

Background: Cardiolipin (CL) deficiency causes multiple defects affecting mitochondrial bioenergetics.
Results: CL deficiency leads to defective mitochondrial Fe-S biogenesis, causing decreased activity of several mitochondrial and cytosolic Fe-S proteins and perturbation of iron homeostasis.
Conclusion: CL is an important regulator of mitochondrial and cellular iron homeostasis.
Significance: Mitochondrial iron homeostasis may be an important physiological modifier that contributes to the clinical phenotypes observed in Barth syndrome patients.
Keywords: Cardiolipin, Iron, Iron Metabolism, Iron-Sulfur Protein, Mitochondria
Abstract
Cardiolipin (CL) is the signature phospholipid of mitochondrial membranes, where it is synthesized locally and plays a critical role in mitochondrial bioenergetic functions. The importance of CL in human health is underscored by the observation that perturbation of CL biosynthesis causes the severe genetic disorder Barth syndrome. To fully understand the cellular response to the loss of CL, we carried out genome-wide expression profiling of the yeast CL mutant crd1Δ. Our results show that the loss of CL in this mutant leads to increased expression of iron uptake genes accompanied by elevated levels of mitochondrial iron and increased sensitivity to iron and hydrogen peroxide. Previous studies have shown that increased mitochondrial iron levels result from perturbations in iron-sulfur (Fe-S) cluster biogenesis. Consistent with an Fe-S defect, deletion of ISU1, one of two ISU genes that encode the mitochondrial Fe-S scaffolding protein essential for the synthesis of Fe-S clusters, led to synthetic growth defects with the crd1Δ mutant. We further show that crd1Δ cells have reduced activities of mitochondrial Fe-S enzymes (aconitase, succinate dehydrogenase, and ubiquinol-cytochrome c oxidoreductase), as well as cytosolic Fe-S enzymes (sulfite reductase and isopropylmalate isomerase). Increased expression of ATM1 or YAP1 did not rescue the Fe-S defects in crd1Δ. These findings show for the first time that CL is required for Fe-S biogenesis to maintain mitochondrial and cellular iron homeostasis.
Introduction
Cardiolipin (CL)3 is a structurally and functionally unique phospholipid that is almost exclusively present in mitochondrial membranes (1, 2). The presence of CL is critical for maintaining mitochondrial function, structure, and membrane fluidity. Perturbation of CL synthesis alters mitochondrial bioenergetics, resulting in reduced membrane potential, inefficient coupling of respiration, and decreased ATP synthesis (3–6). In the inner membrane, CL is tightly associated with several proteins in respiratory complexes I, III, and IV (7). CL is essential for the stability of respiratory chain supercomplexes that, in yeast, are composed of dimeric ubiquinol-cytochrome c oxidoreductase (complex III) and one or two complexes of cytochrome c oxidase (complex IV) (8–10). Perturbation of CL synthesis due to mutations in the CL remodeling enzyme tafazzin causes the severe human genetic disorder known as Barth syndrome (BTHS) (11). Tafazzin (Taz1) deficiency in yeast leads to biochemical and bioenergetic defects similar to those seen in BTHS patients (12–15).
Although perturbation of CL synthesis due to loss of tafazzin leads to cardio- and skeletal myopathy, neutropenia, and growth retardation in BTHS (16), the clinical presentation of this disorder is highly variable, ranging from neonatal death to lack of clinical symptoms (16, 17). To gain insight into CL functions that might explain the pathology and variable phenotypes observed in BTHS, we carried out a genome-wide expression analysis in the yeast CL mutant crd1Δ, which lacks CL synthase. The most striking alterations in gene expression were observed in iron uptake genes. These genes encode components of the yeast high and low affinity iron uptake systems, collectively referred to as the iron regulon (18). Because the gene expression analyses indicating elevated expression of the iron regulon were carried out in iron-replete conditions, we hypothesized that CL might be required for mitochondrial Fe-S biogenesis and/or export of mitochondrial Fe-S co-factors to the cytosol, two processes known to induce up-regulation of the iron regulon (19, 20).
The assembly of Fe-S clusters from ferrous (Fe2+) and sulfide (S2−) ions does not occur spontaneously in living cells, as unchaperoned iron and sulfur are toxic. Rather, cells utilize a complex Fe-S assembly and transport process that is highly conserved from yeast to humans (21). The assembly of Fe-S clusters in the mitochondria begins on the highly conserved scaffolding protein Isu1 and its homolog Isu2 (22). The Nfs1-Isd11 complex delivers sulfur (23, 24), and Yfh1 donates iron (Fe2+) to the Isu scaffold (25). This process also includes ferredoxin (Arh1) and ferredoxin reductase (Yah1), which provide electrons for the reduction of sulfur to sulfide (26, 27). The Fe-S clusters assembled on the scaffold are transferred to the recipient apoproteins, in a process that is assisted by several proteins localized in the mitochondrial matrix (21).
Mitochondria also possess Fe-S export machinery, which transports an unknown Fe-S component from the mitochondria that is matured by the cytosolic machinery into 4Fe-4S clusters (28–30). The mitochondrial Fe-S export machinery includes the ABC transporter Atm1 in the inner membrane and the sulfhydryl oxidase Erv1 in the intermembrane space. Perturbations in mitochondrial Fe-S assembly or Fe-S export machinery are known to induce expression of the Aft1/Aft2-regulated iron uptake genes, leading to increased mitochondrial iron levels. Defects in mitochondrial Fe-S assembly lead to decreased maturation of both mitochondrial and cytosolic Fe-S proteins (20, 28, 31–34). In addition, excess mitochondrial iron causes oxidative damage to Fe-S clusters due to the formation of reactive oxygen species (ROS) (35–37).
In this study, we show that crd1Δ cells exhibit perturbations in iron homeostasis, including increased expression of the iron uptake genes, elevated mitochondrial iron levels, and growth sensitivity to both FeSO4 supplementation and the ROS-inducing agent H2O2. We further demonstrate that the loss of CL leads to decreased activities of both mitochondrial and cytosolic Fe-S enzymes, suggesting that the mechanism underlying altered iron homeostasis is perturbation of Fe-S biogenesis. Consistent with this conclusion, crd1Δ cells exhibit a synthetic genetic interaction with the Fe-S scaffolding protein Isu1. Additionally, the iron homeostasis defects in crd1Δ are not rescued by overexpression of ATM1, the major component of mitochondrial Fe-S export machinery, which is activated by CL (36), nor is it rescued by overexpression of YAP1, which regulates expression of antioxidant genes (38–40). Overexpression of ATM1 and YAP1 might reasonably be expected to overcome defective Fe-S cluster export from mitochondria. However, overexpression did not rescue the mutant defects, suggesting that the loss of CL affects the process of mitochondrial Fe-S biogenesis. This study is the first to demonstrate that CL is required for Fe-S cluster biogenesis and for the maintenance of mitochondrial and cellular iron homeostasis.
EXPERIMENTAL PROCEDURES
Yeast Strains and Growth Media
The yeast Saccharomyces cerevisiae strains used in this work are listed in Table 1. Synthetic defined (SD) medium contained adenine (20.25 mg/liter), arginine (20 mg/liter), histidine (20 mg/liter), leucine (60 mg/liter), lysine (20 mg/liter), methionine (20 mg/liter), threonine (300 mg/liter), tryptophan (20 mg/liter), and uracil (20 mg/liter), yeast nitrogen base without amino acids (Difco), and carbon source (fermentative) glucose (2%) or (respiratory) glycerol (3%) plus ethanol (0.65%) or (respiro-fermentative) galactose (2%). SD-drop out medium contained all of the above-mentioned ingredients except for the indicated amino acid. For growth experiments on excess iron, 1 μm CuSO4 was used, and FeSO4 was solubilized in 0.1 n HCl, filter-sterilized, and added to the culture medium at the indicated concentration. Complex media (YPD or YP-gal) contained yeast extract (1%), peptone (2%), and either glucose (2%) or galactose (2%) as indicated.
TABLE 1.
