

Mechanical dysfunction remains as one of the best predictors of sudden cardiac death (SCD), but despite this close association the causal relationships between electrical instability and mechanical dysfunction are not fully understood. (Bigger et al., 1984; Buxton et al., 1984) Even though the effects of electrical instability on mechanical dysfunction of the heart is well appreciated and relatively clear, the reverse effects that mechanical dysfunction have electrical instability, or mechano-electrical feedback (MEF), are not. (Kohl et al., 2006; Lab, 1996) Stretch activated channels, for example, are one way mechanical activity can directly influence electrical activity at the cellular level under physiologic and pathophysiological conditions. (Hu et al., 1997) Alternatively, intracellular calcium, which regulates mechanical contraction, can significantly influence electrical function of the heart as well. The mechanisms by which intracellular calcium governs electrical instability of the heart is the main focus of this manuscript. Traditionally, calcium mediated arrhythmogenesis is associated with delayed after depolarizations and abnormal impulse formation. (Katra et al., 2007; Liu et al., 2006; Rosen, 1985; Ter Keurs et al., 2006; Wehrens et al., 2003) However, in this manuscript we focus on how cardiac alternans, a mechanisms of reentrant excitation, is another significant way mechanical dysfunction and arrhythmogenesis are causally related.
Cardiac alternans can refer to either mechanical (contractile) or electrical (repolarization) oscillations that occur on an every other beat basis. Cardiac alternans has been recognized for more than 100 years. Shown in Figure 1 (Panel A) is a very early record of arterial pressure measured during a steady state heart rate, where pulse magnitude alternates on every other beat (arrows). (Traube, 1872) At this time, the ECG had not been invented; however, soon after its introduction electrical alternans was reported as well. Shown in Panel B (Figure 1) is an early example of electrical alternans as evidenced by oscillations of the ECG T-wave (arrows) and QRS. (Lewis, 1910) Soon after cardiac alternans was first reported, it was associated with a poor prognosis. (Windle, 1913) Surprisingly, at this same time several important characteristics of cardiac alternans were observed, such as alternans occurring in a structurally normal heart and appearing at faster heart rates. Subsequently, T-wave alternans was more directly related to coronary artery occlusion and shown to be highly reproducible. (Hellerstein et al., 1950)
Figure 1.

Early record of pulse (top) and ECG (bottom) alternans. Reproduced with permission.
1. Alternans and Arrhythmogenesis
Recently, cardiac alternans been mechanistically linked to the genesis of reentrant arrhythmias (Pastore et al., 1999) and shown to be a good marker of risk for sudden cardiac death in patients. (Rosenbaum et al., 1994) Several studies have shown that T-wave alternans is caused primarily by beat-to-beat alternation of repolarization at the cellular level (i.e. alternation of phases 2 and 3 of the action potential), which increase in magnitude with increasing heart rate. (Chinushi et al., 2003; El-Sherif et al., 1996; Pastore et al., 1999; Pruvot et al., 2004) Alternans of the QRS can occur as well, but this typically occurs at faster heart rates, secondary to repolarization alternans. (Pastore et al., 1999; Qian et al., 2003) In brief, action potential alternans can significantly amplify repolarization gradients that are sufficient to cause unidirectional block, a requirement for reentrant excitation, even in the absence of structural or ion channel heterogeneities. Shown in Figure 2 is the induction of ventricular fibrillation (VF) that was caused by repolarization alternans. (Laurita et al., 2001; Pastore et al., 1999) Above a critical heart rate, repolarization occurs with the same phase between all regions (i.e. spatially concordant alternans). During concordant alternans, the spatial gradient of repolarization is not much greater than that during baseline pacing. For faster heart rates, however, repolarization of neighboring regions begin to alternate with opposite phase (i.e. spatially discordant alternans). The development of spatially discordant alternans (not to be confused with electrical-mechanical discordance) is a consistent precursor to reentrant arrhythmogenesis because of its affect on repolarization gradients in the heart. Steep gradients of repolarization form as evidenced by marked crowding of repolarization isochrone lines with a range of repolarization times >100 ms (Figure 2 bottom, beats 1–3). In addition, the orientation of repolarization gradients undergoes nearly a complete reversal in direction from beat to beat, as indicated by the alternating arrow direction. Although repolarization patterns are complex, they are highly reproducible on alternate beats (Figure 2, compare repolarization maps on beat 1 versus beat 3). The pattern of depolarization during spatially discordant repolarization alternans remains stable, without any significant evidence of conduction alternans. Finally, the introduction of a premature beat during discordant alternans results in conduction block (beat 4, depolarization map) into a region having most delayed repolarization from the previous beat (beat 3 repolarization map). The impulse then propagates around both sides of the line of functional block (hatched area) and reenters from outside the mapping field, forming the first spontaneous beat of reentrant VF. This finding demonstrates one way repolarization alternans can transform physiological gradients of repolarization into pathophysiological gradients that are a mechanism of VF. Clearly, this cascade of events that leads to a fatal end critically depends on the development of electrical alternans; so, an important question that remains is what causes alternans in the first place?
Figure 2.

Representative example demonstrating mechanism linking repolarization alternans to the genesis of reentrant VF. Contour maps indicate pattern of depolarization and repolarization across the epicardial surface of guinea pig ventricle (times shown in msec). As pacing rate is increased, discordant alternans develops producing complete reversal in the direction of repolarization gradients from beat-to-beat and, most importantly, steep gradients of repolarization that were not present during concordant alternans (not shown). These gradients formed a suitable substrate for reentry, as slight shorting of stimulus cycle length (beat 4) causes local propagation failure against a repolarization gradient established during the previous beat (upper right corner of beat 3 repolarization map). Discordant alternans produces conditions necessary for unidirectional block after which reentrant VF immediately ensued. The ECG across the top is shown for reference. Reproduced with permission.
2. Intracellular Calcium Regulation
Since electrical and mechanical alternans are frequently observed concurrently at the cellular and whole heart level, (Lu et al., 1968; Murphy et al., 1994; Pruvot et al., 2004) it is likely that calcium, an important regulator of cellular contraction and electrophysiology, is involved. Shown in Figure 3 is a simplified diagram of cellular calcium regulation. Immediately following the rapid depolarization phase of the action potential, calcium entering the cell through the L- type channel (IL,Ca) triggers calcium release from the sarcoplasmic reticulum (SR), in a process referred to as calcium-induced calcium-release (CICR). This is initiated by calcium entering through the IL,Ca channel and binding to nearby SR release channels, which causes calcium release from the SR. A single SR calcium release channel is a macromolecular complex comprised of four ryanodine receptor (RyR) proteins, stabilizing proteins such as FKBP12.6, phosphatases, and many other proteins including calsequestrin, triadin, and junctin. (Gyorke et al., 2004) When calcium is released from the SR, it binds to the troponin complex of the actin filament and initiates contraction. On a beat-to-beat basis, the strength of contraction is proportional to the amount of calcium released. Other factors, such as myofilament sensitivity to calcium and cross-bridge binding are also critically involved. (Gordon et al., 2000) In ventricular cells, IL,Ca and SR calcium release channels are located along the surface membrane and T-tubules to ensure that upon electrical excitation calcium release and, thus, contraction is uniform and synchronous within an entire cell.
