibility to 330YSKF333. However, docking of μ2 may be influenced by Thr-180, assuming the ATP binding site is unoccupied. Because the non-phosphorylatable T180A mutation appears to augment ATP sensitivity of the channel, we can hypothesize that phosphorylation of Thr-180 by CaMKII would discourage ATP binding and thus allow μ2 to dock, thereby permitting endocytosis. Similarly, our model predicts that Thr-224 is unlikely to interact directly with 330YSKF333 and that phosphorylation of Thr-224 is unlikely to affect accessibility of 330YSKF333. However, it does appear that μ2 at 330YSKF333 on one Kir6.2 subunit may interact with Thr-224 on a neighboring Kir6.2 subunit. Thus, our model suggests several potential mechanisms for how Thr-180, Thr-224, and 330YSKF333 on Kir6.2 may interface with the cellular endocytic machinery to permit CaMKII-triggered endocytosis of cardiac KATP channels. These prospects will be the subject of future experimental studies.
Our data also indicate the significance of the Kir6.2 residues Thr-180 and Thr-224 and their phosphorylation state in the regulation of KATP channel gating. The effect of the T180A mutant on KATP channel function and current capacity is particularly intriguing. The immunofluorescence images clearly demonstrate bright fluorescence at the cell membrane thus indicating that the effect of the T180A mutation on reduced NPo of single channel KATP channel recordings does not primarily occur through interference with surface presence of the mutant channel. Rather, it appears the mutation alters channel gating and/or ATP sensitivity. Interestingly, a previous report of the Thr-180 mutation showed abnormal biophysical properties of the channel as well (47). In that study, whole cell KATP channel activity in response to openers and/or DNP, as was done in the current study, was not tested nor was the full-length Kir6.2T180A mutant in the presence of SUR2A tested in the inside-out mode as was done here, and thus a direct comparison with our results is not possible. Because we only saw rare lone channels in the full-length Kir6.2T180A mutant with SUR2A, we were unable to assess the effect of this mutation on rundown. Nonetheless, our study is in line with the findings of other investigators that indicate the Thr-180 residue is a critical determinant of channel gating behavior (47, 58).
The T224A and T224(D/E) mutations in Kir6.2 have also been previously described and found to alter protein kinase A-mediated regulation of KATP channel gating (52, 53). We confirmed that mimicking phosphorylation of Thr-224, by the T224E mutation, shifts the ATP sensitivity of Kir6.2/SUR2A complexes to the right. However, this would oppose the decrease in KATP channel current in the presence of CaMKII activation witnessed here and thus can be excluded as contributor to the phenomenon.
Taken together with previous findings that changes in KATP channel surface abundance have a significant impact on cardiac energetics (23–25), sarcolemmal KATP channel down-regulation via CaMKII-mediated endocytosis reveals a new mechanism for control of myocardial performance and energy use. Indeed, CaMKII inhibition has been investigated as a promising treatment and prevention strategy for heart failure (35); however, diverse tissue expression and pleiotropic vital functions of CaMKII make it a problematic target. Similarly, targeting of KATP channel gating for cardioprotection has proven problematic, as presently available KATP channel openers do not have cardiac specificity, and their therapeutic use is limited due to potent effects on vasculature tone and related hypotension (60, 61). Thus, in identifying an important interaction between CaMKII and cardiac KATP channels, this study provides a new set of potential targets for the promotion of cardiac energy efficiency and stress resistance.
