

Abstract
Background
Gene Ontology (GO) has been widely used in biological databases, annotation projects, and computational analyses. Although the three GO categories are structured as independent ontologies, the biological relationships across the categories are not negligible for biological reasoning and knowledge integration. However, the existing cross-category ontology term similarity measures are either developed by utilizing the GO data only or based on manually curated term name similarities, ignoring the fact that GO is evolving quickly and the gene annotations are far from complete.
Results
In this paper we introduce a new cross-category similarity measurement called CroGO by incorporating genome-specific gene co-function network data. The performance study showed that our measurement outperforms the existing algorithms. We also generated genome-specific term association networks for yeast and human. An enrichment based test showed our networks are better than those generated by the other measures.
Conclusions
The genome-specific term association networks constructed using CroGO provided a platform to enable a more consistent use of GO. In the networks, the frequently occurred MF-centered hub indicates that a molecular function may be shared by different genes in multiple biological processes, or a set of genes with the same functions may participate in distinct biological processes. And common subgraphs in multiple organisms also revealed conserved GO term relationships. Software and data are available online at http://www.msu.edu/˜jinchen/CroGO.
Background
Gene Ontology (GO) is one of the most popular languages for describing and categorizing attributes of biological entities, and utilizes three key categories that are shared by all organisms [1]: molecular function (MF; biochemical function of the gene product), biological process (BP; the biological process to which the gene product contributes) and cellular component (CC; location of the gene product in the cell). To automatically discover novel biological relationships between GO terms, the measurement of term similarities has been extensively studied [2-5], and it remains an active research area in semantic comparison and search [6]. However, most of these similarity methods cannot measure semantic similarities between terms in the different root ontology categories. Although the three root GO categories (MF, BP and CC) are structured as independent ontologies, their biological relationships (especially between BP and MF terms) may provide useful evidence for gene annotation [7]. More importantly, discovering such cross-category associative relationships may help researchers conduct biological reasoning and generate biological hypotheses. For example, if a set of gene products that have the same molecular function often participate in multiple biological processes, then these biological processes may be tightly associated with each other at metabolic level via this molecular function.
To the best of our knowledge, the state-of-art algorithms to identify strong association relationships across GO categories can be classified into two categories: Association Rule Mining (ASR) and Text Mining such as Vector Space Model (VSM). Several algorithms have been developed to identify strong association relationships across GO categories [6-8]; For example, the similarity between terms tb and tj that belong to two different GO categories are shown in Figure 1(a, b). A classic data mining algorithm called association rule mining (ASR) was adopted by Bodenreider et al [8] and Kumar et al [6] to compute cross-category GO term similarity SimASR(t1, t2), where terms t1 and t2 are in category C1 and C2 respectively. Based on these approaches, a ready-for-use inter-category GO structure has been constructed by Myhre et al [7] and is provided as an addition to GO. Note that the ASR-based term associations are directional, i.e., SimASR (t1, t2) may be different to SimASR (t2, t1). However, the "shallow annotation" problem [9] was ignored in the ASR-based measures, because if both t1 and t2 are very close to the root of C1 and C2, chances are high that both SimASR (t1, t2) and SimASR (t2, t1) are high regardless of whether they are biologically related, since the terms near the root contain almost all of the genes after propagation [5]. As a result, term pairs that are at very shallow levels of the GO hierarchy (e.g., "response to stimulus") can yield very high semantic similarities, and such pairs are not distinguishable from high-scoring pairs that are "deep" at GO hierarchical structure [10].
Figure 1.

An example of two GO categories, gene co-function network and gene set association. (a and b) An example of two GO categories, in which each node is a GO term, each edge represents a conceptual relation between two terms, and {g1...g13} is the set of genes annotated to corresponding terms. (c) An example of gene co-function network, in which each node is a gene, each edge represents the functional associations between the genes, and the confidence score at each edge measures the probability of an interaction to represent a true functional linkage between two genes. (d) The gene set association between gene set Gb and Gj.
To avoid the "shallow annotation" problem, Bodenreidar et al proposed a Vector Space Model (VSM)based measure [8], which considers the semantic weight of each gene product. VSM has been widely used in information retrieval applications for calculating the similarities among documents that can be described with vectors [11]. Mathematically, given two vectors v1 and v2 consisting of binary values indicating the presence (1) and absence (0) of an association between a term (t1 or t2) and a gene, the term similarity SimVSM (t1, t2) can be calculated with a weight-adjusted cosine similarity equation, representing the angle between v1 and v2. However, using this approach, the resulting cross-category association relationships are undirected because of SimVSM (t1, t2) ≡ SimVSM (t2, t1). Biologically, if a MF term points to a BP term, the MF term is involved in the BP term [7]; and if a BP term points to a MF term, the BP term is realized by the MF term [6]. Therefore, different directions of the term relationships indicate different biological meanings. Since the VSM-based measure treats v1 and v2 equally, the order of the resulting biological associations are lost. Furthermore, the VSM-based measure heavily relies on the overlapped genes of two target terms, ignoring the fact that the annotations are far from complete, e.g., only 28.1% of human genes have at least one non-IEA annotation [12], leading to inaccurate term similarity scores.
