

Abstract
Cardiac myosin-binding protein C (cMyBP-C) is a thick-filament-associated protein that modulates cardiac contractility through interactions of its N-terminal immunoglobulin (Ig)-like C0-C2 domains with actin and/or myosin. These interactions are modified by the phosphorylation of at least four serines located within the motif linker between domains C1 and C2. We investigated whether motif phosphorylation alters its mechanical properties by characterizing force-extension relations using atomic force spectroscopy of expressed mouse N-terminal cMyBP-C fragments (i.e., C0-C3). Protein kinase A phosphorylation or serine replacement with aspartic acids did not affect persistence length (0.43 ± 0.04 nm), individual Ig-like domain unfolding forces (118 ± 3 pN), or Ig extension due to unfolding (30 ± 0.38 nm). However, phosphorylation did significantly decrease the C0-C3 mean contour length by 24 ± 2 nm. These results suggest that upon phosphorylation, the motif, which is freely extensible in the nonphosphorylated state, adopts a more stable and/or different structure. Circular dichroism and dynamic light scattering data for shorter expressed C1-C2 fragments with all four serines replaced by aspartic acids confirmed that the motif did adopt a more stable structure that was not apparent in the nonphosphorylated motif. These biophysical data provide both a mechanical and structural basis for cMyBP-C regulation by motif phosphorylation.
Introduction
Cardiac myosin-binding protein C (cMyBP-C) is a 140-kD sarcomeric protein that is composed of 11 immunoglobulin (Ig)- and fibronectin-like domains (C0–C10) (1). Genetic mutations in cMyBP-C are one of the leading causes of familial hypertrophic cardiomyopathy (1–3), highlighting its importance for normal cardiac structure and function. cMyBP-C is confined to seven to nine stripes in the A-band (i.e., C-zones) on both sides of the M-line through its C-terminal association with the thick-filament backbone (4–6). The N-terminal Ig-like domains C0, C1, and C2; the Pro-Ala linker between C0 and C1; and the phosphorylatable linker (i.e., the motif) between C1 and C2 are believed to extend away from the thick-filament backbone (4) and bind actin (7) and/or the myosin head (8), S2 (9), and regulatory light chain (10) in a phosphorylation-dependent manner (11–13). This may provide a partial molecular and structural basis for cMyBP-C’s modulation of cardiac contractility. Specifically, both removal of the N-terminal domains and ablation of MyBP-C, through either genetic manipulation or chemical extraction, results in enhanced actomyosin kinetics, as evidenced by faster actin velocities over native thick filaments (14), increased unloaded shortening velocity (15), and rates of tension recovery after a length change (16). Given that cMyBP-C’s C-terminal domain is attached to the thick filament (5), cMyBP-C could inhibit actomyosin force and motion generation by acting as a tether or strut to constrain myosin head mobility and thus the kinetics of motion generation through its N-terminal interactions with domains near the myosin head (10,17). In contrast, cMyBP-C binding to actin may compete for myosin-binding sites (18), modulate the regulatory capacity of troponin-tropomyosin (19–21), or likewise inhibit motion by acting as an elastic tether (13).
cMyBP-C motif phosphorylation in response to β-adrenergic stimulation is a major determinant of increased cardiac contractility (1–3), with phosphorylation occurring on at least four serines (S273, S282, S302, and S307) (2,14,22,23). Upon protein kinase A (PKA) treatment of myocardial preparations, accelerated cross-bridge kinetics contribute to the faster unloaded shortening velocities (24) and tension recoveries in response to a stretch (16,25). Because these enhanced mechanical properties are similar to those observed in the absence of cMyBP-C (see above), motif phosphorylation and its ability to weaken cMyBP-C’s interaction with actin (12,13) and myosin (11) effectively reduce cMyBP-C’s capacity to inhibit actomyosin force and motion generation. Determining how phosphorylation affects cMyBP-C’s structure and function (specifically that of the motif) is critical for understanding why high levels of cMyBP-C phosphorylation are required for normal cardiac performance (26,27), in contrast to low phosphorylation levels that are closely associated with cardiac pathologies and heart failure (26,28,29).
The structure of the 103 amino acid motif was originally thought to resemble an Ig-like fold, based on homology modeling of small-angle x-ray scattering (SAXS) and circular dichroism (CD) data (30). However, in later SAXS analyses, Jeffries et al. (31) proposed that the motif is unstructured, as confirmed by NMR structure determination, except for a triple-helix bundle in the motif’s C-terminus (residues 317–361) (32). The unstructured nature of the motif was reinforced by atomic force spectroscopy studies in which the force-extension relationships for expressed C1-C2 fragments of cMyBP-C provided evidence that the motif is freely extensible (33). This extensibility may be necessary for the N-terminal domains of cMyBP-C to interact with actin and/or myosin while the C-terminal domains are anchored to the thick filament. However, motif phosphorylation may regulate cMyBP-C function either by stabilizing the existing motif structure through new electrostatic interactions or by facilitating the development of new secondary structural components (32).
If the motif is stabilized upon phosphorylation, this change may be apparent through a reduction in extensibility. To test this hypothesis, we used atomic force microscopy (AFM) to characterize the force-extension relations of bacterially expressed mouse N-terminal fragments (i.e., C0-C3). The C0-C3 fragment is ideal for AFM-based extension measurements because the C0-C1 and C2-C3 portions of the fragment act as molecular handles (34), allowing the motif to be stretched free of any interactions with either the glass slide or the AFM probe tip. The force-extension relations of nonphosphorylated C0-C3 showed a characteristic sawtooth, Ig-like domain unfolding pattern, with unfolds occurring at lengths consistent with the motif being freely extensible as previously reported (33). To determine the impact of motif phosphorylation on C0-C3’s molecular mechanics, we phosphorylated fragments by PKA treatment and expressed mutant C0-C3s with the four motif serines (S273, S282, S302, and S307) substituted with either alanines (A) or aspartic acids (D) to mimic the nonphosphorylated or phosphorylated states, respectively (14). Upon PKA phosphorylation or phosphomimetic replacement, the force-extension relations suggested that C0-C3 was less extensible, with a reduction in length consistent with a stable motif. Similarly, circular dichroism (CD) spectroscopic and dynamic light scattering (DLS) data for shorter expressed C1-C2 fragments with the four serines replaced by aspartic acids confirmed that the phosphorylated motif, unlike its nonphosphorylated analog, has properties consistent with a stable structure. These biophysical data provide both mechanical and structural bases for cMyBP-C regulation by motif phosphorylation.
