

Abstract
p73 is a p53 family transcription factor. Due to the presence in the 5′ flanking region of two promoters, there are two N-terminal variants, TAp73, which retains a fully active transactivation domain (TA), and ΔNp73, in which the N terminus is truncated. In addition, extensive 3′ splicing gives rise to at least seven distinctive isoforms; TAp73-selective knockout highlights its role as a regulator of cell death, senescence and tumor suppressor. ΔNp73-selective knockout, on the other hand, highlights anti-apoptotic function of ΔNp73 and its involvement in DNA damage response. In this work, we investigated the expression pattern of murine p73 C-terminal isoforms. By using a RT-PCR approach, we were able to detect mRNAs of all the C-terminal isoforms described in humans. We characterized their in vivo expression profile in mouse organs and in different mouse developmental stages. Finally, we investigated p73 C-terminal expression profile following DNA damage, ex vivo after primary cultures treatment and in vivo after systemic administration of cytotoxic compounds. Overall, our study first elucidates spatio-temporal expression of mouse p73 isoforms and provides novel insights on their expression-switch under triggered conditions.
Keywords: C-terminal isoforms, SAM domain, cancer, development, p73
Introduction
p73 is a p53-related transcription factor with fundamental roles in development,1-4 tumor suppression5-11 and senescence.12-23 Transcription from two different promoters on the TRp73 gene results in generation of TAp73 and ΔNp73 isoforms with opposing pro- and anti-apoptotic functions.24-28 Although p73 shares tumor-suppression functions with p53,29-44 it plays some very distinctive roles in development.45-47 Mice lacking p73 show neurodegeneration, defects in pheromone detection as well as chronic infection and inflammation that lead to a shorter lifespan.3 In vivo studies demonstrated that more than 70% of mice lacking TAp73 develop tumors.48 On the other hand, ΔNp73 isoforms are known to exhibit dominant-negative activity toward the tumor-suppressor functions of both TAp73 and p53 and also act as a negative regulator of DNA damage response.27,49-53 Besides, ΔNp73 interferes with many developmental programs, such as the myogenic differentiation program.54 Moreover, both TAp73 and ΔNp73 KO models show mild degenerative phenotypes, underlying the importance of p73 in brain development.48,55-60 This scenario becomes even more complex by focusing on the C terminus, where many splicing events occur, giving rise to at least seven different isoforms.61 p73α is the only one that contains a fully functional sterile alpha motif (SAM), which has been described as a putative protein-protein interaction domain.62-64 TAp73γ rises from alternative splicing at exon 11 and p73δ, missing exon 11, 12 and 13.65 Although p73γ retains all the exons coding for SAM domain, the splicing event at exon 11 produces a shift of the reading frame, leading to a premature STOP codon.65 Stimulation of human peripheral blood led to identification of two additional isoforms, p73ε and p73ζ, with p73ε lacking exon 11 and 13 and p73ζ excluding exons 11 and 12.66 Elucidation of p73ζ isoform clarifies that this splicing variant includes most of the SAM domain, although it misses a hydrophobic residue that seems to be fundamental for stability and consequent domain functionality.66 Similar observation was pointed out in this study, regarding the isoform p73ε. In this case, the deletion covers the first three amino acids of an α-helix, negatively influencing proper folding of the domain. Even if p73γ encodes for all the exons involved in the SAM domain, due to the splicing at exon 11, the open reading frame is different.65,67 Here, we investigated the tissue spatiotemporal expression profile of all p73 isoforms in mice and their expression switch under stressed conditions.
Results
Identification of mouse p73 C-terminal isoforms
Figure 1A reports a schematic representation of the alternative splicing occurring in the C-terminal region of human p73 gene. Based on this, we tried to understand whether all the isoforms identified in human were also present in the mouse. There are commercial antibodies available, sensitive enough to detect p73 and its N-terminal variants;68,69 however, these antibodies fail to discriminate C-terminal isoforms at endogenous levels in the mouse. For this reason, we monitored mRNA levels. Organs from adult (2-mo-old) C57Bl/6 mice were collected and RNA was extracted (Fig. 2). cDNA derived from kidney was then used for PCR in saturating conditions (40 cycles). PCR was performed by using forward and reverse primers designed, respectively, on exons 10 and 14. As an empty control we used RNase DNase-free water. We were able to detect all the isoforms, although the signal deriving from some of them was much weaker than others (Fig. 1B). To overcome this problem and to further prove the existence of all isoforms, we performed a PCR using isoform-specific primers designed at the specific exon-exon junctions. Through this strategy, we generated a specific PCR for each C-terminal splicing variant. As shown in Figure 1C, all the isoforms were easily detected. Identity of the isoforms was further confirmed by DNA sequencing on PCR products.
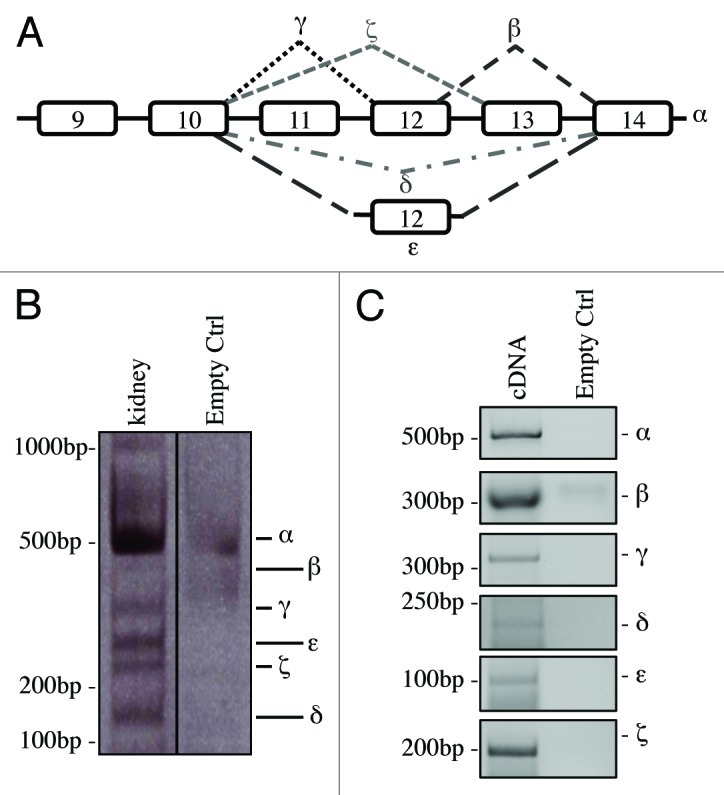
Figure 1. C-terminal isoforms of p73. (A) Schematic representation of splicing of human C-terminal p73. (B) RNA from kidney of adult mouse was reversed transcribed and cDNA was amplified by PCR. Product was run on 10% acrylamide gel. All the isoforms identified in human were detected and distinguished for different nucleotide length. (C) cDNA derived from an adult mouse was also amplified using isoforms-specific primers for each specific splicing variants. PCR products were run on agarose gel. All experiments have been repeated at least three times. Ctrl, control (DNase RNase-free H2O).
