

Abstract
Objectives
To describe a severe form of demyelinating HIV-associated leukoencephalopathy in AIDS patients failing highly active antiretroviral therapy (HAART), its relationship to clinical and neuroimaging findings, and suggest hypotheses regarding pathogenesis.
Design and methods
AIDS patients who failed HAART and displayed severe leukoencephalopathy were included. All cases had detailed neuromedical, neuropsychological, neuroimaging and postmortem neuropathological examination. Immunocytochemical and PCR analyses were performed to determine brain HIV levels and to exclude other viruses.
Results
Seven recent autopsy cases of leukoencephalopathy in antiretroviral-experienced patients with AIDS were identified. Clinically, all were severely immunosuppressed, six (86%) had poorly controlled HIV replication despite combination antiretroviral therapy, and five (71%) had HIV-associated dementia. Neuropathologically, all seven had intense perivascular infiltration by HIV-gp41 immunoreactive monocytes/macrophages and lymphocytes, widespread myelin loss, axonal injury, microgliosis and astrogliosis. The extent of damage exceeds that described prior to the use of HAART. Brain tissue demonstrated high levels of HIV RNA but evidence of other pathogens, such as JC virus, Epstein–Barr virus, cytomegalovirus, human herpes virus type-8, and herpes simplex virus types 1 and 2, was absent. Comparison of the stages of pathology suggests a temporal sequence of events. In this model, white matter damage begins with perivascular infiltration by HIV-infected monocytes, which may occur as a consequence of antiretroviral-associated immune restoration. Intense infiltration by immune cells injures brain endothelial cells and is followed by myelin loss, axonal damage, and finally, astrogliosis.
Conclusions
Taken together, our findings provide evidence for the emergence of a severe form of HIV-associated leukoencephalopathy. This condition warrants further study and increased vigilance among those who provide care for HIV-infected individuals.
Keywords: HIV leukoencephalopathy, antiretroviral therapy
Introduction
Combination antiretroviral therapy (ART) extends the survival of many HIV-infected individuals and decreases the occurrence of several opportunistic conditions [1,2]. For example, the incidence of cytomegalovirus (CMV) disease, Pneumocystis carinii pneumonia, and disseminated Mycobacterium avium complex infection has decreased [3]. Despite these improvements, the incidences of fungal infections and Kaposi’s sarcoma have not changed while those of bacterial infections and non-Hodgkin’s lymphoma have actually increased [2–4]. Similarly, in the central nervous system (CNS), the incidences of many opportunistic conditions have generally decreased but the prevalence of HIV-associated dementia (HAD) and its neuropathiologic correlate, HIV encephalitis (HIVE) seems to have increased [3,5,6]. Perhaps even more concerning, new cases of HAD may be occurring in patients with higher CD4 cell counts [7–9].
Neuropathologic changes characteristically associated with HIVE include formation of multinucleated giant cells (MNGC), microglial nodules (MGN) and astrogliosis in the white matter, basal ganglia and neocortex [10]. Diffuse damage to the white matter occurs more frequently in patients with cognitive impairment and is characterized in life by diffuse or focal hyperintensity on T2-weighted magnetic resonance imaging (MRI) [11] and at autopsy by mild to moderate myelin loss and astrogliosis [10,12]. As ART may improve cognitive impairment [7] it may also reverse such white matter damage. However, HIV-infected patients treated with potent ART more frequently have extensive focal white matter lesions on neuroimaging studies, compared to those not taking antiretroviral drugs [13]. Consistent with these findings, we have observed seven cases of severe HIV-associated leukoencephalopathy in patients who have failed combination ART. The three objectives of this study were to describe this condition, its relationship to clinical and neuroimaging findings, and suggest hypotheses regarding its pathogenesis.
Materials and methods
Subjects
Subjects were enrolled in studies at the UCSD HIV Neurobehavioral Research Center (HNRC) and had mild to severe leukoencephalopathy at autopsy. The UCSD Human Subjects Program Office approved all contributing studies and subjects (or their caregivers) gave informed consent for all procedures. Cases were excluded if neurocognitive impairment was attributed to a condition other than HIV infection (e.g., a history of significant head trauma, neurologic infections, or psychiatric conditions) or if autopsy uncovered evidence of CNS infections, neoplasms, or anoxic brain injury. Seven cases met our criteria for inclusion.
The clinical medical records of all cases were reviewed. Subjects also had ante-mortem, prospectively collected, research data, including standardized neuroimaging [14], neuromedical, and neuropsychological evaluations [15,16]. Laboratory data were obtained between 1 and 11 months prior to death, neuropsychological data between 6 and 12 months, and neuroimaging data between 2 and 48 months. The median interval between death and collection of ante-mortem data was 8 months for all three categories. Dates of HIV infection were obtained by self-report, except in two subjects who were dated seroconverters. We classified antiretroviral drugs by the number of classes [nucleoside reverse transcriptase inhibitor (NRTI), non-NRTI, and protease inhibitor] and the total number taken. Fasting and non-fasting glucose and triglyceride measurements were reviewed and the highest levels recorded.
The HNRC’s neuropsychological evaluations classify subjects into one of four levels of neurocognitive functioning: (i) no impairment; (ii) subsyndromic neuropsychologic-impairment; (iii) minor cognitive motor disorder; or (iv) HAD [16–19].
Neuropathologic examination
All subjects died of acute bronchopneumonia and/or sepsis and all autopsies were performed within 24 h of death [3]. After macroscopic examination, tissue blocks from the midfrontal cortex, temporal cortex, parietal cortex, cingulate cortex, hippocampus, basal ganglia, mesencephalon, pons, medulla and spinal cord were obtained, immersion-fixed in 4% formalin and embedded in paraffin. Paraffin blocks were serially sectioned and stained with hematoxylin and eosin (H & E), Luxol fast blue, and Bodian and Prussian blue for ferric iron. Additional paraffin sections were used for subsequent immunocytochemical analysis for HIV-gp41, CD23, CD68, and glial fibrillary acidic protein (GFAP) [20]. RNA and DNA were extracted from frozen tissue blocks for analysis of HIV and other viruses [21].