Figure 3.
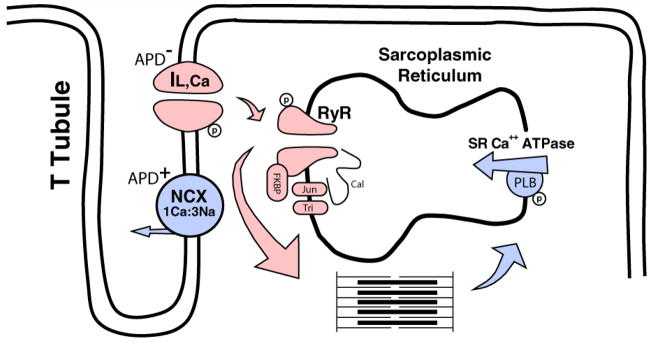
A simplified schematic of normal ventricular myocyte intracellular calcium cycling. Shown are the main proteins and their primary responsibility in the rapid increase of cytoplasm calcium (red) during systole and the subsequent decrease in cytoplasmic calcium levels (blue) during diastole. During steady state conditions, intracellular calcium homeostasis is maintained by an equal amount of calcium entering and exiting the cytoplasm for each beat. APD+ and APD− indicate effects that either lengthen APD or shorten APD, respectively. RyR, ryanodine receptor; Jun, junctin; Tri, triadin; Cal, calsequestrin; PLB, phospholamban; FKBP, FK-506 binding protein.
The amount of calcium released from the SR, which regulates cellular contraction, has a critical dependency on several factors including, primarily, the amount of calcium entering through IL,Ca, (Fabiato, 1985) the amount of calcium load in the SR, (Bassani et al., 1995; Bers et al., 1998; Lukyanenko et al., 1996; Terentyev et al., 2002) and recovery (inactivation and adaptation) of RyR. (Lukyanenko et al., 1998) There are also many other factors that can modulate RyR. For example, PKA-dependent phosphorylation of RyR has been shown to have varied phosphorylation sites (Marx et al., 2000; Xiao et al., 2005) and effects on RyR open probability (Po). (Valdivia et al., 1995) Ca-Calmodulin dependent protein kinase (CaMKII) can also phosphorylate RyR. (Guo et al., 2006) The FK-506 binding protein, FKBP12.6, is also an important modulator of SR calcium release. One FKBP12.6 binds tightly to each RyR protein subunit of the tetramer to prevent sub-conductance states and aberrant activation during diastole. This stabilizes the RyR in the closed and open states. In addition, multiple RyR channels are functionally related and closely coupled (i.e. FKBP12.6-mediated coupled gating) such that synchronous activation and inactivation of multiple RyR channels occurs. (Marx et al., 2001) It has also been suggested that β-stimulation plays an important role in modulating the effect of FKBP12.6;(Doi et al., 2002; Marx et al., 2000; Song et al., 2001) however, this remains controversial. (Xiao et al., 2004) Finally, the proteins calsequestrin, triadin, and junctin on the luminal side of the SR, assemble with RyR and are known to play a critical regulatory role. (Gyorke et al., 2004)
For mechanical relaxation to occur, intracellular calcium is returned to resting diastolic levels by two primary mechanisms. First, calcium is removed from the cell by the sodium calcium (Na+-Ca++) exchanger operating in the forward mode, where 3 Na+ ions enter the cell in exchange for 1 Ca++ ion leaving the cell. To maintain homeostasis, the amount of calcium removed from the cell by the Na+-Ca++ exchanger is equivalent to the amount entering the cell through, primarily, the IL,Ca calcium channel. The remaining cytoplasmic calcium is reclaimed by the SR via SR Ca++ ATPase, which (to some extent) competes with the Na+-Ca++ exchanger for cytoplasmic calcium. Phospholamban (PLB) is an important regulator of SR Ca++ ATPase. PLB normally decreases SR Ca++ ATPase activity, (Ravens et al., 2000) and reduced expression can significantly enhance SR Ca++ ATPase. (Hoit et al., 1999) Catecholamines accelerate relaxation and PKA dependent phosphorylation of PLB is the likely mechanism. (Li et al., 2000) CaMKII can also accelerate SR calcium reuptake by phosphorylating PLB and, possibly, by a mechanism that does not require PLB. (DeSantiago et al., 2002) The numerous molecular processes involved in cytoplasmic calcium homeostasis highlight the complex nature of its regulation and, thus, the many ways in which calcium dysregulation can occur.
3. Voltage and Calcium Coupling
In addition to electrical excitation inducing SR calcium release and mechanical contraction, calcium release can feedback and significantly influence transmembrane potential in several ways. This has been referred to as “bi-directional” coupling of membrane voltage and intracellular calcium. (Shiferaw et al., 2005) For example, in the process of returning calcium to diastolic levels, the Na+-Ca++ exchanger expels one Ca++ ion for 3 Na+ ions entering the cell, resulting in a net depolarizing current. Thus, a larger than normal calcium release would be expected to prolong APD. This type of “coupling” between calcium release and APD is referred to as positive Ca-to-Vm coupling. (Shiferaw et al., 2005) Shown in Figure 4 is an example of positive Ca-to-Vm coupling (i.e. electrical-mechanical concordance). Shown are the ECG, action potentials (AP), and calcium transients (Ca++) recorded simultaneously using optical mapping techniques during pacing induced alternans in a normal Langendorff perfused guinea pig heart. (Pruvot et al., 2004) In the presence of significant T-wave alternans, APD alternates as well. Significant calcium transient amplitude alternans also occurs, indicating alternating calcium release from the SR. In this example of positive Ca-to-Vm coupling the larger calcium release is associated with the long APD, and the small release with the short APD.
Figure 4.
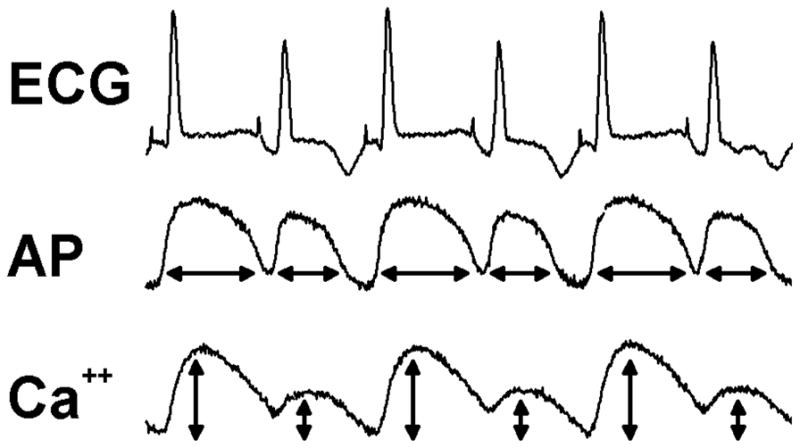
Shown are the ECG, action potentials (AP) and calcium transients (Ca++) recorded simultaneously in the Langendorff perfused guinea pig heart during alternans. Alternans was induced by pacing at a cycle length of 170 ms. The arrows below each action potential and calcium transient represent APD (msec) and normalized CaF amplitude (%) respectively. The development of T-wave alternans was closely paralleled by alternans of APD and calcium transient amplitude with positive Ca-to-Vm coupling (electrical-mechanical concordance).