Yeast strains and plasmids used in this study
| Strain | Genotype | Source or Ref. |
|---|---|---|
| FGY3 | MATa, ura3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1 | 52 |
| FGY2 | MATa, ura 3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1, crd1Δ::URA3 | 52 |
| FGY3 isu1Δ | MATa, ura 3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1, isu1Δ::KanMX4 | This study |
| FGY2 isu1Δ | MATa, ura 3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1, crd1Δ::URA3, isu1Δ::KanMX4 | This study |
| FGY3 isu2Δ | MATa, ura 3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1, isu2Δ::KanMX4 | This study |
| FGY2 isu2Δ | MATa, ura 3-52, lys2-801, ade2-101, trp1-Δ1, his3-Δ200, leu2-Δ1, crd1Δ::URA3, isu2Δ::KanMX4 | This study |
| pCM182 | Low copy number plasmid, TRP1 nutritional marker | ATCC[regs] no. 87656 |
| pCM182-ISU1 | Derivative of pCM182, expresses ISU1 from the Tet-Off promoter | This study |
| pRS415 | Low copy number plasmid, LEU2 nutritional marker | 111 |
| YEp351 | High copy number plasmid, LEU2 nutritional marker | 112 |
| YEp351-YAP1 | Derivative of YEp351, expresses YAP1 from the native promoter | 42 |
Deletion mutants were constructed by replacing the entire open reading frame of the target gene with the KanMX4 cassette by homologous recombination. The KanMX4 cassette was amplified from the pUG6 plasmid using primers consisting of 51 nucleotides identical to the target gene flanking regions at the 5′ end and 21 nucleotides for the amplification of the KanMX4 gene at the 3′ end. The PCR product was transformed by electroporation into cells, and transformants were selected on YPD media containing G418 (300 μg/ml). Disruption of the target gene was confirmed by PCR using primers against the target gene coding sequences.
Plasmid Construction and Cloning
To construct the ISU1-overexpressing plasmid, a 538-bp sequence containing the entire open reading frame of ISU1 was amplified from yeast genomic DNA using BamHI-tagged primer ISU1-BamHIF (5-GGAAAACACAACGGATCCCACATATTTAACC-3) and PstI-tagged primer ISU1-PstIR (5-GATCTTGTTCTGCAGCCGGTTATCTTCTT-3). Similarly, a 2098-bp sequence containing the entire open reading frame of ATM1 was amplified using NotI-tagged primer ATM1-NotIF (5-TTGATAGATGCGGCCGCAACCTGCAAATG-3) and PstI-tagged primer ATM1-PstIR (5-TACATGTCTGCAGCAATATTTACTTACGAGCG-3). The PCR products were ligated to pCM182 (a low copy number plasmid with selectable marker TRP1) downstream of the Tet-Off promoter. All the plasmids were amplified and extracted using standard protocols. The plasmids were transformed into yeast strains using the yeast one-step transformation protocol (41). A high copy number Yep351-YAP1 overexpression plasmid was a kind gift from W. Scott Moye-Rowley (University of Iowa) (42).
Microarray Analysis
Yeast cells were grown to the early stationary phase in YPD, and total RNA was isolated by hot phenol extraction (43). RNA was further purified using an RNeasy kit from Qiagen. Yeast 6.4k microarray slides containing 6240 different yeast expressed sequence tags (double-spotted) were purchased from University Health Network (Toronto, Canada). Synthesis of Cy3- or Cy5-labeled cRNA and hybridization were performed using SlideHyb 1 buffer (Ambion) at the Research Technology Support Facility at Michigan State University (East Lansing, MI). The glass slides were scanned with an Affymetrix 428 array scanner and quantified using GenePix Pro 3.0 software (Axon). Array normalization and statistical analysis were performed using the “limma: Linear Models for Microarray Data” library module (version 2.2.0) of the R statistical package (version 2.2.0) (44–48). Slide intensity data were normalized using the global loess method. The least squares method was used for the linear model fit utilizing the Benjamini and Hochberg method to control the false discovery rate. Each experiment was repeated once with switched Cy3 and Cy5 labeling. The average of the four signal log ratios of each gene was computed and converted to a fold change. The raw data can be downloaded from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov, GPL3464).
Quantitative PCR (qPCR) Analysis
Yeast cultures (10 ml) were grown to the logarithmic growth phase; cells were harvested, and total RNA was isolated using the RNeasy Plus mini kit from Qiagen. The cDNAs were synthesized with Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics), and quantitative PCRs were performed in a 25-μl volume using BrilliantTM SYBR® Green QPCR master mix (Stratagene) in a 96-well plate. Duplicates were included for each reaction. The primers used for qPCR are listed in Table 2. ACT1 was used as the internal control, and the RNA level of the gene of interest was normalized to ACT1 levels. PCRs were initiated at 95 °C for 10 min for denaturation followed by 40 cycles consisting of 30 s at 95 °C and 60 s at 55 °C.
TABLE 2.
Primers used for qPCR analyses
| Gene | Primer | Sequence |
|---|---|---|
| ACT1 | Forward | TCGTGCTGTCTTCCCATCTATCG |
| Reverse | CGAATTGAGAGTTGCCCCAGAAG | |
| AFT1 | Forward | ATGCATCTAAAAGGCCATGC |
| Reverse | ACTGGCTTTTCGGTTTCCTT | |
| FIT1 | Forward | CCACCTCCTCTGAGTCAAGC |
| Reverse | CAGATTGGGCATCCCTAGAA | |
| FIT2 | Forward | ACAAAGGTTGTCACCGAAGG |
| Reverse | TGAACCTGAACCGTTTGTCA | |
| FIT3 | Forward | GCTACATCCTCTAGCACCGC |
| Reverse | GCACCCATCAAACCAGTACC | |
| FET3 | Forward | ACAGTTTCGATCCGGACAAC |
| Reverse | CACCTGGGTTATCGGCTTTA | |
| FTR1 | Forward | GATTCAACCTTGCCAGTGGT |
| Reverse | ATTGTCCAGTTCTGGGTTGC | |
| ARN1 | Forward | CTCGCGATCCTGTTAAGGAG |
| Reverse | GGGAGACCATATGAGTCGGA | |
| ARN2 | Forward | TGTGGGACTTGTCGGTGTTA |
| Reverse | GGGCCATGAAGGTATCAATG | |
| ARN3 | Forward | TGGATTAGCGGGAACGTAAC |
| Reverse | GGAATACAAGCTAGCGGCAG |
Biochemical Assays and Measurement of Mitochondrial Metal Ion Content
Mitochondria were isolated from cell lysates prepared as described previously (49). Briefly, spheroplasts created by lyticase were ruptured by Dounce homogenization, and mitochondria were isolated by differential centrifugation. Total protein concentration was determined with the Bradford assay kit (Pierce) with BSA as the standard. The following assays were performed in isolated mitochondria. Succinate dehydrogenase activity was assayed by determining succinate-dependent reduction of 2,6-dichlorophenolindophenol. The absorbance decrease at A600 was recorded as a reporter of decylubiquinone reduction (50). Ubiquinol-cytochrome c oxidoreductase activity was assayed by monitoring reduction of cytochrome c at A550 (51).
Cell extracts were prepared by resuspending cells in 500 μl of TNTEG buffer (10 mm Tris-Cl, pH 7.4, 2.5 mm EDTA, 150 mm NaCl, 10% v/v glycerol, 0.5% v/v Triton X-100) and subjecting them to mechanical breakage with glass beads. Cell debris and unbroken cells were separated by low speed centrifugation (2000 × g for 5 min at 4 °C). The obtained supernatant was further centrifuged at 13,000 × g for 10 min, and the resulting supernatant was transferred to a new tube. Total protein concentration was determined as mentioned above. The following enzyme assays were performed in whole-cell extracts. Aconitase was assayed by the aconitase-isocitrate dehydrogenase-coupled assay, in which NADPH formation was monitored at A340 (50). Sulfite reductase was assayed by monitoring methylene blue formation at A670 from sulfide produced by NADPH-dependent sulfite reduction (19, 50). Isopropylmalate isomerase was assayed by monitoring formation of isopropylmalate at A235 from dehydration of 3-isopropylmalate (50). Statistical significance of all enzyme assay results was determined by an analysis of variance and Bonferroni's post hoc test in KaleidaGraph. In the isopropylmalate isomerase assay, because the parental strains carry the leu2Δ null mutation, cells were transformed with a low/single copy pRS415 plasmid containing the LEU2 marker.
For the measurement of mitochondrial iron content, mitochondria were further purified via ultracentrifugation through a discontinuous Histodenz (Sigma) gradient (14 and 22%). Mitochondria (0.25 mg of mitochondrial protein) were digested in 70% HNO3 by boiling for 2 min and then diluted to 30% HNO3. Iron content was determined using an inductively coupled plasma-optical emission spectrometer.
RESULTS
Loss of CL Leads to Increased Expression of Iron Uptake Genes
To understand the cellular response to CL deficiency, we performed a genome-wide microarray analysis in cells of the CL synthase mutant crd1Δ, which completely lacks CL (52–54). The microarray analysis revealed increased expression of genes involved in iron homeostasis in the crd1Δ mutant (supplemental Table 1). To confirm the effect of loss of CL on expression of the iron regulon genes, we carried out quantitative PCR analysis of the Aft1-regulated iron uptake genes in crd1Δ. Cells were grown in SD respiratory media to the logarithmic growth phase, and RNA was extracted for mRNA quantitation, as described under “Experimental Procedures.” As seen in Fig. 1, the mRNA levels of AFT1, FIT1–3, FET3, FTR1, and ARN1–3 were up-regulated more than 3-fold in crd1Δ. Up-regulation of AFT1 and the iron regulon genes in the crd1Δ mutant suggested either deficient cellular iron levels or perturbation of mitochondrial Fe-S cluster biogenesis, and/or export of extra-mitochondrial Fe-S co-factors are perturbed in this mutant (20, 28, 55).