This work was supported by National Institutes of Health Grants HL092286 and R01HL113089 (to D. H.-Z.), HL093368 and R01DK092412 (to L. Z.), R01HL079031, R01HL62494, R01HL70250, and R01HL113001 (to M. E. A.), HL084583 and HL083422 (to P. J. M.), and HL058812 (to P. M. S). This work was also supported by VA Merit Review Program Grant 1I0BX000718 (to L. Z.), The Medical Research Council, United Kingdom Grant G0802050 (to A. Sivaprasadarao), The Carver Trust Grant 01-224 (to D. H.-Z. and L. Z.), The Central Society for Clinical Research (to D. H.-Z.), The Fraternal Order of Eagles (to D. H.-Z. and L. Z.), Saving Tiny Hearts Society (to P. J. M.), an American Heart Association Established Investigator Award (to P. J. M.), Agence Nationale de la Recherche (ANR-09-PIRI-0010 and Laboratory of Excellence “Ion Channel Science and Therapeutics” network grant; to M. V.), and Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease (to M. E. A. and P. J. M.).
- SUR
- sulfonylurea receptor
- CaMKII
- calcium/calmodulin-dependent protein kinase II
- DNP
- 2,3 dinitrophenol
- NS
- not significant
- Bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Weiss J. N., Venkatesh N. (1993) Metabolic regulation of cardiac ATP-sensitive K+ channels. Cardiovasc. Drugs Ther. 7, 499–505 [DOI] [PubMed] [Google Scholar]
- 2. Noma A. (1983) ATP-regulated K+ channels in cardiac muscle. Nature 305, 147–148 [DOI] [PubMed] [Google Scholar]
- 3. Aguilar-Bryan L., Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 [DOI] [PubMed] [Google Scholar]
- 4. Ashcroft F. M. (1988) Adenosine 5′-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 11, 97–118 [DOI] [PubMed] [Google Scholar]
- 5. Lederer W. J., Nichols C. G. (1989) Nucleotide modulation of the activity of rat heart ATP-sensitive K+ channels in isolated membrane patches. J. Physiol. 419, 193–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flagg T. P., Enkvetchakul D., Koster J. C., Nichols C. G. (2010) Muscle KATP channels. Recent insights to energy sensing and myoprotection. Physiol. Rev. 90, 799–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seino S., Miki T. (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 81, 133–176 [DOI] [PubMed] [Google Scholar]
- 8. Shyng S., Ferrigni T., Nichols C. G. (1997) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110, 643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zingman L. V., Hodgson D. M., Bast P. H., Kane G. C., Perez-Terzic C., Gumina R. J., Pucar D., Bienengraeber M., Dzeja P. P., Miki T., Seino S., Alekseev A. E., Terzic A. (2002) Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. U.S.A. 99, 13278–13283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hodgson D. M., Zingman L. V., Kane G. C., Perez-Terzic C., Bienengraeber M., Ozcan C., Gumina R. J., Pucar D., O'Coclain F., Mann D. L., Alekseev A. E., Terzic A. (2003) Cellular remodeling in heart failure disrupts K(ATP) channel-dependent stress tolerance. EMBO J. 22, 1732–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwappach B., Zerangue N., Jan Y. N., Jan L. Y. (2000) Molecular basis for K(ATP) assembly. Transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron 26, 155–167 [DOI] [PubMed] [Google Scholar]
- 12. Inagaki N., Gonoi T., Clement J. P., 4th, Namba N., Inazawa J., Gonzalez G., Aguilar-Bryan L., Seino S., Bryan J. (1995) Reconstitution of IKATP. An inward rectifier subunit plus the sulfonylurea receptor. Science 270, 1166–1170 [DOI] [PubMed] [Google Scholar]
- 13. Inagaki N., Gonoi T., Clement J. P., Wang C. Z., Aguilar-Bryan L., Bryan J., Seino S. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16, 1011–1017 [DOI] [PubMed] [Google Scholar]