Negative Ca-to-Vm coupling is possible as well. When calcium is released from the SR, it also inactivates the IL,Ca calcium channel, which would decrease net depolarizing current during the action potential plateau and shorten APD. In this case a larger than normal calcium release would shorten APD and a smaller calcium release would prolong APD. In addition, the slow delayed rectifier K+ current (IKs) is enhanced by intracellular calcium such that a larger calcium transient amplitude would also shorten APD. Finally, in addition to the currents already mentioned, the calcium activated transient outward chloride current, Ito2, and calcium activated nonselective current Ins(Ca) can also influence membrane potential, but their affect on APD is not as clear. An important consequence of both positive and negative coupling is that the balance of currents involved, which may be varied for different species and disease conditions, will determine whether or not the action potential will be prolonged or shortened for a given change in SR calcium release. If, for example, negative Ca-to-Vm coupling dominates, then it is expected that a long APD would be associated with a small SR calcium release. This behavior is also known as electrical-mechanical discordance. For the most part, we see electrical-mechanical concordance, but discordance is possible, especially on a microscopic scale when electrotonic forces constrain local spatial discontinuities of voltage but not calcium. There are several reports of electrical-mechanical discordance under disease conditions. For example, Lee et al (1988) have shown electrical-mechanical discordance (short APD associated with large calcium transient) in ischemic hearts. Murphy et al (1994) have also shown electrical-mechanical discordant alternans, but in normal Landrace pigs that were paced rapidly.
APD can also couple to calcium release (Vm-to-Ca coupling). For example, given a constant heart rate, a prolongation of APD will shorten the following diastolic interval and thus provide less time for IL,Ca to fully recover. As a result, IL,Ca will be less and calcium release from the SR will be less as well. So, if APD alternates on a beat-to-beat basis, calcium release (or mechanical) alternans will occur as well. In this regard, mechanical alternans is just an innocent bystander and MEF is not playing an integral role. While the absence of MEF is not a focus of this manuscript, as a mechanism of alternans it is worth mentioning briefly. For example, it is possible that the rate dependence of repolarizing ion channels is responsible for electrical alternans. This can be demonstrated by APD restitution, which describes how APD responds to heart rate. The basic premise of the “restitution hypothesis” says that repolarization alternans will occurs when the slope of the restitution curve is >1, which has been taken as evidence that sarcolemmal ion channels rather than SR calcium regulation determine repolarization alternans. (Garfinkel et al., 2000; Riccio et al., 1999; Räcke et al., 1994; Watanabe et al., 2001) One problem with this approach, however, is that restitution is likely a product of several ionic mechanisms and intracellular calcium regulation. Therefore, as an approach to identify molecular mechanisms of alternans, restitution of APD can yield conflicting results (Banville et al., 2002) and may not be the best approach. (Laurita, 2004) Ignoring the limitations associated with APD restitution, there are instances when membrane ionic currents are the likely cause of alternans. For example Hua et al (2004) demonstrated very nicely how overexpression of an important repolarizing current, IKr, can suppress alternans. Similarly, patients with prolonged repolarization (LQTS) are susceptible to T-wave alternans. (Platt et al., 1996; Zareba et al., 1994) In either case, a specific change in repolarization, either enhanced or suppressed alternans. These are good examples of why it’s difficult to rule out membrane ion channel kinetics as a mechanism of alternans. However, in the setting of mechanical dysfunction it is unlikely that repolarization channels are the primary mechanism of alternans. A more likely paradigm is intracellular calcium dysfunction and its effects on electrical instability.
4. Mechanisms of Alternans
We (Pruvot et al., 2004; Walker et al., 2003; Wan et al., 2005) and others (Diaz et al., 2004; Picht et al., 2006; Weiss et al., 2006) have hypothesized that repolarization alternans is attributable to beat-to-beat alternans of SR calcium release. To date, there is no single experiment that can prove definitively that repolarization alternans is caused by calcium transient alternans. However, there are several lines of evidence that suggest it is likely. In isolated cells, Chudin et al (1999) demonstrated alternans of the calcium transient in the presence and absence of action potential alternans (i.e. AP clamped). This important observation indicates that calcium transient alternans can occur in the absence of action potential alternans. Interestingly, a similar conclusion could be drawn from measurements in vivo without voltage clamping. Aistrup et al (2006) measured calcium transient alternans in intact myocardium using confocal imaging techniques. They found that at fast heart rates local calcium transient alternans occurred with opposite phase (i.e. spatially discordant) between adjoining cells. Membrane potential was not measured simultaneously; but, assuming that electrotonic forces prevent any significant discontinuities in membrane potential between adjoining cells, then it is likely that APD alternated with similar phase. Thus, at the cellular level calcium transient alternans was occurring, at least in one of the cells, independent of voltage. Similar results have been shown in modeling studies. (Sato et al., 2006) While these data are not definitive proof, they do strongly support the notion that repolarization alternans arises from calcium transient alternans.
To further understand the mechanism of alternans, we have used clues from naturally occurring heterogeneities of repolarization alternans in the whole heart. In the guinea pig, we consistently found that sites showing the first signs of alternans with increasing heart rate were at the base of the LV. Locations where alternans first appeared were defined as alternans prone regions, in contrast to regions where alternans occurred at faster pacing rates (i.e. alternans resistant regions). Shown in Figure 5 is the alternans prone region (hatched area) relative to baseline APD gradients (Panel A) in the normal guinea pig heart. Interestingly, the alternans prone region did not correlate with longer APDs (Panel A,B) or with faster restitution kinetics (not shown), as would be expected if APD restitution is the underlying mechanisms of alternans. (Pruvot et al., 2004) Rather, in a separate study we found that the alternans susceptible region corresponded to regions where calcium release is weakest (LV base). (Katra et al., 2004) Shown in Panel C is the spatial heterogeneity of calcium transient amplitude across the normal guinea pig heart (CaRamp), which is smallest near the LV base. This finding is consistent with previous studies reporting base to apex heterogeneities of LV mechanical function. (Bogaert et al., 2001; Feiring et al., 1988) In addition, this is consistent with the notion that a weaker calcium release may be a mechanism of repolarization alternans in the intact heart, (Diaz et al., 2004) and may explain the development of regional arrhythmogenic repolarization alternans. (Pruvot et al., 2004) These findings further support the notion that calcium transient alternans causes repolarization alternans.
