

Abstract
Point mutations at Arg132 of the cytoplasmic NADP+-dependent isocitrate dehydrogenase 1 (IDH1) occur frequently in gliomas and result in a gain-of-function to produce the “oncometabolite” D-2-hydroxyglutarate (D-2HG). The mutated IDH1 allele is usually associated with a wild type IDH1 allele (heterozygous) in cancer. Here, we identify two gliomas which underwent loss of the wild type IDH1 allele but retained the mutant IDH1 allele following tumor progression from World Health Organization (WHO) grade III anaplastic astrocytomas to WHO grade IV glioblastomas. Intratumoral D-2HG was 14-fold lower in the glioblastomas lacking wild type IDH1 compared with glioblastomas with heterozygous IDH1 mutations. To characterize the contribution of wild type IDH1 to cancer cell D-2HG production, we established an IDH1-Mutated Astrocytoma cell line (IMA) from a WHO grade III anaplastic astrocytoma. Disruption of the wild type IDH1 allele in IMA cells by gene-targeting resulted in an 87-fold decrease in cellular D-2HG levels, demonstrating that both wild type and mutant IDH1 alleles are required for D-2HG production in glioma cells. Expression of wild type IDH1 was also critical for mutant IDH1-associated D-2HG production in the colorectal cancer cell line HCT116. These insights may aid in the development of therapeutic strategies to target IDH1-mutated cancers.
Keywords: IDH1, 2-hydroxyglutarate, glioma, astrocytoma, ATRX
Introduction
Recent exomic sequencing identified frequent mutations in the IDH1, or in its homolog IDH2, in gliomas and other cancers (1–3). Almost all IDH1 mutations result in an amino acid substitution at R132. These mutations impair the physiologic function of IDH1 to convert isocitrate to α-ketoglutarate (αKG) and confer a gain-of-function to convert αKG to D-2-hydroxyglutarate (D-2HG), which accumulates to extremely high levels in tumors with IDH1 mutations (~100-fold increase) (2, 4, 5). D-2HG inhibits αKG-dependent dioxygenases, including Jumonji C domain-containing histone demethylases and Tet 5-methylcytosine (5mC) hydroxylases, resulting in epigenetic alterations and perturbed cellular differentiation that may contribute to tumorigenesis (8, 9). These observations suggest that glioma patients may benefit from therapeutic inhibition of D-2HG production (4).
Since IDH1 functions as a homodimer, a critical question is whether IDH1 mutants exert their biological function as mutant:mutant homodimers and/or as mutant:wild type heterodimers. We showed using coimmunoprecipitation that wild type IDH1 can bind to mutant IDH1 in glioma cells (5). Also, the wild-type:mutant IDH1 heterodimer produces D-2HG at a faster rate than the mutant:mutant homodimer under specific in vitro reaction conditions (10, 11). While wild type IDH1 can produce low levels of D-2HG at a slow rate on its own (11, 12), it is unknown whether wild type IDH1 is required for mutant IDH1 to elicit the extremely high D-2HG found in IDH1-mutated tumor cells.
Here, we report that loss of wild type IDH1 was associated with a dramatic decrease in D-2HG in two IDH1-mutated astrocytomas. We demonstrate in a novel IDH1-mutated astrocytoma cell line and a colon cancer cell line that expression of wild type IDH1 is required to produce high levels of D-2HG. These findings reveal that wild type IDH1 contributes to D-2HG production in the glioma cellular environment.
Materials and Methods
Ethics Statement and Patient Samples
Tumors were obtained from the Brain Tumor Biorepository at Duke with written informed consent from patients and Institutional Review Board approval and analyzed previously for IDH mutation status (2). Short tandem repeat genotyping was performed with the AmpFlSTR® Identifiler® Kit (Applied Biosystems).
Cell lines
IMA was derived from a 26-year old male anaplastic astrocytoma WHO grade III patient treated at Duke in 2009. Tissue was dissociated by 100 μg/ml liberase and cultured in stem cell medium (Knockout minimum essential medium supplemented with 20ng/mL EGF, 50 ng/mL bFGF, B-27 without Vitamin A supplement (1:50, Life Technologies), GlutaMAX (1:100, Life Technologies), non-essential amino acids solution (1:100, Life Technologies), leukocyte inhibitory factor 10ng/mL, heparin 2µg/mL) as floating spheres. After 2.5 months, cells were transferred to 45% stem cell medium, 45% DMEM, 10% fetal bovine serum and grew as monolayers. After 9 months, cells were monoclonedand and IDH1 gDNA and cDNA was sequenced.