- 14. Lefer D. J., Nichols C. G., Coetzee W. A. (2009) Sulfonylurea receptor 1 subunits of ATP-sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 19, 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clement J. P., 4th, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. (1997) Association and stoichiometry of K(ATP) channel subunits. Neuron. 18, 827–838 [DOI] [PubMed] [Google Scholar]
- 16. Babenko A. P., Bryan J. (2003) Sur domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 278, 41577–41580 [DOI] [PubMed] [Google Scholar]
- 17. Zingman L. V., Hodgson D. M., Bienengraeber M., Karger A. B., Kathmann E. C., Alekseev A. E., Terzic A. (2002) Tandem function of nucleotide binding domains confers competence to sulfonylurea receptor in gating ATP-sensitive K+ channels. J. Biol. Chem. 277, 14206–14210 [DOI] [PubMed] [Google Scholar]
- 18. Abraham M. R., Selivanov V. A., Hodgson D. M., Pucar D., Zingman L. V., Wieringa B., Dzeja P. P., Alekseev A. E., Terzic A. (2002) Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J. Biol. Chem. 277, 24427–24434 [DOI] [PubMed] [Google Scholar]
- 19. Carrasco A. J., Dzeja P. P., Alekseev A. E., Pucar D., Zingman L. V., Abraham M. R., Hodgson D., Bienengraeber M., Puceat M., Janssen E., Wieringa B., Terzic A. (2001) Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. U.S.A. 98, 7623–7628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bienengraeber M., Alekseev A. E., Abraham M. R., Carrasco A. J., Moreau C., Vivaudou M., Dzeja P. P., Terzic A. (2000) ATPase activity of the sulfonylurea receptor. A catalytic function for the KATP channel complex. FASEB J. 14, 1943–1952 [DOI] [PubMed] [Google Scholar]
- 21. O'Rourke B., Ramza B. M., Marban E. (1994) Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science 265, 962–966 [DOI] [PubMed] [Google Scholar]
- 22. Sasaki N., Sato T., Marbán E., O'Rourke B. (2001) ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 280, H1882–H1888 [DOI] [PubMed] [Google Scholar]
- 23. Alekseev A. E., Reyes S., Yamada S., Hodgson-Zingman D. M., Sattiraju S., Zhu Z., Sierra A., Gerbin M., Coetzee W. A., Goldhamer D. J., Terzic A., Zingman L. V. (2010) Sarcolemmal ATP-sensitive K+ channels control energy expenditure determining body weight. Cell Metab. 11, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zingman L. V., Zhu Z., Sierra A., Stepniak E., Burnett C. M., Maksymov G., Anderson M. E., Coetzee W. A., Hodgson-Zingman D. M. (2011) Exercise-induced expression of cardiac ATP-sensitive potassium channels promotes action potential shortening and energy conservation. J. Mol. Cell. Cardiol. 51, 72–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Z., Burnett C. M., Maksymov G., Stepniak E., Sierra A., Subbotina E., Anderson M. E., Coetzee W. A., Hodgson-Zingman D. M., Zingman L. V. (2011) Reduction in number of sarcolemmal KATP channels slows cardiac action potential duration shortening under hypoxia. Biochem. Biophys. Res. Commun. 415, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zingman L. V., Alekseev A. E., Hodgson-Zingman D. M., Terzic A. (2007) ATP-sensitive potassium channels. Metabolic sensing and cardioprotection. J. Appl. Physiol. 103, 1888–1893 [DOI] [PubMed] [Google Scholar]
- 27. Tong X., Porter L. M., Liu G., Dhar-Chowdhury P., Srivastava S., Pountney D. J., Yoshida H., Artman M., Fishman G. I., Yu C., Iyer R., Morley G. E., Gutstein D. E., Coetzee W. A. (2006) Consequences of cardiac myocyte-specific ablation of KATP channels in transgenic mice expressing dominant negative Kir6 subunits. Am. J. Physiol. Heart Circ. Physiol. 291, H543–H551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nichols C. G., Lederer W. J. (1991) Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am. J. Physiol. 261, H1675–H1686 [DOI] [PubMed] [Google Scholar]