Immunocytochemical analysis
For HIV immunolocalization in the CNS, paraffin sections were incubated overnight at 4°C with monoclonal antibody against HIV gp41 (1 : 200) (Genetics Systems, Seattle, Washington, USA) [20]. For analysis of macrophage infiltration into the brain, sections were incubated with monoclonal antibodies against CD23 and/or CD68 (1 : 100) (DAKO Laboratories, Carpinteria, California, USA). For analysis of astrogliosis, sections were incubated with a polyclonal antibody against GFAP (1 : 500) (DAKO). Sections were then incubated in biotinylated secondary antibody (1 : 200) (Vector Laboratories, Burlingame, California, USA), followed by Avidin D-HRP (ABC Elite; Vector) and reacted with diaminobenzidine (0.2 mg/ml) in 50 mM Tris buffer (pH 7.4) with 0.001% H2 O2.
HIV RNA quantitation and PCR
Regional quantitation of HIV RNA was performed with the QUANTIPLEX bDNA Signal Amplification System (Chiron Corporation, Emeryville, California, USA), according to the method described by Wiley and colleagues [21]. Briefly, tissues were homogenized in TRIzol Reagent (Gibco-BRL, Life Technologies, Gaithersburg, Maryland, USA). Following overnight precipitation at −80°C, the mixture was centrifuged, the supernatant discarded, and the pellet washed in 0.5 ml 75% ethanol, followed by another brief centrifugation for 5 min. The ethanol was decanted and the pellet was air-dried for 5 min. For QUANTIPLEX quantitation of HIV RNA, 0.44 ml of HIV sample working reagent was added to the pellet. The suspension was vortexed and incubated at 53°C for 20 min followed by vortexing for 10 s to ensure complete solubilization of the RNA pellet. Two hundred micro-liters of the solubilized RNA was placed in duplicate HIV capture wells in a standard QUANTIPLEX HIV RNA assay including standards and controls. Samples were incubated overnight at 53°C and the HIV content was measured by luminometry. If duplicate QUANTI-PLEX measurements varied by more than 35%, the test was considered to be invalid.
To screen for other viral sequences, genomic DNA was extracted [22] and amplified in 35 cycles (93°C for 30 s, 50°C for 30 s, 72°C for 1 min) with a final extension at 72 ° C for 5 min. Primer sequences for Epstein–Barr virus (EBV), CMV, JC virus (JCV), human herpes viruses 6 and 8 (HHV-6, HHV-8) and varicellazoster virus (VZV) were obtained from published data [23]. The herpes simplex virus (HSV) 1 and 2 primer sequence was obtained from Sanders et al. [24]. The quality of extracted DNA was assessed by amplification of the glyceraldehyde 3-phosphate dehydrogenase gene [25]. Controls for PCR detection sensitivity included positive control samples from AIDS patients with confirmed EBV, CMV, JCV, HHV-6, HHV-8 and VZV infections. Samples from non-HIV patients without evidence of opportunistic infections or brain alterations were used as negative controls.
Results
Clinical and demographic characteristics are summarized in Table 1 and gross and microscopic findings in Table 2. Six subjects had a history of homosexual activity, one reported heterosexual contacts only, and two used intravenous drugs (Table 1). All subjects were severely immunosuppressed with CD4 cell counts < 200 × 106/l, most (4/7, 57%) < 25 × 106/l. At the time of their last evaluation, HIV replication was poorly controlled in six patients (86%), with plasma HIV RNA levels greater than the upper limit of quantitation in four. All subjects had high viral loads in brain, including the subject with plasma HIV RNA levels < 400 copies/ml. Ante-mortem cerebrospinal fluid HIV RNA concentrations were measured in six patients (86%) and reflected high brain levels. In two patients, HIV RNA levels in cerebrospinal fluid actually exceeded levels in plasma (Table 2). Most subjects had been diagnosed with HIV-related conditions, including oropharyngeal candidiasis, wasting, Kaposi’s sarcoma, viral hepatitis, distal sensory polyneuropathy and disseminated mycobacterial infection (Table 1). Five subjects were diagnosed with HAD, one with subsyndromic neuropsychologic impairment, and one was neurocognitively normal. Cognitive impairment was not explained by psychotropic medications or other conditions, such as CNS opportunistic infections, psychiatric or mood disorders, or a history of head trauma.
Table 1.
Summarized clinical and laboratory data.
| Case | Age (years)a | Sex | Risk factor | Date of HIV infection | Date of death | Years infectedb | Cognitive diagnosisc | Other ante-mortem diagnoses | ART historyd | Last samplinge | Plasma HIV RNA (copies/ml)f | CSF HIV RNA (copies/ml) | CD4 cell count (× 106/l)h | Glucose (mg/dl)i | Triglycerides (mg/dl)j |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 41 | Male | MSM | 12/85 | 11/4/98 | 12.9 | NPI | Disseminated KS | 2 Classes 4 Agents |
10 | < 400 | 1396 | 114 | 103 | 157 |
| 2 | 48 | Male | MSM | 09/87 | 2/27/99 | 11.5 | Normal | KS, CMV, VZV, DSPN, OPC | 3 Classes 10 Agents |
10 | > 750 000 | 85 800 | 17 | 136 | 1378 |
| 3 | 50 | Male | MSM IDU |
06/85 | 2/21/99 | 13.7 | HAD (stage 2) | HBV, HCV, HSV, VZV, DSPN, OPC, CAP, wasting | 3 Classes 7 Agents |
11 | > 750 000 | 6861 | 85 | 109 | 221 |
| 4 | 41 | Male | MSM | 02/92 | 2/7/01 | 9.0 | HAD (stage 4) | HSV, DSPN, PCP, wasting, myositis, salmonellosis | 3 Classes 12 Agents |
1 | 423 865 | > 750 000 | 10 | 238 | 546 |
| 5 | 34 | Male | MSW | 07/94 | 3/15/99 | 4.7 | HAD (stage 4) | Disseminated MAC, HIV retinopathy, seizures | 2 Classes 6 Agents |
8 | > 750 000 | NP | 24 | 182 | 188 |
| 6 | 40 | Male | MSM | 05/87 | 9/27/00 | 13.4 | HAD (stage 3) | VZV, OPC, wasting | 3 Classes 11 Agents |
3 | > 750 000 | 8264 | 14 | 92 | 312 |
| 7 | 39 | Male | MSM IDU |
06/93 | 12/27/98 | 5.6 | HAD (stage 4) | HBV, DSPN, wasting | 3 Classes 10 Agents |
1 | 313 987 | 483 | 90 | 134 | 1712 |
Mean age was 41.9 years
Median duration of HIV infection was 11.5 years.