There have been many studies which have shown that integrating several different broad types of data can significantly improve the results of bioinformatics methods [13,14], but no such method exists for GO analyses. Starting with the intuition that the incorporation of extra biological information may improve the performance of a cross-category term similarity measure, we propose a new algorithm, Cross-Category Gene Ontology Measurement (CroGO), for calculating the similarity between two cross-category terms by effectively incorporating genome-specific gene co-function network data. Compared to the existing algorithms, CroGO has the following advantages:
1. CroGO incorporates the information from gene co-function networks, which are widely believed to be good complements to GO for understanding the associations between biological concepts. The co-function networks have been constructed using extensive gene expression and protein interaction data containing millions of individual observations from DNA microarrays, physical protein interactions, genetic interactions, literature, and comparative genomics methods [15-17]. Therefore, numerous new cross-ontology associations can be learned by incorporating the co-function networks into a model.
2. Our algorithm determins the directions of term relationships by considering the GO hierarchical structure, while the existing methods either ignores the directions or simply defines the directions by using the different numbers of genes annotated to two terms.
3. The "shallow annotation" problem has been avoided in CroGO by considering the specificity of GO terms, while the ASR-based measures and some statistical measures, e.g., χ2-test, may mix shallow-level term pairs with term pairs that are deep at GO hierarchical structure.
4. The term association network generated with CroGO is genome-specific, from which conserved term associations may suggest vital functional connections, and unique term associations in certain organisms may suggest genome-specific functions even for homolog genes. While the term associations generated with lexical approaches [7,8,18] that test whether one term is a substring in the other term are universal for all kinds of organisms.
Method
To measure the similarity between the terms in different GO categories, CroGO has three steps. First, the association between two sets of genes that are annotated to any two given GO terms is calculated. Second, the gene annotations and gene set associations are integrated to calculate the pair-wise term similarity. Third, the directions of all the pair-wise term relationships are inferred with a GO structure based approach.
Step 1: Gene set association
To measure the association between two gene sets G1 and G2 that are annotated to terms t1 and t2 in GO categories C1 and C2 respectively, we define Gene Set Association (GSA) by taking into consideration the weighted edges in a gene co-function network N. Nodes in N represent genes and edges represent functional interactions between genes, and every edge is associated with a confidence score that measures the probability of interaction. An illustrative example of a gene co-function network with four genes is shown in Figure 1(c). GSA is defined as:
| (1) |
where G1 (or G2) is the set of genes annotated to t1 (or t2), |X| represents the size of set X, G1 ∪ G2 is the union of G1 and G2, and |G1 − G2| is defined as:
| (2) |
where wij is functional similarity score between genes gi and gj in gene co-function network N:
| (3) |
where <gi, gj > is an edge in N, and cof (gi, gj) is the likelihood of the functional interaction between gi and gj in N, and cof (gi, gj) ∈ [0, 1].
In Equation 2, the right part represents the semantic overlapping between G1 and G2. If gene set G1 and G2 are the same, then |G1 − G2| = 0, consequently GSA(G1, G2) = 1; and if G1 and G2 do not have any overlap and there is no linkage between the gene sets in N, then |G1 − G2| = |G1|, consequently GSA(G1,G2) = 0. In summary, the gene set association score GSA(G1, G2) represents the association between two gene sets G1 and G2 based on the shared genes and the gene associations in a co-function network.
Step 2: Pair-wise similarity measure
Given two GO terms t1 and t2 from different GO categories C1 and C2, the term similarity Sim(t1, t2) is defined with the integration of GO structure, gene annotations and co-function network:
| (4) |
where GSA(G1, G2) is calculated with Equation 1, and and are the sets of all the genes involved in category C1 and C2 respectively.
In Equation 4, the first part, GSA(G1, G2), represents the association between the gene sets annotated to the terms t1 and t2, which takes advantage of both the prior knowledge deposited in GO and the experimental gene-gene associations summarized in the gene co-function networks. The second part describes the specificity of both terms by considering the level of generality of t1 and t2 in their own GO categories to avoid the "shallow annotation" problem.
Step 3: Term pair direction assignment
We look for the directions of the relationships between two terms with a pruning approach. First, all-by-all term similarities are calculated with Equation 4 and term pairs with high similarity scores are saved with bi-directions. Then for each term t and a set of terms T that connect to t, we remove the edge direction from t to term t' only if there exists another term t'' such that t is an ancestor of t'' (t', t'' ∈ T ). In the end, if both directions at edge <t, t' > are removed, then edge <t, t' > shall be removed as well.
In the example in Figure 1(a, b), tb and tj are two terms in category C1 and C2, and the genes annotated to them are shown in Figure 1(d), in which the functional similarity scores of any two genes are calculated with a co-function network in Figure 1(c). To calculate the directional association between tb and tj, we first calculate all-by-all similarity scores, e.g., Sim(tb, tj) = 0.651, Sim(tb, th) = 0.465, etc., using Equation 4, and then apply a user defined threshold (say, 0.45) to filter term relationships with low scores. In the third step, the direction from tb to tj is removed, since tj is an ancestor of th, and the direction from tj to tb is retained, because there is no child of tb that also connects to tj and Sim(tb, th) is greater than the threshold. Finally, we conclude that tj points to tb with similarity score 0.651.