FIGURE 1.
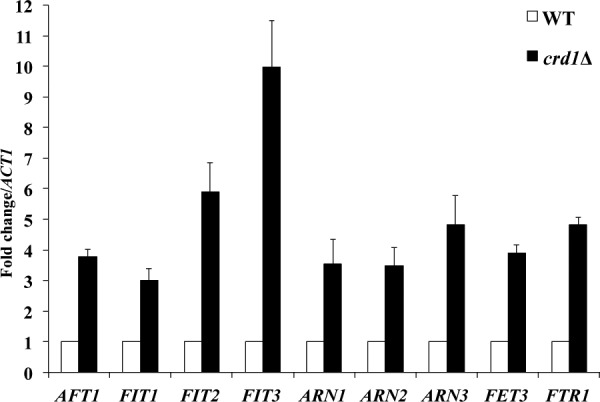
Increased expression of the iron regulon in crd1Δ. The mRNA levels of iron regulon genes were quantified by qPCR from cells grown in SD glycerol-ethanol at 30 °C to the logarithmic phase. Values are reported as fold change in expression over WT. Expression was normalized to the mRNA levels of the internal control ACT1. Data shown are mean ± S.E. (n = 6).
Perturbation of Iron Homeostasis in crd1Δ
We quantified mitochondrial iron using inductively coupled plasma-optical emission spectroscopy and found that iron levels in the crd1Δ mutant were significantly increased by 33% relative to WT levels (Fig. 2). This result suggested that the iron regulon in crd1Δ is up-regulated for a reason other than low cellular iron levels. Previous studies have reported that mutations in yeast genes involved in Fe-S cluster synthesis or in the export of Fe-S co-factors lead to elevated mitochondrial iron levels (20, 32, 34, 56–58).
FIGURE 2.
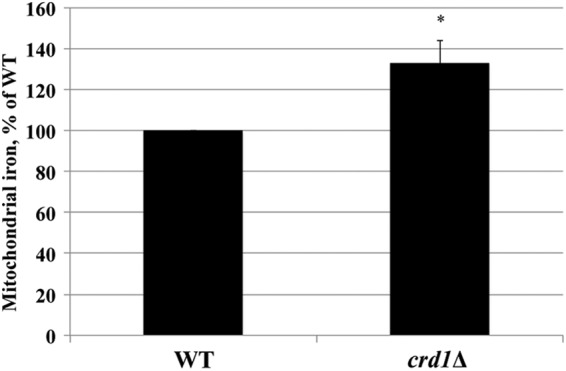
Increased mitochondrial iron levels in crd1Δ. Cells were grown in YP-galactose at 30 °C to the logarithmic phase, and mitochondrial iron levels were determined by an inductively coupled plasma-optical emission spectrometer. Data shown are mean ± S.E. (n = 9). The asterisk indicates a significant difference relative to WT.
Perturbation of mitochondrial Fe-S biogenesis leads to growth sensitivity in the presence of FeSO4 (59–62). As seen in Fig. 3, B and C, crd1Δ cells showed growth sensitivity to 5 and 10 mm FeSO4. This sensitivity to iron supplementation was observed when crd1Δ cells were grown in galactose (respiro-fermentative) and ethanol (respiratory) but not in glucose (fermentable) media (Fig. 3A). This is most likely because cells have a greater demand for iron in respiratory and respiro-fermentative media, so as to synthesize heme and Fe-S containing proteins involved in oxidative phosphorylation (63, 64).
FIGURE 3.
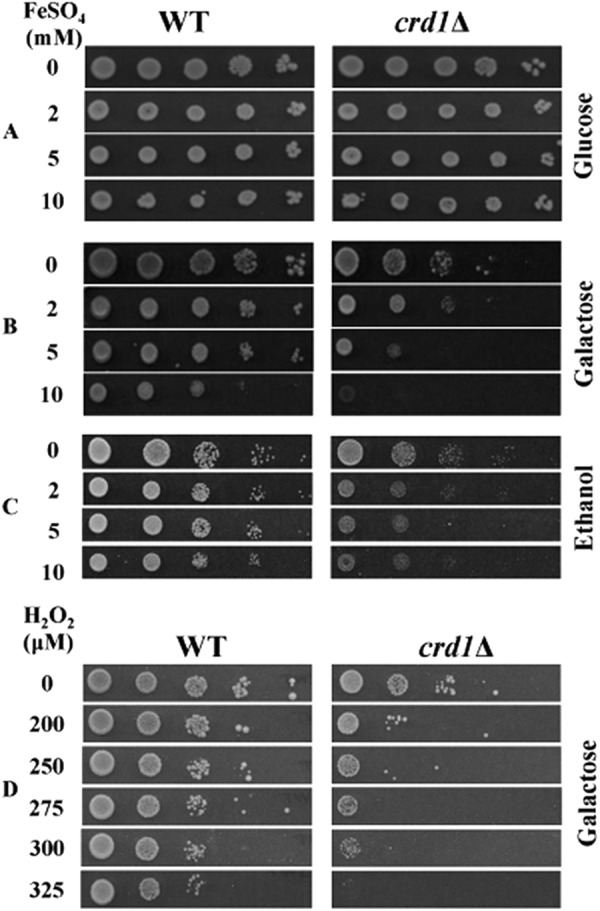
Sensitivity of crd1Δ to iron and H2O2. Cells were precultured in YPD overnight, serially diluted, spotted on SD plates containing glucose + FeSO4 (A), galactose + FeSO4 (B), ethanol + FeSO4 (C), and galactose + H2O2 (D), and incubated at 30 °C for 5 days.
Increased mitochondrial iron levels cause hypersensitivity to oxidative stress (32, 65, 66), which may be reflected in sensitivity to ROS-inducing agents. Consistent with increased oxidative stress, crd1Δ cells exhibited increased sensitivity to the ROS-inducing agent H2O2 (Fig. 3D). In summary, the absence of CL leads to increased mitochondrial iron levels as well as sensitivity to iron supplementation and oxidative stress, consistent with perturbation of iron homeostasis.
Fe-S Deficiencies in crd1Δ
We explored the possibility that the iron homeostasis defects in the crd1Δ mutant resulted from perturbation of Fe-S biogenesis. Perturbation of mitochondrial Fe-S assembly has been shown to cause decreased activity of mitochondrial proteins containing Fe-S clusters (26, 31, 32). To this end, we assayed the activities of the Fe-S enzymes succinate dehydrogenase, ubiquinol-cytochrome c oxidoreductase, and aconitase in crd1Δ. As seen in Table 3, these enzyme activities were decreased by ∼36, 45, and 78%, respectively, in the crd1Δ mutant. In addition, cytochrome c oxidase activity was decreased by 30% in the crd1Δ mutant (data not shown), consistent with previous studies (3, 67–69). The decreased activities of mitochondrial Fe-S proteins in crd1Δ are not due to reduced transcription of SDH2, RIP1, and ACO1 (supplemental Table 1). These results indicate that CL is required for the activity of mitochondrial Fe-S proteins present in the inner membrane and matrix.
TABLE 3.
Decreased mitochondrial and cytosolic Fe-S enzyme activities in crd1Δ
Cells were grown in galactose medium, and activities of Fe-S enzymes were assayed as described under “Experimental Procedures.” Data shown are mean ± S.D. (n ≥4).
| % activity in crd1Δ relative to WT | |
|---|---|
| Mitochondrial Fe-S enzymes | |
| Succinate dehydrogenase | 63.4 ± 9.5% |
| Ubiquinol cytochrome c oxidoreductase | 54.2 ± 13.8% |
| Aconitasea | 22.2 ± 13.9% |
| Cytosolic Fe-S enzymes | |
| Sulfite reductase | 54.2 ± 1.8% |
| Isopropylmalate isomeraseb | 51.8 ± 9.3% |
a These cells were grown at 35 °C. For sulfite reductase and isopropylmalate isomerase assays, cells were grown in media lacking methionine and cysteine or leucine, respectively.
b These cells were grown at 34 °C.