Figure 5.

APD gradient was oriented from the RV base to the LV free wall, orthogonal to the LAD (Panel A). The onset of APD-ALT and CaF-ALT (hatched area) consistently occurred at the LV base (n=7), independent of pacing site. Alternans onset did not occur where APD was longest (i.e. at the RV base), nor where APD was shortest (i.e. toward LV free wall). Similar results were observed in all experiments (Panel B). At a baseline pacing rate of 150 bpm, APD at the alternans prone site (LV base, hatched area) was significantly shorter by 8 msec than APD at the alternans resistant site from the RV base. Reproduced with permission. Panel C: Shown are heterogeneities of calcium transient amplitude measured using ratiometric optical mapping techniques (CaRamp). Calcium transients from the LV apex had a larger CaRamp compared to the LV base (top), and the contour map (middle) revealed a uniform gradient from base to apex. Reproduced with permission.
5. Mechanisms of Calcium Transient Alternans: Calcium Cycling
The reports described above support the idea that calcium transient alternans is a mechanisms of repolarization alternans. Therefore, what are the cellular mechanisms of calcium transient (i.e. SR release) alternans? One simple explanation is that when heart rate is faster than the ability of a cell to release and reuptake (i.e. cycle) calcium, alternans of calcium release will occur. For example, if an action potential is initiated before calcium returns to diastolic levels (i.e. before all calcium released can be reclaimed), then the amount of calcium available in the SR will be reduced. As a result, release on the subsequent beat will be less. Given a smaller release but the same time to reclaim calcium, more calcium will end up in the SR and result in a larger release on the next beat. Thus, the amount of released calcium can only be fully reclaimed on an alternating beat basis, giving rise to calcium alternans. The calcium transients shown earlier in Figure 4 (bottom) demonstrate this paradigm. Shown in Figure 6 is another example of calcium alternans that depends on the rate of calcium cycling, in particular its uptake by the SR. Calcium transients were measured near the epicardium (EPI) and endocardium (ENDO) during an abrupt decrease in pacing cycle length from 600 ms to 300 ms in the canine left ventricular wedge preparation using optical mapping techniques. Following an abrupt change in cycle length (arrow), calcium transient alternans near the endocardium and epicardium is evident. After approximately 4 seconds, steady state calcium alternans is greater near the endocardium compared to the epicardium. As evident from the decay phase of the calcium transient recorded just before the step change, the rate of calcium decline to diastolic levels is significantly slower (larger Tau) near the endocardium compared to the epicardium. Similar heterogeneities of alternans across the transmural wall have been reported by others and linked to SR calcium regulation. (Cordeiro et al., 2007)
Figure 6.

ECG and calcium transients near the epicardium (EPI) and endocardium (ENDO) recoded during an abrupt increase in pacing cycle length from 600 to 300 msec (arrow). Near ENDO and EPI, calcium transient alternans were persistent following 4 seconds of rapid pacing. At ENDO, where the decay of the calcium transient was slower (174 ms) compared to EPI (93 ms), the magnitude of calcium transient alternans was greater. At ENDO, the calcium transient amplitude for beat a was 68% larger than that for beat b. In contrast, at EPI the degree of calcium transient alternans was less obvious (9%). Reproduced with permission.
In addition to linking calcium transient alternans to the rate of calcium cycling, these results can reveal an important clue to the molecular mechanisms of calcium alternans. That is, there must be some intrinsic differences at regions that are alternans prone. To determine the molecular mechanisms of calcium alternans, we have used a targeted approach by investigated protein expression in tissue samples taken from alternans prone and alternans resistant sites. (Laurita et al., 2003; Wan et al., 2005) In the canine wedge preparation, protein expression of SERCA2a and NCX across the transmural wall was investigated. We found less expression of SERCA2a in the subendocardium compared to the subepicardium. These data are consistent with previous studies that have shown significantly less SERCA2a mRNA in normal canine subendocardium compared to the subepicardium (Igarashi-Saito et al., 1999) and a trend towards less SERCA2a expression near the endocardium in normal human hearts. (Prestle et al., 1999) In contrast, NCX (a proteolytic fragment at 70 kDa) appeared the same across all layers, a finding similar to that in humans. (Prestle et al., 1999). However, other reports of NCX protein expression and function across the canine transmural wall show a different result. Zygmunt et al (2000) reported an unequal distribution of NCX across the canine left ventricular wall, where peak reverse mode current is greatest in midmyocardial cells and weakest in endocardial cells. Xiong et al (2005) found greater NCX mRNA and protein in the epicardium compared to the midmyocardium and endocardium in normal canine tissue. In a separate series of experiments in guinea pig, we found that alternans prone regions (endocardium) express significantly less SERCA2a and RyR compared to alternans resistant regions (epicardium), suggesting a molecular basis for cellular alternans and specifically implicating calcium cycling proteins responsible for SR reuptake and release. Likewise, experiments performed in isolated myocytes subjected to pharmacological inhibition of either RyR or SERCA2a suggest that either of these calcium cycling proteins can cause calcium alternans. (Diaz et al., 2004; Hüser et al., 2000; Wan et al., 2005) Taken together, these data suggest that the molecular mechanism of calcium transient alternans is related to not only the reuptake of calcium into the SR (SERCA2a), but also calcium release from the SR (RyR).
6. Mechanisms of Calcium Transient Alternans: SR Calcium Release
Calcium alternans is also associated with SR calcium release, independent of the rate at which calcium is cycled. Diaz, et al (2004) used a voltage-clamp pulse protocol that was intentionally below the activation voltage for IL,Ca, producing a weak CICR response. As a result, most RyR channels will not open by CICR, but rather by calcium waves that will cause a spatially desynchronized calcium release. Moreover, these calcium waves occur only when SR calcium content is above a certain threshold. So, when an SR calcium release does occur, the SR will become depleted below this threshold such that on the subsequent voltage clamp pulse no release will occur. The absence of a release will leave enough calcium in the SR such that on the next voltage clamp pulse a release (albeit desynchronized) will occur, perpetuating the alternans of calcium release pattern. Under this condition, calcium transient alternans occurs at stimulation rates much slower than what is required when CICR is normal. Importantly, this protocol enhances the gain between SR calcium content and SR calcium release so that small alternans of in SR calcium content promote large alternans of SR calcium release. In a theoretical study, Shiferaw et al (2003) incorporated similar local dynamics of calcium release in a computer model and also found that alternans is dependent on a steep, non-linear, relationship between calcium release and SR content. These findings suggest that the SR calcium store is an important mechanism of alternans. (Sipido, 2004) This, however, has been a point of contention. In contrast, others have shown that calcium alternans does not depend solely on SR calcium content. Picht et al (2006) used direct measurements of intra-SR free calcium to show that pacing induced calcium alternans does not require SR calcium content alternans but, rather, may depend more on RyR recovery. (Dumitrescu et al., 2002)
In addition to artificial means of depressing SR calcium release, ischemia (Kurz et al., 1993; Mohabir et al., 1991; Wilson et al., 2006) and metabolic inhibition (Hüser et al., 2000; Kockskamper et al., 2002) have been associated with calcium transient alternans as well. In a series of studies, Qian et al (2003) have shown the development of significant alternans during the acute phases of ischemia. Interestingly, they found that during acute ischemia arrhythmogenic alternans occurred at a much slower heart rate than under normal conditions and that repolarization alternans occurred in the absence of conduction alternans. (Qian et al., 2003) This finding suggests that during ischemia the rate of calcium cycling may not be the mechanism of alternans; rather, it suggests RyR dysfunction as the cause. Local inhibition of glycolysis impairs RyR function and is also associated with calcium transient alternans in the absence of beat-to-beat fluctuations in SR content. (Hüser et al., 2000; Kockskamper et al., 2002) These studies highlight RyR channel dysfunction as a primary mechanism of calcium transient alternans, irrespective of SR calcium load. Taken together, it is clear that RyR dysfunction is an important mechanism of alternans, independent of the rate of calcium cycling. (Pieske et al., 2002) Finally, it is possible that other changes associated with abnormal RyR function can impact calcium transient alternans. For example, detubulation, (Louch et al., 2004) disruption of the regular organization of transverse tubules, (Song et al., 2006) and a leaky RyR. (Lehnart et al., 2006) may desynchronize calcium release in ventricular cells and promote the development of alternans.
