

Abstract
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease in which fibroblast-like synoviocytes (FLS) exhibit an aggressive phenotype. While the mechanisms responsible are not well defined, epigenetics determinants such as DNA methylation might contribute. DNA methyltransferases (DNMTs) are critical enzymes that establish and maintain DNA methylation. We evaluated whether pro-inflammatory cytokines might contribute to differential DNA methylation previously described in RA FLS through altered DNMT expression.
Methods
FLS were obtained from RA and osteoarthritis (OA) synovium at total joint replacement. Gene expression was determined by qPCR and protein expression by Western blot analysis. DNMT activity was measured with a functional assay and global methylation was determined by an immunoassay that detects methyl-cytosine.
Results
Resting expression of DNMT1, −3a, and −3b mRNA were similar in RA and OA FLS. Western blot showed abundant DNMT1 and DNMT3a protein. Exposure to IL-1 decreased DNMT1 and DNMT3a mRNA expression in FLS. Dose responses demonstrated decreased DNMT expression at concentrations as low as 1 pg/ml of IL-1. DNMT mRNA levels decreased rapidly, with significant suppression after 2 to 8 hr of IL-1 stimulation. IL-1 stimulation of OA FLS did not affect methylation of LINE1 sites but led to demethylation of a CHI3L1 locus that is hypomethylated in RA FLS. Chronic IL-1 stimulation also mimicked the effect of a DNMT inhibitor on FLS gene expression.
Conclusion
Exposure to pro-inflammatory mediators alters DNA methylation in FLS by decreasing DNMT expression and function. These data suggest that IL-1 can potentially imprint cells in chronic inflammatory diseases.
Introduction
Rheumatoid arthritis (RA) is an immune-mediated disease marked by synovial inflammation, and joint damage. Pre-determined risk factors, such as genetics, play major role in the development of RA. For instance, genome-wide association studies demonstrate numerous polymorphisms associated with RA susceptibility. Genetics does not fully explain risk of developing RA, as identical twins have a concordance rate of 12-15% (1, 2). Stochastic factors, such as environmental exposures and epigenetic changes, could account for the non-genetic components of disease susceptibility.
Differential DNA methylation at cytosine-guanine (CpG) dinucleotides is a well-characterized epigenetic hallmark for several diseases, most notably cancer (3). More recently, altered DNA methylation has been observed in autoimmune diseases like RA and SLE (4-6). CpG methylation is established and maintained by DNA methyltransferases (DNMTs), which transfer a methyl group from S-adenosylmethionine (SAM) to cytosine (7). Three mammalian DNMTs have been described: DNMT1 maintains the existing methylation patterns during DNA replication while DNMT3a and DNMT3b are responsible for de novo CpG methylation (8).
Fibroblast-like synoviocytes (FLS) in the synovial intimal lining play an integral role in RA by producing key cytokines, small molecule mediators, and proteases (9). Rheumatoid FLS exhibit an aggressive phenotype that could contribute to the inflammatory milieu and damage to the extracellular matrix (10, 11). Several mechanisms that potentially contribute to the rheumatoid phenotype have been proposed, including abnormal tumor suppressor gene function (12), sentrin (13), PTEN (14), or sumoylation. Differential DNA methylation could also alter gene expression and synoviocyte function, and DNA methylation abnormalities have been described (15-17). Our array-based DNA methylation analysis recently revealed a rheumatoid DNA methylome signature involving genes that are critical to cell trafficking, inflammation and cell-extracellular matrix interactions (5).
Based on these observations, we hypothesized that the inflammatory milieu might contribute to differential DNA methylation in RA FLS through altered DNMT expression. These studies demonstrate that IL-1 modulate DNMT levels. DNMT function is also suppressed and DNA methylation patterns are subsequently modified. The data suggest that the inflammatory milieu might at least transiently alter FLS methylation and behavior.
Materials and Methods
Fibroblast-like synoviocytes
FLS were isolated from synovial tissues obtained from 14 RA and 12 OA patients at the time of joint replacement as described previously (9). The diagnosis of RA conformed to the American College of Rheumatology 1987 revised criteria (18). The protocol was approved by the UCSD Human Subjects Research Protection Program. Synoviocytes were used from passage 4 through 8, when FLS were a homogeneous population with <1% CD11b, <1% phagocytic, and <1% FcR II positive cells.
Reagents
The antibodies and reagents used were as follows: recombinant human TNF and IL-1β (R&D Systems, Minneapolis, MN), and LPS, poly(I:C), and 5-aza-2′-deoxycytidine (5Aza-dC) were obtained from Sigma (St Louis, MO, USA). Actinomycin D was purchased from USB Corporation (Cleveland, OH, USA).