Subjects with mild leukoencephalopathy (see Table 2) had only mild or no cognitive impairment while those with more severe disease were also more impaired.
Antiretroviral drugs were classified by the number of classes [nucleoside reverse transcriptase inhibitor (NRTI), non-NRTI, and protease inhibitor] and the total number taken. Subjects took a median of three classes and 10 antiretroviral drugs prior to death.
All subjects had ante-mortem laboratory data available with a mean sampling interval of 6.3 months prior to death.
HIV RNA levels in plasma approached or exceeded the upper limit of quantitation in six patients (86%).
HIV RNA levels in cerebrospinal fluid were measured in six patients and were also very high in most, exceeding plasma levels in two.
The median CD4 T lymphocyte count was 24 × 106/l.
Medical records were reviewed for all glucose and triglyceride measurements, fasting and non-fasting; the highest levels are recorded.
Random glucose levels were elevated in four out of seven (57%) (normal < 110 mg/dl) and triglyceride levels were elevated in six patients (86%), markedly so in three (43%) (normal < 160 mg/dl). ART, Antiretroviral therapy; MSM, men who have sex with men; NPI, neuropsychological impairment; KS, Kaposi’s sarcoma; CMV, cytomegalovirus; VZV, varicellazoster virus; DSPN, distal sensory polyneuropathy; OPC, oropharyngeal candidiasis; IDU, injecting drug user; HBV, hepatitis B virus; HCV, hepatitis C virus; HSV, herpes simplex virus; CAP, community-acquired pneumonia; PCP, Pneumocystis carinii pneumonia; MSW, men who have sex with women; MAC, disseminated Mycobacterium avium complex infection.
Table 2.
Summarized neuropathologic data. Subjects were classified as either mild, moderate or severe stage of white matter neuropathology according to gross brain examination, microscopically by determining demyelination, gliosis, perivascular macrophages (PV), microglial nodules (MG Nod), multinucleated giant cells (MNGC) and Luxol fast blue (LFB) for myelin and iron (Fe) for hemosiderin, where ‘1’ represents least damage and ‘3’ represents most damage. Other findings at autopsy are listed as Kaposi sacrcoma (KS), cytomegalovirus (CMV), Epstein–Barr virus (EBV), Mycobaterium avium complex (MAC), chronic obstructive pulmonary disease (COPD) or actinomycosis. Regions of the brain most affected were noted: frontal (F), cortical (C), basal ganglia (BG), substania nigra (SN), pons (P) or occipital (O). Three subjects were diagnosed as neuropathologically normal on gross examination at autopsy.
| Patient | Gross | Brain viral load (copies/ml) | Demylenation | Gliosis | PV Mac | MG Nod | MNGC | LFB | FE | Stage | Regions most affected | Other autopsy diagnoses |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Normal | 9877 | 2 | 2 | 2 | 1 | 0 | 1 | 2 | Mild | SN, P | KS |
| 2 | Normal | 85 863 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | Mild | F, C, P | KS |
| 3 | Normal | 45 216 | 2 | 1 | 2 | 2 | 2 | 1 | 1 | Mild | F, C, P | CMV |
| 4 | Mild atrophy, diminished white matter | 1 865 600 | 2 | 2 | 1 | 0 | 1 | 1 | 2 | Mild-moderate | F, C, BG, SN | EBV |
| 5 | Atrophic, dilated ventricles | 987 000 | 3 | 2 | 2 | 2 | 2 | 3 | 1 | Moderate | F, C, BG | MAC |
| 6 | Atrophic, dilated ventricles, diffuse lesions | 6 621 600 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | Moderate-severe | F, C, BG | COPD |
| 7 | Atrophic focal lesions | 5 940 000 | 3 | 3 | 3 | 2 | 2 | 3 | 3 | Severe | F, C, BG, O | Actinomycosis |
T2-weighted MRI revealed multifocal white matter hyperintensities in three of the seven cases (Fig. 1b–d). The other four displayed extensive, diffuse, and more confluent white matter hyperintensity (Fig. 1e–h). Lesions were more abundant in the areas of the centrum semiovale (Fig. 1f–h), corpus callosum (Fig. 1g) and optical radiations. Lesions were observed less frequently in the frontal pole and the parietal white matter (Fig. 1e–h).
Fig. 1.

White matter alterations by in vivo MRI in patients with HIV-associated leukoencephalopathy. All are T2-weighted images obtained within 12 months of death. The available images were coronal in some but horizontal in others. (a) Normal MRI illustrating the expected characteristics of the cortical ribbon and white matter. (b–d) Cases with mild neuropathology had focal and diffuse white matter hyperintensities (arrow). (e–h) Cases with moderate to severe neuropathology had extensive white matter hyperintensities (arrows). Lesions were most prominent in the white matter surrounding the lateral ventricles (LV), corpus callosum, and optic radiations but were also observed in the frontal pole and parietal white matter.
Consistent with neuroimaging, gross neuropathologic examination revealed a range of white matter alterations. In mild cases, these alterations included white matter atrophy with focal lesions in the frontal, temporal and parietal cortices (Fig. 2b–d). Cases presenting with more extensive, diffuse white matter damage showed both cortical atrophy and reduced basal ganglia volume (Fig. 2e–h). In the most severe cases, the corpus callosum was diffusely attenuated, the lateral ventricles were dilated, and cavitating lesions were present in the centrum semiovale of the frontoparietal region (Fig. 2g).
Fig. 2.

Gross findings in the white matter in patients with HIV-associated leukoencephalopathy. All images are coronal sections of the right hemibrain at the level of the hippocampus. The order of the images corresponds to the MRI images in Fig. 1. (a) Normal brain tissue illustrating the white matter and cortical ribbon in a fixed specimen. (b–d) Cases with mild neuropathology had focal white matter lesions in the frontoparietal region (arrows). (e–h) In the most severe cases, the corpus callosum (CC) was diffusely attenuated, the lateral ventricles were dilated, and cavitating lesions were present in the centrum semiovale of the frontoparietal region (arrows). Those with extensive white matter damage also showed cortical atrophy (CA) and reduced basal ganglia volume.