7. Alternans Caused by Direct Mechano-Electrical Feedback (MEF)
In addition to the role of intracellular calcium regulation, it is also possible that direct MEF is an important mechanism of alternans. Murphy et al (1996; 1994) observed electrical alternans during simulated pulse alternans, which was achieved by clamping the aorta on every other beat. What’s interesting is that SR calcium release alternans was probably not occurring under this condition. Enhanced T-wave alternans has also been associated with acute volume overload in structurally normal hearts. (Narayan et al., 2007) These findings suggest that some form of MEF other than calcium alternans may be operating. For example, mechanosensitive ion channels, such as stretch activated channels (SAC), are able to significantly change membrane potential on a beat-to-beat basis and may be playing a role. (Kohl, 2001) It is also possible that alternating contraction strength is modulating the binding affinity of troponin C resulting in alternans of cytoplasmic free calcium. (Housmans et al., 1983) As mentioned earlier, beat-to-beat changes in free calcium are likely to affect several calcium sensitive ion channels and, thus, beat-to-beat APD. Despite these possibilities, the exact mechanisms by which mechanical alternans can directly affect electrical alternans is not well understood and worthy of further investigation. However, it is unlikely that direct MEF is the only important mechanism. For instance, several studies using optical mapping techniques, which often utilize contraction inhibition, have shown in the whole heart that electrical alternans can occur in the absence of mechanical alternans. (Banville et al., 2002; Pruvot et al., 2004)
8. Mechanical Dysfunction and Alternans
Given the close association between mechanical dysfunction and SCD, one may expect that alternans is an important link. Moreover, changes in calcium handling and protein expression associated with mechanical dysfunction promote alternans as well. Studies in isolated cells show diminished amplitudes and longer duration of calcium transients in heart failure (HF) compared to normal. (Beuckelmann et al., 1992; Gómez et al., 2001; Jiang et al., 2002; Kubo et al., 2001; O’Rourke et al., 1999) Likewise, reduced SERCA2a expression, (Davies et al., 1995; De La Bastie et al., 1990; Kihara et al., 1991; Tomaselli et al., 1999) as well as the expression of PLB(Flesch et al., 1996; Schwinger et al., 1995; Studer et al., 1994) and FKBP 12.6(Marx et al., 2000; Yano et al., 2000) are diminished in some models of HF. However, reduced expression of SERCA2a is not a finding common across all models. (Belevych et al., 2007) Upregulation of Na+-Ca++ exchanger is almost uniformly reported in human and animal models of HF. (Hasenfuss, 1998; Tomaselli et al., 1999) In HF, increased Na+-Ca++ exchanger may be a compensatory response to decreased SERCA2a expression or a leaky RyR in order to extrude calcium and improve diastolic function. (Hobai et al., 2000) The affect of increase Na+-Ca++ exchanger on alternans may be primarily through Ca-to-Vm coupling. That is, positive coupling of Ca-to-Vm is expected to be greater in HF compared to normal and, thus, could amplify repolarization alternans. In addition, faster stimulation rates result in decreased SR calcium reuptake in HF vs. normal hearts, indicating a decreased ability for calcium cycling to keep up with increasing heart rate. (Pieske et al., 1995) This, in addition to abnormalities of SR calcium release, may explain why alternans is increased in failing myocytes.
Despite a significant number of studies in isolated cells, there exists little information on calcium regulation in the intact falling heart. Using ratiometric optical mapping techniques, we have shown in the canine tachycardia induced HF model (Akar et al., 2003) that calcium transient amplitude is reduced compared to normal. Shown in Figure 7 are intracellular calcium transients recorded at a basic cycle length of 600 ms in normal (N) and failing (HF) wedge preparations. Panel A shows representative ratiometric calcium transients from normal and failing animals plotted on the same scale to directly compare amplitude. Calcium transient amplitude from the HF preparation is substantially reduced with respect to normal. In Panel B, representative ratiometric calcium transients are shown normalized to the same amplitude to compare differences in calcium transient duration at 50% decay (CaD50). CaD50 is prolonged in the HF preparations compared to normal. Summary data indicate that calcium transients recorded from the midmyocardium of failing preparations (n=12) are 21% smaller and 10% longer compared to normal (n=9). While these differences are statistically significant, surprisingly, they are not as great as one would expect based on studies in isolated cells.
Figure 7.

Shown are ratiometric calcium transients recorded from the transmural surface of a canine left ventricular wedge preparation isolated from a normal canine (N) and a canine with rapid pacing-induced heart failure (HF). In Panel A, both signals are plotted in absolute ratio units (RU) so that amplitude can be directly compared. However, in Panel B both signals are normalized to the same amplitude, to compare their duration at 50%. Scale bar shows time in ms and amplitude of transient in ratiometric units (RU).
In the same model of HF, we have found that the magnitude of alternans is increased compared to control wedge preparations. Shown in Figure 8 is the magnitude of APD alternans as a function of pacing rate in HF versus that from normal canine hearts. In Panel A, the magnitude of APD alternans is greater in HF compared to normal at any cycle length tested. As a result, the threshold pacing cycle length was significantly longer (400 ms) compared to normal (280 ms). Similar results were observed over several preparations (Panel B). The reason why APD alternans is elevated in HF compared to normal is not clear. This could be due dysfunctional release or reuptake, as manifest in the calcium transient signals. Enhanced susceptibility to alternans in HF has also been associated with abnormal RyR function. Lehnart et al (2006) has shown that FKBP12.6 deficient mice, which may be more likely to develop HF, are less prone to electrical alternans when pretreated with JTV519, a compound that increases the binding affinity of FKBP12.6 for RyR. However, the role FKBP12.6 plays in HF and arrhythmogenesis is controversial. (Xiao et al., 2004)
Figure 8.
