

Abstract
The extraordinary potential of metagenomic functional analyses to identify activities of interest present in uncultured microorganisms has been limited by reduced gene expression in surrogate hosts. We have developed vectors and specialized E. coli strains as improved metagenomic DNA heterologous expression systems, taking advantage of viral components that prevent transcription termination at metagenomic terminators. One of the systems uses the phage T7 RNA-polymerase to drive metagenomic gene expression, while the other approach uses the lambda phage transcription anti-termination protein N to limit transcription termination. A metagenomic library was constructed and functionally screened to identify genes conferring carbenicillin resistance to E. coli. The use of these enhanced expression systems resulted in a 6-fold increase in the frequency of carbenicillin resistant clones. Subcloning and sequence analysis showed that, besides β-lactamases, efflux pumps are not only able contribute to carbenicillin resistance but may in fact be sufficient by themselves to convey carbenicillin resistance.
Metagenomic libraries are gene libraries constructed from total DNA directly isolated from an environmental source rather than laboratory cultures. The key advantage of these libraries is that they allow the access to unknown environmental genetic resources independently of our ability to cultivate the microorganisms encoding them. Metagenomic libraries can be analyzed by systematically sequencing all the genomic clones obtained, or by functionally screening clones for novel phenotypes conveyed to host bacteria from metagenomic sequences.
Sequence-based gene identification relies on similarities to known gene sequences, while functional screening approaches have the advantage of being able to identify genes whose functions cannot be predicted by sequence analysis alone. However, a significant limitation of functional screening is that detection depends on efficient expression of the cloned genes. In fact, it has been shown that most genes are not usually expressed in the selected host bacterium1,2. Although some novel activities have been detected using E. coli as the surrogate host, increasing the efficiency of metagenomic gene expression in the bacterial hosts could greatly improve our ability to detect metagenomic clones encoding genes with novel functions.
Once environmental DNA samples (metagenomic DNA) are cloned into multicopy expression vectors, their expression can be driven in heterologus systems using promoters adjacent to the cloning site. However, the ability to efficiently drive metagenomic gene expression inversely correlates with the size of the cloned DNA. A major limiting factor is the presence of transcription terminators upstream the gene of interest. High hit rates have been reported for metagenomic libraries with heterologous promoters and very short DNA fragments of 1–3 Kb long3,4,5. However, reducing the size of the cloned DNA fragments implies a lower probability of having a gene of interest in a given clone and, therefore, a higher number of metagenomic clones are required to cover the same length of total metagenomic DNA. The use of small clones may be successful in identifying activities that are selectable and depend on expression of a single gene but is unlikely to be suitable for non-selectable activities given the large number of metagenomic clones that would need to be screened.
In order to overcome the inherent limitation of expressing genes in E. coli, the most frequent surrogate host for metagenomic libraries, a number of shuttle or broad host range vectors have been constructed with differing degrees of success in allowing the transfer and screening of the metagenomic library to different host bacteria6,7,8,9,10,11,12,13. Functional screening of metagenomic libraries using these vectors have shown that different positive clones can be obtained depending on the host bacteria used for the screening14. Although many of these methods represent an improvement, the expression of metagenomic genes still relies on their own expression capacity within the surrogate host.
In this work, we used an alternative approach to increase the potential of functional metagenomic analysis. We report on the construction of specialized vectors and strains, which combine the use of E. coli as the surrogate host with two modified heterologous expression systems that incorporate viral components. One is based on the phage T7 RNA-polymerase that is insensitive to many of the bacterial transcription termination signals15,16. The other expression method involves the use of the N anti-termination protein from the λ phage17 coupled to a positively controlled bacterial regulatory system inducible by salicylate18.
Results
Construction of metagenomic vectors
In order to improve performance of the pCC1FOS cloning vector, we modified it in three different ways: 1) insertion of an oriT to allow transfer of the metagenomic library by conjugation, 2) the addition of a transcription system subject to anti-termination to improve metagenomic gene expression, and 3) incorporation of a promoterless gfp gene to allow detection of metagenomic gene expression.
We started from a previous modification of the pCC1FOS fosmid vector (Epicentre), consisting of the insertion of two CeuI sites flanking the metagenomic DNA cloning site (Eco72I) (M. Ferrer, personal communication).
The oriT from plasmid RP4 was cloned into the unique HpaI site of pCC1FOS-CeuI, thus generating pMPO561, to allow transfer by conjugation in triparental matings19. Next, a DNA fragment containing the psaI promoter regulatory sequence, the nahGHILNJK operon promoter and the lambda phage nutL site (N-utilization leftward site) was synthesized and cloned just upstream of the T7 gene 10 already present in pCC11FOS, to yield pMPO571 (Fig. 1). The nahGHILNJK operon is involved in naphthalene biodegradation and is activated by NahR in response to salicylate18,20,21. The nutL site (N-utilization leftward site) allows the phage transcription anti-termination protein N to assemble with the transcription complex, thus making it insensitive to transcription termination signals22,23. This vector should allow transcription of cloned metagenomic DNA by two distinct RNA polymerases: (i) the T7 RNA polymerase, from the T7 gene 10 promoter, and (ii) the bacterial RNA polymerase modified for processive anti-termination by the lambda phage N protein, from the psal promoter.
Figure 1. Schematic diagram of the fosmids derived from pCC1FOS-CeuI.

Modifications resulting from this work, including oriT, psal, the nutL site and the promoterless gfp shown in the amplified region.