Microscopically, cases with milder white matter alterations showed abundant perivascular infiltration by macrophages and lymphocytes, as indicated by labeling with anti-CD23 and anti-CD68, respectively, with occasional formation of MGN and MNGC surrounding blood vessels and along white matter tracks (Fig. 3b–d). HIV-gp41 immunoreactive cells localized in close association with blood vessels in mild cases (Fig. 4a) but were found in brain parenchyma in moderate cases (Fig. 4b and c) and were observed throughout the white matter in the most severe cases (Fig. 4d). In moderate to severe cases, perivascular macrophages frequently contained hemosiderin (Fig. 3g). The most advanced cases exhibited frequent MGN and MNGC, severe disruption of microvessel walls with evidence of endothelial cell damage, expansion of Virchow’s space, and hypertrophic glial cell processes (Fig. 3e–h).
Fig. 3.

Neuropathological alterations in the white matter of patients with HIV-associated leukoencephalopathy. All images are from the white matter of the frontal lobe and corpus callosum, stained with H&E, approximate magnification 400 ×. (a) Normal brain tissue illustrating white matter (WM) and microvasculature. (b–d) Mild cases had intense lymphocytic (L) and macrophage (M) infiltration around blood vessels, with expansion of the perivascular space and white matter rarefaction. (e–h) More severe cases also had disrupted microvasculature (arrows). Multinucleated giant cells (MNGC), microglial cells (*), and reactive astrocytes (a) surrounded vessels. White matter was diffusely fragmented and diminished.
Fig. 4.

Patterns of HIV-gp41 immunoreactivity in white matter of patients with HIV-associated leukoencephalopathy. All images are from the white matter of the frontal lobe and corpus callosum. Immunostained sections were developed with diaminobenzidine and counter-stained with hematoxylin. Approximate magnification is 400 ×. (a and b) Mild cases had abundant gp41 immunoreactive macrophages (arrows) around blood vessels (BV). (c and d) More severe cases also had abundant gp41 immunostained macrophages and microglia (*) around vessels and in the midst of white matter fibers.
Mild cases showed moderate degrees of demyelination and gliosis (Figs. 5b and 5f). In moderate and severe cases, demyelination was considerably more extensive with prominent astrogliosis and formation of MGN and MNGC (Figs. 5c and 5g). White matter destruction was so extreme in some cases that myelin was completely absent and axons were damaged (Fig. 5d and 5h).
Fig. 5.
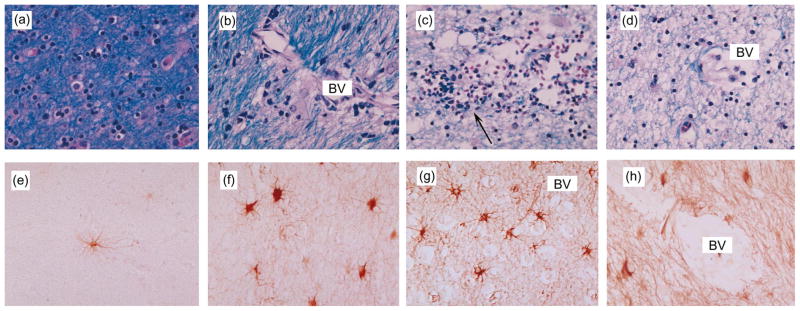
Patterns of demyelination and astrogliosis in white matter of patients with HIV-associated leukoencephalopathy. All images are from the white matter of the frontal lobe and corpus callosum. The first sections were stained with Luxol fast blue and the second four panels were immunostained with antibody against GFAP. Approximate magnification is 400 ×. (a) Normal brain tissue illustrating myelin characteristics. (b–c) Cases with mild-to-moderate demyelination, most apparent around blood vessels (BV) infiltrated by mononuclear cells (arrow). (d) Severe case showing diffuse and very extensive loss of white matter. (e) Normal brain tissue illustrating normal astroglia. (f–g) Mild-to-moderate cases showing perivascular (BV) and diffuse astrogliosis. (h) Severe case showing intense, diffuse astrogliosis.
In all cases, characteristic findings of HIVE (e.g., macrophage infiltration, presence of HIV immunoreactive cells, MNGC and formation of MGN) were associated exclusively with white matter microvasculature but not with the cortical ribbon or deep gray matter (Table 2). Lesions were consistently found in the white matter underlying frontal and cingulate cortices, corpus callosum, basal ganglia, substantia nigra, and pons. In each case, lesions found in the different brain regions were similar in appearance.
PCR analysis confirmed the presence of very high HIV levels in the brains of all cases, primarily in white matter rather than other brain regions (data not shown). HIV RNA levels in the brain were exceedingly high. PCR did not detect the presence of other viruses in the brain, including CMV, EBV, JCV, HHV-6 and 8, VZV, and HSV-1 and 2.
Comparing neuroimaging (Fig. 1) and neuropathologic (Table 2, Figs. 3, 4 and 5) findings to ante-mortem cognitive impairment (Table 1), the two non-demented cases had occasional focal and diffuse lesions on MRI and mild demyelination and perivascular cellular infiltration with low to moderately high HIV RNA levels in brain on neuropathologic examination. In contrast, the five cases with ante-mortem dementia had extensive, diffuse white matter hyperintensities by MRI corresponding to intense infiltration by monocytes with more severe demyelination, astrogliosis, and higher brain concentrations of HIV RNA.
Discussion
This study describes a form of severe, HIV-associated leukoencephalopathy in seven patients who failed highly active antiretroviral therapy (HAART). Our observations are consistent with recent reports of an increased incidence of focal white matter lesions in HAART-treated patients [13] and of an unexpectedly high incidence of ‘not determined’ leukoencephalopathy in AIDS patients [26]. Prior to the HAART era, most reports described leukoencephalopathy cases that were either associated with opportunistic pathogens (such as JCV, CMV or EBV) or did not specifically exclude them [22,27–31]. Berger and colleagues did report leukoencephalopathy cases with multiple sclerosis-like lesions [32] or relapsing and remitting leukoencephalopathy [33]. However, the pathogenesis of our cases may be different for three reasons. First, we found only HIV in the brain, but no other pathogens. Second, the tissue injury in our cases was confined to the white matter, unlike previous cases that also had gray matter abnormalities. Third, the leukoencephalopathy we describe is more severe than that described prior to the use of HAART. In the past, perivascular infiltration by mononuclear cells and myelin loss was less extensive, white matter atrophy was milder, and HIV concentrations in the brain were lower [12]. In comparison, our cases are characterized by intense perivascular macrophage infiltration, extensive demyelination, and evidence of very high levels of HIV replication in the brain.