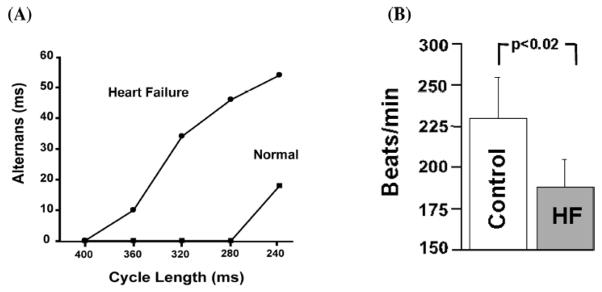
Panel A. Cycle length dependence of Vm-ALT recorded from midmyocardial cells using transmural optical mapping in normal and heart failure canine wedge preparation. Heart failure produced a profound leftward shift in Vm-ALT to cycle length relationship, demonstrating that heart failure myocytes have greater susceptibility to alternans. Panel B summarizes heart rate threshold for Vm- ALT (n = 7). Reproduced with permission.
9. Therapeutic Implications
It is very exciting to imagine the new therapeutic targets that calcium mediated alternans suggests. For example, accelerating SR uptake of calcium could suppress alternans and reduced the risk of SCD in patients with HF. Likewise, improving the efficiency of calcium release from the SR by restoring FKBP12.6 binding may also suppress alternans. (Lehnart et al., 2006) As an added advantage, these targets that can enhance calcium cycling may also improve contractile function. This is in stark contrast to targets that reside on the cell membrane (such as the L-type calcium channel) that when blocked can suppress alternans but may also reduce contraction, which could be unwanted side effect in patients with reduced contractile function. Moreover, pharmacological blockade of membrane K channels not only fails to ameliorate arrhythmias, but often provokes them (i.e. “proarrhythmic effect”). Therefore, new therapeutic targets such as calcium cycling proteins need to be evaluated both for their antiarrhythmic activity and proarrhythmic potential.
10. Conclusion
Understanding the complex feedback relationship between cardiac mechanical and electrophysiological dysfunction may prove imperative for elucidating the mechanisms of SCD. As outlined in this article, cardiac alternans is likely to be an important link. Cardiac alternans is also a unique link because it is a mechanism of abnormal impulse conduction (i.e. reentrant excitation), unlike more traditional calcium-mediated arrhythmias caused by abnormalities of impulse formation (i.e. triggered activity). Thus, calcium-mediated (or MEF) arrhythmias cover a broad range of mechanisms. Just as broad are the ways in which intracellular calcium dysregulation can cause cardiac alternans. There is significant support for the role of SR reuptake, SR release, and SR calcium load. The relative importance of each is likely to depend on the disease and species. Therefore, our knowledge of calcium dysregulation at the cellular and molecular level is essential; however, relating such abnormalities to arrhythmias that occur in the intact heart is equally important. In sum, it is not a coincidence that mechanical dysfunction remains as one of the best predictors of SCD. Therefore, a better appreciation and understanding of MEF-mediated arrhythmogenesis could lead to the identification of novel targets and therapy for treating patients at risk of SCD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
11. Reference List
- Aistrup GL, Kelly JE, Kapur S, Kowalczyk M, Sysman-Wolpin I, Kadish AH, Wasserstrom JA. Pacing-induced heterogeneities in intracellular Ca2+ signaling, cardiac alternans, and ventricular arrhythmias in intact rat heart. Circ Res. 2006;99:e65–73. doi: 10.1161/01.RES.0000244087.36230.bf. [DOI] [PubMed] [Google Scholar]
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–45. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- Banville I, Gray RA. Effect of action potential duration and conduction velocity restitution and their spatial dispersion on alternans and the stability of arrhythmias. J Cardiovasc Electrophysiol. 2002;13:1141–9. doi: 10.1046/j.1540-8167.2002.01141.x. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced Ryanodine Receptor-Mediated Calcium Leak Determines Reduced Sarcoplasmic Reticulum Calcium Content in Chronic Canine Heart Failure. Biophys J. 2007;93:4083–92. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Li L, Satoh H, McCall E. Factors that control sarcoplasmic reticulum calcium release in intact ventricular myocytes. Ann N Y Acad Sci. 1998;853:157–77. doi: 10.1111/j.1749-6632.1998.tb08264.x. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Erdmann E. Ca2+ - currents and intraellular [Ca2+] - transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol. 1992;87:235–243. doi: 10.1007/978-3-642-72474-9_19. [DOI] [PubMed] [Google Scholar]
- Bigger JT, Fleiss JL, Kleiger R, Miller JP, Rolnitzky LM. The relationship among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation. 1984;69:250–258. doi: 10.1161/01.cir.69.2.250. [DOI] [PubMed] [Google Scholar]
- Bogaert J, Rademakers FE. Regional nonuniformity of normal adult human left ventricle. Am J Physiol. 2001;280:H610–H620. doi: 10.1152/ajpheart.2001.280.2.H610. [DOI] [PubMed] [Google Scholar]
- Buxton AE, Marchlinski FE, Waxman HL, Flores BT, Cassidy DM, Josephson ME. Prognostic factors in nonsustained ventricular tachycardia. Am J Cardiol. 1984;53:1275–1279. doi: 10.1016/0002-9149(84)90078-x. [DOI] [PubMed] [Google Scholar]
- Chinushi M, Kozhevnikov D, Caref EB, Restivo M, El-Sherif N. Mechanism of discordant T wave alternans in the in vivo heart. J Cardiovasc Electrophysiol. 2003;14:632–638. doi: 10.1046/j.1540-8167.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- Chudin E, Goldhaber JI, Weiss J, Kogan B. Intracellular Ca2 dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JM, Malone JE, Di Diego JM, Scornik FS, Aistrup GL, Antzelevitch C, Wasserstrom JA. Cellular and subcellular alternans in the canine left ventricle. Am J Physiol Heart Circ Physiol. 2007;293:H3506–16. doi: 10.1152/ajpheart.00757.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- De La Bastie D, Levitsky D, Rappaport L, Mercadier JJ, Marotte F, Wisnewsky C, Brovkovich V, Schwartz K, Lompre AM. Function of the sarcoplasmic reticulum and expression of it’s Ca2 (+)-ATPase gene in pressure overload-induced cardiac hypertrophy in the rat. Circ Res. 1990;66:554–564. doi: 10.1161/01.res.66.2.554. [DOI] [PubMed] [Google Scholar]
- DeSantiago J, Maier LS, Bers DM. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol. 2002;34:975–984. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- Doi M, Yano M, Kobayashi S, Kohno M, Tokuhisa T, Okuda S, Suetsugu M, Hisamatsu Y, Ohkusa T, Kohno M, Matsuzaki M. Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation. 2002;105:1374–1379. doi: 10.1161/hc1102.105270. [DOI] [PubMed] [Google Scholar]
- Dumitrescu C, Narayan P, Efimov IR, Cheng Y, Radin MJ, McCune SA, Altschuld RA. Mechanical alternans and restitution in failing SHHF rat left ventricles. Am J Physiol Heart Circ Physiol. 2002;282:H1320–1326. doi: 10.1152/ajpheart.00466.2001. [DOI] [PubMed] [Google Scholar]