- 29. Li J., Marionneau C., Koval O., Zingman L., Mohler P. J., Nerbonne J. M., Anderson M. E. (2007) Calmodulin kinase II inhibition enhances ischemic preconditioning by augmenting ATP-sensitive K+ current. Channels 1, 387–394 [DOI] [PubMed] [Google Scholar]
- 30. Hu K., Huang C. S., Jan Y. N., Jan L. Y. (2003) ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron 38, 417–432 [DOI] [PubMed] [Google Scholar]
- 31. Brown D. A., Chicco A. J., Jew K. N., Johnson M. S., Lynch J. M., Watson P. A., Moore R. L. (2005) Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J. Physiol. 569, 913–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang R., Khoo M. S., Wu Y., Yang Y., Grueter C. E., Ni G., Price E. E., Jr, Thiel W., Guatimosim S., Song L. S., Madu E. C., Shah A. N., Vishnivetskaya T. A., Atkinson J. B., Gurevich V. V., Salama G., Lederer W. J., Colbran R. J., Anderson M. E. (2005) Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417 [DOI] [PubMed] [Google Scholar]
- 33. Wu Y., Shintani A., Grueter C., Zhang R., Hou Y., Yang J., Kranias E. G., Colbran R. J., Anderson M. E. (2006) Suppression of dynamic Ca2+ transient responses to pacing in ventricular myocytes from mice with genetic calmodulin kinase II inhibition. J. Mol. Cell. Cardiol. 40, 213–223 [DOI] [PubMed] [Google Scholar]
- 34. Li J., Marionneau C., Zhang R., Shah V., Hell J. W., Nerbonne J. M., Anderson M. E. (2006) Calmodulin kinase II inhibition shortens action potential duration by up-regulation of K+ currents. Circ. Res. 99, 1092–1099 [DOI] [PubMed] [Google Scholar]
- 35. Anderson M. E., Brown J. H., Bers D. M. (2011) CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neef S., Maier L. S. (2007) Remodeling of excitation-contraction coupling in the heart. Inhibition of sarcoplasmic reticulum Ca2+ leak as a novel therapeutic approach. Curr. Heart Fail. Rep. 4, 11–17 [DOI] [PubMed] [Google Scholar]
- 37. Maier L. S. (2009) Role of CaMKII for signaling and regulation in the heart. Front. Biosci. 14, 486–496 [DOI] [PubMed] [Google Scholar]
- 38. Kushnir A., Shan J., Betzenhauser M. J., Reiken S., Marks A. R. (2010) Role of CaMKIIδ phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc. Natl. Acad. Sci. U.S.A. 107, 10274–10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zingman L.V., Alekseev A. E., Bienengraeber M., Hodgson D., Karger A. B., Dzeja P. P., Terzic A. (2001) Signaling in channel/enzyme multimers. ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31, 233–245 [DOI] [PubMed] [Google Scholar]
- 40. Bienengraeber M., Olson T. M., Selivanov V. A., Kathmann E. C., O'Cochlain F., Gao F., Karger A. B., Ballew J. D., Hodgson D. M., Zingman L. V., Pang Y. P., Alekseev A. E., Terzic A. (2004) ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 36, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Erickson J. R., Joiner M. L., Guan X., Kutschke W., Yang J., Oddis C. V., Bartlett R. K., Lowe J. S., O'Donnell S. E., Aykin-Burns N., Zimmerman M. C., Zimmerman K., Ham A. J., Weiss R. M., Spitz D. R., Shea M. A., Colbran R. J., Mohler P. J., Anderson M. E. (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grueter C. E., Abiria S. A., Dzhura I., Wu Y., Ham A. J., Mohler P. J., Anderson M. E., Colbran R. J. (2006) L-type Ca2+ channel facilitation mediated by phosphorylation of the β subunit by CaMKII. Mol. Cell 23, 641–650 [DOI] [PubMed] [Google Scholar]
- 43. Moretti S., Armougom F., Wallace I. M., Higgins D. G., Jongeneel C. V., Notredame C. (2007) The M-Coffee web server. A meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res. 35, W645–W648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Light P. (1996) Regulation of ATP-sensitive potassium channels by phosphorylation. Biochim. Biophys. Acta 1286, 65–73 [DOI] [PubMed] [Google Scholar]