Quantitative real-time polymerase chain reaction (qPCR)
mRNA was isolated using RNA Stat 60 (Tel-Test, Friendswood, TX) as described previously (19). cDNA was prepared, and qPCR was performed with TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) to determine the relative mRNA levels using the GeneAmp 7300 Sequence Detection system (Applied Biosystems). Pre-developed specific primers were used to detect DNMT1 (assay no. Hs00154749_m1), DNMT3a (Hs01027166_m1), DNMT3b (Hs00171876_m1), CHI3L1 (Hs00609691_m1), COL1A1 (Hs00164004_m1), and MYEF2 (Hs00972066_m1) (Applied Biosystems). Threshold cycle values in each sample were used to calculate the number of cell equivalents in the test samples. The data were normalized to the values for GAPDH expression (for human samples) (catalog no. 402869; Applied Biosystems) to obtain the relative cell expression (19).
DNMT function
Nuclear extracts were obtained using the nuclear extraction kit (Panomics, Fremont, CA, USA). Extracts from the IL-1β-treated and control cells cultured for 14 d were assayed for DNMT activity using a colorimetric DNMT Activity/Inhibition assay kit (Active Motif, Carlsbad, CA). The reaction was initiated by adding 10 μg of nuclear extracts to the unique cytosine-rich DNA substrate coated ELISA plate and incubated for 2 h at 37°C. Methylated DNA was detected with anti-methyl CpG binding domain 2b antibody and quantified at 450 nm. Enzyme activity (OD/h/mg) was calculated according to the following formula: DNMT activity (OD/h/mg) = 1000 × (sample OD − blank OD)/(10 μg × 2 hour)
Global DNA methylation assay
Global DNA methylation was assayed using an ELISA based commercial kit (MDQ1, Imprint® Methylated DNA Quantification Kit, Sigma Aldrich). Two μl of genomic DNA at a concentration of 100 ng/μl were diluted with 28 μl of lysis and binding buffers. After incubation with the detection antibody, absorbance was read at 450 nm. Quantification of global DNA methylation was performed by calculating the amount of methylated cytosine in the sample relative using an arbitrary scale. All samples were analyzed in duplicate.
Genomic DNA extraction, bisulfite treatment, and pyrosequencing
Genomic DNA was isolated using the MagMAX™ DNA Multi-Sample Kit (Applied Biosystems). DNA quality and quantity was determined with a NanoDrop ND-2000 spectrometer (NanoDrop Technologies, Wilmington, DE, USA). Sodium bisulfite modification of genomic DNA was performed with the Epitect bisulfite kit (Qiagen, Valencia, CA). The primers for PCR amplification and sequencing of CHI3L1 (chr1:203156625) were designed using PyroMark assay design SW 2.0. PCR was performed with biotinylated primers to convert the PCR product to single-stranded DNA templates. Pyrosequencing reactions and methylation quantification were performed by the Stanford Protein and Nucleic Acid Facility (http://pan.stanford.edu).
Statistical analysis
Results are expressed as the mean±SEM. Differences were assessed by one-way and two-way repeated-measures analysis of variance (ANOVA), followed by the Bonferroni post hoc test for comparison of multiple groups or student’s t-test for comparison of 2 groups. The criterion for significance was set at P values less than 0.05 after correction.
Results
Basal expression of DNMTs in FLS
DNMT1, DNMT3a, and DNMT3b expression was initially determined in cultured synoviocytes by qPCR. A modest trend towards lower expression was observed in RA FLS, but the differences were not significant (n=10/group; see Figure 1A). We then examined DNMT protein expression in FLS nuclear extracts by Western blot. Abundant DNMT1 and DNMT3a protein were observed in both RA and OA FLS. Immunoreactive DNMT3b protein was difficult to detect by Western blot analysis using currently available reagents (not shown). As with mRNA levels, there was no significant difference between RA and OA FLS (Figure 1B).
Figure 1.
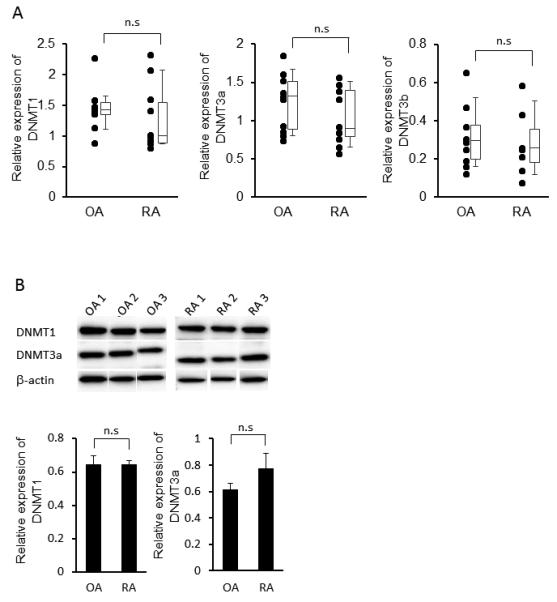
Basal gene and protein expression of DNMT in fibroblast-like synoviocytes (FLS) from rheumatoid arthritis (RA) and osteoarthritis (OA). A, Gene expression of DNMT1, −3a, and −3b in cultured RA and OA FLS, as determined by qPCR and normalized to GAPDH (n=10 for each). No differences in basal DNMT expression were observed between RA and OA. The results are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th to 90th percentiles. B, Representative Western blots (top) and quantitation (n=6) (bottom) of DNMT1 and DNMT3a protein expression in RA and OA FLS. As with mRNA, no differences between RA and OA were observed. The expression of DNMTs expression is normalized to β-actin.