IMA cells were tested and authenticated by Sanger sequencing of IDH1, TP53, and ATRX as well as AmpFISTR genotyping as recently as May, 2012. Differentiation status was evaluated with mouse anti-GFAP (1:100, 556328 BD Biosciences), mouse anti-Tuj1 (1:200, MMS-435P Covance), rabbit anti-Sox2 (1:150, sc-20978 Santa Cruz), and rabbit anti-Nestin (1:200, AB5603 Chemicon) with Oregon Green 488 anti-rabbit (1:200, 11038 Life Technologies) or Alexa Fluor 350 anti-mouse (1:200, A10035 Life Technologies) secondary antibodies by immunofluorescence (Nikon TE2000-E microscope). Cell growth was compared by assessing viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (14). Genomic DNA content was assessed by qPCR as described previously (14). Apoptosis was evaluated with ApoDetect Annexin V-FITC Kit (Life Technologies) by immunofluorescence.
Knockout and overexpression
Knockout generation was performed as described (15). IDH1WT or IDH1R132H was ectopically expressed using lentivirus constructs described previously (16) after one month selection with 0.5 μg/ml Blasticidinand expression was assessed by western blot. Anti-IDHC (Santa Cruz, N-20) and anti-IDH1R132H (Dianova) were utilized with anti-GAPDH (Santa Cruz, FL-335) followed by HRP-conjugated secondary antibody and chemiluminescence detection.
Metabolite quantification
Octyl-αKG synthesis is detailed in Supplementary Fig. S1 (7). D-2HG was quantified by LC-MS/MS (5) and αKG was quantified using the αKG Assay Kit (BioVision, K677–100
Results
Loss of IDH1WT is associated with lowered tumoral D-2HG
Among 494 central nervous system tumors used to survey the frequency of IDH1 mutations, we identified two WHO grade IV secondary glioblastomas with IDH1R132H mutations in which a wild type IDH1 allele was undetectable by Sanger sequencing (2). Both tumors had progressed from WHO grade III anaplastic astrocytomas in which wild type and mutant IDH1 alleles (heterozygous) were present (2). Short tandem repeat marker analysis revealed that loss of heterozygosity of D2S1338 (located at 2q35) occurred after progression of those tumors (Fig. 1A). These results indicate that the chromosomal region containing wild type IDH1 (located at 2q34) was deleted, but mutant IDH1 was retained (IDH1R132H/−) during progression. The IDH1R132H/− tumors had 8-fold lower mean intratumoral D-2HG compared with the tumors from which they progressed (Fig. 1A).
Figure 1.

IDH1R132H/− gliomas have lower D-2HG than IDH1R132H/WT gliomas. A, Two anaplastic astrocytomas WHO grade III (A3) which progressed to glioblastomas WHO grade IV (sGBM) demonstrate loss of the wild type IDH1 allele and decreased levels of D-2HG. Mutant IDH1R132H is CAT (His); wild type IDH1 is CGT (Arg). B, D-2HG levels in human gliomas with wild type IDH1 (WT/WT), heterozygous IDH1 mutation (R132H/WT), or IDH1 mutation with loss of wild type IDH1 (R132H/−). LGG, lower grade glioma WHO grade II or III.
We next compared the D-2HG levels in the IDH1R132H/− tumors to the D-2HG levels in other WHO grade IV glioblastomas for which tissue was available (Fig. 1B and Supplementary Table S1). As expected based on previous findings (4, 5), the IDH1R132H/WT glioblastomas had high D-2HG levels (n=7, mean 103 mg/g protein) compared to the IDH1WT/WT glioblastomas (n=8, mean 0.241 mg/g protein, P<0.005, t test). The IDH1R132H/− glioblastomas had 14-fold lower mean D-2HG compared to IDH1R132H/WT glioblastomas. Similar results were found when comparing the IDH1R132H/−glioblastomas to grade II-III gliomas that we analyzed previously (n=11) (5), which are also displayed in Fig. 1B. The finding that IDH1R132H/− gliomas had lower D-2HG compared to IDH1R132H/WT gliomas led us to hypothesize that efficient D-2HG production in glioma cells may require both wild type IDH1 and mutant IDH1 alleles.