Mitochondrial Fe-S cluster biogenesis is also required for the maturation of cytosolic Fe-S proteins, as cytosolic Fe-S assembly depends on Fe-S co-factors synthesized in the mitochondria (26, 28, 31, 32). To determine the impact of CL deficiency on the activities of cytosolic Fe-S proteins, we measured the activities of sulfite reductase, which catalyzes the conversion of sulfite to sulfide, and isopropylmalate isomerase, which catalyzes the inter-conversion of α-isopropyl malate and β-isopropyl malate (70–73). Sulfite reductase and isopropylmalate isomerase each contain a 4Fe-4S cluster (74–76). As seen in Table 3, sulfite reductase activity was decreased by ∼46% in crd1Δ. Because sulfite reductase is required for the synthesis of methionine and cysteine, a decrease in activity would be expected to lead to methionine auxotrophy. As seen in Fig. 4A, crd1Δ was auxotrophic for methionine at elevated temperature. The crd1Δ mutant also exhibited an ∼49% decrease in activity of the leucine biosynthetic pathway enzyme isopropylmalate isomerase (Table 3) along with leucine auxotrophy at elevated temperature (Fig. 4B). These results indicate that the loss of CL also affects activity of Fe-S proteins in the cytosol. Taken together, these experiments indicate that activities of both mitochondrial and cytosolic Fe-S enzymes are affected by CL deficiency.
FIGURE 4.
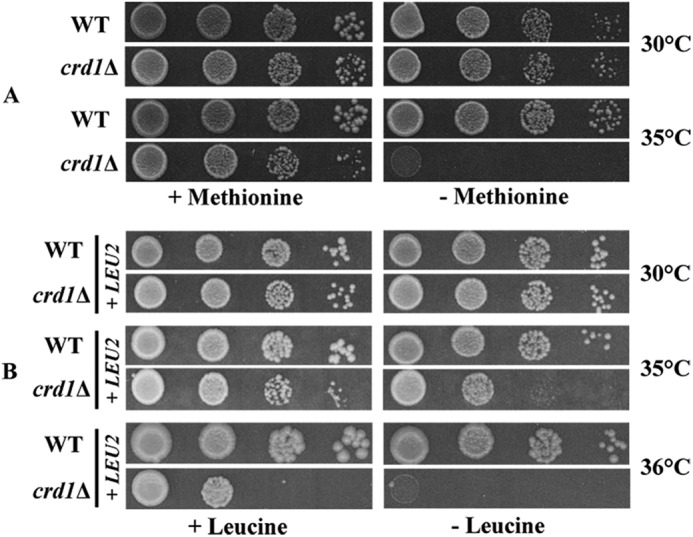
Methionine and leucine auxotrophy in crd1Δ. A, cells were precultured overnight in galactose medium, serially diluted, spotted on SD galactose media plates lacking methionine, and incubated for 3–5 days. B, cells were precultured overnight in galactose medium lacking leucine, serially diluted, spotted on SD glucose plates lacking leucine, and incubated for 3–5 days.
Genetic Interaction between CRD1 and ISU1
If CL is required for the biogenesis of Fe-S clusters in the mitochondria, crd1Δ would be expected to be sensitive to further perturbation of Fe-S biogenesis. Most of the genes involved in Fe-S cluster assembly, including NFS1, ISD11, YAH1, and ARH1, are required for viability (34, 77–79). YFH1 deletion mutants are viable in some genetic backgrounds but exhibit severe growth defects (80, 81). However, ISU1 and ISU2 both encode the mitochondrial Fe-S scaffolding protein and have overlapping functions. Single mutants isu1Δ and isu2Δ do not show growth defects, as the presence of either Isu1 or Isu2 is sufficient for survival, but deletion of both genes is lethal (31, 66). The crd1Δisu1Δ double mutant showed a synthetic growth defect in galactose media, but crd1Δisu2Δ grew normally (Fig. 5A). Isu1 is a more abundant scaffolding protein than Isu2 (82), which likely accounts for the more severe phenotypic defect of the crd1Δisu1Δ mutant. To confirm that genetic defects observed in the crd1Δisu1Δ double mutant are due to deletion of ISU1, we re-introduced ISU1 under the control of the Tet-Off promoter on a low copy plasmid, and this overexpression of ISU1 in the crd1Δisu1Δ double mutant reversed the growth defect (Fig. 5B). The genetic interaction between crd1Δ and isu1Δ is consistent with perturbation of Fe-S biogenesis in crd1Δ and suggests that decreased Fe-S biogenesis resulting from CL deficiency is exacerbated by further loss of the Fe-S scaffold in the presence of the isu1Δ mutation.
FIGURE 5.
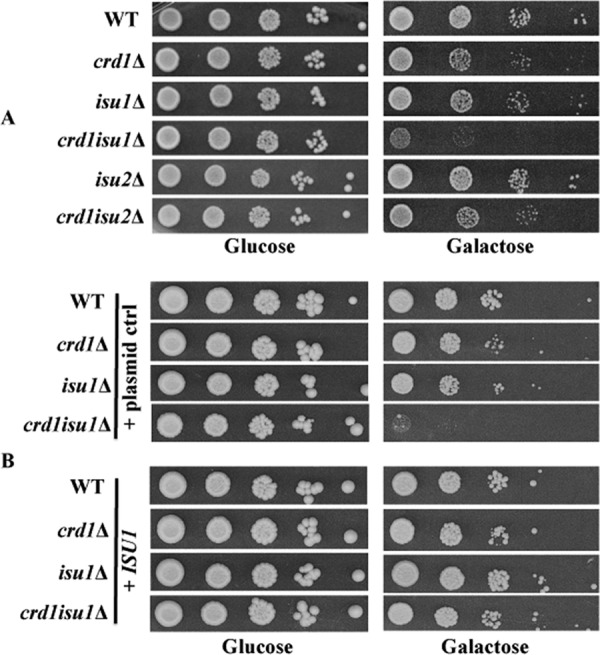
Genetic interaction between crd1Δ and isu1Δ. A, cells were precultured overnight in YPD, serially diluted, spotted on SD glucose or galactose plates, and incubated at 30 °C for 3–5 days. B, cells were precultured overnight in SD glucose lacking tryptophan, serially diluted, spotted on SD glucose or galactose plates lacking tryptophan, and incubated for 3–5 days.
Increased Expression of ATM1 Does Not Rescue Iron Defects in crd1Δ
The inner membrane protein Atm1, which is involved in the export of Fe-S co-factors from mitochondria, is activated by CL (83); the in vitro activity of Atm1 is ∼50% lower in the absence of CL. If up-regulation of the iron regulon resulted from reduced Atm1 activity, then increasing Atm1 levels in crd1Δ cells might be expected to restore the elevated iron regulon to WT levels. However, overexpression of ATM1 in crd1Δ cells did not restore the expression of FET3, FIT2, and FIT3 to WT levels (Fig. 6). In addition, others have shown that loss of ATM1 does not affect the activities of aconitase and succinate dehydrogenase (28). Therefore, it is not likely that Fe-S defects in the CL mutant result from Atm1 deficiency.
FIGURE 6.
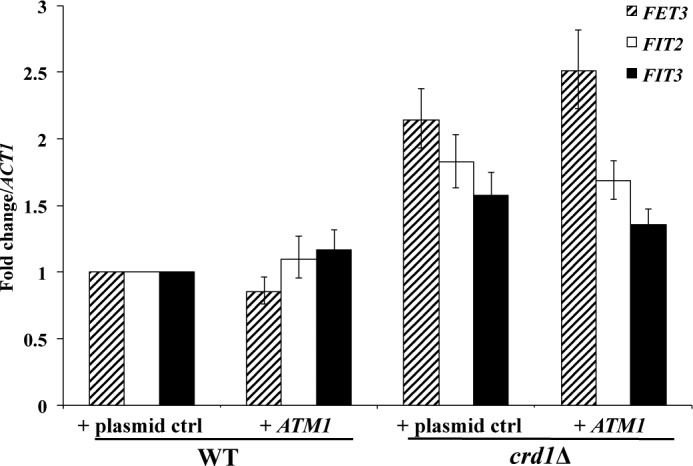
Overexpression of ATM1 in crd1Δ does not restore wild type expression of iron-uptake genes. The mRNA levels of FET3, FIT2, and FIT3 were quantified by qPCR in cells grown in SD glycerol-ethanol at 30 °C to the logarithmic phase. Values are reported as fold change in expression over WT. Expression was normalized to the mRNA levels of the internal control ACT1. Data shown are mean ± S.E. (n = 6).