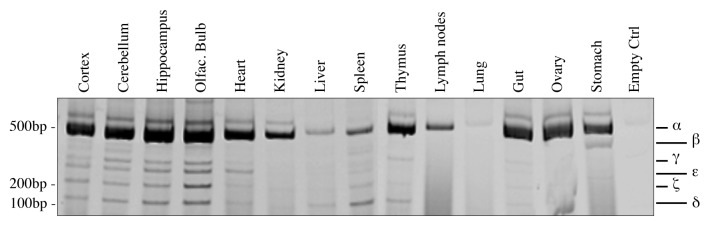
Figure 2. Organs-specific expression of p73 C-terminal isoforms. Screening of the isoform expression in organs of 2-mo-old mice. RT-PCR was performed using primers that amplify all isoforms (exons 10–14, mp73-X10 FWD and mp73-X14 REV). Samples were analyzed as in Figure 1B, and representative result is depicted. Experiments have been reproduced at least three times. Olfac. Bulb, olfactory bulb; ctrl, control.
Analysis of organ-specific expression of mouse p73 C-terminal isoforms
The expression of human p73 variants has been characterized in different tissues and cell lines.65,66,70,71 Since the expression pattern in the mouse has still not been investigated, we analyzed different organs using the same strategy used in Figure 1B and performing PCR in saturating conditions (40 cycles) in order to detect all possible isoforms. We found out that C-terminal variants of p73 are expressed in all the organs tested, even if at different levels, with p73α being the most abundant (Fig. 2). Since we detected also products at unexpected mobility shifts, we sequenced all of them, but we failed to identify brand new isoforms, while we confirmed presence of all the variants previously described in human.
Analysis of isoforms expression at different developmental stages
We then focused on expression upon different developmental stages, since p73 seems to be a key regulator in this process.60,72-75 We started from embryonic up to adult stages (2-mo-old). In this case, we performed a semi-quantitative RT-PCR (30 cycles). Quantification was done in relationship with starting levels (E12). Also in this system, p73α was the most abundant isoform, even if it did not undergo major changes, while p73ζ, but also to a smaller extent, p73ε and p73δ, were upregulated over time (Fig. 3A and B). P73γ, on the other hand, was downregulated during development (Fig. 3A and B). We also monitored levels of TAp73 (25 cycles) and ΔNp73 (30 cycles); we determined that TAp73 was more abundant than ΔNp73, even if, on the other hand, there was no significant regulation during development of any of the N-terminal variants (Fig. 3A and C). This type of analysis also revealed other migrating band at unexpected sizes (Fig. 3A), which were sequenced, but revealed to be not specific.

Figure 3. Expression of C-terminal isoforms during mouse development. Screening of the isoform expression during mouse development starting from embryonic stage E12, reaching adult (2 mo) age. Samples were analyzed as in Figure 1B and a representative result is depicted in (A). Semi-quantitative RT-PCR (30 cycles for C-terminal p73, 20 cycles for GAPDH) was performed and samples were run on a 10% acrylamide gel. Densitometry analysis was performed on at least three gels in order to quantify C-terminal isoforms levels (B) or TAp73 and ΔNp73 levels (C). Experiments have been repeated at least three times. E, embryonic stage; P7, seventh day after birth; TA, TAp73; Δn, ΔNp73; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Analysis of isoforms expression upon DNA damage in vitro
Since p73 is induced upon DNA damage and its loss confers resistance to cell death,48,76-78 we checked whether cytotoxic drug treatments, with cisplatin79,80 or etoposide,81-84 affected expression levels of C-terminal isoforms in the N2a cell line. In semi-quantitative RT-PCR, α-isoform results slightly increased, while p73β and p73ζ appeared strongly decreased (Fig. 4A–C). In another system instead, the expression levels of the isoforms varied slightly. Indeed, in spleen-derived primary splenocytes, p73γ and p73ε were the two most upregulated isoforms upon DNA damage, while p73ζ was downregulated, consistently with the results in N2a cells (Fig. 5A and B). In this scenario, we also monitored levels of N-terminal isoforms. TA levels at 24 h were lower than untreated cells, while ΔN levels were comparable between treated vs. untreated at 24 h. Levels of TA and ΔN were lower at 24 h than at 6 h, probably due to ongoing massive apoptotic events, as demonstrated by PARP cleavage85-89 (Fig. S1).
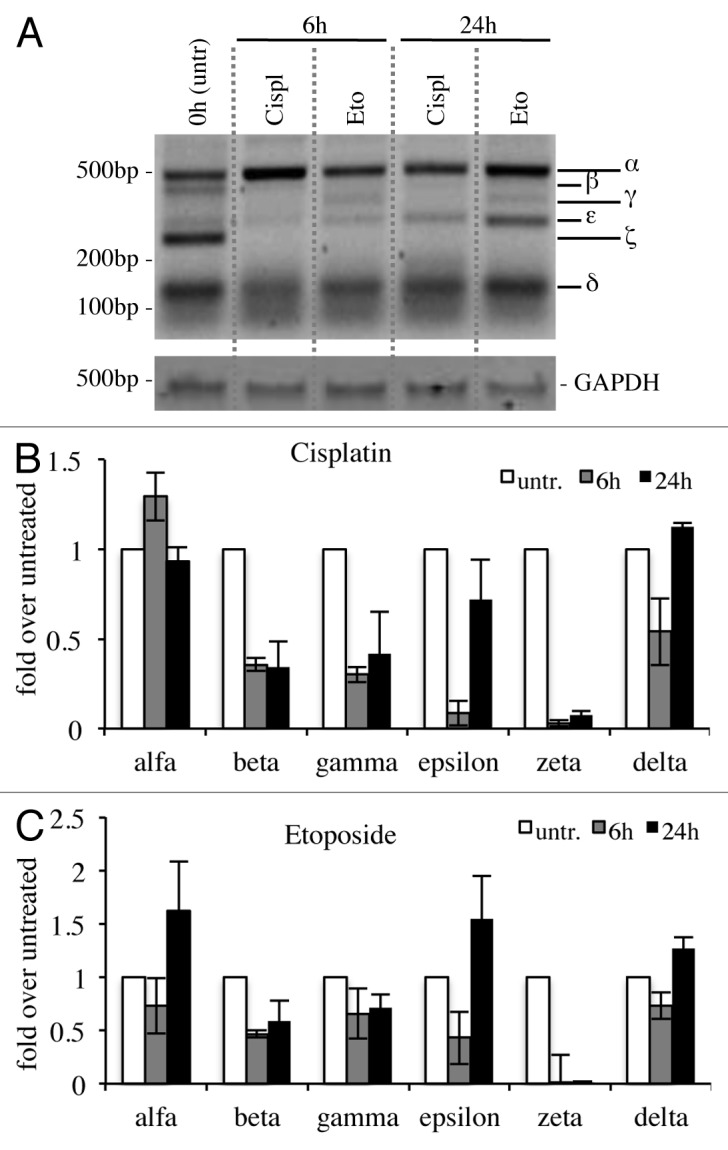
Figure 4. C-terminal isoform expression upon DNA damage in vitro. N2a (neuroblastoma cell line) were treated with 1 μg/ml etoposide or 5 μg/ml cisplatin, collected at the indicated time points and processed. Semi-quantitative RT-PCR (30 cycles for C-terminal p73, 20 cycles for GAPDH) was performed, and samples were run on a 10% acrylamide gel. A representative example (of at least three experiments) is depicted in (A). Densitometry analysis of at least three gels was achieved, relative to untreated cells, upon cisplatin treatment (B) or etoposide treatment (C). Cispl, cisplatin; Eto, etoposide; untr., untreated; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.