Leukoencephalopathy in HIV-infected individuals may result from HIV-, immune-, opportunist-, or drug-based mechanisms. HIV itself could injure white matter by damaging oligodendrocytes or brain endothelial cells or by directly injuring myelin (e.g., via myelinotoxic viral proteins, such as gp120 or Tat [34]). The immune system can injure white matter by several mechanisms, including the release of pro-inflammatory cytokines (such as tumor necrosis factor-alpha [35,36]) or the phagocytosis of myelin by macrophages (as in immune-mediated demyelinating diseases, such as acute disseminated encephalomyelitis) [37]. Reactivated infection with opportunistic viruses, such as HHV-6 or JCV [38,39] can also damage the white matter of AIDS patients. Finally, prescribed (e.g., HIV protease inhibitors) or illicit (e.g., CNS stimulants) drugs may penetrate the blood–brain barrier and injure myelin [40].
While our study does not directly test these hypotheses, opportunist- and illicit drug-based mechanisms are unlikely. Opportunistic pathogens probably did not damage white matter as evidence of common, myelinotoxic viruses was not detected. Five of the seven subjects did not report the use of illicit drugs and did not show the patterns of dopaminergic neuronal damage reported in HIV patients using stimulant drugs [40,41]. However, we cannot rule out drug abuse as a compounding factor in this syndrome.
Leukoencephalopathy more probably resulted from HIV, the immune system, or antiretroviral drugs. The emergence of this condition in the post-HAART era strongly argues that potent ART plays an important role in pathogenesis. As one of HAART’s cardinal effects is the reduction of HIV-related mortality [42], patients whose lives are prolonged by HAART but who also have HIV encephalitis may be susceptible to progressive CNS disease even though HIV replication is suppressed in other tissues [5,43,44]. These seemingly conflicting effects may be attributable to the use of antiretroviral drugs that penetrate poorly into the brain, such as most protease inhibitors, and may explain why HIV encephalitis continues to be observed at a quarter of autopsies [3]. Antiretroviral drugs themselves could be responsible if they directly damaged myelin or oligodendrocytes, causing a toxic leukoencephalopathy [45]. Alternatively, one of several, recently recognized, antiretroviral drug-associated complications might be responsible. Chief among these complications are the emergence of highly adapted HIV strains, immune reconstitution-related tissue injuries, and lipid and metabolic disorders [46–55].
HIV’s high replication and mutation rates allow it to readily adapt to different environmental pressures. These adaptive pressures often vary between tissues and include different resident cell types, immunologic characteristics, and antiretroviral drug concentrations. In the brain, HIV commonly infects microglia and brain macrophages, cell-mediated immunity is usually restricted, and concentrations of most antiretroviral drugs, particularly protease inhibitors, are low. These differences can foster the development of virodemes in the CNS that evolve independently from other tissues, particularly when immunosuppression is pronounced [55–57]. In white matter, these combined pressures may yield strains that can both replicate in the presence of antiretroviral drugs and efficiently infect resident cells, such as brain endothelial cells or oligodendrocytes. In fact, strains that infect these cells have been described. Strelow et al. described an SIVmac variant that was derived from a neuropathogenic strain and efficiently infected brain microvascular endothelial cells [58]. Albright and colleagues were able to productively infect oligodendrocytes with primary and laboratory strains of HIV [59]. Other SIV studies described strains that can specifically damage white matter [60–62]. Therefore, severe leukoencephalopathy may result from antiretroviral-resistant HIV strains that have adapted to cells that reside in white matter. Notably, all of our subjects had advanced immunosuppression and failed at least one antiretroviral regimen, supporting the presence of an independently evolving CNS virodeme that was resistant to antiretroviral drugs.
While failing antiretroviral therapy can select highly adapted HIV strains, successful therapy can substantially improve CD4 T lymphocyte numbers and function. Unfortunately, tissue injury can occur when the newly reconstituted immune system responds overzealously to clinical or subclinical infections that are present when therapy is initiated [46]. In the CNS, such injury has been described for cryptococcal meningitis [49] and CMV retinitis [63]. We did not detect the presence of other CNS pathogens in our subjects but HIV antigens themselves might induce a reconstituted immune response in white matter, resulting in the intense infiltration of lymphocytes and monocytes and blood–brain barrier damage that we observed.
Such immune reconstitution injury would result only from therapy that was successful, at least initially. However, if resistance subsequently developed, infiltrating lymphocytes could begin producing HIV virions and proteins, leading to progressive injury. As immune re-deterioration proceeded, peripheral monocytes infected with antiretroviral drug-resistant HIV strains might preferentially and efficiently migrate into deep white matter. In fact, Liu and colleagues postulated that migration of bone marrow-derived monocytes into deep white matter may be accelerated in patients with advanced HIV disease, based on phylogenetic studies of a demented patient who failed HAART [45].
The role of antiretroviral-induced immune reconstitution injury would be supported if disease onset occurred in treated subjects when CD4 cell lymphocyte counts were high. Antinori and colleagues found that most subjects with ‘not determined’ leukoencephalopathy had CD4 counts above > 200 × 106/l [26]. While all of our subjects had advanced immunosuppression at the time of death, all had at least some ante-mortem sampling. Three had sufficient documentation to allow assessment of CD4 lymphocyte trends. The CD4 cell counts of all three reached < 100/× 106/l before improving in response to a new antiretroviral regimen and then subsequently deteriorating (data not shown). This rising–falling pattern is consistent with initial immune reconstitution injury followed by subsequent antiretroviral failure and disease progression.
In addition to adaptive pressures and immune reconstitution injury, treatment with antiretroviral drugs can result in several related, but distinct, metabolic syndromes characterized by insulin resistance, hyperlipidemia, lipodystrophy (or fat redistribution), and lactic acidosis [47,49,52,63,64]. These conditions have resulted in serious complications, including insulin-requiring diabetes, coronary artery disease, and death [52,65].
The pathogenesis of these disorders is still uncertain but at least two mechanisms seem likely. Certain NRTIs, such as stavudine, can inhibit mitochondrial DNA polymerase, resulting in mitochondrial injury and impaired cell metabolism. Protease inhibitors may disrupt normal lipid metabolism because of HIV protease’s homology with proteins such as LRP (low density lipoprotein-receptor-related protein) and CRABP-1 (cytoplasmic retinoic acid binding protein type 1) [51]. Either mechanism could damage the CNS. The brain is frequently damaged in congenital mitochondrial syndromes, suggesting that it is very sensitive to mitochondrial injury. Alternatively, white matter may be particularly susceptible to disruption of lipid metabolism as myelin is composed primarily of fat.