Establishment of IMA
To examine whether IDH1WT was needed along with IDH1R132H to produce D-2HG in glioma cells, we derived IMA, an anaplastic astrocytoma WHO grade III cell line containing a native, heterozygous IDH1 mutation (IDH1R132H/WT) (Fig. 2A). IMA also contained stable mutations in TP53 (p.G245V) and in ATRX (p.R781X). No copy number alterations were detected in genomic loci that are frequently altered in primary glioblastoma, including chromosomes 9q, 10p, 19q or in the genomic regions containing EGFR, PDGFR, and PTEN. IMA cells formed colonies from single cells when single-cell diluted and had a doubling time of ~72 hours. The cell line expressed Nestin but was negative for GFAP, Sox2, and Tuj1 and had a stellate morphology (Fig. 2B). As expected for a cell line with an IDH1 mutation, IMA had >80-fold elevated D-2HG compared to three IDH1 wild type cell lines (Fig. 2C, Supplementary Fig. S2).
Figure 2.
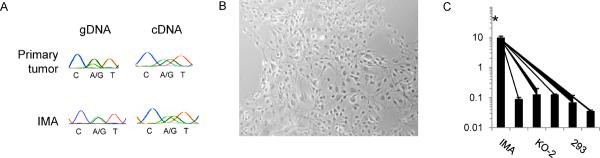
Establishment of an IDH1 mutated anaplastic astrocytoma WHO grade III cell line (IMA). A, Representative sequencing chromatograms for IDH1 codon 132 in gDNA and cDNA. Both the primary tumor and IMA are heterozygous for wild type (CGT) and mutant (CAT) alleles coding for an Arginine (R) to Histidine (H) change at amino acid residue 132. B, Bright field image of adherent IMA cells. C, D-2HG level in lysates of IMA (IDH1R132H/WT), two IDH1 wild type allele knocked out subclones KO-1 and KO-2 (IDH1R132H/−), and HCT116, HOG and 293 cells which do not contain IDH1 mutations. *D-2HG was significantly higher in IMA for pairwise comparisons with each other cell line (P<0.0001 for each, Student's t test). Mean ± s.d. are shown from lysates collected in triplicate.
D-2HG production in IDH1-mutated cells requires IDH1WT
To study the effects of loss of the wild type allele of IDH1, we utilized a recombinant adeno-associated virus targeting system (15) to knock out one IDH1 allele in IMA (Fig. 3A). After screening 1034 clones, we obtained 11 positive clones with PCR-confirmed homologous integration into one allele of the native IDH1 locus (Fig. 3B). To determine which IDH1 allele (mutant or wild type) was disrupted in these clones, a region containing the integrated targeting vector and the adjacent IDH1-R132 locus was PCR amplified and sequenced to determine IDH1 mutation status. All 11 positive clones disrupted the IDH1WT allele and produced only IDH1R132H cDNA (IDH1R132H/−) (Fig. 3C,D).
Figure 3.

Targeted knockout of wild type IDH1 in IMA. A, Targeting vector used to disrupt expression of one allele of IDH1. The targeting vector contains a splice acceptor (SA), internal ribosomal entry sequence (IRES), neomycin selectable marker (neo), and a polyadenylation site (pA), all flanked by right and left homology arms (LHA and RHA, respectively) and inverted terminal repeats (ITR). B, Diagnostic PCR confirms homologous integration of the targeting vector into the IDH1 genomic locus for clones KO-1 and KO-2, but not for the parental IMA cells. Diagnostic PCR primer pairs P1 and P2 each employ one primer that anneals within the targeting vector and a second primer that is outside the homology region to specifically amplify DNA that has incorporated the targeting vector. C, IDH1 R132 gDNA sequencing shows that the targeting vector disrupted the IDH1WT allele and not the IDH1R132H allele in KO-1 and KO-2. The sequence of PCR product P3 reflects the genotype of both the intact and targeted alleles, while P4 reflects the genotype of the intact allele only. D, cDNA sequencing confirms that KO-1 and KO-2 only express IDH1R132H, and not IDH1WT.