An additional modification of pMPO571 to yield pMPO579 involved cloning a promoterless gfp gene with a strong ribosome binding site from the T7 gene 10, downstream of the vector promoters and the metagenomic DNA cloning site (Fig. 1). Metagenomic DNA transcription in the appropriate direction can result in transcription continuing through to this promoterless gfp gene. This allows for the exploitation of Substrate Induced Gene Expression (SIGEX) technology24 to identify regulatory systems, by detecting GFP fluorescence as an indication of active transcription of the fosmid clones and sorting the appropriate clones by fluorescence assisted cell sorting (FACS). For pMPO571 and pMPO579 construction details, see supplementary Methods.
Construction of specialized strains for metagenomic DNA transcription
The MPO553 strain is a EPI300-T1 derivative, with the lacUV5 promoter, the attenuator nasF and the gene-1 of the T7 phage, coding for the T7 RNA polymerase, inserted into its trg locus (Fig. 2. For MPO553 construction details, see supplementary Methods). In the absence of the lacI repressor, such as in EPI300-T1, the lacUV5 promoter constitutively drives gene-1 transcription. The nasF attenuator is a transcription terminator signal within the nitrate assimilatory nasFEDCBA operon, subjected to transcriptional attenuation25, which has been used very successfully to reduce expression vector transcription levels26,27. Therefore, this strain should produce constitutive but low levels of T7 RNA polymerase.
Figure 2. Schematic diagram of the specialized strains derived from E. coli EPI300™-T1, showing the DNA fragment that has been integrated into their trg locus.
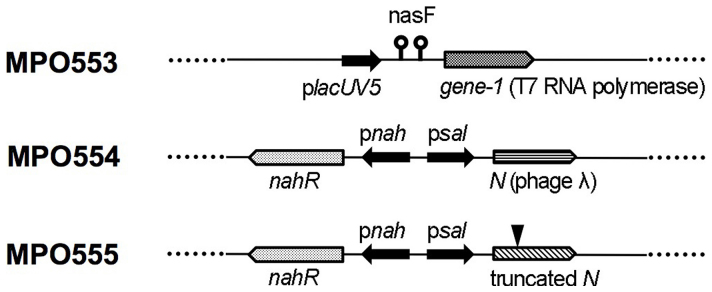
The MPO554 strain has an insertion in trg containing the activator nahR gene and the psal promoter followed by the lambda gene N (Fig. 2. For MPO554 construction details, see supplementary Methods). Upon induction with salicylate, NahR would activate psal in the fosmid vector, thus activating transcription of the metagenomic DNA, and simultaneously psal in the bacterially-inserted construction, thus activating N protein-dependent processive antitermination e at transcripts initiated from psal in the vector. MPO555 is a strain essentially the same as MPO554 but with a frameshift in the gene N.
Functionality tests of the vectors and specialized strains
Triparental matings were prepared in which EPI300-T1 carrying different plasmids were used as donors, rifampicin resistant (RifR) or nalidixic acid resistant (NalR) derivatives of EPI300-T1 were used as recipients, and DH5α bearing pRK2013 used as the helper strain. pCC1FOS-CeuI could not be transferred by conjugation (mating frequency <10−7). However, pMPO579 was transferred to all the recipient strains with frequencies above 10%, representing similar or higher frequencies to those obtained with the well-known mobilizable plasmid pBBR1MCS-3 (not shown).
Transcription levels from the heterologous promoters present in the modified vectors were tested in pMPO579, which contains the promoterless gfp reporter gene downstream of the metagenomic cloning site. To test whether transcription initiated at the heterologous promoters could proceed through transcription terminators, a 2.5 Kb DNA fragment bearing the thnL transcription terminator from Sphingomonas macrogolitabida strain TFA28 was cloned into the Eco72I site of pMPO579 to generate pMPO580.
Fluorescence of the EPI300-T1/pMPO579 strain was low (about 1,300 F.U., similar to the intrinsic fluorescence of EPI300-T1R). The addition of arabinose and salicylate resulted in a small increase in GFP expression (Fig. 3). In the specialized strain that produces the transcriptional activator NahR but not the antitermination protein N (MPO555), the basal level of gfp expression from pMPO579 was also low. Increasing the plasmid copy number by adding arabinose resulted in a modest increase in expression levels, but much higher levels of expression were obtained when both arabinose and salicylate were added to the culture medium together (Fig. 3). This shows that psal in pMPO579 is functional and that MPO555 produces the required transcriptional activator NahR. In contrast, when using pMPO580, which bears the thnL terminator, induction in the presence of arabinose and salicylate, was almost negligible, demonstrating that the transcription terminator was functional. Interestingly, gfp transcription from pMPO580 in MPO554, which expresses both NahR and N genes, was almost as high as that obtained with pMPO579. Taken together, these results indicate that transcription from psal in pMPO580 is terminated at the thnL terminator before reaching the gfp gene, but that transcription can continue onto the gfp gene if production of the anti-termination protein is also induced by salicylate.
Figure 3. Expression of the gfp gene from pMPO579, or its derivative pMPO580 bearing the thnL terminator, in the strains MPO554 that produces the activator NahR and the antitermination protein N, and MPO555 that is similar to MPO554 but bears a frameshift in the gene N.