- El-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome - Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–89. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiring AJ, Rumberger JA, Reiter SJ, Collins SM, Skorton DJ, Rees M, Marcus ML. Sectional and segmental variability of left ventricular function: experimental and clinical studies using ultrafast computed tomography. J Am Coll Cardiol. 1988;12:415–25. doi: 10.1016/0735-1097(88)90415-9. [DOI] [PubMed] [Google Scholar]
- Flesch M, Schwinger RHG, Schiffer F, Frank K, Südkamp M, Kuhn-Regnier F, Arnold G, Böhm M. Evidence for functional relevance of an enhanced expression of the Na+-Ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- Garfinkel A, Kim YH, Voroshilovsky O, Qu ZL, Kil JR, Lee MH, Karagueuzian HS, Weiss JN, Chen PS. Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA. 2000;97:6061–6066. doi: 10.1073/pnas.090492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction - Altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279–289. doi: 10.1016/s0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- Hellerstein HK, Leibow IM. Electrical alternation in experimental coronary artery occlusion. Am J Physiol. 1950;160:366–374. doi: 10.1152/ajplegacy.1950.160.2.366. [DOI] [PubMed] [Google Scholar]
- Hobai IA, O’Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–8. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- Hoit BD, Tramuta DA, Kadambi VJ, Dash R, Ball N, Kranias EG, Walsh RA. Influence of transgenic overexpression of phospholamban on postextrasystolic potentiation. J Mol Cell Cardiol. 1999;31:2007–2015. doi: 10.1006/jmcc.1999.1031. [DOI] [PubMed] [Google Scholar]
- Housmans PR, Lee NK, Blinks JR. Active shortening retards the decline of the intracellular calcium transient in mammalian heart muscle. Science. 1983;221:159–161. doi: 10.1126/science.6857274. [DOI] [PubMed] [Google Scholar]
- Hu H, Sachs F. Stretch-activated ion channels in the heart. J Mol Cell Cardiol. 1997;29:1511–23. doi: 10.1006/jmcc.1997.0392. [DOI] [PubMed] [Google Scholar]
- Hua F, Johns DC, Gilmour RF., Jr Suppression of electrical alternans by overexpression of HERG in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2342–2351. doi: 10.1152/ajpheart.00793.2003. [DOI] [PubMed] [Google Scholar]
- Hüser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol (Lond) 2000;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi-Saito K, Tsutsui H, Takahashi M, Kinugawa S, Egashira K, Takeshita A. Endocardial versus epicardial differences of sarcoplasmic reticulum Ca2+-ATPase gene expression in the canine failing myocardium. Basic Res Cardiol. 1999;94:267–273. doi: 10.1007/s003950050152. [DOI] [PubMed] [Google Scholar]
- Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- Katra RP, Oya T, Hoeker GS, Laurita KR. Ryanodine receptor dysfunction and triggered activity in the heart. Am J Physiol Heart Circ Physiol. 2007;292:H2144–H2151. doi: 10.1152/ajpheart.00924.2006. [DOI] [PubMed] [Google Scholar]
- Katra RP, Pruvot E, Laurita KR. Intracellular calcium handling heterogeneities in intact guinea pig hearts. Am J Physiol Heart Circ Physiol. 2004;286:H648–H656. doi: 10.1152/ajpheart.00374.2003. [DOI] [PubMed] [Google Scholar]
- Kihara Y, Morgan JP. Abnormal Cai2+ handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am J Physiol. 1991;261:H1746–55. doi: 10.1152/ajpheart.1991.261.6.H1746. [DOI] [PubMed] [Google Scholar]
- Kockskamper J, Blatter LA. Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol. 2002;545:65–79. doi: 10.1113/jphysiol.2002.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl P, Bollensdorff C, Garny A. Effects of mechanosensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006;91:307–321. doi: 10.1113/expphysiol.2005.031062. [DOI] [PubMed] [Google Scholar]
- Kohl P, Sachs F. Mechanoelectric feedback in cardiac cells. Philosoph Trans Royal Soc Lond Series A Mathemat Phys Eng Scie. 2001;359:1173–1185. [Google Scholar]
- Kubo H, Margulies KB, Piacentino VI, Gaughan JP, Houser SR. Patients with end-stage congestive heart failure treated with ®-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation. 2001;104:1012–1018. doi: 10.1161/hc3401.095073. [DOI] [PubMed] [Google Scholar]
- Kurz RW, Mohabir R, Ren XL, Franz MR. Ischaemia induced alternans of action potential duration in the intact-heart: dependence on coronary flow, preload and cycle length. Eur Heart J. 1993;14:1410–1420. doi: 10.1093/eurheartj/14.10.1410. [DOI] [PubMed] [Google Scholar]
- Lab MJ. Mechanoelectric feedback (transduction) in heart: Concepts and implications. Cardiovasc Res. 1996;32:3–14. [PubMed] [Google Scholar]
- Laurita KR. Role of action potential duration restitution in arrhythmogenesis. J Cardiovasc Electrophysiol. 2004;15:464–465. doi: 10.1046/j.1540-8167.2004.03701.x. [DOI] [PubMed] [Google Scholar]
- Laurita KR, Katra R, Wible B, Wan X, Koo MH. Transmural heterogeneity of calcium handling in canine. Circ Res. 2003;92:668–675. doi: 10.1161/01.RES.0000062468.25308.27. [DOI] [PubMed] [Google Scholar]
- Laurita KR, Pastore JM, Rosenbaum DS. Mapping arrhythmia substrates related to repolarization: 1. Dispersion of repolarization. In: Rosenbaum DS, Jalife J, editors. Optical mapping of cardiac excitation and arrhythmias. New York, NY: Futura Publishing; 2001. [Google Scholar]
- Lee HC, Mohabir R, Smith N, Franz MR, Clusin WT. Effect of ischemia on calcium-dependent fluorescence transients in rabbit hearts containing Indo-1: Correlation with monophasic action potentials and contraction. Circulation. 1988;78:1047–1059. doi: 10.1161/01.cir.78.4.1047. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, Mancarella S, Coromilas J, Lederer WJ, Kass RS, Marks AR. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci U S A. 2006;103:7906–10. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis T. Notes upon alternation of the heart. Quart J Med. 1910;4:141–144. [Google Scholar]
- Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769–779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- Lu H, Lange G, Brooks C. Comparative studies of electrical and mechanical alternation in heart cells. J Electrocardiol. 1968;1:7–17. doi: 10.1016/s0022-0736(68)80004-4. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Györke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflugers Arch. 1996;432:1047–1054. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Wiesner TF, Gyorke S. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J Physiol. 1998;507 (Pt 3):667–677. doi: 10.1111/j.1469-7793.1998.667bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ Res. 2001;88:1151–8. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Mohabir R, Lee HC, Kurz RW, Clusin WT. Effects of ischemia and hypercarbic acidosis on myocyte calcium transients, contraction, and pHi in perfused rabbit hearts. Circ Res. 1991;69:1525–37. doi: 10.1161/01.res.69.6.1525. [DOI] [PubMed] [Google Scholar]