Figure 5. C-terminal isoforms expression upon DNA damage in primary splenocytes. Primary splenocytes were treated with 1 μg/ml etoposide or 5 μg/ml cisplatin, collected at the indicated time points and processed. Semi-quantitative RT-PCR (30 cycles for C-terminal p73, 20 cycles for GAPDH) was performed and samples were run on a 10% acrylamide gel. Representative result is depicted in (A). Densitometry analysis of at least three blots was achieved, relative to starting point (untreated, 6 h), to monitor C-terminal isoforms levels (B), TAp73 levels (C) and ΔNp73 levels (D) over time. Cispl, cisplatin; Eto, etoposide; untr., untreated; TA, TAp73; Δn, ΔNp73; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Analysis of isoforms expression upon DNA damage in vivo
Finally, we investigated the effects on levels of p73 C-terminal isoforms in vivo upon DNA damage. We treated adult mice intra-peritoneally and analyzed levels of C-terminal isoforms 20 h after treatment. We had different outcomes in relationship to the tissue analyzed, probably due to the ability of the drug to reach different organs but most likely also depending on starting endogenous levels of p73. In some organs, such as in the lung, all the isoforms were induced (Fig. 6A–C), while in other organs, there were no detectable effects of p73 levels, such as in the heart (data not shown). Moreover, in other tissues there were varying effects depending on the treatment that the animal received; in the spleen for example, etoposide was capable of inducing all isoforms (Fig. 6D and E), while cisplatin was causing a shift from α-isoform toward p73γ and p73δ (Fig. 6D and F).

Figure 6. C-terminal isoforms expression upon DNA damage in vivo. C57Bl/6 mice (2-mo-old, n ≥ 6 per group) were treated i.p. with 10 mg/kg etoposide or 5 mg/kg cisplatin or with PBS (control group). Animals were sacrificed after 20 h and tissues were then processed. Semi-quantitative RT-PCR (24 cycles for C-terminal p73, 20 cycles for GAPDH) was performed and samples were run on a 10% acrylamide gel. Example of results deriving from lung is depicted in (A). Densitometry analysis of at least three blots was achieved, showing levels of C-terminal isoforms upon etoposide (B) or cisplatin treatment (C). The same was done for the spleen (D) and quantification upon etoposide (E) or cisplatin (F) is shown. Cispl, cisplatin; Eto, etoposide; untr., untreated; TA, TAp73; Δn, ΔNp73; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
Here, we identified and characterized the tissue-specific expression of C-terminal isoforms of murine p73. This has been partly published regarding human p73, while investigation on its murine counterpart has been totally neglected. The isoform-specific KO mice models focused only on the characterization of p73 N terminus.48,55,56 These tools yield a lot of insight into understanding functions and roles of the specific N-terminal isoforms of p73; however, they leave some unsolved questions, since not all the defects displayed by the full p73−/− mouse3 were represented in one of these two models. For example, both TAp73−/− and ΔNp73−/− show mild neurological defects, while the full p73−/− displays a more penetrant phenotype; besides, the strong immunological defect found in the full p73−/− mice is absent in the TAp73−/− or ΔNp73−/−, leading to the conclusion that other factors might be involved. We may speculate that TAp73 and ΔNp73 could have overlapping functions that allow one isoform to overcome the absence of the other and vice versa. Moreover, C-terminal domains might play a role in this, since in the N-terminal KO models they were still expressed and functional in the remaining N-terminal isoform. As support to this theory, there is the presence of a sterile alpha motif (SAM) at the C terminus of p73. SAM domains are small putative protein-protein interaction domains,90 and in the mouse, this region overlaps exons 12–14 of p73;63 therefore, only α encodes a fully functional SAM domain and hypothetically could have an unique pool of interaction partners. Moreover, it has been shown that p73 SAM domain, but also the extreme C terminus, are able to regulate negatively the transcriptional activity of the protein,91,92 while on the other hand, deletion of SAM domain and extreme C terminus enhances transactivation and DNA-binding activity but inhibits apoptosis.92 Thus, it is striking that in all the tissues analyzed, we identified α as the most abundant. It could possibly have some involvement with control of proliferation, since presence of a fully functional SAM domain apparently inhibits it. This could be an incredibly interesting and still not described aspect of control of p73. In fact, the p73β, rather than α, variant has an interesting potential to transactivate target genes.65 This similarity retains from p63, where the C-terminal domains (TI and SAM) have been proved to act as dominant transcription repression modules.93-99 Many mutations found in the AEC syndrome have been shown to destabilize or modify the structure of one of the helices of this region, leading to a loss of function and a consequent deregulation in transactivation and growth suppression.100-104 These findings clearly state a connection between functionality of the SAM domain and AEC syndrome, opening possible new hints of investigation for its p73 homolog. In absence of triggers, the most preferred isoform transcribed could be p73α, due to its low transactivation potential, while upon a specific stimulus there could be a shift toward other isoforms, as we were able to highlight at developmental stages but also upon DNA damaging agents. Another interesting new aspect was highlighted by our work: no striking differences between TAp73 and ΔNp73 levels were detected during development or upon stresses. Instead C-terminal isoforms resulted to be tightly regulated; for example, during development, p73ε, p73ζ and to a lesser extent p73δ, were specifically, induced while γ was downregulated. Also, upon DNA damage in vitro and in vivo, we highlighted specific regulation of each isoform, suggesting that every C-terminal variant could play specific roles, possibly depending on the tissue or cell system analyzed. In line with this, interesting observations have been made on the C terminus of p63; in fact, mutations leading to premature stop codon in exon 14 of p63 are correlated with limb mammary syndrome and SHFM (split-hand-foot malformation).105-107 For these reasons, a further analysis should be done in order to clarify aspects regarding p73 functions correlated with its C terminus. Moreover, it is now becoming crucial to generate C-terminal isoform-specific KO models, which could also become powerful tools for studying potential human diseases correlated by p73 misfunctions, such as neurodegeneration108-113 and cancer.34,114-120
Our work also underlined the incredible necessity of developing an antibody with enough sensitivity to detect endogenous p73 C-terminal isoforms, as well as an antibody specific for the SAM domain. This would open a wide range of new directions, including screening for interaction partners, due to the putative function of the SAM. It would be intriguing to investigate influences of the SAM on tetramerization of p73, since this domain has been suggested to play a role in regulation of transcription through lipids interaction.121-123 Studying interactions with new partners, with powerful techniques such as TAP tag124,125 or MAPPIT,126,127 could lead to some clarifications of p73’s still unknown functions, related, for example, to strong defects in brain and chronic inflammation.