In the MPO553 strain, which constitutively produces low levels of T7 RNA-polymerase, expression levels were very low in the absence of arabinose (Fig. 4). However, GFP expression dramatically increased to even higher levels than those obtained from the psal promoter when the plasmid copy number was increased by addition of arabinose. This clearly shows that the T7 promoter in pMPO579 is functional and that the specialized MPO553 strain produces T7 RNA-polymerase. The gfp expression level from the plasmid pMPO580, which contains the thnL terminator, was higher than that from pMPO579, indicating that this terminator does not terminate transcription by the T7 RNA-polymerase.
Figure 4. Expression of the gfp gene from pMPO579 or its derivative bearing the thnL terminator pMPO580, in the strain MPO553 that constitutively produces low levels of T7 RNA-polymerase.

A reconstruction of the SIGEX technology24 was performed by mixing cultures of MPO554 RifR and MPO554 NalR carrying the plasmid pMPO579. Only the MPO554 NalR/pMPO579 culture was pre-induced with salicylate to transcribe gfp from the standard psal catabolic promoter. Cultures were mixed in the 1:1, 1:10 and 10:1 ratios of NalR:RifR, purified by FACS and plated for subsequent resistance analysis to establish the ratios of induced vs. non-induced bacteria recovered after sorting. At the 1:1 NalR:RifR ratio, all the bacteria recovered after sorting were NalR (induced); at the 1:10 NalR:RifR ratio, the proportion of induced bacteria increased from 0.01% to approximately 30% after sorting; finally, at the 1:10 NalR:RifR ratio this proportion increased from 0.001% to approximately 15% after sorting. Thus, FACS enriched the induced strain 104-fold.
Construction of a metagenomic library and identification of clones conferring carbenicillin resistance
The plasmid pMPO579 was used to construct a metagenomic library from a shore at Punta San García, Cádiz, Spain contaminated with crude oil from a tanker spill. The library comprises approximately 2 Gigabases distributed across approximately 54,000 different clones, and is maintained in the strain EPI300-T1.
Resistance to β-lactams are predicted to be abundant in soils even in the absence of anthropogenic selection pressure29,30. Because of this and because it is easily selectable, resistance to carbenicillin was chosen as the activity of interest to be identified in the transferred metagenomic clones.
Direct selection of the EPI300-T1 culture hosting the library for carbenicillin-resistant clones resulted in the isolation of just a few tiny colonies after 2 days of incubation with a frequency much lower than 1/54,000. This indicated that none of the fosmids was efficiently conferring resistance to Cb100 to EPI300.
Triparental matings were performed using the EPI300-T1 hosting the bulk metagenomic library as the donor culture, each of the nalidixic acid derivatives from EPI300-T1, MPO553 and MPO554 as recipient strains, and DH5α/pRK2013 as the helper strain. We observed a very high conjugation frequency of over 6%, regardless the recipient strain. This indicates that the original pMPO579 vector and the metagenomic clones can be transferred to other strains with similar efficiency, despite the approximately 40 Kb of metagenomic DNA that each clone is expected to carry.
An approximately 6-fold increase in transconjugants resistant to 100 mg L−1 carbenicillin (CbR) were obtained with the specialized strains (6×10−5 positives/transconjugants) versus the conventional EPI300-T1 NalR strain. In the case of the MPO554 strain, this increase was dependent on the addition of salicylate, which induces transcription activation from psal by NahR and anti-termination by the N protein (not shown).
A total of 6 different fosmids were isolated from the 100 transconjugants derived from NalR derivatives of EPI300-T1, MPO553 or MPO554. All EPI300-T1 CbR transconjugants contained the same fosmid, whose restriction pattern matched that previously identified in the clone directly selected from the library. This fosmid, designated ETN1, was also found after the direct selection of carbenicillin resistant clones from the culture hosting the original library, and among the transconjugants derived from the specialized strains. The use of the strain producing T7 RNA polymerase allowed identification of 3 additional fosmids, TN2, TN3 and TN4. These fosmids were also identified among the transconjugants derived from MPO554, the specialized strain bearing the N anti-termination system. This strain allowed identification of 2 additional fosmids, N5 and N6, which were not detected using the other strains.
To confirm the capacity of each fosmid to confer CbR to each of the recipient strains, the six fosmids were transferred back to the EPI300-T1 RifR derivative and the resulting strains were used as donors in triparental matings together with the NalR derivatives as recipient strains and the DH5α/pRK2013 helper strain. As shown in Table 1, only the fosmid ETN1 conferred CbR to EPI300-T1 RifR. In addition, the carbenicillin resistance provided by this fosmid was apparently limited since only one tenth of the transconjugants exhibited sufficient resistance to generate a colony on LB-agar containing carbenicillin. The fosmids ETN1, TN2, TN3 and TN4 provided CbR to the strain MPO553 that produces T7 RNA-polymerase, and all 6 fosmids conferred CbR to the strain MPO554. These results using the isolated fosmids are in full agreement with those obtained using the bulk metagenomic library and confirm that at least six fosmids of the library potentially encode carbenicillin resistance genes. However, only a small fraction (one out of six) of the fosmids actually expressed the resistance gene by itself and conferred CbR in the EPI300-T1 strain.
Table 1. Mating transfer of the carbenicillin resistant fosmids. Values represent the frequency of carbenicillin resistant transconjugants relative to those transconjugants that had received the fosmid.
| Recipient: | |||||
|---|---|---|---|---|---|
| MPO554 NalR | |||||
| EPI300-T1R NalR | MPO553 NalR | −salicylate | +salicylate | ||
| Donor: EPI300-T1R RifR/ | ETN1 | 0.1 | 0.07 | 0.3 | 0.5 |
| TN2 | <10−5 | 0.05 | <10−5 | 1 | |
| TN3 | <10−5 | 0.2 | <10−5 | 1 | |
| TN4 | <10−5 | 0.2 | <10−5 | 0.9 | |
| N5 | <10−5 | <10−5 | 0.2 | 1 | |
| N6 | <10−5 | <10−5 | 0.1 | 1 | |
Gene identification in fosmids providing carbenicillin resistance
Sequence analysis of each fosmid revealed a number of genes that may confer resistance to carbenicillin, whose locations and orientations are shown in Figure 5.