Decreased DNMT gene expression after cytokine or TLR ligand stimulation
FLS were stimulated with IL-1ß, TNF or Toll-like receptor ligands LPS and poly(I:C) for 24 hr. DNMT expression was then determined by qPCR. DNMT3a mRNA expression was significantly decreased by IL-1ß (2 ng/ml), TNF (50 ng/ml), or LPS (1 μg/ml), while DNMT1 was significantly decreased by IL-1ß and LPS (Figure 2A). Similar trends were observed for DNMT3b, but the differences were not statistically significant (data not shown).
Figure 2.
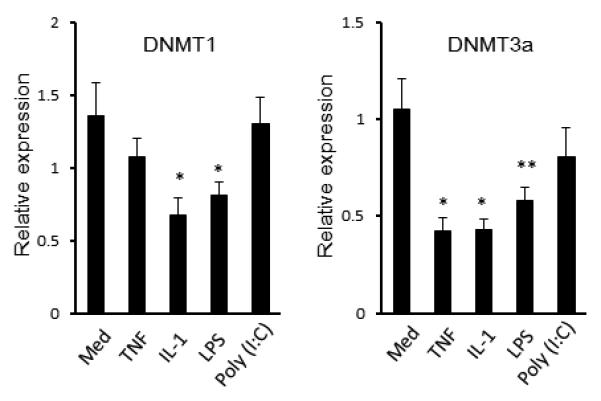
Effect of inflammatory mediators on DNMTs mRNA expression in FLS. FLS were stimulated for 24 h with TNF (50 ng/ml), interleukin-1β (IL-1β) (2 ng/ml), LPS (1 μg/ml), or poly(I:C) (20 μg/ml). DNMTs gene expression was determined by qPCR and normalized to GAPDH. IL-1 and LPS significantly decreased DNMT1 and DNMT3a, while TNF also decreased DNMT3a. (* = P < 0.01, ** = P < 0.05 versus untreated control). The mean and SEM of 6 separate FLS lines is shown.
Regulation of DNMTs by IL-1ß
Because IL-1ß caused the greatest reduction in DNMT levels, subsequent studies focused on this cytokine. Figure 3A illustrates that concentrations as low as 1 pg/ml decreased DNMT gene expression. We then examined the kinetics of DNMT suppression in FLS. Cells were stimulated with 2 ng/ml of IL-1ß and DNMT expression was evaluated for up to 24 hr. As shown in Figure 3B, DNMT1 and DNMT3a mRNA levels began to decrease in 8 and 2 hours after exposure to IL-1ß, respectively. When cells exposed to IL-1ß were washed and recultured in medium alone, DNMT expression gradually increased and reached baseline within 14 days (Figure 3C).
Figure 3.
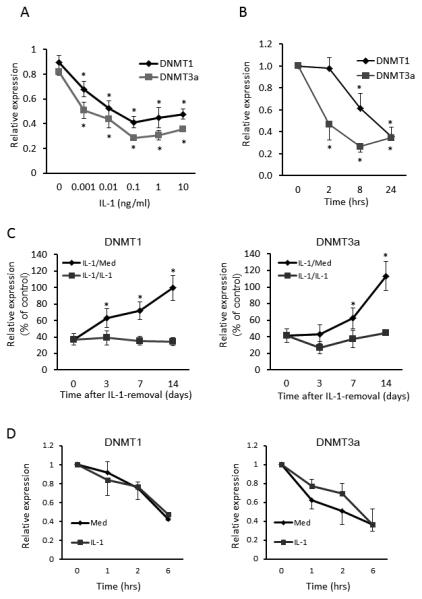
Regulation of DNMT gene expression in FLS by IL-1ß. A. Dose-dependent reduction of DNMTs expression by IL-1β (left panel). FLS were stimulated for 24 h with the indicated concentration of IL-1β. B. Time course of IL-1β-induced DNMTs reduction (right panel). FLS were incubated with 2 ng/ml of IL-1β for up to 24 h. Very low concentrations of IL-1 (1 pg/ml) significantly decreased gene expression Bars show the mean and SEM of 4 separate FLS lines. * = P < 0.01, ** = P < 0.05 versus untreated control. C. Recovery of DNMT mRNA expression after IL-1β removal. FLS were cultured with IL-1β (1 ng/ml) for 14 days and then either washed and recultured with fresh medium (IL-1/Med) or in medium plus IL-1β (IL-1/IL-1) for up to 14 additional days. Note that full recovery required up to 14 days. Bars show the mean and SEM of 3 separate FLS lines. *= P < 0.05 versus IL-1/IL-1 group. D. Effect of IL-1β on DNMT mRNA stability. FLS were stimulated with or without IL-1β (2 ng/ml) for 30 min. Cells were washed, incubated with 10 μg/ml actinomycin D, and harvested at indicated time points. mRNA expression of DNMT1 and DNMT3a was determined by qPCR and normalized to ß-actin mRNA and to time 0. No effect on mRNA half life was observed, suggesting that the effects on gene expression are related to transcriptional events. Bars show the mean and SEM of 3 separate FLS lines.