To conclude, this work identifies the C-terminal isoforms transcribed in the mouse, upon endogenous and challenging conditions. It underlines the extreme importance of studying these isoforms more in detail, since they could play a fundamental and still-not-investigated role in pathologies such as cancer, degeneration and development.
Materials and Methods
Cells cultures, primary cells and reagents
Cells were cultured at 37°C in 5% CO2 in culture medium. N2a were purchased from ATCC (#CCL-131) and maintained in a mix of 45% DMEM high glucose, 45% Optimem (Gibco) and 10% fetal bovine serum, 250 mM L-glutamine, 1 U/ml penicillin/streptomycin (all Gibco). Splenocytes were generated as already been described128 and cultured in RPMI 1640 medium (Gibco), supplemented with 10% FCS, 250 mM L-glutamine, 50 mM 2-mercaptoethanol, penicillin/streptomycin (1 U/ml), non-essential amino acids and 1 mM pyruvate (all Invitrogen).
Western blotting
Western blotting was performed as previously described.129 In brief, proteins were extracted with RIPA buffer containing cocktail inhibitors (Roche), and concentration was determined using a Bradford dye-based assay (Biorad). Total protein (30 μg) was subjected to SDS-PAGE followed by immunoblotting with appropriate antibodies at the recommended dilutions. The blots were then incubated with peroxidase-linked secondary antibodies followed by enhanced-chemiluminescent detection using Super Signal chemiluminescence kit (Thermo Scientific). Antibodies: mouse monoclonal anti PARP (1:1,000; Alexis), mouse monoclonal anti GAPDH (1:10,000; Sigma-Aldrich).
DNA damage in vivo
C57Bl/6 mice (2-mo-old) were injected i.p and sacrificed 20 h after treatment. Organs were collected and frozen on dry ice. Tissue homogenization was performed in 750 μl of TRIzol using a tissue grinder (Precellys). Mice were bred and subjected to listed procedures under the project license released from the United Kingdom Home Office.
RNA extraction, reverse transcription and PCR analysis
RNA was extracted using TRIzol (Invitrogen) and following manufacturer’s guidelines. After extraction, RNA was quantified with NanoDrop 2000 (Thermo Scientific) and 5 μg were treated with DNase I (Sigma) in order to eliminate DNA contamination. cDNA was reversed transcribed using RevertAid H Minus First Strand cDNA synthesys kit (Fermentas) and gene-specific primers (RT FWD and RT REV for C-terminal p73, GAPDH as internal control). Semi-quantitative PCR was performed using GoTaq DNA Polymerase (Promega) and the following cycle conditions: 5 min at 95°C; 30 sec at 95°C, 1 min at 58°C, 1 min at 72°C (24–40 cycles) and 10 min at 72°C; cycle number varied in relationship with the organ/cell type analyzed. PCR product was run on a 10% acrylamide gel (BioRad) and stained afterwards for 10 min in a 0.5 μg/ml ethidium bromide solution. Densitometry analysis was achieved using ImageJ software.
Primers
RT FWD 5′-GCTTGTGCCCCAGCCTTTG-3′
RT REV 5′-CCCCTCCAGATGGTCATACG-3′
mp73-X10 FWD 5′-GAGATCTTGATGAAAGTCAAGG-3′
mp73-X10–11 FWD 5′-CAGAGGCCGAGTCACCTG-3′
mp73-X10–12 FWD 5′-TACAGAGGCCGCTCCGGG-3′
mp73-X10–13 FWD 5′-CAGAGGCCTTTTTTGACAGGG-3′
mp73-X10–14 FWD 5′-CCTACAGAGGCCGACCTTGG-3′
mp73-X14 REV 5′-GCATTTCCGTGTGCGCCAC-3′
mp73-X12–14 REV 5′-GCCTCGTCAGGACCTTGGG-3′
mp73-X13–14 REV 5′-CCTGAAGCAGAGCCATGACTG-3′
mTAp73 FWD 5′-GCACCTACTTTGACCTCCCC-3′
mTAp73 REV 5′-GCACTGCTGAGCAAATTGAAC-3′
mDNp73 FWD 5′-ATGCTTTACGTCGGTGACCC-3′
mDNp73 REV 5′-GCACTGCTGAGCAAATTGAAC-3′
GAPDH FWD 5′-CAAGGTCATCCATGACAACTTTG-3′
GAPDH REV 5′-GTCCACCACCCTGTTGCTGTAG-3′
RT FWD and RT REV along with GAPDH FWD and REV were used to reverse transcribe cDNA. Primers mp73-X10 FWD and mp73-X14 REV were used to amplify by PCR all C-terminal isoforms. Primers mp73-X10–11 FWD and mp73-X13–14 REV were used to amplify p73α specifically. Primers mp73-X10–11 FWD and mp73-X12–14 REV were used to amplify p73β specifically. Primers mp73-X10–12 FWD and mp73-X13–14 REV were used to amplify p73γ specifically. Primers mp73-X10–12 FWD and mp73-X12–14 REV were used to amplify p73ε specifically. Primers mp73-X10–13 FWD and mp73-X14 REV were used to amplify p73ζ specifically. Primers mp73-X10–14 FWD and mp73-X14 REV were used to amplify p73δ specifically.