Figure 5. Schematic diagram showing the clones that confer carbenicillin resistance.

All putative orfs and their orientations are shown although only those putatively involved in carbenicillin resistance are highlighted. Genes are not drawn to scale.
ETN1 encodes a class-C β-lactamase that is oriented opposite to the heterologous promoters. This β-lactamase is most similar to homologues found in different Pseudomonas species. Immediately downstream of the blaC gene is a 103 amino acid open reading frame containing a 4 nucleotide overlap with the blaC 3′end. The overlap may be indicative of translational coupling, suggesting that the short Orf could be related to the β-lactamase function. The blaC gene either with or without the short orf was subcloned into pMPO579 in the same orientation as the heterologous promoters. Although more tolerant than the strain without the plasmid, most of the transconjugants receiving any of these subclones were unable to produce a colony in the presence of Cb100, which indicated that the blaC alone cannot account for all the carbenicillin resistance provided by the original fosmid.
ETN1 also codes for the 3 components of an efflux pump of the resistance-nodulation-cell division (RND) permease family. The fosmid also contains a fourth component encoding an additional outer membrane protein. Since efflux pumps have been shown to contribute to the tolerance of bacteria to different toxic compounds such as aromatic compounds or antibiotics31, this efflux pump could also contribute to the carbenicillin resistance.
In the TN2 fosmid a class-B β-lactamase gene was found 6 Kb downstream of the heterologous promoters in the same orientation and a blaR gene encoding a LysR-type transcriptional activator just upstream in the opposite direction. The remaining sequence codes for hypothetical proteins or proteins with unrelated functions. In spite of blaR, this blaB gene could not be autonomously expressed in E. coli, though it was transcribed from any of the heterologous promoters present in the vector.
TN3 and TN4 have largely common inserts. In fact, the TN4 insert is included within the insert of TN3. Neither of these fosmids contain Orfs putatively coding for a β-lactamase enzyme. The only genes potentially involved in Cb resistance in these fosmids code for the components of an RND-type efflux pump that are located in the same orientation 16 Kb or 13 Kb from the heterologous promoters, depending on the fosmid. A DNA fragment encoding just the 3 components of the efflux pump was subcloned from TN4 into the pMPO579 vector in the appropriate orientation and the resulting plasmid, pMPO586, transferred to MPO554. The subclone was able to provide resistance to Cb100 although only when salicylate is present, just as with the original fosmid. This indicates that this efflux pump is able to provide carbenicillin resistance by itself if adequately expressed from a heterologous promoter in E. coli.
N5 and N6 also share most of their insert DNA, which is cloned in opposite orientations in each fosmid. In N6, a class-B β-lactamase gene was found 11 Kb downstream from the heterologous promoters and transcribed in the same orientation. Presumably, autonomous expression of this gene is not efficient enough, and requires heterologous transcription from the psal promoter to provide full resistance to Cb100 in E. coli. Intriguingly, in N5 this gene is located in the opposite orientation but N5 also requires salicylate induction to confer carbenicillin resistance. A possible explanation is that heterologous transcription in N5 results in expression of a gene coding for an activator of the blaB gene. A candidate activator gene, located 27 Kb away from the psal promoter is shown in Fig. 5. Alternatively, some additional open reading frames in the same orientation as psal could be responsible for carbenicillin resistance in N5. There are 5 other orfs in the same orientation as psal but none of the predicted amino acid sequences provide any clue regarding their potential function.
Discussion
Functional metagenomic technology is a powerful tool that has been applied to the discovery of novel natural products and enzymes of biotechnological interest32. The potential of functional metagenomics has been hampered by inefficient metagenomic gene expression in heterologous hosts. Although gene expression is a multi-step process involving transcription, translation and post-translational modifications, transcription is the most frequently regulated process and is probably the main limiting step of metagenomic gene expression in surrogate hosts. Because of that, the aim of this work was to improve metagenomic gene transcription. Here we have shown that heterologous gene transcription is an effective way of increasing the efficiency of metagenomic gene expression provided that transcription can proceed along the metagenomic DNA. To prevent transcription termination at metagenomic termination signals, we have taken advantage of two phage expression systems that are much less sensitive to the bacterial transcription termination signals.
The N anti-termination system is inducible allowing metagenomic gene expression to be regulated. In addition, absolute expression levels can be modulated since salicylate-inducible system drives different expression levels in proportion to the salicylate concentration33. This feature may be of particular relevance when expressing functions that might be deleterious to the surrogate host34.
On the other hand, the T7 expression system constructed in the strain MPO553 is constitutive, although polymerase expression levels are kept low by the nasF attenuator inserted between the lacUV5p and the gene-1. Expression levels from the T7 promoter are still 3-fold higher than those obtained from the psal promoter. However, in the case that overexpression of the screened function has a deleterious effect on bacterial growth, expression can be reduced by not increasing the plasmid copy number (Fig. 4).