Materials and Methods
cMyBP-C protein fragment expression
cMyBP-C N-terminal fragments were bacterially expressed from mouse cardiac cDNA using a pET expression system (Novagen, Madison, WI) (35). Five different constructs were expressed for atomic force spectroscopy: C0-C3 control (amino acids 1–539) and four C0-C3 constructs with either the first three serines or all four serines (S273, S282, S302, and S307) replaced with aspartic acids (D) as a phosphomimetic or alanines (A) as a nonphosphorylated analog. The mutant constructs are referred to as either C0-C3XXX or C0-C3XXXX, where X is either A or D and the number of Xs refers to the number of serine replacements. The purity of these expressed samples was reported recently (19). Two shorter C1-C2 fragments containing the motif, C1-C2AAAA and C1-C2DDDD, were expressed for CD and DLS spectroscopy. These shorter fragments were used to maximize the signal of the motif over the two flanking Ig domains as compared with C0-C3 with its four Ig domains.
PKA phosphorylation
One group of C0-C3 control fragments (200 μl at 0.5 mg/ml) was phosphorylated by incubation with 40 units of PKA (P2645; Sigma-Aldrich, St. Louis, MO) at 30°C for 20 min, and then at 4°C overnight in 2 mM DTT, 50 mM MOPS, 1 mM MgCl2, and 2 mM ATP, pH 7.0. As a control, C0-C3 was incubated in the same buffer without PKA, and no apparent effect on the force-extension relations was observed by atomic force spectroscopy (data not shown). This phosphorylation protocol resulted in nearly 100% phosphorylation at S273, S282, and S302, and lesser phosphorylation (i.e., 83%) at S307 as determined previously by mass spectrometry (13). Due to the incomplete phosphorylation of S307 observed after in vitro PKA treatment of C0-C3 fragments (13) or in mouse wild-type native thick filaments (14), a comparison of the mechanical properties of C0-C3DDD and C0-C3DDDD may indicate whether phosphorylation of the fourth site has any functional impact.
Atomic force spectroscopy
Expressed C0-C3 was diluted to a concentration of 10 nM in dilution buffer (25 mM KCl, 1 mM EGTA, 10 mM DTT, 25 mM imidazole, and 4 mM MgCl2, pH 7.4) and stored on ice until use. Before tests were conducted, 50 μl of this protein solution was pipetted onto a glass slide (76 × 25 mm, 1 mm thick; Gold Seal Products, Portsmouth, NH) and allowed to adsorb for 10 min, resulting in a sparse distribution of proteins on the glass surface with minimal aggregation. The slide was then washed three times by solution exchange with 50 μl of dilution buffer. A 150 μl solution bubble was formed over the sample on the glass slide. Experiments were carried out at room temperature (24°C ± 1°C). The effect of ionic strength on force-extension behavior for only the C0-C3AAA and C0-C3DDD constructs was tested in dilution buffers containing 25, 180, and 300 mM KCl.
Force-extension measurements (Fig. 1) were carried out on an AFM (MFP-3D; Asylum Research, Santa Barbara, CA) using uncoated silicon nitride probes (200 μm triangular with 15 nm tip radius; Budget Sensors, Sofia, Bulgaria). Before tests were conducted, the spring constant of each cantilever (average 39 ± 8 pN/nm) was calculated with the use of commercial software provided by the manufacturer. Briefly, the calibration procedure involved calculating the inverse optical lever sensitivity of the cantilever by pressing it against the glass slide surface, performing a thermal sweep, and calculating the cantilever spring constant using the equipartition method (36). Protein extension experiments were carried out by bringing the AFM probe into contact (defined by 5 nm of cantilever deflection) with the slide surface and maintaining contact for 1 s. The probe was then retracted at a velocity of 700 nm/s to a distance of 300 nm. This velocity is within the physiological range of cardiac muscle fiber shortening, and the extension distance was estimated to be far enough to ensure full unfolding of the C0-C3 fragment (assumed to be 194 nm given the 539 amino acid fragment and 0.36 nm/residue). Force and extension were sampled at 6 kHz. The force-extension procedure was repeated automatically over 100–400 μm2 slide surface areas in a square grid pattern of 100–900 points. All data were collected within 90 min after the protein fragments were warmed to room temperature.
Figure 1.

Typical AFM-acquired force-extension traces. C0-C3, composed of Ig-like domains (ovals), Pro-Ala linker, and motif linker (M), is first extended to its initial contour length (LC). Its length is then increased by a series of Ig-like domain unfolding events (n = 1–4; ovals to lines) once unfolding forces (FUnfold) are reached (the order of domain unfolds is assumed for illustrative purposes only). At C0-C3’s final contour length (LC(Unfolded)), detachment (Det.) occurs. Solid curves through the data are wormlike-chain model fits (Eq. 1). (A–F) Nonphosphorylated fragments (A–C) have LC consistent with a freely extensible motif, whereas fragments phosphorylated by either PKA or aspartic acid substitution at three or four sites (D–F) exhibit visibly shorter LC, presumably because the motif (M) adopts a stable structure with phosphorylation.
Data analysis
Data files for a given experimental group consisted of ∼7000 force-extension traces that were first scanned using a mechanical fingerprinting routine (34) implemented in MATLAB (The MathWorks, Natick, MA) to compare and rank each trace based on its similarity to a characteristic sawtooth pattern associated with the unfolding of Ig-like domains (37). This pattern was described by force rising as the fragment was extended (Fig. 1 A), followed by a sudden drop that corresponded to the unfolding of one of the Ig-like domains (38). Such events occurred multiple times as each Ig-like domain unfolded, with the final peak rising and dropping to zero force due to the fragment detaching from either the AFM probe or the glass surface. Useable traces were defined as those containing at least three unfolding peaks, to ensure extension of the motif. Traces in which the unfolding peak spacing was clearly <30 nm were discarded to avoid analyzing data that might have originated from multiple fragments attaching to the probe as a result of dimerization or aggregation (see Fig. S1 in the Supporting Material for a summary of possible molecule-probe adhesions and the predicted resulting unfolding behavior).