Supplementary Material
Acknowledgments
This work has been supported by the Medical Research Council, United Kingdom; MIUR, MinSan, RF73, RF57, ACC12; Odysseus Grant (G.0017.12) from the Flemish government and Flanders Institute for Biotechnology, Belgium.
Glossary
Abbreviations:
- TA
TAp73
- ΔN
ΔNp73
- SAM
sterile alfa motif
- eto
etoposide
- cispl
cisplatin
- untr
untreated
- PCR
polymerase chain reaction
- RT
reverse transcription
- FWD
forward
- REV
reverse
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PBS
phosphate buffered saline
- PARP
poly (ADP-ribose) polymerase
- ORF
open reading frame
- AEC
ankyloblepharon-ectodermal dysplasia-clefting
- SHFM
split-hand-foot malformation
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22787
References
- 1.Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259–65. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- 2.Talos F, Abraham A, Vaseva AV, Holembowski L, Tsirka SE, Scheel A, et al. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ. 2010;17:1816–29. doi: 10.1038/cdd.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 4.Kommagani R, Whitlatch A, Leonard MK, Kadakia MP. p73 is essential for vitamin D-mediated osteoblastic differentiation. Cell Death Differ. 2010;17:398–407. doi: 10.1038/cdd.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das SSK, Somasundaram K. Therapeutic potential of an adenovirus expressing p73 β, a p53 homologue, against human papilloma virus positive cervical cancer in vitro and in vivo. Cancer Biol Ther. 2006;5:210–7. doi: 10.4161/cbt.5.2.2402. [DOI] [PubMed] [Google Scholar]
- 6.Müller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH, Schilling T. One, two, three--p53, p63, p73 and chemosensitivity. Drug Resist Updat. 2006;9:288–306. doi: 10.1016/j.drup.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Graupner V, Alexander E, Overkamp T, Rothfuss O, De Laurenzi V, Gillissen BF, et al. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011;18:1130–9. doi: 10.1038/cdd.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.John K, Alla V, Meier C, Pützer BM. GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ. 2011;18:874–86. doi: 10.1038/cdd.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang M, Chiu SY, Hsu W. SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death Differ. 2011;18:1005–15. doi: 10.1038/cdd.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slatter TL, Ganesan P, Holzhauer C, Mehta R, Rubio C, Williams G, et al. p53-mediated apoptosis prevents the accumulation of progenitor B cells and B-cell tumors. Cell Death Differ. 2010;17:540–50. doi: 10.1038/cdd.2009.136. [DOI] [PubMed] [Google Scholar]
- 11.Knoll S, Fürst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, et al. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle. 2011;10:3554–65. doi: 10.4161/cc.10.20.17856. [DOI] [PubMed] [Google Scholar]
- 12.Klanrit P, Flinterman MB, Odell EW, Melino G, Killick R, Norris JS, et al. Specific isoforms of p73 control the induction of cell death induced by the viral proteins, E1A or apoptin. Cell Cycle. 2008;7:205–15. doi: 10.4161/cc.7.2.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramadan S, Terrinoni A, Catani MV, Sayan AE, Knight RA, Mueller M, et al. p73 induces apoptosis by different mechanisms. Biochem Biophys Res Commun. 2005;331:713–7. doi: 10.1016/j.bbrc.2005.03.156. [DOI] [PubMed] [Google Scholar]
- 14.Toh WH, Nam SY, Sabapathy K. An essential role for p73 in regulating mitotic cell death. Cell Death Differ. 2010;17:787–800. doi: 10.1038/cdd.2009.181. [DOI] [PubMed] [Google Scholar]
- 15.Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26:2009–14. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNF-α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19:274–83. doi: 10.1038/cdd.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY) 2010;2:748. doi: 10.18632/aging.100229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010;2:535–7. doi: 10.18632/aging.100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horikawa I, Fujita K, Harris CC. p53 governs telomere regulation feedback too, via TRF2. Aging (Albany NY) 2011;3:26–32. doi: 10.18632/aging.100271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulic V. Be quiet and you’ll keep young: does mTOR underlie p53 action in protecting against senescence by favoring quiescence? Aging (Albany NY) 2011;3:3–4. doi: 10.18632/aging.100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hulbert AJ. Longevity, lipids and C. elegans. Aging (Albany NY) 2011;3:81–2. doi: 10.18632/aging.100288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darzynkiewicz Z. Another “Janus paradox” of p53: induction of cell senescence versus quiescence. Aging (Albany NY) 2010;2:329–30. doi: 10.18632/aging.100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poyurovsky MV, Prives C. P53 and aging: A fresh look at an old paradigm. Aging (Albany NY) 2010;2:380–2. doi: 10.18632/aging.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oswald C, Stiewe T. In good times and bad: p73 in cancer. Cell Cycle. 2008;7:1726–31. doi: 10.4161/cc.7.12.6148. [DOI] [PubMed] [Google Scholar]
- 25.Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901–11. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- 26.Toh WH, Nam SY, Sabapathy K. An essential role for p73 in regulating mitotic cell death. Cell Death Differ. 2010;17:787–800. doi: 10.1038/cdd.2009.181. [DOI] [PubMed] [Google Scholar]
- 27.Schuster A, Schilling T, De Laurenzi V, Koch AF, Seitz S, Staib F, et al. ΔNp73β is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle. 2010;9:2629–39. doi: 10.4161/cc.9.13.12110. [DOI] [PubMed] [Google Scholar]
- 28.Ravni A, Tissir F, Goffinet AM. DeltaNp73 transcription factors modulate cell survival and tumor development. Cell Cycle. 2010;9:1523–7. doi: 10.4161/cc.9.8.11291. [DOI] [PubMed] [Google Scholar]
- 29.Spinnler C, Hedström E, Li H, de Lange J, Nikulenkov F, Teunisse AF, et al. Abrogation of Wip1 expression by RITA-activated p53 potentiates apoptosis induction via activation of ATM and inhibition of HdmX. Cell Death Differ. 2011;18:1736–45. doi: 10.1038/cdd.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trinh DL, Elwi AN, Kim SW. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget. 2010;1:396–404. doi: 10.18632/oncotarget.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, et al. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle. 2011;10:3346–58. doi: 10.4161/cc.10.19.17630. [DOI] [PubMed] [Google Scholar]