Results
In the test experiments, we focused on identifying the relationships between the BP and MF terms. To show the significance of CroGO, we first compared its results with both the ASR-based and VSM-based measures on a small gold-standard set generated with known reaction-to-pathway relationships on yeast. Then we constructed the MF-BP cross-ontology term association network of yeast, and evaluated it with evidences from the manually-curated yeast pathway database. Finally, we studied the conservation of cross-ontology associations by comparing the yeast and human term association networks.
The GO data and gene annotations was downloaded from GO website in February, 2012, in which only the annotations with non-IEA evidences were used [1]. The gene co-function network was obtained from YeastNet [15], which has 102,803 linkages among 5,483 genes. The co-function score of each linkage was normalized between 0 and 1. CroGO was developed with Java JDK 1.6 and JUNG library [19].
Performance comparison on gold-standard set
To compare the performance of CroGO with the existing measures with confirmed biological knowledge, we first generated a small "gold-standard" set based on the known reaction-to-pathway relationships [7] in yeast in three steps: 1) we associate a BP term to a metabolic pathway if the pathway corresponds directly to a GO-defined biological process; 2) a metabolic pathway is associated to several Enzyme Commission (EC) numbers if the enzymes catalyze the pathway; and 3) we link a EC number to a MF term with the official GO translations [20,21]. In the end, a small gold-standard set of reliable MF-BP associations was obtained via the known metabolic pathways and EC numbers. In YeastCyc [22], 71 out of the total 187 pathways match exactly to BP terms and have at least one EC number associated to them. From these pathways, 175 MF-BP pairs were identified and saved as the yeast gold-standard set (Additional file 1). We also randomly selected term pairs to construct a random set, which is 10 times larger than the gold-standard set.
We calculated pair-wise term similarities for the term pairs in the gold-standard set and the term pairs in the random set using CroGO, and compared its performance with the ASR and VSM based measures by drawing a receiver operating characteristic (ROC) curve [23] for each measure. The ROC curves in Figure 2(a) showed clearly that CroGO has the best performance. As shown in Table 1, when the false positive threshold was set to 15%, the true positive rate of CroGO is 88%, while the true positive rates of the ASR and VSM based measures are both 83%. This analysis also showed that 102 more MF-BP pairs were recognized by CroGO than the ASR and VSM based measures when the number of true positives equals the number of false positives. This indicates that by incorporating the co-function network, CroGO has produced better coverage than the other measures by recognizing more gene associations between genes which are annotated to the gold-standard connected GO terms. In addition, the same experiments were applied on human data, and the results is consistent to the yeast data (see details in the conserved association section).
Figure 2.
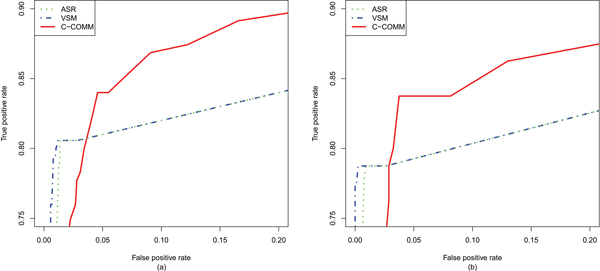
ROC curves for the experimental results on the gold-standard sets. ROC curves for the experimental results on the gold-standard sets of yeast (a) and human (b) calculated with CroGO (red), VSM (blue) and ASR (green). ASR and VSM based measures have very similar trends, and are overlapping for most of the visible portion of the ROC curves.
Table 1.
The performance study on yeast and human gold-standard sets.
| Organism | Measure | No. of term pairs (when FP = TP) | TP rate (when FP = 5%) |
TP rate (when FP = 10%) |
TP rate (when FP = 15%) |
|---|---|---|---|---|---|
| Yeast | ASR | 50 | 81% | 82% | 83% |
| VSM | 50 | 81% | 82% | 83% | |
| CroGO | 152 | 84% | 87% | 88% | |
| Human | ASR | 21 | 79% | 80% | 81% |
| VSM | 21 | 79% | 80% | 81% | |
| CroGO | 67 | 84% | 85% | 87% | |
The performance study of CroGO, VSM and ASR based measures on yeast and human gold-standard sets.
Genome-specific MF-BP association network
To demonstrate the practical use of CroGO and provide researchers a platform to enable a more consistent use of GO, we linked biological concepts by generating a genome-specific term association network. Mathematically, we constructed bipartite graph Ω (M, B, E) where M and B are sets of nodes representing MF and BP terms respectively; edge e <m, b > in E indicates that m is involved in b; and edge e <b, m > indicates b is realized by m (b ∈ B and m ∈ M). We constructed the highly reliable MF-BP association network Ωyeast (M, B, E) by comparing all the MF and BP terms with CroGO and adopting a strict z-score cutoff (in this paper we required z − score > 8.0). Therefore, all the edges in the MF-BP association network are statistically significant. In the end, network Ωyeast (M, B, E) has 613 MF terms, 843 BP terms and 1,485 edges. As shown in Additional file 2 the yeast association network Ωyeast (M, B, E) consists of many small disconnected graphs.
