

Abstract
In this work, the site saturation mutagenesis of tyrosine 195, tyrosine 260 and glutamine 265 in the cyclodextrin glycosyltransferase (CGTase) from Paenibacillus macerans was conducted to improve the specificity of CGTase for maltodextrin, which can be used as a cheap and easily soluble glycosyl donor for the synthesis of 2-O-d-glucopyranosyl-l-ascorbic acid (AA-2G). Specifically, the site-saturation mutagenesis of three sites—tyrosine 195, tyrosine 260, and glutamine 265—was performed, and it was found that the resulting mutants (containing the mutations Y195S [tyrosine → serine], Y260R [tyrosine → arginine], and Q265K [glutamine → lysine]) produced higher AA-2G yields than the wild type and the other mutant CGTases when maltodextrin was used as the glycosyl donor. Furthermore, double and triple mutations were introduced, and four mutants (containing Y195S/Y260R, Y195S/Q265K, Y260R/Q265K, and Y260R/Q265K/Y195S) were obtained and evaluated for the capacity to produce AA-2G. The Y260R/Q265K/Y195S triple mutant produced the highest titer of AA-2G at 1.92 g/liter, which was 60% higher than that (1.20 g/liter) produced by the wild-type CGTase. The kinetics analysis of AA-2G synthesis by the mutant CGTases confirmed the enhanced maltodextrin specificity, and it was also found that compared with the wild-type CGTase, all seven mutants had lower cyclization activities and higher hydrolysis and disproportionation activities. Finally, the mechanism responsible for the enhanced substrate specificity was explored by structure modeling, which indicated that the enhancement of maltodextrin specificity may be related to the changes of hydrogen bonding interactions between the side chain of residue at the three positions (195, 260, and 265) and the substrate sugars. This work adds to our understanding of the synthesis of AA-2G and makes the Y260R/Q265K/Y195S mutant a good starting point for further development by protein engineering.
INTRODUCTION
Vitamin C (VC), usually existing in its reduced form, l-ascorbic acid (l-AA), is a well-known antioxidant used in various industries to maintain organoleptic quality and protect other components from oxidation. It also plays vital roles in collagen formation, carnitine synthesis, iron absorption, and prevention of chronic diseases (1–4). However, these applications are limited due to the instability of VC in oxidative environments, especially in the presence of heat, light, Cu2+, and ascorbate oxidase (5–7). Therefore, various VC derivatives have been synthesized to improve its oxidative stability (8–10). Among all VC derivatives, 2-O-d-glucopyranosyl-l-ascorbic acid (AA-2G) is considered superior, with its high degree of nonreducibility, excellent antioxidant ability, and ready release of l-AA and glucose (7).
AA-2G is synthesized by enzymatic transglycosylation using enzymes such as α-glucosidase (11), cyclodextrin glycosyltransferase (CGTase) (12–14), amylase (15), sucrose phosphorylase (16), and α-isomaltosyl glucosaccharide-forming enzyme (K. Mukai, K. Tsusaki, M. Kubota, S. Fukuda, and T. Miyake, European patent application 1553186). Among these enzymes, CGTase is considered the most suitable for the production of AA-2G due to its stability at high temperatures (17). AA-2G biosynthesis is catalyzed by CGTase via transglycosylation by which a glycosyl residue from a glycosyl donor is transferred to the C-2 position of VC with an α-1,2-linkage. Many saccharides, but not glucose, can be used as glycosyl donors (14). It has been demonstrated that α- and β-cyclodextrins are the best glycosyl donors for AA-2G production (14, 17, 18). In our previous work, the CGTase from Paenibacillus macerans was immobilized to produce AA-2G, and the highest yield (about 21 g/liter) was achieved with β-cyclodextrin as a glycosyl donor (13). However, due to the high cost of α-cyclodextrin and low solubility of β-cyclodextrin in aqueous solution, both of them are unsuitable for large-scale production of AA-2G (8). On the other hand, although the substrate specificity of CGTase for maltodextrin is much lower than those of α- and β-cyclodextrin (17, 18), maltodextrin has great potential in the industrial production of AA-2G due to its low cost and high solubility in aqueous solution. To achieve this objective, the binding specificity of CGTase for maltodextrin should be improved via protein engineering strategies.