Effect of IL-1 on DNMT mRNA stability
To determine the mechanism of IL-1-mediated suppression of DNMT expression, FLS were cultured with medium or IL-1 for 30 min and 10 μg/ml of actinomycin D was then added to inhibit mRNA synthesis. DNMT mRNA levels were measured at various time points by qPCR, and ß-actin was used as an internal control. The decay rates of DNMTs mRNA were similar in medium or IL-1 stimulated cells (See Figure 3D). Therefore, alterations in mRNA stability do not account for the effect of IL-1 and effects on transcription are more likely responsible.
Decreased DNMT function after IL-1 stimulation
A cytosine methylation functional assay was then performed to determine whether IL-1ß suppresses DNA methylation activity in FLS nuclear extracts. Figure 4A shows that basal DNMT function in RA and OA FLS is similar. When FLS were stimulated with 1 ng/ml of IL-1ß, DNMT functional activity was significantly decreased by IL-1ß (See Figure 4B).
Figure 4.
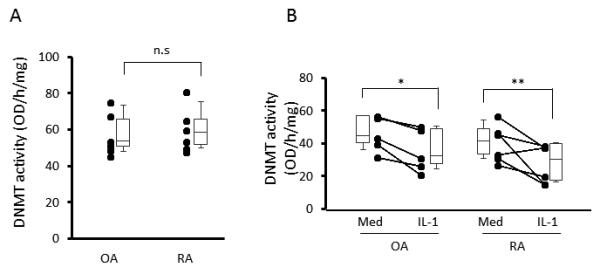
Effect of IL-1β on DNMT enzymatic activity in FLS. DNMT function was measured using the DNMT Activity/Inhibition assay kit. A. Nuclear extract was isolated from resting RA and OA FLS (n=6, each) and basal function was determined. No differences between RA and OA were observed. B. FLS treated with or without 1 ng/ml IL-1β (n=6, each). IL-1 decreased DNMT function in RA and OA FLS as determined by a decrease in total CpG methylation by nuclear extracts in a cell free system. The results are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th to 90th percentiles. n.s.=not significant, * = P < 0.05, ** = P < 0.01 versus untreated control.
DNA methylation levels in cultured FLS stimulated with IL-1
To evaluate whether IL-1ß might alter global DNA methylation (total methylcytosine) or differential methylation of individual loci, we cultured FLS in the presence of IL-1ß (1 ng/ml) for 14 days. An assay of total methylcytosine content of genomic DNA demonstrated that prolonged IL-1ß exposure decreased global methylation of FLS (See Figure 5A). In a second experiment to determine how long it takes for the effect of IL-1 to reverse, additional FLS lines were cultured with IL-1ß for 14 days, washed, and then divided into two groups: 1) cells cultured with medium (IL-1/Med); or 2) cells cultured with fresh IL-1ß (IL-1/IL-1). After 14 days, global DNA methylation in IL-1/Med FLS was significantly higher than that in continuous IL-1/IL-1 FLS (See Figure 5B). These data suggest that DNMT suppression by IL-1 maintains long-term hypomethylation in FLS but that the effect is at least partially reversed when cells are removed from the inflammatory milieu.
Figure 5.
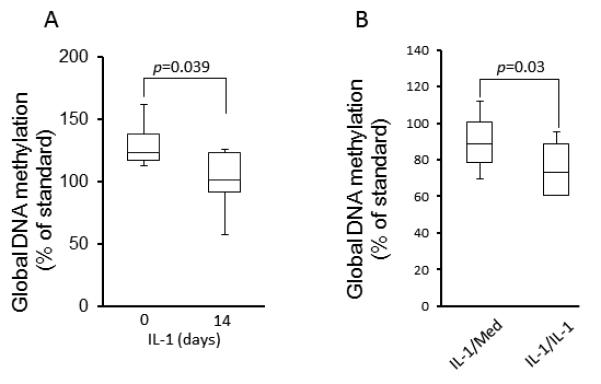
Effect of IL-1β on global DNA methylation in FLS. A. FLS were cultured in the presence of IL-1β (1 ng/ml) for 14 days and genomic DNA was then isolated. Global DNA methylation was determined by ELISA and quantified with a standard curve using methylated CpGs in a control sample (see Material and Methods). Global methylation was significantly decreased by continuous stimulation with IL-1 for 14 days. Bars show the mean and SD of 6 separate FLS lines. B. Recovery of global DNA methylation in FLS after IL-1β removal. A second set of FLS lines were cultured with IL-1β (1 ng/ml) for 14 days and then either washed and recultured with fresh medium (IL-1/Med) or in medium plus IL-1β (IL-1/IL-1) for up to 14 additional days. FLS stimulated continuously with IL-1 28 days had significantly greater amounts of methylated CpG than cells that had the IL-1 removed and cultured in medium for the last 14 days of the 28 day experiment. Bars show the mean and SEM of 6 separate FLS lines.