Traces that showed at least four peaks (three domain unfolds and subsequent detachment) separated by ∼30 nm (33) were manually selected from the best fits and used to characterize the unfolding force of Ig-like domains (FUnfold), along with persistence and contour lengths (see below). Events were classified as peaks if their amplitudes were >30 pN, which was three times the root mean-square noise of the AFM system. Force-extension behavior was characterized by using a MATLAB script to fit the wormlike chain model (39) to each peak in the force-extension trace using least squares. The wormlike chain model relates the force measured up to each unfolding event (Fi) to applied extension (z) as follows:
| (1) |
Equation 1 was fit with Ai (persistence length) and Bi (contour length) as independent variables to each force peak, i = 1,...,n as shown in Fig. 1, where n is the number of force peaks, including n-1 Ig unfolding events followed by detachment. Each of these curve fits contains information about the initial contour length of the protein fragment, LC, and the incremental contour length, ΔLC, resulting from the unfolding of an Ig domain. Because the incremental contour length, defined as ΔLCi = Bi+1 − Bi for i = 1,…,n-1, showed no significant dependence on phosphorylation (Table 1), it was assumed that all Ig unfolds could be represented by the global average incremental contour length, <ΔLC>. A value for initial contour length, LC, could thus be calculated from each curve fit by subtracting <ΔLC> multiplied by the number of unfolded domains, (i-1), as follows: LC = Bi − [(i-1)<ΔLC>]. For each force-extension trace, the persistence length of the fragment was determined from the initial force peak, LP = A1.
Table 1.
Mechanical properties of C0-C3 fragments obtained by AFM
| LC (nm)a | ΔLC (nm) | LP (nm) | |
|---|---|---|---|
| Control | 67 ± 2 (n = 64) | 31 ± 1 (n = 34) | 0.31 ± 0.04 (n = 18) |
| PKA | 38 ± 3 (n = 29)b | 30 ± 2 (n = 20) | 0.34 ± 0.06 (n = 7) |
| AAA | 58 ± 1 (n = 55) | 30 ± 2 (n = 42) | 0.47 ± 0.09 (n = 12) |
| DDD | 36 ± 2 (n = 70)b | 30 ± 1 (n = 52) | 0.39 ± 0.14 (n = 15) |
| AAAA | 62 ± 1 (n = 44) | 30 ± 1 (n = 35) | 0.65 ± 0.07 (n = 9) |
| DDDD | 41 ± 2 (n = 43)b | 30 ± 1 (n = 34) | 0.47 ± 0.10 (n = 9) |
Average mean ± SE values, with the number of observations in parentheses. Data were analyzed across each variable using a one-way ANOVA (ap < 0.0001), and Tukey’s post-hoc test was used to compare each group with control (bp < 0.0001).
CD spectroscopy
CD spectra of the nonphosphorylated analog, C1-C2AAAA, and the fully phosphorylated phosphomimetic, C1-C2DDDD, were recorded on a spectrometer (model 400; AVIV Biomedical, Lakewood, NJ) at 15°C in 10 mM borate buffer containing 1 mM DTT, pH 8, at concentrations ranging from 0.1 to 0.3 mg/ml using a quartz cuvette with a 0.1 cm path length. Readings were collected at 1 nm intervals from 185 to 260 nm with a 150 s averaging time. Background corrections were done by subtracting spectra of the borate buffer from the C1-C2AAAA and C1-C2DDDD spectra. Protein concentrations were determined by UV absorbance at 280 nm of the measured samples. The CD spectra were analyzed using three separate algorithms (CDSSTR, CONTIL, and SELCON; for average values, see Table 3).
Table 3.
Secondary structure content in C1-C2AAAA and C1-C2DDDD
| Construct | β-strand | α-helix | turns/undefined |
|---|---|---|---|
| C1-C2AAAA | 29–43 | 17–20 | 58–68 |
| C1-C2DDDD | 29–44 | 17–21 | 59–68 |
| C1-C2a | 35–41 | 6–8 | 50–60 |
Range of secondary structure percentages determined by CD spectroscopy and by averaging percentage secondary structure from separate data collections that were analyzed using multiple data sets and algorithms (52,53,61–63).
Data from Jeffries et al. (30).
In addition, the temperature stability at 206.5 nm was recorded for 0.2 mg/ml of C1-C2AAAA and C1-C2DDDD in 10 mM borate buffer containing 1 mM DTT, pH 8. For these experiments the temperature was increased from 15°C to 100°C at a fixed heating rate (0.3°C/min), with ellipticity recorded every 2.0°C. The effect of ionic strength on temperature stability was measured in 10 mM borate buffer containing 1 mM DTT, pH 8 and 250 mM KCl. Plots of temperature versus ellipticity (see Fig. 5) were used to estimate the thermal stability of C1-C2AAAA and C1-C2DDDD fragments.
Figure 5.

Temperature dependence of the far-UV CD spectra of C1-C2AAAA and C1-C2DDDD. (A) Temperature dependence of the ellipticity of C1-C2AAAA in 10 mM borate, 1 mM DTT, pH 8. (B) Temperature dependence of the ellipticity of C1-C2DDDD in borate buffer as in A. (C) Temperature dependence of the ellipticity of C1-C2AAAA in borate buffer containing 250 mM KCl. (D) Temperature dependence of the ellipticity of C1-C2DDDD in borate buffer containing 250 mM KCl. The protein concentration for C1-C2AAAA and C1-C2DDDD was 0.2 mg/ml. The data were collected at 206.5 nm in intervals starting at 15°C.
DLS
DLS experiments were performed on a Protein Solutions DynaPro MS/X instrument (Charlottesville, VA) at 25°C and 35°C on 7–30 μM C1-C2AAAA and C1-C2DDDD in 10 mM borate buffer containing 1 mM DTT, pH 8. Before analysis, the samples were centrifuged at 14,000 x g for 10 min to remove large interfering particles. Aliquots (12 μl) of C1-C2AAAA or C1-C2DDDD were placed in a temperature-controlled quartz cell, measurements were acquired for 10 s, and 20 data sets were collected. The data were analyzed using Dynamic V6.0 software (Protein Solutions, Lakewood, NJ).
Results
Force-extension characteristics of nonphosphorylated C0-C3 fragments
The mechanical properties of nonphosphorylated C0-C3 control fragments at the molecular level were characterized by AFM-based force-extension traces (Fig. 1). Individual traces exhibited a characteristic sawtooth pattern with up to five peaks, four of which corresponded to unfolding events associated with each of the four Ig-like domains (C0-C3). The final peak corresponded to the detachment (average force: 185 ± 9 (SE) pN) of the protein from either the probe or the glass surface. From these traces, values for the Ig-domain unfolding force for each peak, FUnfold, were determined (Fig. 1 A). Values for the initial contour length, LC, the incremental contour length resulting from each unfolding event, ΔLC, and the persistence length, LP, were estimated from fits of the force-extension trace (median R2 = 0.972) to the wormlike chain model (see Materials and Methods).