CGTase belongs to the α-amylase family (glycosyl hydrolase family 13) and can catalyze four reactions: cyclization, coupling, disproportionation, and hydrolysis (19, 20). The crystal structure of Bacillus circulans strain 251 CGTase in complex with γ-cyclodextrin reveals the specific differences between the binding mode of linear and cyclic compounds (21). The hydrogen-bonding interactions between the residues and the sugars at the +2 and +3 subsites were different between the two binding modes, indicating that the amino acids around the +2 and +3 subsites played crucial roles in binding with the linear and cyclodextrin compounds (21). It was found that Tyr195, Phe259, and Glu264 (equivalent to Tyr195, Tyr260, and Gln265 in the CGTase from Paenibacillus macerans in this work) were very near the active sites and around the +2 and +3 substrate binding sites (22), and they may play crucial roles in substrate binding.
On the basis of the above structure analysis, in this work we selected tyrosine 195, tyrosine 260, and glutamine 265 in the CGTase from P. macerans as the site saturation mutagenesis sites to improve the binding capacity of CGTase with maltodextrin. Double and triple mutations were also constructed based on the single-site mutagenesis results. The transformation conditions (temperature and pH) were optimized to further improve the yield of AA-2G. In addition, the reaction kinetics of the CGTase mutants was investigated to confirm the enhanced maltodextrin specificity, and the influence of mutation on the cyclization, disproportionation, and hydrolysis activities of the CGTase was explored to clarify which reaction was mainly involved in AA-2G synthesis. Finally, the mechanism responsible for the increased maltodextrin specificity was unraveled at molecular level by modeling the complex structure of CGTase-maltononaose.
MATERIALS AND METHODS
Bacterial strains, plasmids, and materials.
P. macerans strain JFB05-01 (CCTCC M203062) was used as the source of genomic DNA. Escherichia coli JM109 was used as the host for plasmid construction. E. coli BL21(DE3) was used as the host for the expression of α-CGTase. The pMD19-T vector used for cgt gene cloning was purchased from TaKaRa (Dalian, China). The pET-20(+) vector was used for expression of α-CGTase in E. coli BL21(DE3).
PrimeSTAR HS DNA polymerase, restriction endonucleases, PCR reagents, the genomic extraction kit, and the MutanBEST kit used for site-directed mutagenesis were purchased from TaKaRa (Dalian, China). DNA sequencing was performed by Sangon (Shanghai, China). AA-2G was purchased from Wako Pure Chemical (Wako, Japan), and l-AA was purchased from Jiangshan Pharmaceutical (Jiangsu, China). Maltodextrin was purchased from Sangon (Shanghai, China). All other chemicals and reagents were of analytical grade.
Construction of the recombinant plasmid cgt/pET-20b(+).
The genomic DNA of P. macerans strain JFB05-01 was extracted with a genomic extraction kit and used as the DNA template for PCR. The primers were designed by Primer Premier 5 from the published cgt gene (GenBank accession no. AF047363.1). The BamHI and XhoI restriction sites (underlined) were introduced into the forward primer (5′-CGCGGATCCGTCACCCGATACGAGCGTGGACA-3′) and the reverse primer (5′-CCGCTCGAGATTTTGCCAGTCCACCGTCACC-3′), respectively. The gel-purified PCR product was digested with BamHI and XhoI and then ligated into the similarly restriction-digested expression vector pET-20b(+) to construct recombinant plasmid cgt/pET-20b(+), which contained the pelB signal peptide upstream and six histidine codons downstream. The plasmid containing the correct insert was confirmed by DNA sequencing and used for site-directed mutagenesis.