Increased Expression of Antioxidant Genes Does Not Rescue Iron Defects in crd1Δ
Published studies have shown that Fe-S clusters in proteins such as aconitase are particularly sensitive to degradation by superoxide (35, 37). In a previous study, we showed that the loss of CL leads to decreased stability of respiratory supercomplexes (9), which is expected to cause increased ROS formation. Consistent with this, protein carbonylation, a sensitive marker of intracellular ROS, was significantly increased in crd1Δ (84). Therefore, we addressed the possibility that the iron-associated growth defects in crd1Δ cells may result from increased ROS. To do so, we determined the effect on crd1Δ cells of increasing antioxidant production via overexpression of YAP1, which regulates expression of a number of antioxidant genes required for tolerance to oxidants (39, 85). In response to H2O2, Yap1 positively regulates genes that affect glutathione metabolism (GSH1, GLR1, and ZWF1), catalase (CTT1), cytosolic thioredoxins (TRR1 and TRX2), glutathione peroxidases (GPX1 and GPX2), and superoxide dismutases (SOD1 and SOD2) (39, 40, 86, 87). As seen in Fig. 7, overexpression of YAP1 in crd1Δ did not alleviate methionine auxotrophy or growth sensitivity to iron and H2O2. Thus, there is no evidence that the iron-related growth phenotypes in crd1Δ arise from oxidative stress.
FIGURE 7.
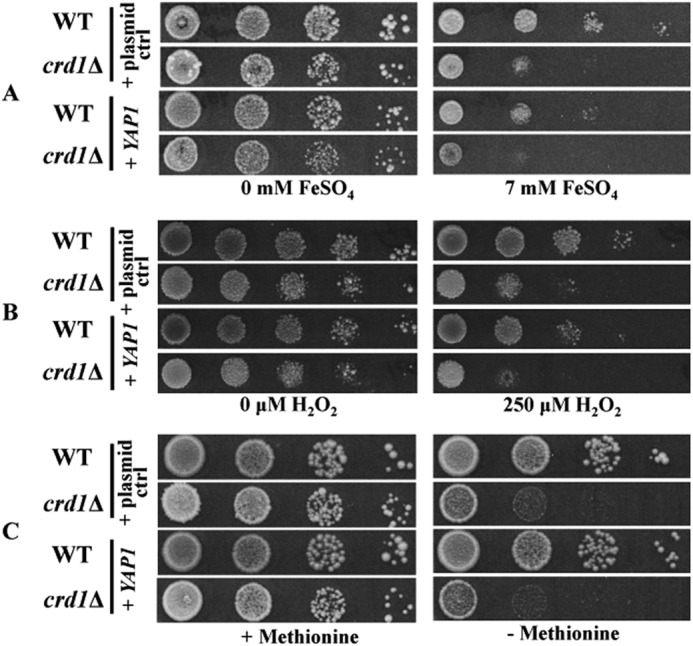
Overexpression of YAP1 in crd1Δ does not alleviate the iron-related growth phenotypes. Cells were precultured in SD media overnight, serially diluted and spotted on SD galactose plates supplemented with FeSO4 (top panels), supplemented with H2O2 (middle panels), and lacking methionine (bottom panels). Plates were incubated for 5 days.
DISCUSSION
In this study, we show for the first time that CL deficiency leads to altered mitochondrial and cellular iron homeostasis, as seen in increased expression of the iron regulon genes, elevated mitochondrial iron levels, and sensitivity to iron supplementation and ROS-inducing agents. Our findings indicate that the most likely mechanism underlying the iron homeostasis defects is that of perturbation of Fe-S biogenesis, as is evident from decreased activities of both mitochondrial and cytosolic Fe-S enzymes, concomitant auxotrophies for the amino acid products of these enzymes, and synthetic interaction of crd1Δ with the mitochondrial Fe-S scaffolding mutant isu1Δ. The observed decrease in Fe-S enzyme activity is not likely to result solely from a loss of direct enzyme activation by CL in the mitochondrial inner membrane. Although the mitochondrial enzymes succinate dehydrogenase and ubiquinol-cytochrome c oxidoreductase are membrane-bound and may be activated by CL, aconitase is a matrix enzyme that is unlikely to be directly regulated by CL. Furthermore, we observed reduced activity of cytosolic Fe-S enzymes sulfite reductase and isopropylmalate isomerase, which are not in contact with the mitochondrial membrane.
How does CL deficiency cause perturbation of mitochondrial Fe-S biogenesis? Several potential mechanisms can be ruled out. First, the Fe-S defects are most likely not due to decreased Atm1 activity, as overexpression of ATM1 did not rescue the iron regulon defects (Fig. 6). Furthermore, as previous studies indicated that aconitase and succinate dehydrogenase activities are not affected by decreased expression of ATM1, it is unlikely that decreased activities of aconitase and succinate dehydrogenase observed in the CL mutant result from Atm1 deficiency (28). A second potential mechanism, disruption of Fe-S clusters by increased ROS in the CL mutant, is also unlikely, as overexpression of YAP1 in crd1Δ did not alleviate methionine auxotrophy or growth sensitivity to iron and H2O2 (Fig. 7). Furthermore, we observed elevated expression of the iron regulon genes even in fermentative growth conditions, during which protein carbonylation in CL mutants is not increased (84). In addition, expression of antioxidant genes is not increased during these growth conditions in crd1Δ (supplemental Table 1). Therefore, there is no evidence that ROS contributes to the observed iron phenotypes of crd1Δ cells.
A third possibility, perturbation of glutathione (GSH) metabolism, is also not a likely cause of Fe-S defects in the CL mutant. GSH plays a critical role in maintaining an intracellular reducing environment and regulates cellular iron homeostasis (88, 89). Although perturbation of GSH metabolism does lead to elevated mitochondrial iron levels and decreased cytosolic Fe-S biogenesis, depletion of GSH does not affect activities of the mitochondrial Fe-S proteins succinate dehydrogenase and aconitase, which are reduced in crd1Δ (88). However, depletion of GSH, a tripeptide of glutamate, cysteine, and glycine, is a predicted outcome of Fe-S deficiency. First, the glutamate precursor α-ketoglutarate is likely to be depleted as a result of aconitase deficiency. Second, decreased activity of the Fe-S enzyme glutamate synthase would lead to a decrease in the conversion of glutamine and α-ketoglutarate to glutamate (90, 91). Third, decreased activity of sulfite reductase is expected to affect synthesis of methionine, the sulfur donor for synthesis of cysteine. Therefore, it is probable that glutathione deficiency is a downstream effect of Fe-S defects in CL-deficient cells.
Previous studies have indicated that yeast cells exhibit iron deficiency resulting from defective vacuolar protein sorting or activation of Fet3, which may activate the iron regulon (92, 93). However, defective vacuolar function is not a likely cause of up-regulation of the iron regulon in crd1Δ cells because, in this study, mitochondria from crd1Δ cells contain elevated, rather than decreased, levels of iron.
The most likely explanation for perturbation of Fe-S biogenesis in CL-deficient cells is that alterations in the mitochondrial membrane perturb the stability and integrity of the protein complexes that drive mitochondrial protein import (94, 95). We have shown that crd1Δ cells exhibit defective import of precursor proteins into mitochondria (3, 94). Cells lacking CL may be compromised in the import of a protein or nutrient important for Fe-S biogenesis in the matrix. Recent studies have shown that Zim17, a heat-shock protein, interacts with both Ssc1 and PAM to promote their activities (96, 97). Mitochondria from the ZIM17 mutant exhibit decreased protein import due to aggregation of Pam16, Ssc1, and Ssq1 proteins (96–99). Aggregation of both Ssc1 and Ssq1 results in decreased Fe-S biogenesis, leading to up-regulation of the Aft1-controlled iron regulon. The loss of CL may affect mitochondrial import or processing of Fe-S biosynthetic proteins or, alternatively, affect nutrient import through inner membrane carrier proteins. Experiments to address this mechanism are in progress.
We propose the following model for the role of CL in maintaining mitochondrial and cellular iron homeostasis (Fig. 8). CL deficiency leads to decreased mitochondrial import of Fe-S proteins resulting in defects in Fe-S cluster biogenesis and maturation of Fe-S proteins and thus reduced activities of mitochondrial and cytosolic Fe-S enzymes. The cellular response to the decrease in Fe-S biogenesis is up-regulation of the iron regulon, leading to elevated mitochondrial iron levels.
FIGURE 8.
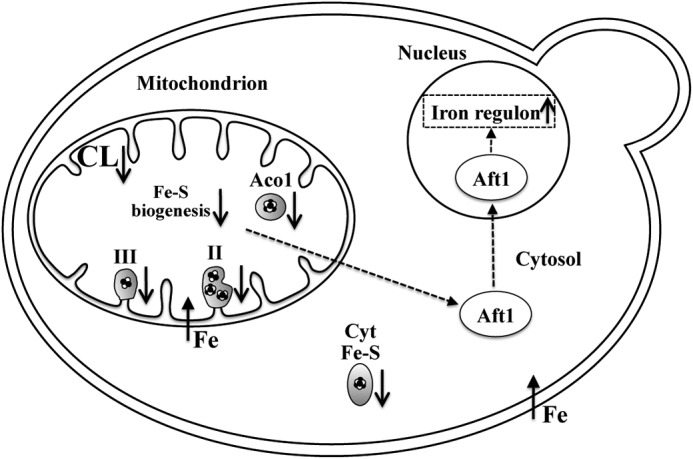
Perturbation of Fe-S biogenesis in crd1Δ. In the proposed model, loss of CL leads to decreased Fe-S biogenesis and maturation of Fe-S proteins, resulting in reduced activities of succinate dehydrogenase (II), ubiquinol-cytochrome c oxidoreductase (III), and aconitase (Aco1) in the mitochondria, as well as cytosolic Fe-S enzymes (Cyt Fe-S) sulfite reductase and isopropylmalate isomerase. Decreased Fe-S biogenesis is sensed by Aft1, which activates expression of the iron regulon genes, leading to increased mitochondrial iron levels.