Potential contribution of IL-1ß to the RA methylome signature
It is not known how changes in DNMT expression regulate only specific loci. However, it probably relates to expression of co-factors such as transcription factors in cell types that, under specific conditions, improve accessibility to individual CpG loci. An unbiased genome-wide evaluation of DNA methylation in FLS demonstrated that RA and OA FLS segregate based on DNA methylation (5). To evaluate the contribution IL-1-induced methylation changes at relevant loci we measured methylation at the previously identified RA hypomethylated loci of the CH13L1 gene. We therefore determined whether IL-1ß could reproduce this component of the RA-specific methylation pattern in OA FLS. OA FLS were stimulated with IL-1ß for 14 days and pyrosequencing was performed on the known hypomethylated locus as well as two loci in the same gene that are not differentially methylated in RA. Figure 6A shows that IL-1 selectively demethylated DNA at the RA hypomethylated locus (chr1:203156625), but had no effect on the other loci (chr1:203156628 and 203156635). As an additional control, LINE1 pyrosequencing was performed on medium and IL-1-stimulated cells. Three selected CpGs of the LINE1 element, which are heavily methylated but not differentially methylated in RA, were not affected by IL-1ß (not shown).
Figure 6.
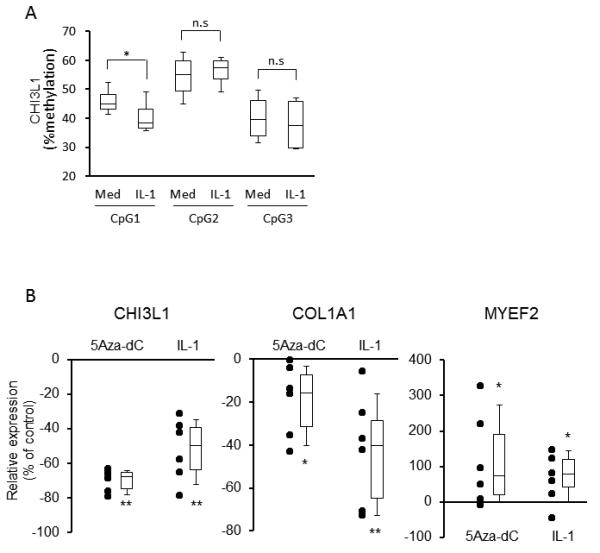
Effect of IL-1β on DNA methylation pattern and gene expression in FLS. A. OA FLS were cultured in the presence of IL-1β (1 ng/ml) for 14 days and genomic DNA was then isolated. After bisulfite conversion, DNA methylation status of CHI3L1 loci (CpG1, chr1:203156625; CpG2, chr1:203156628; CpG3, chr1:203156635) was determined by pyrosequencing. IL-1 stimulation led to hypomethylation of the RA-associated locus (CpG1), but not the other two loci in the same gene. Bars show the mean and SEM of 6 separate FLS lines. The composite results are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th to 90th percentiles. n.s.=not significant, *= P < 0.05 versus untreated control. B. OA FLS were cultured in the presence of 5Aza-dC (5 μM) or IL-1β (1 ng/ml) for 14 days (n=6, each). Gene expression of CHI3L1, COL1A1, and MYEF2 was determined by qPCR and normalized to GAPDH. 5Aza-dC and IL-1 significantly increased MYEF2, while significantly decreased CHI3L1 and COL1A1. The results are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th to 90th percentiles. n.s.=not significant, * = P < 0.05, ** = P < 0.01 versus untreated control. IL-1 and 5Aza-dC were not significantly different.
Effect of IL-1 and a DNMT inhibitor on gene expression
We then evaluated whether the effects of IL-1 on DNA methylation might alter gene expression. Because IL-1 decreases DNMT function, we compared the effect of chronic IL-1 exposure to the DNMT inhibitor 5-Aza-dC. Five genes that are hypomethylated in RA FLS (5). Six OA FLS were treated with the demethylating agent 5Aza-dC or IL-1ß for 14 days to induce hypomethylation, and qPCR was performed. Gene expression of CHI3L1 and COL1A1 was decreased while MYEF2 expression was increased by 5Aza-dC and IL-1 (See Figure 6B). Expression of two additional genes (SYNJ2 and ITGA4) were not changed by either IL-1 or 5Aza-dC (data not shown). These results suggest that long-term exposure to IL-1ß mimics the effect of a DNMT inhibitor on differentially methylated genes.
Discussion
Rheumatoid FLS display aggressive behavior and contribute to joint damage (10). Appropriate fibroblast responses to inflammatory cytokines like IL-1 and TNF can account for some of these observations. However, RA cells are imprinted and maintain these features long after they are removed from the synovial environment. For example, RA FLS, unlike OA or normal synoviocytes, adhere to and invade cartilage explants in SCID mice (20). RA FLS are able to grow under anchorage-independent conditions, are less susceptible to contact inhibition, and are resistant to apoptosis (21, 22). RA synoviocytes can also migrate and transfer the invasive phenotype from one joint to another (11). Several mechanisms have been implicated including abnormal PTEN or sentrin expression, aberrant sumoylation, or somatic mutations (13, 14, 23-30).