We evaluated the whole network performance of Ωyeast (M, B, E) with an GO enrichment approach. A biological process usually contains multiple biochemical reactions. The genes in two adjacent reactions should have similar BP annotations, because the product of one reaction is the substrate of the other one. With this criterion, we evaluate the performance of CroGO by testing whether a given MF term that is linked to a reaction r connects to all the BP terms that are enriched in the adjacent reactions of r. By extracting reaction information from YeastCyc, we found 82 valid MF terms in Ωyeast (M, B, E), and among them 56 MF terms (67.1%) were connected to enriched BP terms. For performance comparison, we constructed two extra yeast MF-BP association networks using the ASR and VSM based approaches with the same z-score cutoff. This test showed that in the VSM-based result, only 25 out of the valid 43 MF terms (58.1%) were connected to enriched BP terms; and due to the "shallow annotation" problem, there is no valid MF term in the ASR-based result. Again, these results indicate that CroGO is superior to the existing measures in constructing term association networks.
An edge <t1, t2 > in the term association network can be classified into one of the three categories: "identical" (G1 = G2), "non-overlap" (G1 ∩ G2 = ∅) and "overlap but not identical" (G1 ∩ G2 ≠ ∅ and G1 ≠ G2), where G1 (or G2) is the set of genes annotated to term t1 (or t2). Figure 3(a) shows that 356 term relationships (24%) in the "non-overlap" category can only be found by CroGO because of the incorporation of extra biological information from a co-function network. The top 20 term associations in the "overlap but not identical" category and the top 20 term associations in "non-overlap" category are listed in Table 2 and 3. In these term associations, 24 were supported by the existing biological studies or lexical matching on term definition, and the rest 16 are new conceptual connections that cannot be found in any literature. For example, MF term "endopeptidase activator activity" is assigned to PRE1 and PUP3 that hydrolyze nonterminal peptide bonds in polypeptides; and BP term "proteasome core complex assembly" means the aggregation, arrangement and bonding together of a mature, active 20S proteasome core particle complex that does not contain any regulatory particles, and it is annotated to PRE2 and PRE9. Clearly these two terms do not have any common genes. However, their annotated genes are tightly connected with each other in the co-function network because the gene products were found in the same protein complexes from affinity purification/mass spectrometry data [15], and an endogenous activator of the protease was also identified [24]. Therefore, the two remote concepts are connected by CroGO, suggesting that endopeptidase activators are involved in proteasome core complex assembly.
Figure 3.
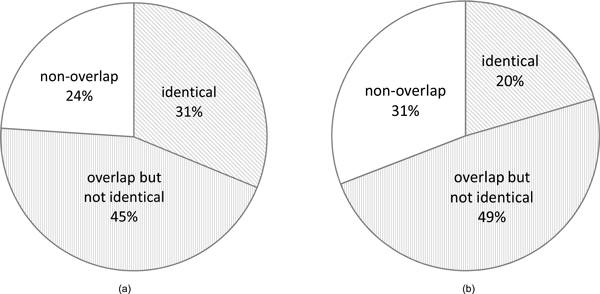
The edges distribution in the genome-specific term association network of yeast (a) or human (b). The edges distribution in the genome-specific term association network of yeast (a) or human (b). The three categories are "identical", "non-overlap" and "overlap but not identical". It indicates a significant part ("non-overlap") of the networks can only be identified by CroGO because of the incorporation of extra biological information from the co-function networks.
Table 2.
Top 20 term associations in category "overlap but not identical" that were identified by CroGO.
| MF Name | BP Name | Evidence |
| polynucleotide adenylyltransferase activity | ncRNA polyadenylation | NEW |
| TFIIF-class binding TF activity | regulation of transcription-coupled nucleotide-excision repair | REF [37] |
| TFIIF-class binding TF activity | positive regulation of transcription elongation from Pol I promoter | REF [38] |
| TFIIF-class binding TF activity | regulation of transcription elongation from Pol I promoter | REF [38] |
| TFIIF-class binding TF activity | positive regulation of histone H3-K36 trimethylation | NEW |
| TFIIF-class binding TF activity | regulation of histone H3-K36 trimethylation | NEW |
| TFIIF-class binding TF activity | positive regulation of histone H3-K36 methylation | NEW |
| TFIIF-class binding TF activity | regulation of nucleotide-excision repair | REF [39] |
| TFIIF-class binding TF activity | regulation of histone H2B ubiquitination | REF [40] |
| TFIIF-class binding TF activity | positive regulation of phosphorylation of Pol II C-terminal domain serine 2 residues | NEW |
| TFIIF-class binding TF activity | regulation of phosphorylation of Pol II C-terminal domain serine 2 residues | NEW |
| TFIIF-class binding TF activity | regulation of histone H2B conserved C-terminal lysine ubiquitination | NEW |
| IMP dehydrogenase activity | GTP biosynthetic process | REF [41] |
| hydrogen ion transporting ATP synthase activity, rotational mechanism | ATP biosynthetic process | LEXICAL |
| RNA-directed RNA polymerase activity | tRNA transcription from Pol III promoter | LEXICAL |
| RNA-directed RNA polymerase activity | tRNA transcription | NEW |
| protein prenyltransferase activity | protein geranylgeranylation | REF [42] |
| second spliceosomal transesterification activity | generation of catalytic spliceosome for second transesterification step | NEW |
| oxoglutarate dehydrogenase activity | 2-oxoglutarate metabolic process | LEXICAL |
| peptide alpha-N-acetyltransferase activity | N-terminal protein amino acid acetylation | REF [43] |
Top 20 term associations in category "overlap but not identical" that were identified by CroGO. In the list, 8 term associations are supported by the existing biological studies, 3 are supported by the lexical matching on term definition, and the rest 7 are new conceptual connections that cannot be found in any literature. Only 3 of the term associations can be identified by the VSM or ASR based measures.