There were no apparent differences in the force-extension traces for the nonphosphorylated C0-C3 control and the two mutant nonphosphorylated analogs, C0-C3AAA and C0-C3AAAA (Fig. 1, A–C). Specifically, LC-values were not different (p > 0.05; Fig. 2, Table 1), with an average of 62 ± 1.2 (SE) nm for all three constructs. This LC should reflect the length of the extended C0-C3 fragment (see Fig. 1 A). The ΔLC associated with each Ig-domain unfolding event was not different for the three constructs, averaging 30 ± 0.29 (SE) nm (Table 1). This value is in close agreement with previously reported measurements of Ig-domain unfolding in titin (38,40) and more recently in whole cMyBP-C (33). The unfolding force, FUnfold, associated with these unfolding events, which ranged between 32 and 273 pN, showed an increasing trend with number of unfolding events (Fig. 3). Although this trend was reported previously (33) and may arise from differences in structure between cMyBP-C domains (31,40), there may in fact be no differences in FUnfold for the C0-C3 Ig domains, because pooling FUnfold data in sequential order from different traces without knowing which Ig domain is unfolding can lead statistically to an apparent trend in FUnfold (41). Finally, the persistence lengths, LP, determined from the force-extension curves leading up to the first peak (see Materials and Methods) were no different for three nonphosphorylated constructs (Table 1), with an average value of 0.47 ± 0.04 (SE) nm, which is on the order of a single amino acid (0.36 nm). This suggests that the LP for the nonphosphorylated C0-C3 constructs should be dominated by highly disordered regions, most likely the motif and the Pro-Ala linker.
Figure 2.

Effect of phosphorylation on initial contour length. Phosphorylation, either by PKA treatment or by replacement with three or four aspartic acids, significantly (∗p < 0.01) reduced the initial contour length by 21–29 nm relative to unphosphorylated analogs (see Table 1).
Figure 3.

Unfolding force as a function of unfold number. All groups showed a similar trend of increasing FUnfold (mean ± SD) with successive unfolding events. Each data point was derived from between two and 41 events. There were no significant differences in the slope of FUnfold versus unfold number for any construct.
Force-extension characteristics of phosphorylated and phosphomimetic C0-C3 fragments
To test the hypothesis that motif phosphorylation alters the structural/mechanical properties of the C0-C3 fragment, and presumably the motif itself, we characterized force-extension relations in PKA-treated C0-C3 fragments that were 83% phosphorylated at all four serines (13), and in mutant phosphomimetic analogs, C0-C3DDD and C0-C3DDDD. Force-extension traces for the PKA-treated, C0-C3DDD, and C0-C3DDDD constructs displayed the same sawtooth pattern as the nonphosphorylated constructs (Fig. 1, D–F). Wormlike chain fits to these traces showed no effect of PKA treatment or phosphomimetic replacement, regardless of the number of serines replaced, on either ΔLC or LP (Table 1). FUnfold showed an increasing trend with unfold event number, as in the nonphosphorylated constructs, and there was no difference in this trend across all constructs tested or in comparison with the nonphosphorylated controls (Fig. 3). The similarity in ΔLC and FUnfold between phosphorylated and nonphosphorylated C0-C3 constructs might be expected given that these are properties that should be dominated by the four Ig domains rather than by the motif itself.
The most striking difference in the force-extension traces for the PKA-treated and phosphomimetic constructs compared with their respective controls was a significant 21–29 nm decrease in LC (Fig. 2, Table 1). These reductions in initial contour length were visibly apparent in the force-extension traces as a decreased extension at which the first unfolding event occurred (Fig. 1). In addition, there was no significant difference in the amount of LC reduction with the addition of a fourth aspartic acid in the C0-C3DDDD construct compared with C0-C3DDD (Fig. 2, Table 1).
Effect of ionic strength on force-extension characteristics
To determine whether the reduction in LC with phosphorylation was dependent on ionic strength, we compared force-extension traces for C0-C3AAA and C0-C3DDD at 25, 180, and 300 mM KCl. There were no associated changes in LP or ΔLC with ionic strength for the C0-C3DDD compared with its C0-C3AAA control (Table 2). Although increasing ionic strength had no significant effect on the force at which proteins disassociated from the AFM probe, it did decrease the frequency of attachment events (data not shown). The average LC-values under all three KCl conditions were significantly shorter for C0-C3DDD than for C0-C3AAA (Table 2). LC frequency distributions for C0-C3AAA (Fig. 4) were described by single Gaussian distributions (R2 = 0.77–0.95) at all KCl concentrations, with values of 57 ± 7 nm, 66 ± 10 nm, and 66 ± 6 nm for 25, 180, and 300 mM KCl, respectively. The LC distributions for C0-C3DDD, however, were best described by the sum of two Gaussians (R2 = 0.82–0.97) at all ionic strengths (Fig. 4). One distribution had LC comparable to that of C0-C3AAA (i.e., 65 ± 3 nm, 60 ± 5 nm, and 66 ± 6 nm for 25, 180, and 300 mM KCl, respectively), and the second distribution was a shorter, less extensible population (i.e., 34 ± 13 nm, 37 ± 6 nm, and 37 ± 3 nm for 25, 180, and 300 mM KCl, respectively). The relative fractions of the extensible and inextensible populations in C0-C3DDD, however, showed a trend with increasing ionic strength, ranging from almost completely inextensible at 25 mM KCl to approximately evenly divided at 300 mM KCl.
Table 2.
Effect of ionic strength on the mechanical properties of C0-C3 fragments obtained by AFM
| KCl (mM) | Construct | LC (nm) | ΔLC (nm) | LP (nm) |
|---|---|---|---|---|
| 25 | AAA | 58 ± 1 (n = 55) | 30 ± 1 (n = 42) | 0.47 ± 0.09 (n = 12) |
| DDD | 36 ± 2 (n = 70)a | 30 ± 1 (n = 52) | 0.39 ± 0.14 (n = 15) | |
| 180 | AAA | 64 ± 2 (n = 54) | 31 ± 1 (n = 40) | 0.52 ± 0.07 (n = 13) |
| DDD | 48 ± 2 (n = 91)a | 28 ± 1 (n = 67) | 0.43 ± 0.06 (n = 18) | |
| 300 | AAA | 62 ± 2 (n = 28) | 31 ± 1 (n = 21) | 0.58 ± 0.07 (n = 7) |
| DDD | 48 ± 3 (n = 37)a | 28 ± 1 (n = 35) | 0.46 ± 0.09 (n = 9) |
Average mean ± SE values, with the number of observations in parentheses; ap < 0.001 between AAA and DDD.
Figure 4.