- 45. Hu K., Duan D., Li G. R., Nattel S. (1996) Protein kinase C activates ATP-sensitive K+ current in human and rabbit ventricular myocytes. Circ. Res. 78, 492–498 [DOI] [PubMed] [Google Scholar]
- 46. Aziz Q., Thomas A. M., Khambra T., Tinker A. (2012) Regulation of the ATP-sensitive potassium channel subunit, Kir6.2, by a Ca2+-dependent protein kinase C. J. Biol. Chem. 287, 6196–6207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Light P. E., Bladen C., Winkfein R. J., Walsh M. P., French R. J. (2000) Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. U.S.A. 97, 9058–9063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ito K., Sato T., Arita M. (2001) Protein kinase C isoform-dependent modulation of ATP-sensitive K+ channels during reoxygenation in guinea-pig ventricular myocytes. J. Physiol. 532, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu Y., Gao W. D., O'Rourke B., Marban E. (1996) Synergistic modulation of ATP-sensitive K+ currents by protein kinase C and adenosine. Implications for ischemic preconditioning. Circ. Res. 78, 443–454 [DOI] [PubMed] [Google Scholar]
- 50. Wu Y., Luczak E. D., Lee E. J., Hidalgo C., Yang J., Gao Z., Li J., Wehrens X. H., Granzier H., Anderson M. E. (2012) CaMKII effects on inotropic but not lusitropic force frequency responses require phospholamban. J. Mol. Cell. Cardiol. 53, 429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tucker S. J., Gribble F. M., Zhao C., Trapp S., Ashcroft F. M. (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387, 179–183 [DOI] [PubMed] [Google Scholar]
- 52. Lin Y. F., Jan Y. N., Jan L. Y. (2000) Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 19, 942–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Béguin P., Nagashima K., Nishimura M., Gonoi T., Seino S. (1999) PKA-mediated phosphorylation of the human K(ATP) channel. Separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 18, 4722–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mankouri J., Taneja T. K., Smith A. J., Ponnambalam S., Sivaprasadarao A. (2006) Kir6.2 mutations causing neonatal diabetes prevent endocytosis of ATP-sensitive potassium channels. EMBO J. 25, 4142–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herrmann T. L., Morita C. T., Lee K., Kusner D. J. (2005) Calmodulin kinase II regulates the maturation and antigen presentation of human dendritic cells. J. Leukoc. Biol. 78, 1397–1407 [DOI] [PubMed] [Google Scholar]
- 56. Perry C., Le H., Grichtchenko I. I. (2007) ANG II and calmodulin/CaMKII regulate surface expression and functional activity of NBCe1 via separate means. Am. J. Physiol. Renal Physiol. 293, F68–F77 [DOI] [PubMed] [Google Scholar]
- 57. Ferrero J. M., Jr, Sáiz J., Ferrero J. M., Thakor N. V. (1996) Simulation of action potentials from metabolically impaired cardiac myocytes. Role of ATP-sensitive K+ current. Circ. Res. 79, 208–221 [DOI] [PubMed] [Google Scholar]
- 58. Antcliff J. F., Haider S., Proks P., Sansom M. S., Ashcroft F. M. (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 24, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trapp S., Haider S., Jones P., Sansom M. S., Ashcroft F. M. (2003) Identification of residues contributing to the ATP binding site of Kir6.2. EMBO J. 22, 2903–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moreau C., Jacquet H., Prost A. L., D'hahan N., Vivaudou M. (2000) The molecular basis of the specificity of action of K(ATP) channel openers. EMBO J. 19, 6644–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ashcroft F. M., Gribble F. M. (2000) New windows on the mechanism of action of K(ATP) channel openers. Trends Pharmacol. Sci. 21, 439–445 [DOI] [PubMed] [Google Scholar]