Table 3.
Top 20 term associations in category "non-overlap" that were identified by CroGO.
| MF Name | BP Name | Evidence |
|---|---|---|
| endopeptidase activator activity | proteasome core complex assembly | NEW |
| TFIIF-class binding TF activity | regulation of histone H3 K79 methylation | NEW |
| RNA-directed RNA polymerase activity | DNA-dependent transcriptional start site selection | LEXICAL |
| RNA-directed RNA polymerase activity | transcriptional start site selection at Pol II promoter | LEXICAL |
| single base insertion or deletion binding | chiasma assembly | REF [26] |
| double-strand/single-strand DNA junction binding | chiasma assembly | REF [26] |
| double-stranded telomeric DNA binding | gene conversion at mating-type locus, DNA double-strand break processing | NEW |
| G-quadruplex DNA binding | gene conversion at mating-type locus, DNA double-strand break processing | NEW |
| very long-chain fatty acid-CoA ligase activity | long-chain fatty-acyl-CoA metabolic process | REF [27] |
| very long-chain fatty acid-CoA ligase activity | fatty-acyl-CoA metabolic process | REF [27] |
| very long-chain fatty acid-CoA ligase activity | acyl-CoA metabolic process | REF [27] |
| single base insertion or deletion binding | meiotic heteroduplex formation | NEW |
| guanine/thymine mispair binding | chiasma assembly | NEW |
| TFIIE-class TF binding | negative regulation of ribosomal protein gene transcription from Pol II promoter in response to chemical stimulus | REF [28] |
| TFIIE-class binding TF activity | negative regulation of ribosomal protein gene transcription from Pol II promoter in response to chemical stimulus | REF [28] |
| Hsp90 protein binding | positive regulation of telomere maintenance via telomerase | NEW |
| Hsp90 protein binding | positive regulation of telomere maintenance | REF [29] |
| Hsp90 protein binding | positive regulation of homeostatic process | REF [30] |
| aldehyde dehydrogenase activity | beta-alanine metabolic process | REF [31] |
| aldehyde dehydrogenase activity | beta-alanine biosynthetic process | REF [31] |
Top 20 term associations in category "non-overlap" that were identified by CroGO. In the list, 11 term associations are supported by the existing biological studies, 2 are supported by the lexical matching on term definition, and the rest 7 are new conceptual connections that cannot be found in any literature. None of the term associations can be identified by the VSM or ASR based measures.
An interesting topological pattern found in the yeast MF-BP association network is the frequently occurred MF-centered hub. As the case study shown in Figure 4, the function "anaphase-promoting complex binding" is involved in seven different biological processes in yeast from metaphase to anaphase during mitosis, including "activation of anaphase-promoting complex activity involved in meiotic cell cycle", suggesting that anaphase-promoting complex is key for mitotic cyclins and anaphase inhibitory protein degradation, thereby triggering sister chromatid separation and exit from mitosis [25]. In summary, by connecting remote concepts, researchers are able to conduct advanced biological reasoning and generate interesting biological hypotheses.
Figure 4.
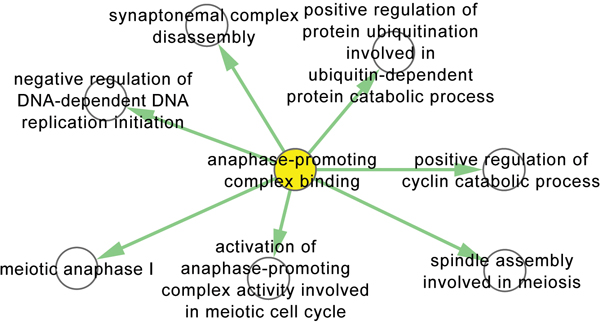
A case study of a MF-centered topological pattern found in the yeast MF-BP association network. A case study of a MF-centered topological pattern found in the yeast MF-BP association network. The yellow and white nodes are MF and BP term respectively.
Conserved MF-BP associations
To explore which part of the MF-BP association network is conservative and which part is varying from one organism to another, we constructed the human MF-BP association network Ωhuman (M, B, E) by comparing all the MFs and BPs that are annotated to at least one human gene and adopting the same z-score cutoff (8.0). Ωhuman (M, B, E) has 1,209 MF terms, 2,250 BP terms and 5,138 edges, among which 1,583 edges are between terms that have no overlap on their annotated genes (see Figure 3(b)), indicating that at least 30% of the edges are solely contributed by the human co-function network HumanNet [17] (which has 476,399 linkages among 16,243 genes). Unlike the yeast MF-BP association network, the human MF-BP association network Ωhuman (M, B, E) showed in Additional file 3 has a large subgraph occupying ~ 50% of the total edges.