It remains unclear how CL deficiency contributes to the observed pathology in BTHS. Interestingly, some of the clinical symptoms found in BTHS patients are also seen in patients with Fe-S biogenesis defects. Mutations in the human ISCU gene, which is homologous to yeast ISU1, lead to deficiencies in succinate dehydrogenase and aconitase in skeletal muscle, causing cardiomyopathy, lactic acidosis, muscle weakness, and exercise intolerance (100–103). Depletion of several proteins of the Fe-S biosynthetic machinery severely affects mitochondrial inner membrane structure and cristae morphology (104, 105), similar to what has been observed in the lymphoblasts of BTHS patients (106). In addition, deficiency of frataxin, which is involved in mitochondrial Fe-S biogenesis, is characterized by hypertrophic cardiomyopathy and heart failure (107). Interestingly, overexpression of frataxin leads to increased mitochondrial membrane potential, elevated ATP levels, resistance to oxidative stress, and life span extension (108, 109), defects which are characteristic of CL deficiency (7). In transgenic mice, overexpression of frataxin counteracted cardiotoxic stress, preventing cardiomyopathy and cardiac failure (110). We suggest that mitochondrial iron homeostasis may be an important physiological modifier that contributes to the phenotypes observed in BTHS patients.
Acknowledgments
We thank Roland Lill for the generous gifts of plasmids and valuable suggestions; Quan He, Tamara Hendrickson, Liangjun Zhao, Amit Joshi, Guiling Li, Vaishnavi Raja, and Sonia Gupta for helpful discussions; David Njus for the use of equipment; Jeff Landgraf and the Research Technology Support Facility at Michigan State University for assistance in carrying out microarray experiments and data analysis; and Kiran Koya for help with qPCR and microarray statistical analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 HL 084218 (to M. L. G.), R01 ES03817 from NIEHS (to D. R. W.), and T32 HL007576-25 (to J. L. F.). This work was also supported by the Barth Syndrome Foundation (to M. L. G.) and graduate enhancement research funds from Wayne State University (to V. A. P.).

This article contains supplemental Table 1.
- CL
- cardiolipin
- BTHS
- Barth syndrome
- Fe-S
- iron-sulfur
- SD
- synthetic defined
- YPD
- yeast extract, peptone, and dextrose
- Aft1
- activator of ferrous transport
- qPCR
- quantitative real time PCR
- ROS
- reactive oxygen species.
REFERENCES
- 1. Schlame M., Ren M. (2009) The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788, 2080–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schlame M., Rua D., Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 [DOI] [PubMed] [Google Scholar]
- 3. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 [DOI] [PubMed] [Google Scholar]
- 4. Koshkin V., Greenberg M. L. (2000) Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347, 687–691 [PMC free article] [PubMed] [Google Scholar]
- 5. Koshkin V., Greenberg M. L. (2002) Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 364, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joshi A. S., Zhou J., Gohil V. M., Chen S., Greenberg M. L. (2009) Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M., Mileykovskaya E., Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 9. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 10. Cruciat C. M., Brunner S., Baumann F., Neupert W., Stuart R. A. (2000) The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275, 18093–18098 [DOI] [PubMed] [Google Scholar]
- 11. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., Toniolo D. (1996) A novel X-linked gene, G4.5, is responsible for Barth syndrome. Nat. Genet. 12, 385–389 [DOI] [PubMed] [Google Scholar]
- 12. Brandner K., Mick D. U., Frazier A. E., Taylor R. D., Meisinger C., Rehling P. (2005) Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes. Implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 [DOI] [PubMed] [Google Scholar]
- 14. Gu Z., Valianpour F., Chen S., Vaz F. M., Hakkaart G. A., Wanders R. J., Greenberg M. L. (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant. A model for Barth syndrome. Mol. Microbiol. 51, 149–158 [DOI] [PubMed] [Google Scholar]
- 15. Ma L., Vaz F. M., Gu Z., Wanders R. J., Greenberg M. L. (2004) The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Δ mutant. Implications for Barth syndrome. J. Biol. Chem. 279, 44394–44399 [DOI] [PubMed] [Google Scholar]
- 16. Barth P. G., Wanders R. J., Vreken P., Janssen E. A., Lam J., Baas F. (1999) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060). J. Inherit. Metab. Dis. 22, 555–567 [DOI] [PubMed] [Google Scholar]
- 17. Bleyl S. B., Mumford B. R., Thompson V., Carey J. C., Pysher T. J., Chin T. K., Ward K. (1997) Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 61, 868–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kosman D. J. (2003) Molecular mechanisms of iron uptake in fungi. Mol. Microbiol. 47, 1185–1197 [DOI] [PubMed] [Google Scholar]
- 19. Rutherford J. C., Ojeda L., Balk J., Mühlenhoff U., Lill R., Winge D. R. (2005) Activation of the iron regulon by the yeast Aft1/Aft2 transcription factors depends on mitochondrial but not cytosolic iron-sulfur protein biogenesis. J. Biol. Chem. 280, 10135–10140 [DOI] [PubMed] [Google Scholar]
- 20. Hausmann A., Samans B., Lill R., Mühlenhoff U. (2008) Cellular and mitochondrial remodeling upon defects in iron-sulfur protein biogenesis. J. Biol. Chem. 283, 8318–8330 [DOI] [PubMed] [Google Scholar]
- 21. Lill R., Mühlenhoff U. (2006) Iron-sulfur protein biogenesis in eukaryotes. Components and mechanisms. Annu. Rev. Cell Dev. Biol. 22, 457–486 [DOI] [PubMed] [Google Scholar]
- 22. Mühlenhoff U., Gerber J., Richhardt N., Lill R. (2003) Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 22, 4815–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mühlenhoff U., Balk J., Richhardt N., Kaiser J. T., Sipos K., Kispal G., Lill R. (2004) Functional characterization of the eukaryotic cysteine desulfurase Nfs1p from Saccharomyces cerevisiae. J. Biol. Chem. 279, 36906–36915 [DOI] [PubMed] [Google Scholar]
- 24. Zheng L., White R. H., Cash V. L., Dean D. R. (1994) Mechanism for the desulfurization of l-cysteine catalyzed by the nifS gene product. Biochemistry 33, 4714–4720 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y., Lyver E. R., Knight S. A., Pain D., Lesuisse E., Dancis A. (2006) Mrs3p, Mrs4p, and frataxin provide iron for Fe-S cluster synthesis in mitochondria. J. Biol. Chem. 281, 22493–22502 [DOI] [PubMed] [Google Scholar]
- 26. Lange H., Kaut A., Kispal G., Lill R. (2000) A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1050–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J., Saxena S., Pain D., Dancis A. (2001) Adrenodoxin reductase homolog (Arh1p) of yeast mitochondria required for iron homeostasis. J. Biol. Chem. 276, 1503–1509 [DOI] [PubMed] [Google Scholar]
- 28. Kispal G., Csere P., Prohl C., Lill R. (1999) The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 18, 3981–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lange H., Lisowsky T., Gerber J., Mühlenhoff U., Kispal G., Lill R. (2001) An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rissler M., Wiedemann N., Pfannschmidt S., Gabriel K., Guiard B., Pfanner N., Chacinska A. (2005) The essential mitochondrial protein Erv1 cooperates with Mia40 in biogenesis of intermembrane space proteins. J. Mol. Biol. 353, 485–492 [DOI] [PubMed] [Google Scholar]
- 31. Gerber J., Neumann K., Prohl C., Mühlenhoff U., Lill R. (2004) The yeast scaffold proteins Isu1p and Isu2p are required inside mitochondria for maturation of cytosolic Fe/S proteins. Mol. Cell. Biol. 24, 4848–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mühlenhoff U., Richhardt N., Ristow M., Kispal G., Lill R. (2002) The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol Genet. 11, 2025–2036 [DOI] [PubMed] [Google Scholar]
- 33. Kaut A., Lange H., Diekert K., Kispal G., Lill R. (2000) Isa1p is a component of the mitochondrial machinery for maturation of cellular iron-sulfur proteins and requires conserved cysteine residues for function. J. Biol. Chem. 275, 15955–15961 [DOI] [PubMed] [Google Scholar]