We focused on two IDH1R132H/− subclones, KO-1 and KO-2. The morphology, TP53 and ATRX mutation status, and Nestin/Sox2/GFAP/Tuj1 status of IDH1R132H/− cells was indistinguishable from the parental IMA cells. No growth defect was observed for IDH1R132H/− cells, and the percentage of apoptotic cells was less than 2% in IDH1R132H/− and parental cells (Supplementary Fig. S2A,B). Compared to parental IMA (IDH1R132H/WT), D-2HG was 87-fold lower in those two IDH1R132H/− subclones (Fig. 2C). Ectopic re-expression of IDH1WT in KO-1 (IDH1R132H/−) cells rescued D-2HG levels to 52-fold higher than an empty vector control (Fig. 4A,B).
Figure 4.
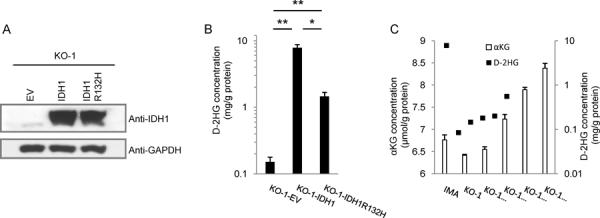
Efficient D-2HG production in IDH1 mutant cells requires wild type IDH1. A, KO-1 (IDH1R132H/−) cells were infected with lentivirus expressing wild type IDH1WT or IDH1R132H. After one month of selection, cell lysates were prepared for western blots, using cells infected with empty vector (EV) lentivirus as a control. B, D-2HG levels in lysates from indicated KO-1 (IDH1R132H/−) cells with ectopic expression of vector control, wild type IDH1WT, or IDH1R132H. C, KO-1 (IDH1R132H/−) cells were treated with the indicated concentrations of cell permeable octyl-αKG for 20 hours, followed by measurement of cellular αKG and D-2HG levels. Mean ± s.d. are shown for samples analyzed in triplicate and are representative of two independent experiments. *P<0.05, **P<0.0001, Student's t test.
To confirm these findings in a second cell line, we investigated the colorectal carcinoma cell line HCT116 (13). Knock-in of an IDH1R132H allele to produce heterozygous IDH1R132H/WT (13) resulted in >100-fold higher D-2HG than the parental IDH1WT/WT HCT116 cells. We were also able to obtain an IDH1R132H/WT subclone of HCT116 in which allelic variation occurred such that only the IDH1R132H allele, but not the IDH1WT allele, was expressed based on cDNA sequencing and immunoblot with IDH1 and IDH1R132H antibodies (Supplymentary Fig. 3A,B). In contrast to the HCT116 cells expressing both mutant and wild type IDH1 alleles, the subclone expressing IDH1R132H alone did not exhibit any elevation of D-2HG levels (Supplymentary Fig. 3C). Thus, both wild type and mutant IDH1 alleles are necessary to achieve elevated D-2HG levels in two cancer cell lines, IMA and HCT116.
We hypothesized that two mechanisms could contribute to a requirement for IDH1WT to overproduce D-2HG: (1) knocking out IDH1WT impairs the conversion of isocitrate to αKG, the substrate for D-2HG production, leading to intracellular αKG levels that are too low for efficient conversion to D-2HG; or (2) efficient D-2HG production requires the heterodimer formed by wild type and mutant IDH1. To exclude the first possibility, we incubated KO-1 (IDH1R132H/−) cells with cell-permeable octyl-αKG and measured the cellular concentrations of αKG and D-2HG. Without octyl-αKG treatment, the KO-1 cells had slightly lower αKG levels than the parental IMA cells, but restoring cellular αKG to the αKG levels in the parental IMA cells could not rescue D-2HG production in the KO-1 cells (Fig. 4C). This finding suggests that the wild type:mutant IDH1 heterodimer is a major mediator of D-2HG production in glioma cells.
Discussion
In this report we characterize IMA, an anaplastic astrocytoma WHO grade III cell line with IDH1R132H/WT mutation. This line joins an anaplastic oligodendroglioma line and an anaplastic oligoastrocytoma line as glioma cell lines that faithfully maintain an IDH1 mutation from the primary tumor (17, 18). WHO grade II–III astrocytomas were recently found to frequently (>60%) contain IDH1, TP53, and ATRX co-mutation (19). IMA provides a model to study the role of these mutations in astrocytoma pathogenesis. IMA exhibited Nestin staining, a marker for neuronal progenitor cells, consistent with IDH1-mutated gliomas exhibiting gene expression profiles that are more similar to neuronal progenitor cells (“proneural”) than other gliomas (20).