To evaluate the performance of CroGO on human data, we generated the human gold-standard set consisting of 80 MF-BP pairs (Additional file 4) from the human pathway data humanCyc [32] and a random set which was 10 times larger. The ROC curves in Figure 2(b) showed that CroGO has the best performance. When the false positive threshold was set to 15%, the true positive rate of CroGO is 87%, while the true positive rates of the ASR and VSM based measures are both 81%. This analysis also showed that 46 more MF-BP pairs were able to be recognized by CroGO than the ASR and VSM based measures when the number of true positives equals the number of false positives, indicating CroGO has outperforms the other measures on human data as well as on the yeast data.
Additional file 2 and Additional file 3 show that the MF-BP relations in human are much more complex than in yeast, yet their common MF-BP relationships still reveal interesting conserved patterns in the long evolution process. Figure 5 shows an example in which five different types of DNA binding proteins are involved in the biological processes "meiotic mismatch repair" and "chiasma assembly" in both human and yeast. As an evidence, the genes and concepts that connect "DNA binding", "meiotic mismatch repair" and "chiasma assembly" are drawn with BioGraph [33] in Additional file 5. Mismatch repair proteins are a highly diverse group of proteins that interact with numerous DNA structures during DNA repair and replication [34]. In this protein group, three MSH proteins form an active protein complex to play an essential role in DNA repair by fixing mistakes that are made when DNA is copied in preparation for cell division [35]. MSH6, a DNA mismatch repair homolog of human MutS protein in yeast, plays a role in binding double-stranded DNA and in four-way junction DNA Binding [36]. Probably because of the importance of these DNA-binding proteins, their functions and their roles in the two biological processes are conserved during evolution.
Figure 5.

An example that five different types of DNA binding proteins are involved in biological processes meiotic mismatch repair and chiasma assembly in both human and yeast. An example that five different types of DNA binding proteins are involved in biological processes meiotic mismatch repair and chiasma assembly in both human and yeast. The yellow and white nodes are MF and BP term respectively.
Conclusions
In each GO category, the ontology is structured as a directed acyclic graph to reflect the complex hierarchy of biological events and locations, but the thee root GO categories are structured as independent ontologies. By connecting MF term to BP terms, the low-granularity MFs can be related to higher-granularity BPs, providing useful evidence for improved biological reasoning.
The similarity measure between GO terms in different categories has been the focus of other studies. However, existing measurements are either developed by utilizing only the GO data, or have been based on the similarity between term names.
Starting with the intuition that the incorporation of extra biological information may improve the performance of a cross-category term similarity measure, we propose a new algorithm called CroGO for calculating the similarity between two cross-category terms by incorporating gene co-function network data. Compared to the existing algorithms, CroGO can clearly identify more biologically verified cross-category term relationships, since it utilizes extra biological information that is good complement to GO for understanding the associations between biological concepts. And the ROC curves on small gold-standard sets of human and yeast indicate CroGO can identify term associations more precisely.
To demonstrate the practical use of CroGO and provide researchers a platform to enable a more consistent use of GO, the genome-specific term association networks of yeast and human were generated. In these networks, we found that the frequently occurred MF-centered hub is an interesting topological pattern as it may indicate a molecular function could be shared by different genes in multiple biological processes, or a set of genes with the same functions may be a common component belonging to distinct biological processes. From the topological view, the human association network is much more complex than the yeast term association network. And their common MF-BP relationships reveal evolutionary conserved patterns, indicating important functional associations.
Note that CroGO uses a gene co-function network as part of its input. Therefore, in the condition that such gene co-function does not exist, CroGO is not applicable. In the future, we will extend CroGO to automatically generate the co-function network from existing of gene expression or protein-protein interaction data that a user specified. Second of all, different directions of the term relationships indicate different biological meanings. A relationship from a MF term to a BP term means the MF term is involved in the BP term; and the reverse direction indicates the BP term is realized by the MF term. We will study the directions between BP and MF terms in the future work. And we also would like to extend CroGO to study the relationships between all the three GO categories and apply it on other biological/medical ontologies. Furthermore, we will develop more advanced network biology approaches, such that genome, proteome, metabolome and other -omics data can be jointly analyzed to understand cross-ontology relationships.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JC conceived the project. JP, JC and YW designed the algorithm and experiments. JP implemented the algorithm and finished the experiments.
Declarations
The publication costs for this article were funded by the corresponding author's institution.
This article has been published as part of BMC Bioinformatics Volume 14 Supplement 2, 2013: Selected articles from the Eleventh Asia Pacific Bioinformatics Conference (APBC 2013): Bioinformatics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/14/S2.
Supplementary Material
Gold-standard set on Yeast. 175 gold-standard set of MF-BP relationships in yeast.
MF-BP Association Network of yeast. MF-BP Association Network of yeast. The nodes represent terms and the edges represent the term associations discovered by CroGO. The yeast MF-BP association network consists many small disconnected graphs.
MF-BP Association Network of human. MF-BP Association Network of human. The nodes represent terms and the edges represent the associations discovered by CroGO. The human MF-BP association network has a large subgraph occupying 50% of total edges.