- 34. Li J., Kogan M., Knight S. A., Pain D., Dancis A. (1999) Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J. Biol. Chem. 274, 33025–33034 [DOI] [PubMed] [Google Scholar]
- 35. Moreno-Cermeño A., Obis E., Bellí G., Cabiscol E., Ros J., Tamarit J. (2010) Frataxin depletion in yeast triggers up-regulation of iron transport systems before affecting iron-sulfur enzyme activities. J. Biol. Chem. 285, 41653–41664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bulteau A. L., Dancis A., Gareil M., Montagne J. J., Camadro J. M., Lesuisse E. (2007) Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 42, 1561–1570 [DOI] [PubMed] [Google Scholar]
- 37. Vasquez-Vivar J., Kalyanaraman B., Kennedy M. C. (2000) Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 275, 14064–14069 [DOI] [PubMed] [Google Scholar]
- 38. Inoue Y., Matsuda T., Sugiyama K., Izawa S., Kimura A. (1999) Genetic analysis of glutathione peroxidase in oxidative stress response of Saccharomyces cerevisiae. J. Biol. Chem. 274, 27002–27009 [DOI] [PubMed] [Google Scholar]
- 39. Lee J., Godon C., Lagniel G., Spector D., Garin J., Labarre J., Toledano M. B. (1999) Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J. Biol. Chem. 274, 16040–16046 [DOI] [PubMed] [Google Scholar]
- 40. Sugiyama K., Izawa S., Inoue Y. (2000) The Yap1p-dependent induction of glutathione synthesis in heat shock response of Saccharomyces cerevisiae. J. Biol. Chem. 275, 15535–15540 [DOI] [PubMed] [Google Scholar]
- 41. Chen D. C., Yang B. C., Kuo T. T. (1992) One-step transformation of yeast in stationary phase. Curr. Genet. 21, 83–84 [DOI] [PubMed] [Google Scholar]
- 42. Wemmie J. A., Szczypka M. S., Thiele D. J., Moye-Rowley W. S. (1994) Cadmium tolerance mediated by the yeast AP-1 protein requires the presence of an ATP-binding cassette transporter-encoding gene, YCF1. J. Biol. Chem. 269, 32592–32597 [PubMed] [Google Scholar]
- 43. Köhrer K., Domdey H. (1991) Preparation of high molecular weight RNA. Methods Enzymol. 194, 398–405 [DOI] [PubMed] [Google Scholar]
- 44. Smyth G. (2005) in Bioinformatics and Computational Biology Solutions Using R and Bioconductor (Gentleman R., Carey V. J., Huber W., Irizarry R. A., Dudoit S., eds) pp. 397–420, Springer-Verlag Inc., New York [Google Scholar]
- 45. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 46. Smyth G. K., Michaud J., Scott H. S. (2005) Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075 [DOI] [PubMed] [Google Scholar]
- 47. Smyth G. K., Speed T. (2003) Normalization of cDNA microarray data. Methods 31, 265–273 [DOI] [PubMed] [Google Scholar]
- 48. R Development Core Team (2005) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Institute for Statistics and Mathematics, University of Vienna, Vienna, Austria [Google Scholar]
- 49. Diekert K., de Kroon A. I., Kispal G., Lill R. (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65, 37–51 [DOI] [PubMed] [Google Scholar]
- 50. Pierik A. J., Netz D. J., Lill R. (2009) Analysis of iron-sulfur protein maturation in eukaryotes. Nat. Protoc. 4, 753–766 [DOI] [PubMed] [Google Scholar]
- 51. Atkinson A., Smith P., Fox J. L., Cui T. Z., Khalimonchuk O., Winge D. R. (2011) The LYR protein Mzm1 functions in the insertion of the Rieske Fe/S protein in yeast mitochondria. Mol. Cell. Biol. 31, 3988–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jiang F., Rizavi H. S., Greenberg M. L. (1997) Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol. Microbiol. 26, 481–491 [DOI] [PubMed] [Google Scholar]
- 53. Chang S. C., Heacock P. N., Mileykovskaya E., Voelker D. R., Dowhan W. (1998) Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J. Biol. Chem. 273, 14933–14941 [DOI] [PubMed] [Google Scholar]
- 54. Tuller G., Hrastnik C., Achleitner G., Schiefthaler U., Klein F., Daum G. (1998) YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae. FEBS Lett. 421, 15–18 [DOI] [PubMed] [Google Scholar]
- 55. Chen O. S., Crisp R. J., Valachovic M., Bard M., Winge D. R., Kaplan J. (2004) Transcription of the yeast iron regulon does not respond directly to iron but rather to iron-sulfur cluster biosynthesis. J. Biol. Chem. 279, 29513–29518 [DOI] [PubMed] [Google Scholar]
- 56. Garland S. A., Hoff K., Vickery L. E., Culotta V. C. (1999) Saccharomyces cerevisiae ISU1 and ISU2. Members of a well conserved gene family for iron-sulfur cluster assembly. J. Mol. Biol. 294, 897–907 [DOI] [PubMed] [Google Scholar]
- 57. Knight S. A., Sepuri N. B., Pain D., Dancis A. (1998) Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J. Biol. Chem. 273, 18389–18393 [DOI] [PubMed] [Google Scholar]
- 58. Kispal G., Csere P., Guiard B., Lill R. (1997) The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 418, 346–350 [DOI] [PubMed] [Google Scholar]
- 59. Ramazzotti A., Vanmansart V., Foury F. (2004) Mitochondrial functional interactions between frataxin and Isu1p, the iron-sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 557, 215–220 [DOI] [PubMed] [Google Scholar]
- 60. Foury F., Pastore A., Trincal M. (2007) Acidic residues of yeast frataxin have an essential role in Fe-S cluster assembly. EMBO Rep. 8, 194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leidgens S., De Smet S., Foury F. (2010) Frataxin interacts with Isu1 through a conserved tryptophan in its β-sheet. Hum. Mol. Genet. 19, 276–286 [DOI] [PubMed] [Google Scholar]
- 62. Foury F. (1999) Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett. 456, 281–284 [DOI] [PubMed] [Google Scholar]
- 63. Kaplan J., McVey Ward D., Crisp R. J., Philpott C. C. (2006) Iron-dependent metabolic remodeling in S. cerevisiae. Biochim. Biophys. Acta 1763, 646–651 [DOI] [PubMed] [Google Scholar]
- 64. Garber Morales J., Holmes-Hampton G. P., Miao R., Guo Y., Münck E., Lindahl P. A. (2010) Biophysical characterization of iron in mitochondria isolated from respiring and fermenting yeast. Biochemistry 49, 5436–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Babcock M., de Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M., Kaplan J. (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276, 1709–1712 [DOI] [PubMed] [Google Scholar]
- 66. Schilke B., Voisine C., Beinert H., Craig E. (1999) Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 96, 10206–10211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fry M., Green D. E. (1980) Cardiolipin requirement by cytochrome oxidase and the catalytic role of phospholipid. Biochem. Biophys. Res. Commun. 93, 1238–1246 [DOI] [PubMed] [Google Scholar]
- 68. Robinson N. C. (1993) Functional binding of cardiolipin to cytochrome c oxidase. J. Bioenerg. Biomembr. 25, 153–163 [DOI] [PubMed] [Google Scholar]
- 69. Sedlák E., Robinson N. C. (1999) Phospholipase A2 digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry 38, 14966–14972 [DOI] [PubMed] [Google Scholar]
- 70. Hsu Y. P., Schimmel P. (1984) Yeast LEU1. Repression of mRNA levels by leucine and relationship of 5′-noncoding region to that of LEU2. J. Biol. Chem. 259, 3714–3719 [PubMed] [Google Scholar]
- 71. Skala J., Capieaux E., Balzi E., Chen W. N., Goffeau A. (1991) Complete sequence of the Saccharomyces cerevisiae LEU1 gene encoding isopropylmalate isomerase. Yeast 7, 281–285 [DOI] [PubMed] [Google Scholar]
- 72. Masselot M., De Robichon-Szulmajster H. (1975) Methionine biosynthesis in Saccharomyces cerevisiae. I. Genetical analysis of auxotrophic mutants. Mol. Gen. Genet. 139, 121–132 [DOI] [PubMed] [Google Scholar]
- 73. Masselot M., Surdin-Kerjan Y. (1977) Methionine biosynthesis in Saccharomyces cerevisiae. II. Gene-enzyme relationships in the sulfate assimilation pathway. Mol. Gen. Genet. 154, 23–30 [DOI] [PubMed] [Google Scholar]
- 74. Yoshimoto A., Sato R. (1968) Studies on yeast sulfite reductase. I. Purification and characterization. Biochim. Biophys. Acta 153, 555–575 [DOI] [PubMed] [Google Scholar]
- 75. Crane B. R., Siegel L. M., Getzoff E. D. (1997) Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation. Heme activation via reduction-gated exogenous ligand exchange. Biochemistry 36, 12101–12119 [DOI] [PubMed] [Google Scholar]