- 32.Bao W, Chen M, Zhao X, Kumar R, Spinnler C, Thullberg M, et al. PRIMA-1Met/APR-246 induces wild-type p53-dependent suppression of malignant melanoma tumor growth in 3D culture and in vivo. Cell Cycle. 2011;10:301–7. doi: 10.4161/cc.10.2.14538. [DOI] [PubMed] [Google Scholar]
- 33.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598–607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2010;2:924–35. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget. 2010;1:583–95. doi: 10.18632/oncotarget.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA, de Jong S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011;2:e148. doi: 10.1038/cddis.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hölzel M, Burger K, Mühl B, Orban M, Kellner M, Eick D. The tumor suppressor p53 connects ribosome biogenesis to cell cycle control: a double-edged sword. Oncotarget. 2010;1:43–7. doi: 10.18632/oncotarget.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoubala M, Murray-Zmijewski F, Khoury MP, Fernandes K, Perrier S, Bernard H, et al. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011;18:248–58. doi: 10.1038/cdd.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–57. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muppani N, Nyman U, Joseph B. TAp73alpha protects small cell lung carcinoma cells from caspase-2 induced mitochondrial mediated apoptotic cell death. Oncotarget. 2011;2:1145–54. doi: 10.18632/oncotarget.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget. 2011;2:654–6. doi: 10.18632/oncotarget.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steelman LS, Martelli AM, Nicoletti F, McCubrey JA. Exploiting p53 status to enhance effectiveness of chemotherapy by lowering associated toxicity. Oncotarget. 2011;2:109–12. doi: 10.18632/oncotarget.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sorscher SM, Hill AE, Belizaire R, Sorscher EJ. Spontaneous inactivating p53 mutations and the “selfish cell”. Aging (Albany NY) 2011;3:181. doi: 10.18632/aging.100294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane DP, Madhumalar A, Lee AP, Tay BH, Verma C, Brenner S, et al. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle. 2011;10:4272–9. doi: 10.4161/cc.10.24.18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zawacka-Pankau J, Kostecka A, Sznarkowska A, Hedström E, Kawiak A. p73 tumor suppressor protein: a close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle. 2010;9:720–8. doi: 10.4161/cc.9.4.10668. [DOI] [PubMed] [Google Scholar]
- 46.Tissir F, Goffinet AM. p73 and p63: Estranged relatives? Cell Cycle. 2011;10:1351. doi: 10.4161/cc.10.9.15383. [DOI] [PubMed] [Google Scholar]
- 47.Sayan AE, D’Angelo B, Sayan BS, Tucci P, Cimini A, Cerù MP, et al. p73 and p63 regulate the expression of fibroblast growth factor receptor 3. Biochem Biophys Res Commun. 2010;394:824–8. doi: 10.1016/j.bbrc.2010.03.084. [DOI] [PubMed] [Google Scholar]
- 48.Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–91. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grob TJ, Novak U, Maisse C, Barcaroli D, Lüthi AU, Pirnia F, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8:1213–23. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 50.Slade N, Zaika AI, Erster S, Moll UM. DeltaNp73 stabilises TAp73 proteins but compromises their function due to inhibitory hetero-oligomer formation. Cell Death Differ. 2004;11:357–60. doi: 10.1038/sj.cdd.4401335. [DOI] [PubMed] [Google Scholar]
- 51.Vernersson-Lindahl E, Mills AA. DeltaNp73beta puts the brakes on DNA repair. Genes Dev. 2010;24:517–20. doi: 10.1101/gad.1914210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niikura Y, Ogi H, Kikuchi K, Kitagawa K. BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD) Cell Death Differ. 2010;17:1011–24. doi: 10.1038/cdd.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bantel H, Simon HU. DeltaNp73beta is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle. 2010;9:2710–1. doi: 10.4161/cc.9.14.12592. [DOI] [PubMed] [Google Scholar]
- 54.Hüttinger-Kirchhof N, Cam H, Griesmann H, Hofmann L, Beitzinger M, Stiewe T. The p53 family inhibitor DeltaNp73 interferes with multiple developmental programs. Cell Death Differ. 2006;13:174–7. doi: 10.1038/sj.cdd.4401809. [DOI] [PubMed] [Google Scholar]
- 55.Tissir F, Ravni A, Achouri Y, Riethmacher D, Meyer G, Goffinet AM. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci USA. 2009;106:16871–6. doi: 10.1073/pnas.0903191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549–60. doi: 10.1101/gad.1873910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kon N, Zhong J, Kobayashi Y, Li M, Szabolcs M, Ludwig T, et al. Roles of HAUSP-mediated p53 regulation in central nervous system development. Cell Death Differ. 2011;18:1366–75. doi: 10.1038/cdd.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holembowski L, Schulz R, Talos F, Scheel A, Wolff S, Dobbelstein M, et al. While p73 is essential, p63 is completely dispensable for the development of the central nervous system. Cell Cycle. 2011;10:680–9. doi: 10.4161/cc.10.4.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agostini M, Tucci P, Steinert JR, Shalom-Feuerstein R, Rouleau M, Aberdam D, et al. microRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc Natl Acad Sci USA. 2011;108:21099–104. doi: 10.1073/pnas.1112063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agostini M, Tucci P, Killick R, Candi E, Sayan BS, Rivetti di Val Cervo P, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci USA. 2011;108:21093–8. doi: 10.1073/pnas.1112061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/S0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 62.Chi SW, Ayed A, Arrowsmith CH. Solution structure of a conserved C-terminal domain of p73 with structural homology to the SAM domain. EMBO J. 1999;18:4438–45. doi: 10.1093/emboj/18.16.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thanos CD, Bowie JU. p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Sci. 1999;8:1708–10. doi: 10.1110/ps.8.8.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arrowsmith CH. Structure and function in the p53 family. Cell Death Differ. 1999;6:1169–73. doi: 10.1038/sj.cdd.4400619. [DOI] [PubMed] [Google Scholar]
- 65.De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A, Falco M, Annicchiarico-Petruzzelli M, et al. Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med. 1998;188:1763–8. doi: 10.1084/jem.188.9.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Laurenzi VD, Catani MV, Terrinoni A, Corazzari M, Melino G, Costanzo A, et al. Additional complexity in p73: induction by mitogens in lymphoid cells and identification of two new splicing variants epsilon and zeta. Cell Death Differ. 1999;6:389–90. doi: 10.1038/sj.cdd.4400521. [DOI] [PubMed] [Google Scholar]