Effect of ionic strength on LC. The C0-C3AAA and C0-C3DDD constructs were tested in buffers with different concentrations of KCl to determine the influence of ionic strength on motif fold stability. Whereas LC for C0-C3AAA had a similar frequency distribution at all KCl concentrations, C0-C3DDD separated into distinct inextensible (short LC) and extensible (LC comparable to C0-C3AAA) populations at higher concentrations.
Motif structural changes with phosphomimetic replacement
CD spectroscopy
CD spectroscopy was used to assess the secondary structure of C1-C2AAAA and C1-C2DDDD at 15°C. CD spectra for C1-C2AAAA and C1-C2DDDD exhibited a broad minima around 215–218 nm and a positive band around 195 nm (data not shown). This was consistent with both proteins being comprised of predominantly β-sheet secondary structure (Table 3).
To further investigate structural differences between C1-C2AAAA and C1-C2DDDD, we recorded the temperature dependence of their CD spectra. We chose a wavelength of 206.5 nm to monitor the temperature dependence of the ellipticity, because the contribution from β-sheet secondary structure at this wavelength is minimal compared with the α-helix and coil components (42). Significant differences in temperature dependence of the CD at 206.5 nm for the two proteins were observed (Fig. 5, A and B). C1-C2AAAA exhibited a weak transition, with a midpoint of ∼41°C, characterized by a slight decrease in negative ellipticity (Fig. 5 A). In contrast, C1-C2DDDD demonstrated a large increase in negative ellipticity with a transition at ∼51°C, which was indicative of an increase in coil or helical structure (Fig. 5 B). The negative CD transition observed for C1-C2DDDD was concomitant with an increase in scattering (data not shown), which suggests a corresponding increase in hydrodynamic size. The temperature dependence of the ellipticity at 206.5 for C1-C2DDDD is most consistent with an increase in helical structure induced by dimerization and/or oligomerization at high temperature. In contrast, no temperature-dependent secondary structure formation was observed for C1-C2AAAA.
To determine the effect of ionic strength on C1-C2AAAA and C1-C2DDDD, we assessed the temperature dependence of the ellipticity at 206.5 nm in the presence of 250 mM KCl (Fig. 5, C and D). In high salt, C1-C2AAAA produced a temperature-dependent decrease in negative ellipticity (Fig. 5 C), which coincided with an increase in both scattering intensity and CD dynode voltage followed by a loss of CD dynode voltage (data not shown). Together, these data suggest a temperature-dependent loss of structure and/or formation of β-type structure with aggregation, followed by heat-induced denaturation and precipitation. In contrast, significant changes in the temperature dependence of the ellipticity at 206.5 nm were observed for C1-C2DDDD (Fig. 5 D), with increased negative ellipticity at ∼46°C preceding a decrease in negative ellipticity. The increased negative ellipticity coincided with an increase in scattering intensity, which suggested a temperature-dependent increase in helical structure accompanying the dimerization of C1-C2DDDD. This was immediately followed by a decrease in negative ellipticity and scattering, suggesting precipitation of the newly formed dimeric structure. A similar atypical CD thermal denaturation curve was observed for the Nudix protein (hydrolase DR_0079 (43)).
DLS
The C1-C2AAAA construct exhibited a hydrodynamic radius (RH) of 31.0 Å with a predicted molecular mass of 49 kD, as compared with C1-C2DDDD with a 38.0 Å RH and predicted molecular mass of 75 kD. A slightly larger radius of gyration (Rg) of 35.2 Å was previously determined for nonphosphorylated mouse C1-C2 by SAXS analysis (31). It should be noted that DLS is converted to hydrodynamic radius by means of the Stokes-Einstein equation assuming a hard sphere. The fact that both C1-C2AAAA and C1-C2DDDD are rod-shaped results in RH values and apparent hydrodynamic molecular masses larger than the actual molecular masses. Whereas the RH value for C1-C2DDDD is consistent with three globular domains in a rigid, cylindrical arrangement, the lower value for C1-C2AAAA likely reflects a distribution of bent conformations arising from extra flexibility in the nonphosphorylated motif. DLS from C1-C2AAAA and C1-C2DDDD was used to further monitor the temperature-dependent buildup of oligomeric intermediates (C1-C2DDDD in Fig. 6). At 25°C and protein concentrations up to ∼30 μM, C1-C2AAAA and C1-C2DDDD were found to be monomeric. However, Fig. 6 shows a plot of RH for C1-C2DDDD that clearly demonstrates the transient buildup of dimers and the formation of oligomers at temperatures > 35°C. Similar results were found for C1-C2AAAA.
Figure 6.

Temperature dependence of DLS intensity and predicted hydrodynamic radius of C1-C2DDDD. The DLS scattering intensity and hydrodynamic radius traces for 7 μM C1-C2DDDD at 25°C (black circles) and at 35°C (gray circles) in 10 mM borate, 1 mM DTT, pH 8, are shown. Both the scattering intensity and hydrodynamic radius remain constant at 25°C but gradually increase at 35°C due to self-association.
Discussion
Phosphorylation of the cMyBP-C motif leads to enhanced cardiac contractility in response to physiologic stress (1–3). The important role of motif phosphorylation, which operates at least partially through changes in the binding affinity of cMyBP-C for actomyosin (11–13) and through potential changes in the mechanical behavior of cMyBP-C once it is bound to actin and/or myosin, directed our focus to potential alterations in the molecular structure and mechanical properties of the phosphorylated motif. Our characterization of force-extension relations in the AFM and CD/DLS analyses using bacterially expressed mouse cMyBP-C N-terminal fragments indicated that phosphorylation stabilized the motif’s structure such that it was no longer flexible and freely extensible. This is supported by the following evidence: 1), phosphorylation of C0-C3’s by PKA-treatment resulted in a 29 nm reduction in LC (Fig. 2, Table 1); 2), phosphomimetic replacement of either the first three or all four serines in the motif (S273, S282, S302, and S307) also resulted in shorter LC; and 3), CD and DLS analyses suggested that the phosphomimetic replacement of all four serines in the C1-C2DDDD caused the motif to develop additional helical structure and become more rigid, resulting in a larger hydrodynamic radius compared with the nonphosphomimetic C1-C2AAAA control. These findings may provide a structural and mechanical basis for the impact that motif phosphorylation has on cMyBP-C function.
Force-extension of nonphosphorylated C0-C3
Single-molecule AFM studies have provided unprecedented insight into the molecular mechanics of titin (44), a sarcomeric protein with elastic properties that resists stretch in relaxed striated muscle (45). Like cMyBP-C, titin is a multidomain protein composed of numerous tandem Ig-like domains (45). Elongation of these domains under constant velocity extension in the AFM produces sawtooth force-extension curves that characterize the unfolding of its individual Ig-like domains (38,40). Thus, the earlier titin AFM studies provided a methodological and analytical framework that was recently applied to whole cMyBP-C (33) and was the basis of the studies presented here.