Gold-standard set on Human. 80 gold-standard set of MF-BP relationships in human.
genes and concepts that connect "DNA binding", "meiotic mismatch repair" and "chiasma assembly". The genes and concepts that connect "DNA binding", "meiotic mismatch repair" and "chiasma assembly". The figure was generated with BioGraph [40].
Contributor Information
Jin Chen, Email: jinchen@msu.edu.
Yadong Wang, Email: ydwang@hit.edu.cn.
Acknowledgements
This project has been funded by the U.S. Department of Energy (Chemical Sciences, Geosciences and Biosciences Division, grant no. DE-FG02-91ER20021 to J.C; the National High Technology Research and Development Program of China grant no. 2012AA020404 and the National Natural Science Foundation of China grant no. 60973078 to Y. W, and China Scholarship Council to J. P.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik P. Semantic similarity in a taxonomy: An information-based measure and its application to problems of ambiguity in natural language. J Art Intel Res. 1999;11:95–130. [Google Scholar]
- Schlicker A, Domingues FS, Rahnenfuhrer J, Lengauer T. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics. 2006;7:302. doi: 10.1186/1471-2105-7-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Gao L, Tu K, Guo Z. Broadly predicting specific gene functions with expression similarity and taxonomy similarity. Gene. 2005;352:75–81. doi: 10.1016/j.gene.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Du ZD, Payattakool R, Yu PS, Chen CF. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23(10):1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- Kumar A, Smith B, Borgelt C. Dependence relationships between Gene Ontology terms based on TIGR gene product annotations. Proceedings of the 3rd International Workshop on Computational Terminology (CompuTerm 2004) 2004. pp. 31–38.
- Myhre S, Tveit H, Mollestad T, Lægreid A. Additional gene ontology structure for improved biological reasoning. Bioinformatics. 2006;22(16):2020–2027. doi: 10.1093/bioinformatics/btl334. [DOI] [PubMed] [Google Scholar]
- Bodenreider O, Aubry M, Burgun A. Non-lexical approaches to identifying associative relations in the gene ontology. Proceedings of the Pacific Symposium on Biocomputing. 2005. pp. 91–102. [PMC free article] [PubMed]
- Sevilla JL, Segura V, Podhorski A, Guruceaga E, Mato JM, Martinez-Cruz LA, Corrales FJ, Rubio A. Correlation between gene expression and GO semantic similarity. IEEE ACM T Comput Bi. 2005;2(4):330–338. doi: 10.1109/TCBB.2005.50. [DOI] [PubMed] [Google Scholar]
- Mistry M, Pavlidis P. Gene ontology term overlap as a measure of gene functional similarity. BMC bioinformatics. 2008;9:327. doi: 10.1186/1471-2105-9-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Yates R, Ribeiro-Neto B. Modern Information Retrieval. Addison-Wesley New York; 1999. [Google Scholar]
- Rhee SY, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nat Rev Genet. 2008;9(7):509–515. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- Bebek G, Koyutürk M, Price N, Chance M. Network biology methods integrating biological data for translational science. Brief Bioinform. 2012;13(3):446–459. doi: 10.1093/bib/bbr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage R, Ghahramani Z, Griffin J, Bernard J, Wild D. Discovering transcriptional modules by Bayesian data integration. Bioinformatics. 2010;26(12):i158–i167. doi: 10.1093/bioinformatics/btq210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Li ZH, Marcotte EM. An Improved, Bias-Reduced Probabilistic Functional Gene Network of Baker's Yeast, Saccharomyces cerevisiae. PLoS ONE. 2007;2(10):e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Ambaru B, Thakkar P, Marcotte EM, Rhee SY. Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol. 2010;28(2):149–156. doi: 10.1038/nbt.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Blom U, Wang P, Shim J, Marcotte E. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21(7):1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogren P, Cohen K, Acquaah-Mensah G, Eberlein J, Hunter L. The compositional structure of Gene Ontology terms. Proceedings of the Pacific Symposium on Biocomputing. 2004. pp. 214–225. [DOI] [PMC free article] [PubMed]
- O'Madadhain J, Fisher D, Smyth P, White S, Boey Y. Analysis and visualization of network data using JUNG. J Stat Software. 2005;10(2):1–25. [Google Scholar]
- Hill DP, Davis AP, Richardson JE, Corradi JP, Ringwald M, Eppig JT, Blake JA. Program description: Strategies for biological annotation of mammalian systems: implementing gene ontologies in mouse genome informatics. Genomics. 2001;74:121–8. doi: 10.1006/geno.2001.6513. [DOI] [PubMed] [Google Scholar]
- Camon E, Barrell D, Dimmer E, Lee V, Magrane M, Maslen J, Binns D, Apweiler R. An evaluation of GO annotation retrieval for BioCreAtIvE and GOA. BMC Bioinformatics. 2005;6(Suppl 1):S17. doi: 10.1186/1471-2105-6-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Foerster H, Fulcher C, Hopkinson R, Ingraham J, Kaipa P, Krummenacker M, Paley S, Pick J, Rhee S, Christophe T, Zhang P, Karp PD. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006;34(suppl 1):D511–D516. doi: 10.1093/nar/gkj128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heagerty P, Zheng Y. Survival model predictive accuracy and ROC curves. Biometrics. 2005;61:92–105. doi: 10.1111/j.0006-341X.2005.030814.x. [DOI] [PubMed] [Google Scholar]