Epigenetic changes, which are heritable alterations in gene expression or phenotype independent of DNA sequences, regulate cell functions relevant to synoviocyte behavior in RA. DNA methylation, histone modification, and microRNAs play a critical role in normal embryonic development and cell transformation in cancer (31). DNA methylation participates by repressing regulatory genes after methylation of the cytosines in CpG motifs. Demethylation is associated with aberrant gene expression and cell transformation (3, 32).
In contrast to cancer, the role of methylation in non-oncologic diseases is less well characterized. Global DNA hypomethylation has been described in RA FLS and peripheral mononuclear cells (15, 33). Certain loci in LINE1 element and gene promoters, such as CXCL12, are hypomethylated in cultured FLS (15, 16), while other gene promoters like DR3 are hypermethylated (17). Furthermore, our recent genome-wide DNA methylation analysis revealed a unique methylome signature that differentiated RA FLS from OA or normal FLS (5). The differentially methylated loci included numerous genes implicated in the pathogenesis of RA, including signaling molecules, adhesion receptors, and proteases (5).
Because the synovial microenvironment might contribute to imprinting, we evaluated whether cytokines and other mediators in the rheumatoid joint could regulate the machinery responsible for DNA methylation. Three DNA methyltransferases, namely DNMT1, −3a, and −3b determine DNA methylation patterns in the mammalian genome (34). DNMT3a and DNMT3b mainly regulate de novo methylation. In contrast, DNMT1 maintains methylation of CpG loci, especially during cell proliferation. The DNMTs can potentially contribute to gene repression through methylation dependent and independent mechanisms (35).
Studies evaluating the regulation of DNMT gene expression in RA are limited. DNMT1 protein levels were reportedly lower in RA FLS than in OA FLS; the levels in RA FLS remained low in culture and were not suppressed further by TNF, IL-1ß, or PDGF (15). In our experiments, basal DNMT expression at the protein and mRNA level was similar in RA and OA FLS. The reasons for the different results are not certain, but are consistent with our observation that RA FLS are differentially methylated but do not exhibit global hypomethylation in a genome-wide scan. It is possible that our culture conditions permit cells to recover more fully from the synovial microenvironment by using later passage cells. The observation that re-exposure to cytokines or TLR ligands suppresses DNMT expression supports this possibility.
Our data also established that pro-inflammatory cytokines, such as IL-1ß and TNF and the TLR4 ligand LPS suppress DNMT gene expression in RA and OA FLS. The effect is rapid and is associated with diminished DNMT function as determined by the ability to methylate CpG loci in a cell free system. However, global changes in DNA methylation based on changes in DNMT expression or using DNMT inhibitors are gradual, usually taking up to 2 weeks equilibrate. This is the first study to show that these inflammatory mediators regulate not only DNMTs expression, but also their function and global DNA methylation.
DNMT regulation in other cell types has been reported to involve transcriptional activation of the DNMT genes (36, 37), miRNAs, RNA binding proteins, alteration of catalytic activity, half-life, or substrate targeting by post-translational modification (38). Environmental factors or disease states can alter DNMT expression and amplify DNA methylation abnormalities. In our FLS studies, IL-1ß did not alter DNMTs mRNA stability indicating that genes are probably suppressed at the transcriptional level. Further studies defining the relationship between IL-1ß and DNMT transcriptional mechanisms are needed to establish how they are regulated in FLS.
One interesting aspect of DNMTs and CpG regulation is that a limited number of DNMT enzymes can potentially regulate methylation of only specific loci. This observation is probably due to altered chromosome, promoter, and/or locus accessibility to DNMTs after environmental stress. Histone modification as well as transcription factors that can direct DNMTs to certain regions of the genome are possible mechanisms that explain this effect. For example, Rel/p65 directly recruits DNMT1 to specific tumor suppressor gene loci (39). Thus, a cytokine like IL-1, which activates a discrete set of transcription factors, could have locus-specific effects in addition to global effects.
To explore this possibility, we evaluated how IL-1 affects individual CpG loci in FLS. LINE1 elements, which are not differentially methylated in the RA signature, were not affected by IL-1. A key RA-associated element in CHI3L1 was demethylated by IL-1ß in OA FLS, converting this locus to the RA pattern. Other RA-specific loci in CHI3L1 were not affected, possibly because these loci are not accessible to DNMTs after IL-1 stimulation or other inflammatory mediators contribute in a locus-specific manner. Experiments are currently in progress to determine which transcription factors and binding motifs contribute to this selectivity in IL-1 stimulated FLS.
Perhaps more important, the effect of IL-1 does not appear to be permanent, as continuous exposure to IL-1 is required to maintain hypomethylation. Differential methylation of the RA genome in FLS, however, is stable and is maintained from passage 3 through at least passage 7 (manuscript in preparation). Thus, a subset of CpG imprinting observed in long term cultured FLS occurs through mechanisms that are independent of transient cytokine effects. Longer term exposure to IL-1 or combinations of cytokines in vivo is one possible explanation that could lead to permanent changes.