Force-extension traces for nonphosphorylated C0-C3 constructs exhibited the expected sawtooth pattern, with a maximum of four peaks associated with Ig-domain unfolding events followed by a peak corresponding to molecular detachment. The initial contour length, LC, should reflect the total length of the three or four stable Ig domains, the Pro-Ala linker, and the motif. Assuming a 4 nm folded length for each Ig-like domain (38,46–48) and 0.36 nm/residue for the two freely extensible linkers (Pro-Ala linker: 50 amino acids/18 nm; motif: 103 amino acids/37 nm) (33), we predict a 67–71 nm LC for the C0-C3 fragment. This value is similar to the 58–67 nm LC measured for the nonphosphorylated C0-C3 control and alanine-replacement mutants (Table 1). The lower experimentally determined value most likely reflects the fact that C0-C3’s attachment points to the glass slide and AFM probe were not vertically aligned, causing the molecule to be stretched on a diagonal rather than along its longitudinal axis (46). In addition, the attachment points may not be at the extreme N- and C-termini, which by itself would lead to a shorter estimated LC (see Fig. S1). These two factors would also contribute to the observed final contour lengths of 172 ± 19 nm for traces, with four unfolds being shorter than the 195 nm predicted value for the fully unfolded 539 amino acid C0-C3 control fragment.
Phosphorylation reduces LC of C0-C3 by stabilizing the motif
The C0-C3 force-extension traces provide a mechanical description of the structural domains and their extensibility. Because both motif phosphorylation as a result of PKA treatment and phosphomimetic replacement induced a 21–29 nm reduction in LC, we believe this mechanical alteration originated within the motif, because no other amino acids were modified or mutated in this study. A less extensible motif upon phosphorylation could arise from the motif becoming stabilized by intramotif electrostatic interactions due to the addition of the negatively charged phosphates, or adopting a new tertiary structure. The recently published NMR motif structure is characterized by several solvent-exposed arginines (R326, R342, and R347) and lysines (K346 and K349) in the C-terminal triple-helix bundle (32). These positively charged residues could electrostatically interact with the negatively charged phosphoserines (or aspartic acid phosphomimetics) in the N-terminal portion of the motif (Fig. 7), resulting in a more stable motif (49). This could account for the measured 21–29 nm decrease in LC with phosphorylation, as similar electrostatic interactions have been proposed for the observed reduction in LC within the titin PEVK region after PKCα serine phosphorylation (50).
Figure 7.

Model of motif equilibrium between two structural states. (A) When it is nonphosphorylated, the motif is conformationally dynamic (blurred image) and compact but readily extensible (kinetic arrow toward extended structure) under low force (F) applied to the end of the motif (upward arrow). Balls with + signs represent positive charged residues, and balls with 0 signs represent nonphosphorylated serines. (B) Upon addition of negatively charged phosphates to motif serines (balls with – signs), the motif structure becomes stabilized by electrostatic interactions (kinetic arrow toward compact structure) but may still be forcibly extended (F, upward arrow).
If electrostatic interactions stabilize the motif structure upon phosphorylation, then changes in ionic strength should affect these interactions. In support of this notion, we found that raising the KCl concentration from 25 to 300 mM reduced the likelihood of LC reduction observed for a given C0-C3DDD molecule (Table 2). Whereas the distribution of LC for C0-C3AAA remained near 60 nm with ionic strength, LC distributions for C0-C3DDD were bimodal at all KCl concentrations with a population at 60 nm, similar to the C0-C3AAA control, and a shorter LC population near 40 nm (Fig. 4), with the relative proportion of these two populations being dependent on salt concentration. Specifically, at 25 mM KCl, the C0-C3DDD LC was predominantly of the shorter population, shifting toward longer LC at higher salts. Therefore, the motif appears to be in equilibrium between two states, with the probability of being in either state depending on both the applied load, phosphorylation state of the motif, and ionic conditions (Fig. 7).
In addition to a 21–29 nm reduction in LC with motif phosphorylation, occasionally the C0-C3DDD and C0-C3DDDD force-extension traces (n = 4) had up to five unfolding peaks (Fig. 8) even though there were only four Ig domains. The appearance of traces containing a fifth unfolding peak suggests that one of the peaks resulted from the extension of the stabilized, phosphorylated motif. Because we cannot assign a particular unfolding event to a specific domain, and given that these unfolding events are mechanically indistinguishable in terms of ΔLC and unfolding force, FUnfold, we propose that the unfolding force of the phosphorylated motif lies within the full range of Ig-domain unfolding forces (50–200 pN) as characterized in the nonphosphorylated controls (Fig. 3). Although the phosphorylated motif may be mechanically indistinguishable from an Ig-like fold in both incremental contour length, ΔLC, and unfolding force, FUnfold, by no means do we propose that the phosphorylated motif has adopted an Ig-like fold, with its associated hydrogen bonding between β-strands (51), given the lack of any change in secondary structure upon phosphorylation (Table 3). Given the similar mechanical characteristics to Ig domain unfolding, then phosphorylated motif unfolding-like behavior most likely also occurred in the more commonly observed force-extension traces with three or four unfolding events (Fig. 1, D–F), with the point of probe attachment to the protein, rather than the lack of motif unfolding, determining the number of peaks. Although trivial explanations could account for a fifth unfolding event, the fact that the final contour length, LC(Unfolded), for these four traces (172 ± 19 nm; Fig. 8) is similar to that of the C0-C3AAA and C0-C3AAAA control traces (170 ± 12 nm) exhibiting four unfolding peaks (LC(Unfolded); Fig. 1, A–C) provides strong evidence that these force-extension traces originated from a single molecule. This would imply that one of the five peaks in these force-extension traces represents the destabilization of the phosphorylated motif. An important avenue of future investigation will be to determine how phosphorylation at a specific site or sites influences the mechanical stability of the phosphorylated motif.
Figure 8.