The results demonstrate that both the wild type and the mutant IDH1 alleles are necessary to elicit highly elevated cellular D-2HG levels (>4mg D-2HG/g protein) in IMA and HCT116. Characterization of two glioblastomas that lost the wild type IDH1 allele and had relatively low D-2HG suggests that both wild type and mutant IDH1 alleles are also required for high levels of D-2HG production in vivo. These results indicate that the wild type:mutant IDH1 heterodimer is critical to support faster αKG → D-2HG turnover and/or to support “coupled” isocitrate → D-2HG conversion in glioma cells, as has been observed for the purified IDH1 proteins in vitro in specific chemical environments (10, 11). Future studies that define the chemical milieu of glioma cells will be needed to determine which chemical conditions, such as the concentration of NADPH or isocitrate substrates, may favor D-2HG production by the wild type:mutant IDH1 heterodimer. As modulation of D-2HG levels has been proposed as a potential therapeutic strategy for IDH1-mutated tumors, these results warrant a consideration of the role of wild type IDH1 in the development of new treatments.
Supplementary Material
Acknowledgments
We thank Cathy Payne, Lisa Ehinger, Diane Satterfield, and Ping fan (Duke University) for technical assistance and Bert Vogelstein (Johns Hopkins University) for knockout vectors.
Grant Support: This study was supported by NIH R01 CA140316 (to H.Yan), American Cancer Society RSG-10-126-01-CCE (to H.Yan), The V Foundation (to H.Yan), The ABC2 Foundation (to H.Yan), the James S. McDonnell Foundation (to H.Yan), Voices against Brain Cancer (to H. Yan), The Pediatric Brain Tumor Foundation (to D.Bigner),.
Footnotes
G. Jin and Z. J. reitman contributed equally to this work.
Authors' Contributions
Conception and design: G. Jin, Z. J. Reitman, Y. He, H. Yan
Development of methodology: G. Jin, C. G. Duncan
Acquisition of data: G. Jin, C. G. Duncan, I. Spasojevic, B. A. Rasheed, R. E. McLendon, R. Yang
Analysis and interpretation of data: G. Jin, Z. J. Reitman, C. G. Duncan, Y. He, H. Yan
Writing, review, and/or revision of the manuscript: Z. J. Reitman, G. Jin, H. Yan, G. Y. Lopez
Administrative, technical, or material support: R. E. McLendon, D. M. Gooden, D. D. Bigner
Study supervision: H. Yan
Conflict of Interest Statement: H. Yan receives research funding and a consulting fee from Sanofi for research related to IDH1 mutations. H. Yan and D. Bigner are entitled to a share of royalties from the University from Personal Genome Diagnostics and Agios Pharmaceuticals related to IDH1 mutation. The terms of these arrangements are managed by the University in accordance with their conflict of interest policies.
References
- 1.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin G, Reitman ZJ, Spasojevic I, Batinic-Haberle I, Yang J, Schmidt-Kittler O, et al. 2-hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PLoS One. 2011;6:e16812. doi: 10.1371/journal.pone.0016812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012 doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012 doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Annals of neurology. 2011;69:455–63. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- 11.Pietrak B, Zhao H, Qi H, Quinn C, Gao E, Boyer JG, et al. A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of alphaHG. Biochemistry. 2011;50:4804–12. doi: 10.1021/bi200499m. [DOI] [PubMed] [Google Scholar]
- 12.Matsunaga H, Futakuchi-Tsuchida A, Takahashi M, Ishikawa T, Tsuji M, Ando O. IDH1 and IDH2 have critical roles in 2-hydroxyglutarate production in D-2-hydroxyglutarate dehydrogenase depleted cells. Biochem Biophys Res Commun. 2012;423:553–6. doi: 10.1016/j.bbrc.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012 doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin G, Cook S, Cui B, Chen WC, Keir ST, Killela P, et al. HDMX regulates p53 activity and confers chemoresistance to 3-bis(2-chloroethyl)-1-nitrosourea. Neuro-oncology. 2010;12:956–66. doi: 10.1093/neuonc/noq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nature protocols. 2007;2:2734–46. doi: 10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- 16.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3270–5. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10) Neuro Oncol. 2010;12:745–55. doi: 10.1093/neuonc/noq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012;14:184–91. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–22. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.