- Murphy CF, Horner SM, Dick DJ, Coen B, Lab MJ. Electrical alternans and the onset of rate-induced pulsus alternans during acute regional ischaemia in the anaesthetised pig heart. Cardiovasc Res. 1996;32:138–147. [PubMed] [Google Scholar]
- Murphy CF, Lab MJ, Horner SM, Dick DJ, Harrison FG. Regional electromechanical alternans in anesthetized pig hearts: modulation by mechanoelectric feedback. Am J Physiol. 1994;267:H1726–H1735. doi: 10.1152/ajpheart.1994.267.5.H1726. [DOI] [PubMed] [Google Scholar]
- Narayan SM, Drinan DD, Lackey RP, Edman CF. Acute volume overload elevates T-wave alternans magnitude. J Appl Physiol. 2007;102:1462–1468. doi: 10.1152/japplphysiol.00965.2006. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Kass DA, Tomaselli GF, Kääb S, Tunin R, Marbán E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I - Experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res. 2006;99:740–748. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- Pieske B, Kockskamper J. Alternans goes subcellular: a “disease” of the ryanodine receptor? Circ Res. 2002;91:553–555. doi: 10.1161/01.res.0000036862.37203.f4. [DOI] [PubMed] [Google Scholar]
- Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- Platt SB, Vijgen JM, Albrecht P, Van Hare GF, Carlson MD, Rosenbaum DS. Occult T wave alternans in long QT syndrome. J Cardiovasc Electrophysiol. 1996;7:144–148. doi: 10.1111/j.1540-8167.1996.tb00509.x. [DOI] [PubMed] [Google Scholar]
- Prestle J, Dieterich S, Preuss M, Bieligk U, Hasenfuss G. Heterogeneous transmural gene expression of calcium-handling proteins and natriuretic peptides in the failing human heart. Cardiovasc Res. 1999;43:323–31. doi: 10.1016/s0008-6363(99)00119-4. [DOI] [PubMed] [Google Scholar]
- Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- Qian YW, Sung RJ, Lin SF, Province R, Clusin WT. Spatial heterogeneity of action potential alternans during global ischemia in the rabbit heart. Am J Physiol Heart Circ Physiol. 2003;285:H2722–33. doi: 10.1152/ajpheart.00369.2003. [DOI] [PubMed] [Google Scholar]
- Räcke H-F, Koppers D, Lemke P, Casaretto H, Hauswirth O. Fosinoprilate prolongs the action potential: Reduction of iK and enhancement of the L-type calcium current in guinea pig ventricular myocytes. Cardiovasc Res. 1994;28:201–208. doi: 10.1093/cvr/28.2.201. [DOI] [PubMed] [Google Scholar]
- Ravens U, Dobrev D. Regulation of sarcoplasmic reticulum Ca2+-ATPase and phospholamban in the failing and nonfailing heart. Cardiovasc Res. 2000;45:245–252. doi: 10.1016/s0008-6363(99)00338-7. [DOI] [PubMed] [Google Scholar]
- Riccio ML, Koller ML, Gilmour RF., Jr Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res. 1999;84:955–963. doi: 10.1161/01.res.84.8.955. [DOI] [PubMed] [Google Scholar]
- Rosen MR. Cellular electrophysiology of digitalis toxicity. J Am Coll Cardiol. 1985;5:22A–34A. doi: 10.1016/s0735-1097(85)80460-5. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–241. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A. Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ Res. 2006;99:520–527. doi: 10.1161/01.RES.0000240542.03986.e7. [DOI] [PubMed] [Google Scholar]
- Schwinger RHG, Böhm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- Shiferaw Y, Sato D, Karma A. Coupled dynamics of voltage and calcium in paced cardiac cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:021903. doi: 10.1103/PhysRevE.71.021903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–3686. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR. Understanding cardiac alternans: the answer lies in the Ca2+ store. Circ Res. 2004;94:570–572. doi: 10.1161/01.RES.0000124606.14903.6F. [DOI] [PubMed] [Google Scholar]
- Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–10. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng HP. β-adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na-Ca exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Ter Keurs HE, Wakayama Y, Miura M, Shinozaki T, Stuyvers BD, Boyden PA, Landesberg A. Arrhythmogenic Ca(2+) release from cardiac myofilaments. Prog Biophys Mol Biol. 2006;90:151–171. doi: 10.1016/j.pbiomolbio.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–20. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Traube L. Ein Fall von Pulsus Bigeminus nebst Bemerkungen uber die Leberschwellungen bei Klappenfehlern und uber acute Leberatrophie. Berlin Klin Wochenschr. 1872;9:185–188. [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker ML, Wan X, Kirsch GE, Rosenbaum DS. Hysteresis effect implicates calcium cycling as a mechanism of repolarization alternans. Circulation. 2003;108:2704–9. doi: 10.1161/01.CIR.0000093276.10885.5B. [DOI] [PubMed] [Google Scholar]
- Wan X, Laurita KR, Pruvot E, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Watanabe MA, Fenton FH, Evans SJ, Hastings HM, Karma A. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol. 2001;12:196–206. doi: 10.1046/j.1540-8167.2001.00196.x. [DOI] [PubMed] [Google Scholar]
- Wehrens XHT, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. doi: 10.1161/01.RES.0000224540.97431.f0. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Wan X, Rosenbaum DS. Cellular alternans: a mechanism linking calcium cycling proteins to cardiac arrhythmogenesis. Ann N Y Acad Sci. 2006;1080:216–234. doi: 10.1196/annals.1380.018. [DOI] [PubMed] [Google Scholar]
- Windle JD. The incidence and prognostic value of the pulsus alternans in myocardial and arterial disease. Quart J Med. 1913;6:453–462. [Google Scholar]
- Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–55. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–95. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- Xiong W, Tian Y, DiSilvestre D, Tomaselli GF. Transmural heterogeneity of Na+-Ca2+ exchange: evidence for differential expression in normal and failing hearts. Circ Res. 2005;97:207–9. doi: 10.1161/01.RES.0000175935.08283.27. [DOI] [PubMed] [Google Scholar]
- Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- Zareba W, Moss AJ, Le Cessie S, Hall WJ. T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:1541–1546. doi: 10.1016/0735-1097(94)90653-x. [DOI] [PubMed] [Google Scholar]
- Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]