None of our cases had gross evidence of fat redistribution or cardiac complications at autopsy. Table 1 shows that four subjects had evidence of at least transient ante-mortem hyperglycemia (normal < 110). Non-fasting triglyceride concentrations were mildly elevated in three subjects and markedly elevated in three others (normal < 160 mg/dl). Although non-fasting triglycerides are an imperfect marker of antiretroviral-associated lipid disorders, these data support that at least three subjects may have had abnormal lipid metabolism.
In conclusion, our findings support the emergence of a severe form of HIV-associated leukoencephalopathy. Intense perivascular macrophage infiltration, extensive demyelination, and very high levels of HIV replication characterize this condition. We identified three stages of severity, which were correlated with ante-mortem clinical, neuropsychological, and neuroimaging findings. These stages suggest a temporal sequence of pathologic events. In this model, leukoencephalopathy begins with perivascular infiltration by HIV-infected monocytes and blood–brain barrier damage, leading to local HIV replication, moderate myelin loss, but limited gliosis. As disease progresses, more myelin is lost and is eventually accompanied by axonal damage and extensive astrogliosis. While the cause is unclear, the condition’s occurrence in the post-HAART era in highly treatment-experienced individuals strongly argues that ART plays a pivotal role in pathogenesis. HIV-associated leukoencephalopathy may simply be a consequence of ART that extends the survival of patients with HIVE without completely controlling HIV replication in the brain. Alternatively, its etiology may be more complex, involving other HIV-, immune-, and drug-mediated effects. Recognition of this condition warrants increased vigilance among HIV care providers and further investigation.
Acknowledgments
Sponsorship: The HIV Neurobehavioral Research Center (HNRC) is supported by Center awards MH45294, MH59745 and DA12065.
Appendix
The San Diego HIV Neurobehavioral Research Center (HNRC) group is affiliated with the University of California, San Diego, the Naval Medical Center, San Diego, and the San Diego VA Medical Center, and includes: I. Grant, Director; J. H. Atkinson, and J. A. McCutchan, Co-Directors; T. D. Marcotte, Center Manager; M. R. Wallace, Co-Investigators Naval Medical Center, San Diego; R. J. Ellis, P.I. Neuro-medical Component; R. K. Heaton, P.I. Neurobehavioral Component; T. Jernigan, P.I. Imaging Component; E. Masliah, P.I. Neuropathology Component; D. R. Masys, P.I. Data Management Component; I. Abramson, P.I. Statistics Unit; M. Frybarger, Data Manager.
References
- 1.Richman DD. HIV chemotherapy. Nature. 2001;410:995–1001. doi: 10.1038/35073673. [DOI] [PubMed] [Google Scholar]
- 2.Calabrese LH. Changing patterns of morbidity and mortality in HIV disease. Clev Clin J Med. 2001;68:105–112. doi: 10.3949/ccjm.68.2.105. [DOI] [PubMed] [Google Scholar]
- 3.Masliah E, DeTeresa R, Mallory M, Hansen L. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS. 2000;14:69–74. doi: 10.1097/00002030-200001070-00008. [DOI] [PubMed] [Google Scholar]
- 4.Jellinger KA, Setinek U, Drlicek M, Bohm G, Steurer A, Lintner F. Neuropathology and general autopsy findings in AIDS during the last 15 years. Acta Neuropathol (Berl) 2000;100:213–220. doi: 10.1007/s004010000245. [DOI] [PubMed] [Google Scholar]
- 5.Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ. Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS. 1999;13:1249–1253. doi: 10.1097/00002030-199907090-00015. [DOI] [PubMed] [Google Scholar]
- 6.McGuire D, Marder K. Pharmacological frontiers in the treatment of AIDS dementia. J Psychopharmacol. 2000;14:251–257. doi: 10.1177/026988110001400310. [DOI] [PubMed] [Google Scholar]
- 7.Sacktor N, Lyles RH, Skolasky R, et al. HIV-associated neurologic disease incidence changes: Multicenter AIDS Cohort Study, 1990–1998. Neurol. 2001;56:257–260. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- 8.Suarez S, Baril L, Stankoff B, et al. Outcome of patients with HIV-1-related cognitive impairment on highly active antiretroviral therapy. AIDS. 2000;15:195–200. doi: 10.1097/00002030-200101260-00008. [DOI] [PubMed] [Google Scholar]
- 9.Chang L, Ernst T, Leonido-Yee M, et al. Highly active antiretroviral therapy reverses brain metabolite abnormalities in mild HIV dementia. Neurol. 1999;53:782–789. doi: 10.1212/wnl.53.4.782. [DOI] [PubMed] [Google Scholar]
- 10.Budka H, Wiley CA, Kleihues P, et al. HIV-associated disease of the nervous system: Review of nomenclature and proposal for neuropathology-based terminology. Brain Pathol. 1991;1:143–152. doi: 10.1111/j.1750-3639.1991.tb00653.x. [DOI] [PubMed] [Google Scholar]
- 11.Aylward EH, Brettschneider PD, McArthur JC, et al. Magnetic resonance imaging measurement of gray matter volume reductions in HIV dementia. Am J Psych. 1995;152:987–994. doi: 10.1176/ajp.152.7.987. [DOI] [PubMed] [Google Scholar]
- 12.Smith TW, DeGirolami U, Henin D, Bolgert F, Hauw JJ. Human immunodeficiency virus (HIV) leukoencephalopathy and the microcirculation. J Neuropathol Exp Neurol. 1990;49:357–370. doi: 10.1097/00005072-199007000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Ammassari A, Cingolani A, Pezzotti P, et al. AIDS-related focal brain lesions in the era of highly active antiretroviral therapy. Neurol. 2000;55:1194–1200. doi: 10.1212/wnl.55.8.1194. [DOI] [PubMed] [Google Scholar]
- 14.Jernigan TL, Archibald S, Hesselink JR, et al. and the HNRC Group. Magnetic resonance imaging morphometic analysis of cerebral volume loss in human immunodeficiency virus infection. Arch Neurol. 1993;50:250–255. doi: 10.1001/archneur.1993.00540030016007. [DOI] [PubMed] [Google Scholar]