- Yukawa M, Sakon M, Kambayashi J, Shiba E, Kawasaki T, Ariyoshi H, Mori T. Proteasome and its novel endogeneous activator in human platelets. Biochem Biophys Res Commun. 1991;178:256–262. doi: 10.1016/0006-291X(91)91807-O. [DOI] [PubMed] [Google Scholar]
- Hill DP, Davis AP, Richardson JE, Corradi JP, Ringwald M, Eppig JT, Blake JA. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetka M, Kawasaki I, Strome S, Müller F. Synapsis and chiasma formation in Caenorhabditis elegans require HIM-3, a meiotic chromosome core component that functions in chromosome segregation. Genes & Dev. 1999;13(17):2258–2270. doi: 10.1101/gad.13.17.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Moulson C, Pei Z, Miner J, Watkins P. Fatty acid transport protein 4 is the principal very long chain fatty acyl-CoA synthetase in skin fibroblasts. J Biol Chem. 2007;282(28):20573–20583. doi: 10.1074/jbc.M700568200. [DOI] [PubMed] [Google Scholar]
- Nikolov D, Burley S. RNA polymerase II transcription initiation: a structural view. P Natl Acad Sci USA. 1997;94:15–22. doi: 10.1073/pnas.94.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin N, Charbonneau M. Hsp90 levels affect telomere length in yeast. Mol Genet Genomics. 2001;265:126–134. doi: 10.1007/s004380000398. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz D, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Bio. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Zetka M, Kawasaki I, Strome S, Müller F. Synapsis and chiasma formation in Caenorhabditis elegans require HIM-3, a meiotic chromosome core component that functions in chromosome segregation. Genetics. 2003;163(1):69–77. doi: 10.1101/gad.13.17.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Foerster H, Fulcher C, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee S, Shearer A, Tissier C, Walk TC, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008;36(suppl 1):D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liekens A, De Knijf J, Daelemans W, Goethals B, De Rijk P, Del-Favero J. BioGraph: unsupervised biomedical knowledge discovery via automated hypothesis generation. Genome Biology. 2011;12(6):R57. doi: 10.1186/gb-2011-12-6-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Borts R. Meiotic recombination intermediates and mismatch repair proteins. Cytogenet Genome Res. 2004;107(3-4):232–248. doi: 10.1159/000080601. [DOI] [PubMed] [Google Scholar]
- Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Familial Cancer. 2005;4(4):323–333. doi: 10.1007/s10689-005-8351-6. [DOI] [PubMed] [Google Scholar]
- Subramanian D, Griffith J. Interactions between p53, hMSH2-hMSH6 and HMG I (Y) on Holliday junctions and bulged bases. Nucleic Acids Res. 2002;30(11):2427–2434. doi: 10.1093/nar/30.11.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell A, Bardwell L, Iyer N, Svejstrup J, Feaver W, Kornberg R, Friedberg E. Yeast nucleotide excision repair proteins Rad2 and Rad4 interact with RNA polymerase II basal transcription factor b (TFIIH) Mol Cell Biol. 1994;14(6):3569–3576. doi: 10.1128/mcb.14.6.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L, Tanese N, Tjian R. The TATA-binding protein and associated factors are integral components of the RNA polymerase I transcription factor, SL1. Cell. 1992;68(5):965–976. doi: 10.1016/0092-8674(92)90039-F. [DOI] [PubMed] [Google Scholar]
- Wang Z, Buratowski S, Svejstrup J, Feaver W, Wu X, Kornberg R, Donahue T, Friedberg E. The yeast TFB1 and SSL1 genes, which encode subunits of transcription factor IIH, are required for nucleotide excision repair and RNA polymerase II transcription. Mol Cell Biol. 1995;15(4):2288–2293. doi: 10.1128/mcb.15.4.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, Reinberg D. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125(4):703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Exinger F, Lacroute F. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr Genet. 1992;22:9–11. doi: 10.1007/BF00351735. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Rossi G, Ferro-Novick S. Bet2p and Mad2p are components of a prenyltransferase that adds geranylgeranyl onto Ypt1p and Sec4p. Nature. 1993;366:84–86. doi: 10.1038/366084a0. [DOI] [PubMed] [Google Scholar]
- Arnesen T. Protein N-terminal acetylation: NAT 2007-2008 Symposia. BMC Proceedings. 2009;3(Suppl 6):S1. doi: 10.1186/1753-6561-3-s6-s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gold-standard set on Yeast. 175 gold-standard set of MF-BP relationships in yeast.
MF-BP Association Network of yeast. MF-BP Association Network of yeast. The nodes represent terms and the edges represent the term associations discovered by CroGO. The yeast MF-BP association network consists many small disconnected graphs.
MF-BP Association Network of human. MF-BP Association Network of human. The nodes represent terms and the edges represent the associations discovered by CroGO. The human MF-BP association network has a large subgraph occupying 50% of total edges.
Gold-standard set on Human. 80 gold-standard set of MF-BP relationships in human.
genes and concepts that connect "DNA binding", "meiotic mismatch repair" and "chiasma assembly". The genes and concepts that connect "DNA binding", "meiotic mismatch repair" and "chiasma assembly". The figure was generated with BioGraph [40].