- 67.Coutandin D, Löhr F, Niesen FH, Ikeya T, Weber TA, Schäfer B, et al. Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death Differ. 2009;16:1582–9. doi: 10.1038/cdd.2009.139. [DOI] [PubMed] [Google Scholar]
- 68.Rosenbluth JM, Johnson K, Tang L, Triplett T, Pietenpol JA. Evaluation of p63 and p73 antibodies for cross-reactivity. Cell Cycle. 2009;8:3702–6. doi: 10.4161/cc.8.22.10036. [DOI] [PubMed] [Google Scholar]
- 69.Sayan AE, Paradisi A, Vojtesek B, Knight RA, Melino G, Candi E. New antibodies recognizing p73: comparison with commercial antibodies. Biochem Biophys Res Commun. 2005;330:186–93. doi: 10.1016/j.bbrc.2005.02.145. [DOI] [PubMed] [Google Scholar]
- 70.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr., Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–9. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 71.De Laurenzi V, Raschellá G, Barcaroli D, Annicchiarico-Petruzzelli M, Ranalli M, Catani MV, et al. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem. 2000;275:15226–31. doi: 10.1074/jbc.275.20.15226. [DOI] [PubMed] [Google Scholar]
- 72.Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012;3:e285. doi: 10.1038/cddis.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gonzalez-Cano L, Herreros-Villanueva M, Fernandez-Alonso R, Ayuso-Sacido A, Meyer G, Garcia-Verdugo JM, et al. p73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis. 2010;1:e109. doi: 10.1038/cddis.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saifudeen Z, Diavolitsis V, Stefkova J, Dipp S, Fan H, El-Dahr SS. Spatiotemporal switch from DeltaNp73 to TAp73 isoforms during nephrogenesis: impact on differentiation gene expression. J Biol Chem. 2005;280:23094–102. doi: 10.1074/jbc.M414575200. [DOI] [PubMed] [Google Scholar]
- 75.V. Dötsch FB D. Coutandin, E. Candi and G. Melino. p63 and p73, the Ancestors of p53. Cold Spring Harb Perspect Biol. 2010;00:a004887. doi: 10.1101/cshperspect.a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D, et al. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ. 2011;18:5–15. doi: 10.1038/cdd.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toh WH, Siddique MM, Boominathan L, Lin KW, Sabapathy K. c-Jun regulates the stability and activity of the p53 homologue, p73. J Biol Chem. 2004;279:44713–22. doi: 10.1074/jbc.M407672200. [DOI] [PubMed] [Google Scholar]
- 78.Seviour EG, Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging (Albany NY) 2010;2:900–7. doi: 10.18632/aging.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prendergast AM, Cruet-Hennequart S, Shaw G, Barry FP, Carty MP. Activation of DNA damage response pathways in human mesenchymal stem cells exposed to cisplatin or γ-irradiation. Cell Cycle. 2011;10:3768–77. doi: 10.4161/cc.10.21.17972. [DOI] [PubMed] [Google Scholar]
- 80.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 81.De Zio D, Bordi M, Tino E, Lanzuolo C, Ferraro E, Mora E, et al. The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage. Cell Death Differ. 2011;18:516–27. doi: 10.1038/cdd.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zalckvar E, Yosef N, Reef S, Ber Y, Rubinstein AD, Mor I, et al. A systems level strategy for analyzing the cell death network: implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 2010;17:1244–53. doi: 10.1038/cdd.2010.7. [DOI] [PubMed] [Google Scholar]
- 83.Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. 2010;35:556–64. doi: 10.1016/j.tibs.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 84.Meley D, Spiller DG, White MR, McDowell H, Pizer B, Sée V. p53-mediated delayed NF-κB activity enhances etoposide-induced cell death in medulloblastoma. Cell Death Dis. 2010;1:e41. doi: 10.1038/cddis.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng H, et al. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ. 2011;18:1702–10. doi: 10.1038/cdd.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boehler C, Dantzer F. PARP-3, a DNA-dependent PARP with emerging roles in double-strand break repair and mitotic progression. Cell Cycle. 2011;10:1023–4. doi: 10.4161/cc.10.7.15169. [DOI] [PubMed] [Google Scholar]
- 87.Straten P, Andersen MH. The anti-apoptotic members of the Bcl-2 family are attractive tumor-associated antigens. Oncotarget. 2010;1:239–45. doi: 10.18632/oncotarget.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Vuurden DG, Hulleman E, Meijer OL, Wedekind LE, Kool M, Witt H, et al. PARP inhibition sensitizes childhood high grade glioma, medulloblastoma and ependymoma to radiation. Oncotarget. 2011;2:984–96. doi: 10.18632/oncotarget.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cantó C, Auwerx J. Interference between PARPs and SIRT1: a novel approach to healthy ageing? Aging (Albany NY) 2011;3:543–7. doi: 10.18632/aging.100326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schultz J, Ponting CP, Hofmann K, Bork P. SAM as a protein interaction domain involved in developmental regulation. Protein Sci. 1997;6:249–53. doi: 10.1002/pro.5560060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu G, Chen X. The C-terminal sterile alpha motif and the extreme C terminus regulate the transcriptional activity of the alpha isoform of p73. J Biol Chem. 2005;280:20111–9. doi: 10.1074/jbc.M413889200. [DOI] [PubMed] [Google Scholar]
- 92.Ozaki T, Naka M, Takada N, Tada M, Sakiyama S, Nakagawara A. Deletion of the COOH-terminal region of p73alpha enhances both its transactivation function and DNA-binding activity but inhibits induction of apoptosis in mammalian cells. Cancer Res. 1999;59:5902–7. [PubMed] [Google Scholar]
- 93.Deutsch GB, Zielonka EM, Coutandin D, Weber TA, Schäfer B, Hannewald J, et al. DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell. 2011;144:566–76. doi: 10.1016/j.cell.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487–99. doi: 10.1038/cdd.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yalcin-Ozuysal O, Fiche M, Guitierrez M, Wagner KU, Raffoul W, Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010;17:1600–12. doi: 10.1038/cdd.2010.37. [DOI] [PubMed] [Google Scholar]