- 15.Masliah E, Heaton RK, Marcotte TD, et al. and the HNRC Group. Dendritic injury is a pathological substrate for human immunodefficiency virus-related cognitive disorders. Ann Neurol. 1997;42 :963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- 16.Heaton RK, Grant I, Butters N, et al. and the HNRC Group. The HNRC 500 - neuropsychology of HIV infection at different disease stages. J Int Neuropsych Soc. 1995;1:231–251. doi: 10.1017/s1355617700000230. [DOI] [PubMed] [Google Scholar]
- 17.Ellis RJ, Deutsch R, Heaton RK, et al. and the HNRC Group. Neurocognitive impairment is an independent risk factor for death in HIV infection. Arch Neurol. 1997;54:416–424. doi: 10.1001/archneur.1997.00550160054016. [DOI] [PubMed] [Google Scholar]
- 18.AAN. Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus-type 1 (HIV-1) infection. Report of a Working Group of the American Academy of Neurology AIDS Task Force. Neurol. 1991;41:778–785. doi: 10.1212/wnl.41.6.778. [DOI] [PubMed] [Google Scholar]
- 19.Heaton RK, Velin RA, McCutchan JA, et al. Neuropsychological impairment in human immunodeficiency virus-infection: implications for employment. Psychosomatic Med. 1994;56:8–17. doi: 10.1097/00006842-199401000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol. 1992;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- 21.Wiley CA, Soontornniyomkij V, Radhakrishnan L, et al. Distribution of brain HIV load in AIDS. Brain Pathol. 1998;8:277–284. doi: 10.1111/j.1750-3639.1998.tb00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller RF, Lucas SB, Hall-Craggs MA, et al. Comparison of magnetic resonance imaging with neuropathological findings in the diagnosis of HIV and CMV associated CNS disease in AIDS. J Neurol Neurosur Psych. 1997;62:346–351. doi: 10.1136/jnnp.62.4.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffais R, Andre PM, Thibon M. Synthesis of digoxigenin-labelled DNA probe by polymerase chain reaction: application to Epstein-Barr virus and Chlamydia trachomatis. Res Virol. 1990;141:331–335. doi: 10.1016/0923-2516(90)90004-3. [DOI] [PubMed] [Google Scholar]
- 24.Sanders VJ, Waddell AE, Felisan SL, Li X, Conrad AJ, Tourtellotte WW. Herpes simplex virus postmortem multiple sclerosis brain tissue. Arch Neuro. 1996;53:125–133. doi: 10.1001/archneur.1996.00550020029012. [DOI] [PubMed] [Google Scholar]
- 25.Achim C, Wang R, Miners DK, Wiley CA. Brain viral burden in HIV infection. J Neuropathol Exp Neurol. 1994;53:284–294. doi: 10.1097/00005072-199405000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Antinori A, Ammassari A, Cinque P, et al. Shift of Prevalence and Selected Characteristics in HIV-1-Related Neurologic Disorders in HAART Era: Data from the Italian Register Investigative NeuroAIDS (IRINA). VIII Conference on Retroviruses and Opportunistic Infections; 2001. [abstract number 8] [Google Scholar]
- 27.Rostad SW, Sumi SM, Shaw CM, Olson K, McDougall JK. Human immunodeficiency virus (HIV) infection in brains with AIDS-related leukoencephalopathy. AIDS Res Hum Retroviruses. 1987;3 :363–373. doi: 10.1089/aid.1987.3.363. [DOI] [PubMed] [Google Scholar]
- 28.Luer W, Gerhards J, Poser S, Weber T, Felgenhauer K. Acute diffuse leukoencephalitis in HIV-1 infection. J Neurol Neurosurg Psych. 1994;57:105–107. doi: 10.1136/jnnp.57.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurol. 1994;44:481–487. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- 30.Anders KH, Becker PS, Holden JK, et al. Multifocal necrotizing leukoencephalopathy with pontine predilection in immunosuppressed patients: a clinicopathologic review of 16 cases. Hum Pathol. 1993;24:897–904. doi: 10.1016/0046-8177(93)90140-c. [DOI] [PubMed] [Google Scholar]
- 31.Rhodes RH. Histopathologic features in the central nervous system of 400 acquired immunodeficiency cases: implications of rates of occurrence. Hum Pathol. 1993;24:1189–1198. doi: 10.1016/0046-8177(93)90215-3. [DOI] [PubMed] [Google Scholar]
- 32.Berger JR, Sheremata WA, Resnick L, Atherton S, Fletcher MA, Norenberg M. Multiple sclerosis-like illness occurring with human immunodeficiency virus infection. Neurology. 1989;39 :324–329. doi: 10.1212/wnl.39.3.324. [DOI] [PubMed] [Google Scholar]
- 33.Berger JR, Tomatore C, Major EO, et al. Relapsing and remitting human immunodeficiency virus-associated leukoencephalomyelopathy. Ann Neurol. 1992;31:34–38. doi: 10.1002/ana.410310107. [DOI] [PubMed] [Google Scholar]
- 34.Arese M, Ferrandi C, Primo L, Camussi G, Bussolino F. HIV-1 Tat protein stimulates in vivo vascular permeability and lymphomononuclear cell recruitment. J Immunol. 2001;166:1380–1388. doi: 10.4049/jimmunol.166.2.1380. [DOI] [PubMed] [Google Scholar]
- 35.Wesselingh SL, Power C, Glass JD, et al. Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- 36.Sato-Matsumura KC, Berger J, Hainfellner JA, Mazal P, Budka H. Development of HIV encephalitis in AIDS and TNF-alpha regulatory elements. J Neuroimmunol. 1998;91:89–92. doi: 10.1016/s0165-5728(98)00161-1. [DOI] [PubMed] [Google Scholar]
- 37.Hartung H-P, Grossman RI. ADEM: Distinct disease or part of the MS spectrum? Neurol. 2001;56:1257–1260. doi: 10.1212/wnl.56.10.1257. [DOI] [PubMed] [Google Scholar]
- 38.Mock DJ, Powers JM, Goodman AD, et al. Association of human herpesvirus 6 with the demyelinative lesions of progressive multifocal leukoencephalopathy. J Neurovirol. 1999;5:363–373. doi: 10.3109/13550289909029477. [DOI] [PubMed] [Google Scholar]