Gene expression in RA FLS in our genome-wide scan correlates with the methylation status of differentially methylated loci (5). Thus, therapeutic interventions that modulate DNMT function currently used in cancer could be useful in RA (40). The potential benefit could potentially be offset by de-repression of key regulatory or pro-inflammatory genes. For example, DNMT inhibitors promote tumor progression in some animal models (41, 42), and previous studies in RA FLS indicate that DNMT inhibition alters gene expression. The mechanism for selectively modifying only certain genes by inhibiting DNMT function is complex and involves a number of gene-targeting co-factors. DNMTs recognize distinct consensus sequences, but these motifs have low specificity (43). DNMTs can directly interact with transcription factors to preserve the methylation status of promoter regions, which might explain why only specific loci are affected by cytokines like IL-1 (43, 44).
The functional relevance of decreased DNMT expression was confirmed by demonstrating that a DNMT inhibitor and IL-1 have similar effects on FLS gene expression. We originally expected that demethylation would increase expression for all genes, as we observed with MYEF2. However, lower expression of CHI3L1 and COL1A1 suggests that IL-1 and 5Aza-dC alters methylation of their transcriptional regulators. The fact that both methods of decreasing DNMT activity had parallel effects supports the concept that IL-1-mediated demethylation alters FLS function.
In conclusion, long-term exposure to the pro-inflammatory cytokine IL-1 can potentially contribute to the RA FLS DNA methylation pattern through altered DNMT expression. Only certain loci are affected by IL-1, suggesting that other inflammatory mediators are involved or that the cells are irreversibly imprinted through other mechanisms or prior to the onset of arthritis. Furthermore, IL-1-mediated demethylation through DNMT suppression mimicked the effect of a DNMT inhibitor on FLS gene expression. These data suggest that the rheumatoid synovial cytokine milieu can contribute to epigenetic modification of FLS in RA.
Acknowledgments
The authors thank Josh Hillman for outstanding technical assistance and Dr. Scott Ball and Dr. William Bugbee for providing clinical samples. Dr. Olivier Harismendy and the Clinical and Translational Research Institute BioComputational Center provided assistance designing pyrosequencing experiments.
Funding This project was supported by a grant from the American College of Rheumatology Research and Education Foundation as well as grant number UL1RR031980 from the NIH National Center for Advancing Translational Science.
Footnotes
Competing interests None
Authors’ Contributions KN, DB and GF designed methods and experiments, analyzed the data, and interpreted the results. KN also carried out the laboratory experiments. KN and GF wrote the paper. All authors have contributed to, seen and approved the manuscript.
Patient consent Written, informed consent was obtained from participants at the time of sample collection.
Ethics approval All study protocols related to human studies were approved by the Institutional Review Board of the University of California San Diego
References
- 1.Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, Ollier WE. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. 1993;32:903–907. doi: 10.1093/rheumatology/32.10.903. [DOI] [PubMed] [Google Scholar]
- 2.Jarvinen P, Aho K. Twin studies in rheumatic diseases. Semin Arthritis Rheum. 1994;24:19–28. doi: 10.1016/0049-0172(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 3.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 4.Trenkmann M, Brock M, Ospelt C, Gay S. Epigenetics in Rheumatoid Arthritis. Clinical Reviews in Allergy and Immunology. 2009;39:10–19. doi: 10.1007/s12016-009-8166-6. [DOI] [PubMed] [Google Scholar]
- 5.Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2012 doi: 10.1136/annrheumdis-2012-201526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol. 2011;7:263–271. doi: 10.1038/nrrheum.2011.16. [DOI] [PubMed] [Google Scholar]
- 7.Goll MG, Bestor TH. Eukaryotic cytosine methylatranferases. Annual Review of Biochemistry. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 8.Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 9.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis & Rheumatism. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 11.Lefevre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, Korb A, Schnaker E-M, Tarner IH, Robbins PD, Evans CH, Sturz H, Steinmeyer J, Gay S, Scholmerich J, Pap T, Muller-Ladner U, Neumann E. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tak PP, Zvaifler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82. doi: 10.1016/s0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 13.Franz JK, Pap T, Hummel KM, Nawrath M, Aicher WK, Shigeyama Y, Müller-Ladner U, Gay RE, Gay S. Expression of sentrin, a novel antiapoptotic molecule, at sites of synovial invasion in rheumatoid arthritis. Arthritis & Rheumatism. 2000;43:599–607. doi: 10.1002/1529-0131(200003)43:3<599::AID-ANR17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis & Rheumatism. 2009;60:3613–3622. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- 16.Karouzakis E, Rengel Y, Jungel A, Kolling C, Gay RE, Michel BA, Tak PP, Gay S, Neidhart M, Ospelt C. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. 2011;12:643–652. doi: 10.1038/gene.2011.45. [DOI] [PubMed] [Google Scholar]