The metagenomic library constructed with DNA from a contaminated coastal soil resulted in 54,000 independent clones. Although, metagenomic libraries with higher number of clones have been reported using the original vector, the number of clones depends on the quality of the DNA samples and, although we did not test whether during pCC1-FOS modifications we had inadvertently altered the cos site of the vector, we did not alter this sequence and, a priori, there is no reason to think that the modified vector had lower packaging efficiency than the the original vector. Testing heterologous gene expression involved transferring the metagenomic library by mating to the specialized strains. This way, a total of 6 clones conferring carbenicillin resistance were isolated from a metagenomic library containing 2 Gb of metagenomic DNA. Although this ratio of positive clones may look low, it is comparable to those obtained with other metagenomic libraries5,30. In addition, the diversity and abundance of antibiotic resistance determinants can also be very different depending on the DNA source. It is important to bear in mind that our metagenomic DNA comes from a soil contaminated by an oil spill, and therefore the biodiversity and relative abundance of antibiotic resistance genes may be lower than that of a non-contaminated soil. Theoretically, this additional mating step might impose some bias against fosmids that in some way reduce the growth rate of the bacterial host, thereby reducing the recovered diversity after the mating. However, at least in the case of carbenicillin resistance determinants, this additional step did not prevent the isolation of the fosmid already identified by direct selection using the original library, but rather increased recovered diversity since it allowed the identification of 5 additional fosmids. In any case, in order to prevent this potential bias, it is possible to directly construct the metagenomic library using a specialized strain such as MPO554 as the surrogate host.
Although the N anti-termination system allowed identification of some fosmids that the T7 expression system could not, both systems allowed metagenomic gene transcription from promoters located at least 16 Kb upstream, and through a number of genes in the opposite orientation to the promoter (see fosmid TN3). Although other factors may influence gene expression levels and may preclude identification of other positive clones present in the metagenomic library, it is clear that transcription is a major limiting factor for the functional analyses of metagenomic libraries, which can be circumvented by the phage-based heterologous expression systems.
The vector also includes a promoterless gfp downstream of the metagenomic cloning site to exploit the SIGEX technology, which has considerable potential for the identification of inducible genes and regulatory systems. In spite of the potential problems that may prevent the identification of a regulatory system by using this type of screen35, there are ways of avoiding them36. Our reconstruction of SIGEX technology indicates that a bacterial subpopulation expressing genes from a conventional catabolic promoter such as psal can be detected and enriched 104-fold by FACS. This enrichment is sufficiently high to detect positive clones that were present in a metagenomic library with a frequency of 10−6 or even lower, therefore, enough to detect any positive clone that is present in a metagenomic library.
Besides the relatively low permeability of the outer membrane of gram negative bacteria, there are 3 main mechanisms for conferring tolerance to β-lactam antibiotics37. Among the resistant clones we have found genes coding for β-lactamases and for efflux pumps (Fig. 5) but not penicillin binding proteins (insensitive transpeptidases). Of the efflux pumps, those belonging to the RND (Resistance-Nodulation-Division) are the only ones that exist in a tripartite form traversing both the outer and inner membrane and can pump molecules from the cytoplasm, from the periplasm or even when associated to the inner membrane31. Although many RND pumps can accommodate β-lactams, just a few have been definitely linked to β-lactam resistance38. They are usually associated to other forms of resistance such as a β-lactamase although beta-lactamase-negative, ampicillin-resistant (BLNAR) Haemophilus influenza isolates have been described that can tolerate up to 16 mg L−1 of ampicillin39. In these cases, overexpression of the AcrAB efflux pump was associated with mutations in PBP3, one of the 5 penicillin binding proteins of H. influenzae, that lowered affinities of beta-lactams to PBP3. Previous metagenomic analyses of β-lactam resistance resulted in large numbers of clones bearing bla genes but no efflux pumps have previously been linked to bla genes or to beta-lactam resistance40.
The way earlier metagenomic libraries were constructed may have prevented the identification of other resistance determinants in the past given that we have shown that some metagenomic RND pumps are not only contributing to carbenicillin resistance but are able to confer E. coli resistance to 100 mg L−1 of carbenicillin by themselves (fosmids TN3 and TN4).
Methods
Bacterial strains, growth conditions, plasmids and oligonucleotides
The bacterial strains and plasmids used in this study are described in Table 2. Luria-Bertani (LB) medium or LB-agar was used as the standard growth medium, and bacteria were grown at 37°C except for the screening and selection of fosmids conferring carbenicillin resistance that were carried out at 30°C. When needed, antibiotics were used at the following concentrations: 12.5 μg mL−1 chloramphenicol, 100 μg mL−1 carbenicillin, 15 μg mL−1 nalidixic acid, 20 μg mL−1 rifampicin, 25 μg mL−1 kanamycin and 100 μg mL−1 ampicillin. The copy number of the fosmids in EPI300™-T1 and derivative strains was increased by the addition of 1 mM arabinose. 5 mM salicylate was used as an inducer for transcription level analyses and 1 mM salicylate for the screens.