- 76. Balk J., Aguilar Netz D. J., Tepper K., Pierik A. J., Lill R. (2005) The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol. Cell. Biol. 25, 10833–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wiedemann N., Urzica E., Guiard B., Müller H., Lohaus C., Meyer H. E., Ryan M. T., Meisinger C., Mühlenhoff U., Lill R., Pfanner N. (2006) Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 25, 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Barros M. H., Nobrega F. G. (1999) YAH1 of Saccharomyces cerevisiae. A new essential gene that codes for a protein homologous to human adrenodoxin. Gene 233, 197–203 [DOI] [PubMed] [Google Scholar]
- 79. Manzella L., Barros M. H., Nobrega F. G. (1998) ARH1 of Saccharomyces cerevisiae. A new essential gene that codes for a protein homologous to the human adrenodoxin reductase. Yeast 14, 839–846 [DOI] [PubMed] [Google Scholar]
- 80. Lesuisse E., Santos R., Matzanke B. F., Knight S. A., Camadro J. M., Dancis A. (2003) Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 12, 879–889 [DOI] [PubMed] [Google Scholar]
- 81. Zhang Y., Lyver E. R., Knight S. A., Lesuisse E., Dancis A. (2005) Frataxin and mitochondrial carrier proteins, Mrs3p and Mrs4p, cooperate in providing iron for heme synthesis. J. Biol. Chem. 280, 19794–19807 [DOI] [PubMed] [Google Scholar]
- 82. Andrew A. J., Song J. Y., Schilke B., Craig E. A. (2008) Posttranslational regulation of the scaffold for Fe-S cluster biogenesis, Isu. Mol. Biol. Cell 19, 5259–5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kuhnke G., Neumann K., Mühlenhoff U., Lill R. (2006) Stimulation of the ATPase activity of the yeast mitochondrial ABC transporter Atm1p by thiol compounds. Mol. Membr. Biol. 23, 173–184 [DOI] [PubMed] [Google Scholar]
- 84. Chen S., He Q., Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wemmie J. A., Steggerda S. M., Moye-Rowley W. S. (1997) The Saccharomyces cerevisiae AP-1 protein discriminates between oxidative stress elicited by the oxidants H2O2 and diamide. J. Biol. Chem. 272, 7908–7914 [DOI] [PubMed] [Google Scholar]
- 86. Lucau-Danila A., Lelandais G., Kozovska Z., Tanty V., Delaveau T., Devaux F., Jacq C. (2005) Early expression of yeast genes affected by chemical stress. Mol. Cell. Biol. 25, 1860–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jamieson D. J. (1998) Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast 14, 1511–1527 [DOI] [PubMed] [Google Scholar]
- 88. Sipos K., Lange H., Fekete Z., Ullmann P., Lill R., Kispal G. (2002) Maturation of cytosolic iron-sulfur proteins requires glutathione. J. Biol. Chem. 277, 26944–26949 [DOI] [PubMed] [Google Scholar]
- 89. Kumar C., Igbaria A., D'Autreaux B., Planson A. G., Junot C., Godat E., Bachhawat A. K., Delaunay-Moisan A., Toledano M. B. (2011) Glutathione revisited. A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 30, 2044–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Vanoni M. A., Curti B. (1999) Glutamate synthase. A complex iron-sulfur flavoprotein. Cell. Mol. life Sci. 55, 617–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shakoury-Elizeh M., Tiedeman J., Rashford J., Ferea T., Demeter J., Garcia E., Rolfes R., Brown P. O., Botstein D., Philpott C. C. (2004) Transcriptional remodeling in response to iron deprivation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Radisky D. C., Snyder W. B., Emr S. D., Kaplan J. (1997) Characterization of VPS41, a gene required for vacuolar trafficking and high affinity iron transport in yeast. Proc. Natl. Acad. Sci. U.S.A. 94, 5662–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Davis-Kaplan S. R., Ward D. M., Shiflett S. L., Kaplan J. (2004) Genome-wide analysis of iron-dependent growth reveals a novel yeast gene required for vacuolar acidification. J. Biol. Chem. 279, 4322–4329 [DOI] [PubMed] [Google Scholar]
- 94. Kutik S., Rissler M., Guan X. L., Guiard B., Shui G., Gebert N., Heacock P. N., Rehling P., Dowhan W., Wenk M. R., Pfanner N., Wiedemann N. (2008) The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J. Cell Biol. 183, 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tamura Y., Endo T., Iijima M., Sesaki H. (2009) Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J. Cell Biol. 185, 1029–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sanjuán Szklarz L. K., Guiard B., Rissler M., Wiedemann N., Kozjak V., van der Laan M., Lohaus C., Marcus K., Meyer H. E., Chacinska A., Pfanner N., Meisinger C. (2005) Inactivation of the mitochondrial heat shock protein zim17 leads to aggregation of matrix hsp70s followed by pleiotropic effects on morphology and protein biogenesis. J. Mol. Biol. 351, 206–218 [DOI] [PubMed] [Google Scholar]
- 97. Díaz de la Loza Mdel C., Gallardo M., García-Rubio M. L., Izquierdo A., Herrero E., Aguilera A., Wellinger R. E. (2011) Zim17/Tim15 links mitochondrial iron-sulfur cluster biosynthesis to nuclear genome stability. Nucleic Acids Res. 39, 6002–6015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Burri L., Vascotto K., Fredersdorf S., Tiedt R., Hall M. N., Lithgow T. (2004) Zim17, a novel zinc finger protein essential for protein import into mitochondria. J. Biol. Chem. 279, 50243–50249 [DOI] [PubMed] [Google Scholar]
- 99. Yamamoto H., Momose T., Yatsukawa Y., Ohshima C., Ishikawa D., Sato T., Tamura Y., Ohwa Y., Endo T. (2005) Identification of a novel member of yeast mitochondrial Hsp70-associated motor and chaperone proteins that facilitates protein translocation across the inner membrane. FEBS Lett. 579, 507–511 [DOI] [PubMed] [Google Scholar]
- 100. Haller R. G., Henriksson K. G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N. H., Gunder M., Ayyad K., Blomqvist C. G. (1991) Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 88, 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hall R. E., Henriksson K. G., Lewis S. F., Haller R. G., Kennaway N. G. (1993) Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 92, 2660–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mochel F., Knight M. A., Tong W. H., Hernandez D., Ayyad K., Taivassalo T., Andersen P. M., Singleton A., Rouault T. A., Fischbeck K. H., Haller R. G. (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kollberg G., Tulinius M., Melberg A., Darin N., Andersen O., Holmgren D., Oldfors A., Holme E. (2009) Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 132, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 104. Sheftel A. D., Wilbrecht C., Stehling O., Niggemeyer B., Elsässer H. P., Mühlenhoff U., Lill R. (2012) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol. Biol. Cell 23, 1157–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Biederbick A., Stehling O., Rösser R., Niggemeyer B., Nakai Y., Elsässer H. P., Lill R. (2006) Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell. Biol. 26, 5675–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Acehan D., Xu Y., Stokes D. L., Schlame M. (2007) Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 87, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Dürr A., Cossee M., Agid Y., Campuzano V., Mignard C., Penet C., Mandel J. L., Brice A., Koenig M. (1996) Clinical and genetic abnormalities in patients with Friedreich's ataxia. New Engl. J. Med. 335, 1169–1175 [DOI] [PubMed] [Google Scholar]
- 108. Ristow M., Pfister M. F., Yee A. J., Schubert M., Michael L., Zhang C. Y., Ueki K., Michael M. D., 2nd, Lowell B. B., Kahn C. R. (2000) Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 97, 12239–12243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Runko A. P., Griswold A. J., Min K. T. (2008) Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 582, 715–719 [DOI] [PubMed] [Google Scholar]
- 110. Schulz T. J., Westermann D., Isken F., Voigt A., Laube B., Thierbach R., Kuhlow D., Zarse K., Schomburg L., Pfeiffer A. F., Tschöpe C., Ristow M. (2010) Activation of mitochondrial energy metabolism protects against cardiac failure. Aging 2, 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhong Q., Gohil V. M., Ma L., Greenberg M. L. (2004) Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J. Biol. Chem. 279, 32294–32300 [DOI] [PubMed] [Google Scholar]
- 112. Hill J. E., Myers A. M., Koerner T. J., Tzagoloff A. (1986) Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2, 163–167 [DOI] [PubMed] [Google Scholar]