- 39.Monno L, Di Stefano M, Zimatore GB, et al. Measurement of viral sequences in cerebrospinal fluid of AIDS patients with cerebral white-matter lesions using polymerase chain reaction. AIDS. 1998;12:581–590. doi: 10.1097/00002030-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Bell J, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS: Effect of neocortical involvement. Brain. 1998;121:2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- 41.Turchan J, Anderson C, Hauser KF, et al. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neuroscience. 2001;2:3. doi: 10.1186/1471-2202-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman D, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. NEJM. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan JE, Hansen D, Dworkin MS, et al. Epidemiology of the human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy. Clin Infect Dis. 2000;30(Suppl 1):S5–14. doi: 10.1086/313843. [DOI] [PubMed] [Google Scholar]
- 44.Gasnault J, Taoufik Y, Goujard C, et al. Prolonged survival without neurological improvement in patients with AIDS-related progressive multifocal leukoencephalopathy on potent combined antiretroviral therapy. J Neurovirol. 1999;5:421–429. doi: 10.3109/13550289909029483. [DOI] [PubMed] [Google Scholar]
- 45.Filley CM, Kleinschmidt-DeMasters BK. Toxic Leukoencephalopathy. New Engl J Med. 2001;345:425–432. doi: 10.1056/NEJM200108093450606. [DOI] [PubMed] [Google Scholar]
- 46.Cheng VCC, Yuen K-Y, Chan W-M, Wong SSY, Ma ESK, Chan RMT. Immunorestitution Disease Involving the Innate and Adaptive Response. Clin Infect Dis. 2000;30:882–892. doi: 10.1086/313809. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain. J Neurovirol. 2000;(Suppl 1):S70–S81. [PubMed] [Google Scholar]
- 48.DeSimone JA, Pomerantz RJ, Babinchak TJ. Inflammatory reactions in HIV-1-infected persons after initiation of highly active antiretroviral therapy. Ann Intern Med. 2000;133:447–454. doi: 10.7326/0003-4819-133-6-200009190-00013. [DOI] [PubMed] [Google Scholar]
- 49.Foudraine NA, Hovenkamp E, Notermans DW, et al. Immunopathology as a result of highly active antiretroviral therapy in HIV-1-infected patients. AIDS. 1999;13:177–184. doi: 10.1097/00002030-199902040-00005. [DOI] [PubMed] [Google Scholar]
- 50.Carr A. HIV Protease Inhibitor-Related Lipodystrophy Syndrome. Clin Infect Dis. 2000;30:S135–S142. doi: 10.1086/313854. [DOI] [PubMed] [Google Scholar]
- 51.Carr A, Samaras K, Chisholm DJ, Cooper DA. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet. 1998;351:1881–1883. doi: 10.1016/S0140-6736(98)03391-1. [DOI] [PubMed] [Google Scholar]
- 52.Friedl AC, Jost CH, Schalcher C, et al. Acceleration of confirmed coronary artery disease among HIV-infected patients on potent antiretroviral therapy. AIDS. 2000;14:2790–2792. doi: 10.1097/00002030-200012010-00021. [DOI] [PubMed] [Google Scholar]
- 53.Vigouroux C, Gharakhanian S, Salhi Y, et al. Adverse metabolic disorders during highly active antiretroviral treatments (HAART) of HIV disease. Diabetes Metab. 1999;25:383–392. [PubMed] [Google Scholar]
- 54.Smit TK, Wang B, Ng T, Osborne R, Brew B, Saksena NK. Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): evidence for neurotropic HIV variants. Virol. 2001;279:509–526. doi: 10.1006/viro.2000.0681. [DOI] [PubMed] [Google Scholar]
- 55.Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJ, Richman DD. In vivo compartmentalization of human immunodeficiency virus: evidence for the examination of pol sequences from autopsy tissues. J Virol. 1997;71:2059–2071. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haddad DN, Birch C, Middleton T, Dwyer DE, Cunningham AL, Saksena NK. Evidence for late stage compartmentalization of HIV-1 resistance mutation between lymph node and peripheral blood mononuclear cells. AIDS. 2000;14:2273–2281. doi: 10.1097/00002030-200010200-00008. [DOI] [PubMed] [Google Scholar]
- 57.van’t Wout AB, Ran LJ, Kuiken CL, Kootstra NA, Pals ST, Schuite-maker H. Analysis of the temporal relationship between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy. J Virol. 1998;72:488–496. doi: 10.1128/jvi.72.1.488-496.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strelow LI, Watry DD, Fox HS, Nelson JA. Efficient infection of brain microvascular endothelial cells by an in vivo-selected neuroinvasive SIVmac variant. J Neurovirol. 1998;4:269–280. doi: 10.3109/13550289809114528. [DOI] [PubMed] [Google Scholar]
- 59.Albright AV, Strizki J, Harouse JM, Lavi E, O’Connor M, Gonzalez-Scarano F. HIV-1 infection of cultured human adult oligodendrocytes. Virol. 1996;217:211–219. doi: 10.1006/viro.1996.0108. [DOI] [PubMed] [Google Scholar]
- 60.Raghavan R, Cheney PD, Raymond LA, et al. Morphological correlates of neurological dysfunction in macaques infected with neurovirulent simian immunodeficiency virus. Neuropathol Appl Neurobiol. 1999;25:285–294. doi: 10.1046/j.1365-2990.1999.00185.x. [DOI] [PubMed] [Google Scholar]
- 61.Raghavan R, Stephens EB, Joag SV, et al. Neuropathogenesis of chimeric simian/human immunodeficiency virus infection in pig-tailed and rhesus macaques. Brain Pathol. 1997;7:851–861. doi: 10.1111/j.1750-3639.1997.tb00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardner MB, Dandekar S. Neurobiology of simian and feline immunodeficiency virus infections. Curr Topics Microbiol Immunol. 1996;202:135–150. doi: 10.1007/978-3-642-79657-9_10. [DOI] [PubMed] [Google Scholar]
- 63.Jacobson MA, Zegans M, Pavan PR, et al. Cytomegalovirus retinitis after initiation of highly active antiretroviral therapy. Lancet. 1997;349:1443–1445. doi: 10.1016/S0140-6736(96)11431-8. [DOI] [PubMed] [Google Scholar]
- 64.Telenti A, Paolo Rizzardi G. Limits to potent antiretroviral therapy. Rev Med Virol. 2000;10:385–393. doi: 10.1002/1099-1654(200011/12)10:6<385::aid-rmv296>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 65.Baril L, Beucler I, Valantin MA, et al. Low lipolytic enzyme activity in patients with severe hypertriglyceridemia on highly active antiretroviral therapy. AIDS. 2001;15:415–417. doi: 10.1097/00002030-200102160-00016. [DOI] [PubMed] [Google Scholar]