Table 2. Bacterial strains and plasmids used in this work.
| Strain | Genotype/Phenotype | Reference/Source |
|---|---|---|
| E. coli EPI300-T1R | F− mcrA Δ(mrr-hsdRMS-mcrBC) (StrR) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL nupG trfA tonA dhfr | Epicentre |
| E. coli MPO553 | EPI300-T1R Δtrg::PlaUV5-nasF attenuator-gene1 | This work |
| E. coli MPO554 | EPI300-T1R Δtrg:: nahR/Psal–gene N | This work |
| E. coli MPO555 | EPI300-T1R Δtrg:: nahR/Psal– truncated gene N | This work |
| E. coli DH5α | ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 (rk– mk+) supE44 thi-1 gyrA relA1 | 43 |
| Plasmid | Genotype/Phenotype | Reference/Source |
| pCC1FOS-CeuI | CmR, pCC1FOS (Epicentre) with two CeuI sites flanking the cloning site Eco72I | M. Ferrer |
| pMPO27 | ApR, expression vector with rrnBT1T2-Pm-nasF attenuator::MCS, ColE1 replication origin | 26 |
| pCNB4-S2 | ApR KmR pUT/mini-Tn5 nahR/Psal→sylS2 | 44 |
| pMPO556 | ApR PlacUV5 cloned into pBluescript II SK+ | This work |
| pMPO557 | ApR, nasF attenuator cloned into pMPO556 | This work |
| pMPO558 | ApR KmR, kanamycin resistance gene from pKD4 cloned into pMPO557 | This work |
| pMPO559 | ApR KmR CmR, chloramphenicol resistance gene from pKD3 cloned into pGP1-2 | This work |
| pMPO561 | CmR, pCC1FOS-CeuI derived fosmid with oriT | This work |
| pMPO563 | ApR, nahR-Pnah-Psal cloned into pBluescript II SK+ | This work |
| pMPO564 | ApR CmR, chloramphenicol resistance gene from pKD3 cloned into pMPO563 | This work |
| pMPO565 | ApR CmR, gene N cloned into pMPO564 | This work |
| pMPO571 | CmR, pMPO561 derived fosmid with Psal-nutL cloned downstream T7 promoter and the cloning site Eco72I | This work |
| pMPO575 | ApR CmR, pMPO565 derived plasmid with a frameshift in gene N | This work |
| pMPO579 | CmR, pMPO571 derived fosmid with a promoterless gfp cloned downstream the cloning site Eco72I | This work |
| pMPO580 | CmR, pMPO579 derived fosmid with the thnL transcription terminator from Sphingomonas macrogolitabida strain TFA cloned in the Eco72I site | This work |
| pMPO586 | CmR CbR, efflux pump from TN4 subcloned in pMPO579 | This work |
| pMPO634 | KmR, plasmid containing gfp gene | 45 |
| RP4 | KmR ApR (Tn1) TcR IncP-1 Tra+ Cma− | 46 |
| pNK736 | ApR, plasmid containing PlacUV5 | 47 |
| pBluescript II SK+ | ApR, phagemid vector | Stratagene |
| pBluescript II KS+ | ApR, phagemid vector | Stratagene |
| pBBR1MCS-3 | TcR, mobilisable plasmid | 48 |
| pRK2013 | KmR, helper in triparental matings | 19 |
| pGP1-2 | KmR, plasmid containing gene-1 (T7 RNA polymerase) | 49 |
| pKD3 | ApR CmR OriRγ | 50 |
| pKD4 | ApR KmR OriRγ | 50 |
| pKD46 | ApR, oriR101 repA101(ts) araBp-gam-bet-exo | 50 |
| pCP20 | ApR CmR Ts (30°C) | 51 |
DNA manipulations were performed according to standard procedures41. Details on the construction of fosmids and strains in this work are described in Supplementary Methods.
Oligonucleotides are described in Supplementary Table S1.
Conjugative matings
Vectors and metagenomic libraries were transferred by triparental matings19 with DH5α/pRK2013 as the helper strain. Conjugative matings were performed on LB-agar without antibiotic selection overnight at 30°C. The mating mixtures were then plated on LB-agar with the necessary antibiotics for the transconjugants selection, carbenicillin for the screening and arabinose for increasing the copy number of the fosmid, and incubated at 30°C for 48 h.
Mating frequencies were estimated as the ratio of transconjugant clones of the recipient strain (chloramphenicol+rifampicin resistant or chloramphenicol+nalidixic acid resistant clones) to total clones of the recipient strain (either rifampicin or nalidixic acid resistant, depending on the recipient strain).
Functionality tests of the vectors and specialized strains
The transcription levels from the heterologous promoters present in the modified vectors were analysed by detecting the fluorescence levels from the promoterless gfp gene, which is used as a reporter gene. Analyses were performed in the multifunctional microplate reader POLARstar Omega (BMG LABTECH GmbH, Germany), with a microplate COSTAL 96 and the following parameters: excitation filter 485BP1, emission filter EM520 and gain 1000.
Flow cytometry and sorting
Mixed bacterial cell suspensions were analyzed by flow cytometry using a FACScalibur cytometer equipped with an argon laser emitting at 488 nm. Bacteria were detected by side scatter (488/10 nm band pass filter) and the fluorescence intensity was detected using the FL1 channel (530/30 nm band pass filter). The Cellquest Pro software (Becton-Dickinson) was used for data acquisition and analysis. For cell sorting, flow cytometer water lines were pre-treated with a bleach solution (FACSClean) and autoclaved phosphate saline buffer (PBS) was used as sheath fluid. The exclusion mode was used to sort GFP-expressing cells. Sorting gates were sufficiently separated to avoid false positives/negatives.