- 96.Huang Y, Ratovitski EA. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging (Albany NY) 2010;2:959–68. doi: 10.18632/aging.100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Straub WE, Weber TA, Schäfer B, Candi E, Durst F, Ou HD, et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis. 2010;1:e5. doi: 10.1038/cddis.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget. 2011;2:1203–17. doi: 10.18632/oncotarget.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lena AM, Cipollone R, Amelio I, Catani MV, Ramadan S, Browne G, et al. Skn-1a/Oct-11 and ΔNp63α exert antagonizing effects on human keratin expression. Biochem Biophys Res Commun. 2010;401:568–73. doi: 10.1016/j.bbrc.2010.09.102. [DOI] [PubMed] [Google Scholar]
- 100.Ghioni P, Bolognese F, Duijf PH, Van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol. 2002;22:8659–68. doi: 10.1128/MCB.22.24.8659-8668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, et al. ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 2011;18:887–96. doi: 10.1038/cdd.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Borrelli S, Candi E, Hu B, Dolfini D, Ravo M, Grober OM, et al. The p63 target HBP1 is required for skin differentiation and stratification. Cell Death Differ. 2010;17:1896–907. doi: 10.1038/cdd.2010.59. [DOI] [PubMed] [Google Scholar]
- 103.Talos F, Wolff S, Beyer U, Dobbelstein M, Moll UM. Brdm2 - an aberrant hypomorphic p63 allele. Cell Death Differ. 2010;17:184–6. doi: 10.1038/cdd.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leonard MK, Kommagani R, Payal V, Mayo LD, Shamma HN, Kadakia MP. ΔNp63α regulates keratinocyte proliferation by controlling PTEN expression and localization. Cell Death Differ. 2011;18:1924–33. doi: 10.1038/cdd.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001;69:481–92. doi: 10.1086/323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Forster N, Ellisen LW. Notch signaling mediates p63-induced quiescence: a new facet of p63/Notch crosstalk. Cell Cycle. 2011;10:3632–3. doi: 10.4161/cc.10.21.18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim DA, Lee BL, Suh EK. Ionizing radiation-induced TAp63α phosphorylation at C-terminal S/TQ motifs requires the N-terminal transactivation (TA) domain. Cell Cycle. 2011;10:840–9. doi: 10.4161/cc.10.5.15008. [DOI] [PubMed] [Google Scholar]
- 108.Lee J, Hwang YJ, Boo JH, Han D, Kwon OK, Todorova K, et al. Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ. 2011;18:1726–35. doi: 10.1038/cdd.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Penas C, Font-Nieves M, Forés J, Petegnief V, Planas A, Navarro X, et al. Autophagy, and BiP level decrease are early key events in retrograde degeneration of motoneurons. Cell Death Differ. 2011;18:1617–27. doi: 10.1038/cdd.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 111.Nisoli I, Chauvin JP, Napoletano F, Calamita P, Zanin V, Fanto M, et al. Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ. 2010;17:1577–87. doi: 10.1038/cdd.2010.31. [DOI] [PubMed] [Google Scholar]
- 112.Ciavardelli D, Silvestri E, Del Viscovo A, Bomba M, De Gregorio D, Moreno M, et al. Alterations of brain and cerebellar proteomes linked to Aβ and tau pathology in a female triple-transgenic murine model of Alzheimer’s disease. Cell Death Dis. 2010;1:e90. doi: 10.1038/cddis.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stanga S, Lanni C, Govoni S, Uberti D, D’Orazi G, Racchi M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: is HIPK2 the link? Aging (Albany NY) 2010;2:545–54. doi: 10.18632/aging.100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18:1441–9. doi: 10.1038/cdd.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–24. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ahmed A, Yang J, Maya-Mendoza A, Jackson DA, Ashcroft M. Pharmacological activation of a novel p53-dependent S-phase checkpoint involving CHK-1. Cell Death Dis. 2011;2:e160. doi: 10.1038/cddis.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smit MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging (Albany NY) 2010;2:735–41. doi: 10.18632/aging.100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R, et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011;18:1046–56. doi: 10.1038/cdd.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2010;2:445–52. doi: 10.18632/aging.100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Agostini M, Tucci P, Melino G. Cell death pathology: perspective for human diseases. Biochem Biophys Res Commun. 2011;414:451–5. doi: 10.1016/j.bbrc.2011.09.081. [DOI] [PubMed] [Google Scholar]
- 121.Rufini A, Agostini M, Grespi F, Tomasini R, Sayan BS, Niklison-Chirou MV, et al. p73 in Cancer. Genes Cancer. 2011;2:491–502. doi: 10.1177/1947601911408890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rufini S, Lena AM, Cadot B, Mele S, Amelio I, Terrinoni A, et al. The sterile alpha-motif (SAM) domain of p63 binds in vitro monoasialoganglioside (GM1) micelles. Biochem Pharmacol. 2011;82:1262–8. doi: 10.1016/j.bcp.2011.07.087. [DOI] [PubMed] [Google Scholar]
- 123.Barrera FN, Poveda JA, González-Ros JM, Neira JL. Binding of the C-terminal sterile alpha motif (SAM) domain of human p73 to lipid membranes. J Biol Chem. 2003;278:46878–85. doi: 10.1074/jbc.M307846200. [DOI] [PubMed] [Google Scholar]
- 124.Drakas R, Prisco M, Baserga R. A modified tandem affinity purification tag technique for the purification of protein complexes in mammalian cells. Proteomics. 2005;5:132–7. doi: 10.1002/pmic.200400919. [DOI] [PubMed] [Google Scholar]
- 125.Knuesel M, Wan Y, Xiao Z, Holinger E, Lowe N, Wang W, et al. Identification of novel protein-protein interactions using a versatile mammalian tandem affinity purification expression system. Mol Cell Proteomics. 2003;2:1225–33. doi: 10.1074/mcp.T300007-MCP200. [DOI] [PubMed] [Google Scholar]
- 126.Eyckerman S, Lemmens I, Lievens S, Van der Heyden J, Verhee A, Vandekerckhove J, et al. Design and use of a mammalian protein-protein interaction trap (MAPPIT) Sci STKE. 2002;2002:pl18. doi: 10.1126/stke.2002.162.pl18. [DOI] [PubMed] [Google Scholar]
- 127.Tavernier J, Eyckerman S, Lemmens I, Van der Heyden J, Vandekerckhove J, Van Ostade X. MAPPIT: a cytokine receptor-based two-hybrid method in mammalian cells. Clin Exp Allergy. 2002;32:1397–404. doi: 10.1046/j.1365-2745.2002.01520.x. [DOI] [PubMed] [Google Scholar]
- 128.Grespi F, Ottina E, Yannoutsos N, Geley S, Villunger A. Generation and evaluation of an IPTG-regulated version of Vav-gene promoter for mouse transgenesis. PLoS One. 2011;6:e18051. doi: 10.1371/journal.pone.0018051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Amelio I, Lena AM, Viticchiè G, Shalom-Feuerstein R, Terrinoni A, Dinsdale D, et al. miR-24 triggers epidermal differentiation by controlling actin adhesion and cell migration. J Cell Biol. 2012;199:347–63. doi: 10.1083/jcb.201203134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.