- 17.Takami N, Osawa K, Miura Y, Komai K, Taniguchi M, Shiraishi M, Sato K, Iguchi T, Shiozawa K, Hashiramoto A, Shiozawa S. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis & Rheumatism. 2006;54:779–787. doi: 10.1002/art.21637. [DOI] [PubMed] [Google Scholar]
- 18.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis & Rheumatism. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 19.Boyle D, Rosengren S, Bugbee W, Kavanaugh A, Firestein G. Quantitative biomarker analysis of synovial gene expression by real-time PCR. Arthritis Res Ther. 2003;5:R352–R360. doi: 10.1186/ar1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, Gay S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 21.Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. The Journal of Clinical Investigation. 1989;83:1267–1276. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baier A, Meineckel I, Gay S, Pap T. Apoptosis in rheumatoid arthritis. Curr Opin Rheumatol. 2003;15:274–279. doi: 10.1097/00002281-200305000-00015. [DOI] [PubMed] [Google Scholar]
- 23.Meinecke I, Pap G, Mendoza H, Drange S, Ender S, Strietholt S, Gay RE, Seyfert C, Ink B, Gay S, Pap T, Peters MA. The small ubiquitin-like modifier mediates the resistance of prosthesis-loosening fibroblast-like synoviocytes against fas-induced apoptosis. Arthritis & Rheumatism. 2009;60:2065–2070. doi: 10.1002/art.24633. [DOI] [PubMed] [Google Scholar]
- 24.You X, Boyle D, Hammaker D, Firestein G. PUMA-mediated apoptosis in fibroblast-like synoviocytes does not require p53. Arthritis Research & Therapy. 2006;8:R157. doi: 10.1186/ar2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proceedings of the National Academy of Sciences. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Igarashi H, Hashimoto J, Tomita T, Yoshikawa H, Ishihara K. TP53 mutations coincide with the ectopic expression of activation-induced cytidine deaminase in the fibroblast-like synoviocytes derived from a fraction of patients with rheumatoid arthritis. Clinical & Experimental Immunology. 2010;161:71–80. doi: 10.1111/j.1365-2249.2010.04163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inazuka M, Tahira T, Horiuchi T, Harashima S, Sawabe T, Kondo M, Miyahara H, Hayashi K. Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology. 2000;39:262–266. doi: 10.1093/rheumatology/39.3.262. [DOI] [PubMed] [Google Scholar]
- 28.RÈme, Travaglio, Gueydon, Adla, Jorgensen, and Sany Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clinical & Experimental Immunology. 1998;111:353–358. doi: 10.1046/j.1365-2249.1998.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bang H, Egerer K, Gauliard A, Lüthke K, Rudolph PE, Fredenhagen G, Berg W, Feist E, Burmester G-R. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis & Rheumatism. 2007;56:2503–2511. doi: 10.1002/art.22817. [DOI] [PubMed] [Google Scholar]
- 30.Da Sylva T, Connor A, Mburu Y, Keystone E, Wu G. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Research & Therapy. 2005;7:R844–R851. doi: 10.1186/ar1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotech. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 32.Christophersen N. S. y., Helin K. Epigenetic control of embryonic stem cell fate. The Journal of Experimental Medicine. 2010;207:2287–2295. doi: 10.1084/jem.20101438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu CC, Fang TJ, Ou TT, Wu CC, Li RN, Lin YC, Lin CH, Tsai WC, Liu HW, Yen JH. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol Lett. 2011;135:96–99. doi: 10.1016/j.imlet.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10:631–647. [PubMed] [Google Scholar]
- 35.Szyf M, Detich N. DNA methylation and cancer therapy. Springer US: 2005. Regulation of DNA Methyltransferases in Cancer DNA Methylation and Cancer Therapy; pp. 125–141. [Google Scholar]
- 36.Bigey P, Ramchandani S, Theberge J, Araujo FD, Szyf M. Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene. 2000;242:407–418. doi: 10.1016/s0378-1119(99)00501-6. [DOI] [PubMed] [Google Scholar]
- 37.Jinawath A, Miyake S, Yanagisawa Y, Akiyama Y, Yuasa Y. Transcriptional regulation of the human DNA methyltransferase 3A and 3B genes by Sp3 and Sp1 zinc finger proteins. Biochem. J. 2005;385:557–564. doi: 10.1042/BJ20040684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–656. doi: 10.1038/embor.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Mayo MW, Nagji AS, Smith PW, Ramsey CS, Li D, Jones DR. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene. 2012;31:1143–1154. doi: 10.1038/onc.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 41.Gao Q, Steine EJ, Barrasa MI, Hockemeyer D, Pawlak M, Fu D, Reddy S, Bell GW, Jaenisch R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proceedings of the National Academy of Sciences. 2011;108:18061–18066. doi: 10.1073/pnas.1114946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chik F, Szyf M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis. 2010;32:224–232. doi: 10.1093/carcin/bgq221. [DOI] [PubMed] [Google Scholar]
- 43.Hervouet E, Vallette F. o. M., Cartron P.-F. o. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics. 2009;4:487–499. doi: 10.4161/epi.4.7.9883. [DOI] [PubMed] [Google Scholar]
- 44.Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H. l. n., Loussouarn D, Martin S. p. A., Vallette F. o. M., Cartron P.-F. o. Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial Cells. PLoS ONE. 2010;5:e11333. doi: 10.1371/journal.pone.0011333. [DOI] [PMC free article] [PubMed] [Google Scholar]