Construction of a metagenomic library
Bacteria were extracted by direct addition of 400 mL of disruption buffer (0.2 M NaCl, 50 mM Tris-HCl pH 8.0) to 160 g of the soil sample and mixing overnight with shaking. The suspension was centrifuged at low speed (400 g for 3 minutes) and the supernatant poured over 10 mL of a Nycodenz resin solution (1.3 g mL−1). Centrifugation in a gradient of the Nycodenz resin (Axis-Shield) was carried out at 10,000 g × for 40 minutes at 4°C. The bacteria-containing a band at the interface between the Nycodenz and the aqueous layer were recovered and mixed with PBS. The cells were pelleted by centrifugation at 10,000 g for 20 minutes, and re-suspended in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). DNA was then extracted with the G NOME DNA kit (MP Biomedicals). Using this method, we obtained 24 μg of DNA from 160 g of soil, with an average size of approximately 40 kb.
The metagenomic library was constructed according to the CopyControl™ Fosmid Library Production kit protocol (Epicentre), except that pMPO579 was used instead of pCC1FOS. pMPO579 was linearised by restriction with the PmlI enzyme (New England Biolabs, isoschizomer of Eco72I), dephosphorylated with Shrimp Alkaline Phosphatase (USB) and concentrated to 300 mg L−1 with a Centrifugal Filter Device (Microcon, Millipore).
Gene identification in fosmids conferring carbenicillin resistance
Each fosmid was sequenced with a Roche 454 GS FLX Ti sequencer (454 Life Sciences, Branford, CT, USA) at Lifesequencing S.L. (Valencia, Spain). Sequences were assembled using the Newbler GS De Novo Assembler v.2.3 (Roche). The assembled sequences were compared to those in the databases using the Blastx and Blastn programs42. Those orfs potentially involved in carbenicillin resistance are highlighted in Figure 5.
Author Contributions
L.T.-G. designed and performed most of the experiments and analyzed the results, C.M. designed and performed the experiments to validate SIGEX technology, M.C.L.-M. participated in the construction of fosmids and strains, and E.S. designed the general strategy, supervised the work, analyzed data and wrote the paper. All authors reviewed the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
We wish to thank Manuel Ferrer for the kind gift of pCC1FOS-CeuI, Encarnación Carrillo for her coments/criticisms, Guadalupe Martín and Nuria Pérez for technical help and all members of the Santero laboratory for their insights and helpful suggestions. This work was supported by grants CSD2007-0005 and BIO2011-24003, co-funded by the Spanish Ministerio de Educación y Ciencia and the European Regional Development Fund, and a fellowship from the FPU program (Ministerio de Educación y Ciencia, Spain), awarded to L.T.-G.
Accession codes: The metagenomic sequences contained within the fosmids have been deposited at theNational Center for Biotechnology Information database (GenBank) under accession numbers JX406851 (ETN1), JX406852 (TN2), JX406853 (TN3), JX406854 (N5) and JX406855 (N6).
References
- Ekkers D. M., Cretoiu M. S., Kielak A. M. & van Elsas J. D. The great screen anomaly—a new frontier in product discovery through functional metagenomics. Appl. Microbiol. Biotechnol. 93, 1005–1020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama T. & Miyazaki K. Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr. Opin. Biotechnol 20, 616–622 (2009). [DOI] [PubMed] [Google Scholar]
- Lämmle K. et al. Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 127, 575–592 (2007). [DOI] [PubMed] [Google Scholar]
- Simon C., Herath J., Rockstroh S. & Daniel R. Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol. 75, 2964–2968 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer M. O., Dantas G. & Church G. M. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosio M. et al. Artificial chromosomes for antibiotic-producing actinomycetes. Nature Biotechnol. 18, 343–345 (2000). [DOI] [PubMed] [Google Scholar]
- Martinez A. et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl. Environ. Microb. 70, 2452–2463 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain T. et al. Novel bacterial artificial chromosome vector pUvBBAC for use in studies of the functional genomics of Listeria spp. Appl. Environ. Microb. 74, 1892–1901 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aakvik T. et al. A plasmid RK2-based broad-host-range cloning vector useful for transfer of metagenomic libraries to a variety of bacterial species. FEMS Microbiol. Lett. 296, 149–158 (2009). [DOI] [PubMed] [Google Scholar]
- Craig J. W., Chang F. Y. & Brady S. F. Natural products from environmental DNA hosted by Ralstonia metallidurans. ACS Chem. Biol. 4, 23–28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakirde K. S. et al. Gram negative shuttle BAC vector for heterologous expression of metagenomic libraries. Gene 475, 57–62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C. & Daniel R. Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. 77, 1153–1161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeschel S. C. et al. Novel broad host range shuttle vectors for expression in Escherichia coli, Bacillus subtilis and Pseudomonas putida. J. Biotechnol. 161, 71–79 (2012). [DOI] [PubMed] [Google Scholar]
- Craig J. W., Chang F. Y., Kim J. H., Obiajulu S. C. & Brady S. F. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse Proteobacteria. Appl. Environ. Microbiol. 76, 1633–1641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. & Moffatt B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 (1986). [DOI] [PubMed] [Google Scholar]
- Jeng S.-T., Gardner J. F. & Gumport R. I. Transcription termination by bacteriophage T7 RNA polymerase at rho-independent terminators. J. Biol. Chem. 265, 3823–3830 (1990). [PubMed] [Google Scholar]
- Greenblatt J. et al. Structure and mechanism in transcriptional antitermination by the bacteriophage lambda N protein. Cold Spring Harb Symp Quant Biol. 63, 327–336 (1998). [DOI] [PubMed] [Google Scholar]