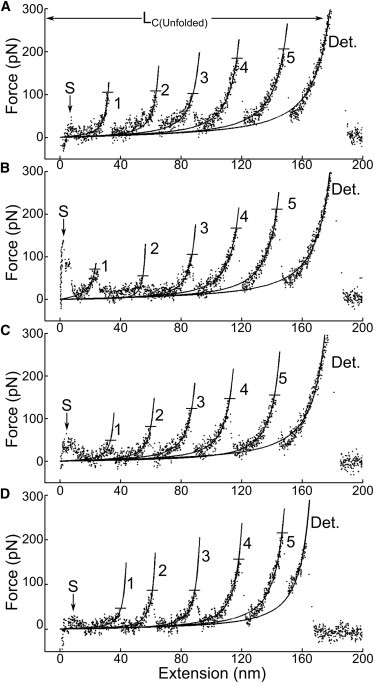
(A–D) Force-extension traces (with wormlike chain model fits as solid lines) of phosphomimetic DDDD (A and B) and DDD (C and D) constructs showing a clear separation between the AFM probe and glass surface (S) followed by five unfolding events (1–5) and subsequent detachment (Det.). Measured FUnfold values for each peak are highlighted by horizontal bars. The final contour lengths of these traces (LC(Unfolded)) are consistent with the 549 amino acid sequence of the C0-C3 fragment, and are comparable to the final contour lengths of nonphosphorylated fragments having only four unfolds.
A possible mechanism for the phosphorylation-dependent reduction in LC is that the motif adopts new secondary structural elements. Therefore, we performed CD and DLS experiments to structurally characterize the motif and its stability before and after phosphorylation. Based on CD spectroscopy, both C1-C2AAAA and C1-C2DDDD at 15°C have extensive β-sheet secondary structure, which is consistent with the presence of two Ig-like domains, and smaller amounts of α-helix are also present (Table 3), as previously measured by CD for C1-C2 (30). Interestingly, fitting the CD spectra using the CONTIL algorithm alone suggested that C1-C2DDDD has an ∼10% higher α-helical content than C1-C2AAAA. This is consistent with previous NMR measurements showing that substitution of an aspartic acid at S273 extends a helical structure between residues 265 and 270 (32). One must use caution when placing emphasis on small differences in secondary structure using CONTIL, because errors in protein concentration can lead to calculated differences in helical content (52,53). However, this apparent difference in helical content is consistent with the temperature-dependent CD differences in C1-C2AAAA and C1-C2DDDD (Fig. 5, A and B). The DLS data provide further evidence for stability within the motif because C1-C2DDDD is predicted to be more rigid than C1-C2AAAA. Because the only change between C1-C2AAAA and C1-C2DDDD is the introduction of negative charge in the motif, together these structural data support the AFM results suggesting that phosphorylation stabilizes the motif’s structure.
Physiological relevance of phosphorylation-dependent changes in motif structure and mechanics
The ability of cMyBP-C to modulate actomyosin force and motion generation must relate to its binding to actin and/or myosin, which has been localized to the N-terminal C0-C2 domains (9,13,20,21,54–56) and at a minimum, the first 29 kDa, which encompasses domains C0-C1 and 17 amino acids of the motif (13,14). Therefore, the changes in motif structure and mechanics reported here provide a molecular basis for the phosphorylation-dependent modulation of cMyBP-C’s interaction with its binding partners.
To be an effective modulator of actomyosin function within the sarcomere, the cMyBP-C N-terminal actomyosin-binding domains should extend at least 15–20 nm from the thick-filament surface to reach their farthest binding partner, the actin-containing thin filament (57), as suggested by electron tomographic reconstructions (4). The mean-squared end-to-end length, R, of a disordered polymer may be approximated by <R2> = 2LCLP (58). The persistence lengths measured here (average of 0.4 nm) thus predict globular conformations for the motif and Pro-Ala linker, with lengths of 3.8 and 4.5 nm respectively, which added to four 4 nm Ig domains predicts a 25.2 nm length for C0-C3, consistent with the dimensions of this fragment predicted by SAXS (31). The additional flexibility and extensibility of the nonphosphorylated motif may allow the N-terminal 29 kD to find its binding partners by exploring its sarcomeric space via an effective fly-casting mechanism (59), an ability that likely would be impaired by the increase in motif rigidity (as seen by CD) and decrease in motif extensibility (as seen by AFM) after phosphorylation. Extensibility of the nonphosphorylated motif may also be required for N-terminal domains to remain attached to actin and/or myosin during sarcomeric shortening (33), while the C-terminal domains remain anchored to the thick filament. Finally, the conformationally dynamic nature of the motif (Fig. 7) may be required to expose actomyosin-binding sites or portions of sites that exist within the motif (33). Thus, reducing cMyBP-C’s motif extensibility by phosphorylation may provide a means of regulating cMyBP-C’s affinity for actin and/or myosin (60), either by limiting its fly-casting capacity or by stabilizing the motif structure so that part of the actomyosin-binding interface is compromised. In fact, differential changes in actin- versus myosin-binding affinity with motif phosphorylation might even allow cMyBP-C to shift between its two binding partners (55) depending on the extent of phosphorylation.
In this study we have defined a general physical mechanism for regulation of cMyBP-C function via phosphorylation; however, important areas of future investigation include testing shorter protein fragments to better isolate the mechanical properties of the motif by eliminating confounding contributions from flanking domains, and determining the effects of specific sites of phosphorylation and the number of these sites on cMyBP-C’s molecular mechanics.
Acknowledgments
We thank A. Quinn at the Microscopy Imaging Center of the University of Vermont for assistance with the AFM, and S. Beck and A. Weith for technical assistance.
This work was supported by grants from the National Institutes of Health (HL007944 to A.J.M.; HL059408 to D.M.W., J.G., and J.R.; and HL007647 to M.P.). The project described was supported by Award Number S10RR025498 from the National Center for Research Resources for purchase of the AFM.
Contributor Information
Paul R. Rosevear, Email: paul.rosevear@uc.edu.
David M. Warshaw, Email: david.warshaw@uvm.edu.
Supporting Material
References
- 1.Winegrad S. Cardiac myosin binding protein C. Circ. Res. 1999;84:1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 2.Barefield D., Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris S.P., Lyons R.G., Bezold K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luther P.K., Winkler H., Liu J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA. 2011;108:11423–11428. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okagaki T., Weber F.E., Reinach F.C. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J. Cell Biol. 1993;123:619–626. doi: 10.1083/jcb.123.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luther P.K., Bennett P.M., Moss R.L. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J. Mol. Biol. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moos C., Mason C.M., Dubin J.H. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J. Mol. Biol. 1978;124:571–586. doi: 10.1016/0022-2836(78)90172-9. [DOI] [PubMed] [Google Scholar]
- 8.Witt C.C., Gerull B., Thierfelder L. Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J. Biol. Chem. 2001;276:5353–5359. doi: 10.1074/jbc.M008691200. [DOI] [PubMed] [Google Scholar]
- 9.Gruen M., Gautel M. Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 10.Ratti J., Rostkova E., Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 2011;286:12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruen M., Prinz H., Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453:254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- 12.Shaffer J.F., Kensler R.W., Harris S.P. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 2009;284:12318–12327. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weith A., Sadayappan S., Warshaw D.M. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J. Mol. Cell. Cardiol. 2012;52:219–227. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Previs M.J., Beck Previs S., Warshaw D.M. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmann P.A., Greaser M.L., Moss R.L. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J. Physiol. 1991;439:701–715. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelzer J.E., Patel J.R., Moss R.L. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ. Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann P.A., Hartzell H.C., Moss R.L. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J. Gen. Physiol. 1991;97:1141–1163. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saber W., Begin K.J., VanBuren P. Cardiac myosin binding protein-C modulates actomyosin binding and kinetics in the in vitro motility assay. J. Mol. Cell. Cardiol. 2008;44:1053–1061. doi: 10.1016/j.yjmcc.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mun J.Y., Gulick J., Craig R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) J. Mol. Biol. 2011;410:214–225. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Razumova M.V., Shaffer J.F., Harris S.P. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: evidence for long-lived cross-bridges. J. Biol. Chem. 2006;281:35846–35854. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 21.Whitten A.E., Jeffries C.M., Trewhella J. Cardiac myosin-binding protein C decorates F-actin: implications for cardiac function. Proc. Natl. Acad. Sci. USA. 2008;105:18360–18365. doi: 10.1073/pnas.0808903105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gautel M., Zuffardi O., Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia W., Shaffer J.F., Leary J.A. Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis. J. Proteome Res. 2010;9:1843–1853. doi: 10.1021/pr901006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadayappan S., Gulick J., Robbins J. Cardiac myosin binding protein-C phosphorylation in a β-myosin heavy chain background. Circulation. 2009;119:1253–1262. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong C.W., Stelzer J.E., Moss R.L. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ. Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacques A.M., Copeland O., Marston S.B. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J. Mol. Cell. Cardiol. 2008;45:209–216. doi: 10.1016/j.yjmcc.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Sadayappan S., Osinska H., Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Copeland O., Sadayappan S., Marston S.B. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 2010;49:1003–1011. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 29.El-Armouche A., Pohlmann L., Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell. Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Jeffries C.M., Whitten A.E., Trewhella J. Small-angle X-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J. Mol. Biol. 2008;377:1186–1199. doi: 10.1016/j.jmb.2008.01.080. [DOI] [PubMed] [Google Scholar]
- 31.Jeffries C.M., Lu Y., Trewhella J. Human cardiac myosin binding protein C: structural flexibility within an extended modular architecture. J. Mol. Biol. 2011;414:735–748. doi: 10.1016/j.jmb.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 32.Howarth J.W., Ramisetti S., Rosevear P.R. Structural insight into unique cardiac myosin-binding protein-C motif: a partially folded domain. J. Biol. Chem. 2012;287:8254–8262. doi: 10.1074/jbc.M111.309591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karsai A., Kellermayer M.S., Harris S.P. Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophys. J. 2011;101:1968–1977. doi: 10.1016/j.bpj.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietz H., Rief M. Detecting molecular fingerprints in single molecule force spectroscopy using pattern recognition. Jpn. J. Appl. Phys. 2007;46:5540–5542. [Google Scholar]
- 35.Sadayappan S., Gulick J., Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cleveland J.P., Manne S., Hansma P.K. A nondestructive method for determining the spring constant of cantilevers for scanning force microscopy. Rev. Sci. Instrum. 1993;64:403–405. [Google Scholar]
- 37.Carrion-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 38.Rief M., Gautel M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 39.Bustamante C., Marko J.F., Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe K., Muhle-Goll C., Granzier H. Different molecular mechanics displayed by titin’s constitutively and differentially expressed tandem Ig segments. J. Struct. Biol. 2002;137:248–258. doi: 10.1006/jsbi.2002.4458. [DOI] [PubMed] [Google Scholar]
- 41.Zhmurov A., Dima R.I., Barsegov V. Order statistics theory of unfolding of multimeric proteins. Biophys. J. 2010;99:1959–1968. doi: 10.1016/j.bpj.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeer A.W., Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchko G.W. Circular dichroism studies on the Deinococcus radiodurans Nudix hydrolase DR_0079: an atypical thermal melt. Protein Pept. Lett. 2010;17:831–835. doi: 10.2174/092986610791306689. [DOI] [PubMed] [Google Scholar]
- 44.Linke W.A., Grützner A. Pulling single molecules of titin by AFM—recent advances and physiological implications. Pflugers Arch. 2008;456:101–115. doi: 10.1007/s00424-007-0389-x. [DOI] [PubMed] [Google Scholar]
- 45.LeWinter M.M., Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrion-Vazquez M., Marszalek P.E., Fernandez J.M. Atomic force microscopy captures length phenotypes in single proteins. Proc. Natl. Acad. Sci. USA. 1999;96:11288–11292. doi: 10.1073/pnas.96.20.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H., Fernandez J.M. Mechanical design of the first proximal Ig domain of human cardiac titin revealed by single molecule force spectroscopy. J. Mol. Biol. 2003;334:75–86. doi: 10.1016/j.jmb.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 48.Li H., Linke W.A., Fernandez J.M. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 49.Woods A.S., Ferré S. Amazing stability of the arginine-phosphate electrostatic interaction. J. Proteome Res. 2005;4:1397–1402. doi: 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson B.R., Bogomolovas J., Granzier H. The effects of PKCα phosphorylation on the extensibility of titin’s PEVK element. J. Struct. Biol. 2010;170:270–277. doi: 10.1016/j.jsb.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu H., Isralewitz B., Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Provencher S.W., Glöckner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 53.van Stokkum I.H., Spoelder H.J., Groen F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 54.Kensler R.W., Shaffer J.F., Harris S.P. Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 2011;174:44–51. doi: 10.1016/j.jsb.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu Y., Kwan A.H., Jeffries C.M. The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J. Mol. Biol. 2011;413:908–913. doi: 10.1016/j.jmb.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 56.Orlova A., Galkin V.E., Trewhella J. The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J. Mol. Biol. 2011;412:379–386. doi: 10.1016/j.jmb.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Squire J.M., Luther P.K., Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J. Mol. Biol. 2003;331:713–724. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 58.Rubinstein M., Colby R.H. Oxford University Press; Oxford/New York: 2003. Polymer Physics. [Google Scholar]
- 59.Shoemaker B.A., Portman J.J., Wolynes P.G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kulikovskaya I., McClellan G., Winegrad S. Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 2003;122:761–774. doi: 10.1085/jgp.200308941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 62.Whitmore L., Wallace B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Böhm G., Muhr R., Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.