- Schell M. A. Molecular Biology of the LysR Family of Transcriptional Regulators. Annu. Rev. Microbiol. 47, 597–626 (2003). [DOI] [PubMed] [Google Scholar]
- Figurski D. H. & Helinski D. R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 76, 1648–1652 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K.-M. & Gunsalus I. C. Plasmid Gene Organization: Naphthalene/Salicylate Oxidation. PNAS 79, 874–878 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell M. A. & Wender P. E. Identification of the nahR gene product and nucleotide sequences required for its activation of the sal operon. J. Bacteriol. 166, 9–14 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodwell J. R. & Greenblatt J. The nut site of bacteriophage lambda is made of RNA and is bound by transcription antitermination factors on the surface of RNA polymerase. Genes & Dev. 5, 2141–2151 (1991). [DOI] [PubMed] [Google Scholar]
- Greenblatt J., Mah T. F., Legault P., Mogridge J., Li J. & Kay L. E. Structure and mechanism in transcriptional antitermination by the bacteriophage lambda N protein. Cold Spring Harb Symp Quant Biol. 63, 327–36 (1998). [DOI] [PubMed] [Google Scholar]
- Uchiyama T., Abe T., Ikemura T. & Watanabe K. Substrate-induced gene expression screening of environmental metagenome libraries for isolation of catabolic genes. Nature Biotechnol. 23, 88–93 (2005). [DOI] [PubMed] [Google Scholar]
- Chai W. & Stewart V. RNA sequence requirements for NasR-mediated, nitrate-responsive transcription antitermination of the Klebsiella oxytoca M5al nasF operon leader. J. Mol. Biol. 292, 203–16 (1999). [DOI] [PubMed] [Google Scholar]
- Royo J. L., Manyani H., Cebolla A. & Santero E. A new generation of vectors with increased induction ratios by overimposing a second regulatory level by attenuation. Nucleic Acids Res. 33, e169 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina C., Camacho E. M., Flores A., Mesa-Pereira B. & Santero E. Improved Expression Systems for Regulated Expression in Salmonella Infecting Eukaryotic Cells. PLoS One 6, e23055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Sánchez A., Floriano B., Andújar E., Hernáez M. J. & Santero E. Tetralin-induced and ThnR-regulated aldehyde dehydrogenase and beta-oxidation genes in Sphingomonas macrogolitabida strain TFA. Appl. Environ. Microbiol. 76, 110–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demanèche S. et al. Antibiotic-resistant soil bacteria in transgenic plant fields. Proc. Natl. Acad. Sci. USA 105, 3957–3962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen H. K., Moe L. A., Rodbumrer J., Gaarder A. & Handelsman J. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J. 3, 243–251 (2009). [DOI] [PubMed] [Google Scholar]
- Nikaido H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 77, 1–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J. et al. Functional metagenomic strategies for the discovery of novel enzymes and biosurfactants with biotechnological applications from marine ecosystems. J. Appl. Microbiol. 111, 787–799 (2011). [DOI] [PubMed] [Google Scholar]
- Royo J. L. et al. In vivo gene regulation in Salmonella spp. by a salicylate-dependent control circuit. Nat. Methods 4, 937–942 (2007). [DOI] [PubMed] [Google Scholar]
- Warren R. L., Freeman J. D., Levesque R. C., Smailus D. E., Flibotte S. & Holt R. A. Transcription of foreign DNA in Escherichia coli. Genome Res. 18, 1798–1805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvão T. C., Mohn W. & de Lorenzo V. Exploring the microbial biodegradation and biotransformation gene pool. Trends Biotechnol. 23, 497–506 (2005). [DOI] [PubMed] [Google Scholar]
- Yun J. & Ryu S. Screening for novel enzymes from metagenome and SIGEX, as a way to improve it. Microb. Cell Fact. 4, 8–12 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke M., Lovering A. & Strynadka N. β-Lactam antibiotic resistance: a current structural perspective. Curr. Opin. Microbiol. 8, 525–533 (2005). [DOI] [PubMed] [Google Scholar]
- Poole K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56, 20–51 (2005). [DOI] [PubMed] [Google Scholar]
- Kaczmarek F. S. et al. Genetic and molecular characterization of β-lactamase-negative ampicillin-resistant Haemophilus influenzae with unusually high resistance to ampicillin. Antimicrob. Agents Chemother. 48, 1630–1639 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R. & Edwards R. Insights into antibiotic resistance through metagenomic approaches. Future Microbiol. 7, 73–89 (2012). [DOI] [PubMed] [Google Scholar]
- Sambrook J. & Russell D. W. Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. (2001).
- Altschul S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983). [DOI] [PubMed] [Google Scholar]
- Cebolla A., Sousa C. & de Lorenzo V. Rational design of a bacterial transcriptional cascade for amplifying gene expression capacity. Nucleic Acids Res. 29, 759–766 (2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Gallardo L. et al. Molecular and biochemical characterization of the tetralin degradation pathway in Rhodococcus sp. strain TFB. Microbial Biotechnol. 2, 262–273 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway B. W. Plasmids that mobilize bacterial chromosome. Plasmid 2, 1–19 (1979). [DOI] [PubMed] [Google Scholar]
- Simons R. W., Hoopes B. C., McClure W. R. & Kleckner N. Three promoters near the termini of IS10: pIN, pOUT, and pIII. Cell 34, 673–682 (1983). [DOI] [PubMed] [Google Scholar]
- Kovach M. E. et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–6 (1995). [DOI] [PubMed] [Google Scholar]
- Tabor S. & Richardson C. C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of especific genes. Proc. Natl. Acad. Sci. USA 82, 1074–1078 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K. A. & Wanner B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P. P. & Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information