Site-directed mutagenesis.
Site-directed mutagenesis was performed with a MutanBEST kit. One-step PCR method was carried out by PrimeSTAR HS DNA polymerase using the plasmid cgt/pET-20b(+) as the template DNA. The following oligonucleotides were used to carry out site saturation mutagenesis of Tyr260, Gln265, and Tyr195: 5′-GGGGAATGGTATCTTGGCGCGG-3′ (Tyr260 forward primer), 5′-GAACGTAAATACCGGATGATCGCC-3′ (Tyr260 reverse primer), 5′-CGCGGATCAAACCGACGGAGA-3′ (Gln265 forward primer), 5′-CCAAGATACCATTCCCCGAACG-3′ (Gln265 reverse primer), 5′-TACAAGAACCTCTACGACCTGGC-3′ (Tyr195 forward primer), and 5′-AATACCGTCTTCAATCGTGGAAAAATC-3′ (Tyr195 reverse primer). The underlined nucleotides (corresponding to tyrosine 260, glutamine 265, and tyrosine 195) were replaced by the following amino acid codons: phenylalanine (TTC), leucine (CGT), isoleucine (ATC), methionine (ATG), valine (GTT), serine (TCT), proline (CCG), threonine (ACC), alanine (GCG), tyrosine (TAC), histidine (CAC), glutamine (CAG), aspartic acid (AAC), glutamic acid (GAA), cysteine (TGC), tryptophan (TGG), arginine (CGT), glycine (GGT), asparagine (AAC), and lysine (AAA). For the construction of double and triple mutants, the reaction product of the first PCR mutagenesis was used as the template for the next PCR. Then, all of the PCR products were treated with Blunting Kination Enzyme mix (TaKaRa) and Ligation Solution I, ligated into circular plasmids, and then transformed into E. coli JM109. These plasmids were confirmed by DNA sequencing and the correct ones were transformed into E. coli BL21(DE3) for expression.
Preparation and purification of CGTase and its mutants.
The mutant CGTases were prepared according to what previously reported (23). The recombinant E. coli BL21(DE3) were inoculated into 20 ml Luria-Bertani (LB) medium containing 100 μg/ml ampicillin and grown at 37°C overnight. Then the seed culture was inoculated into the flask with a ratio of 4% (vol/vol) for fermentation. The fermentation medium (pH 7.0) contained the following (g/liter): glucose, 8; lactose, 0.5; peptone 12; yeast extract, 24; K2HPO4, 16.43; KH2PO4, 2.31; CaCl2, 0.28; glycerol, 4; ampicillin, 0.1. The flask culture was incubated on a rotary shaker (200 rpm) at 25°C for 90 h. The expression of CGTases was induced with 0.01 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) when the optical density at 600 nm (OD600) reached 0.6. The broth was centrifuged at 10,000 ×g and 4°C for 5 min, and then the supernatant was purified and used for the subsequent transformation. The crude enzyme solution was purified on a nickel-nitrilotriacetic acid (Ni-NTA) agarose column (Qiagen, Chatsworth, CA) as described in the literature (24).
Biosynthesis and analysis of AA-2G.
The purified wild-type and mutant CGTases were diluted with acetic acid-sodium acetate buffer (pH 5.5) to a protein concentration of 1 mg/ml and then were mixed with the substrates (l-AA and maltodextrin), the final concentration of which was 5% (wt/vol). The mixture was incubated at 37°C for 24 h in the dark under oxygen-free conditions. Glucoamylase (10 U/ml) was finally added to the reaction mixture, which was incubated at 65°C and pH 5.5 for 24 h to hydrolyze the reaction intermediate AA-2Gn (where n is the number of glycosyl groups attached to l-AA) to AA-2G. AA-2G was analyzed using the method described previously (14). On the basis of the initial reaction conditions (temperature, 37°C; pH 5.5), the influences of reaction temperature (20, 28, 36, 44, and 52°C) and pH (acetic acid-sodium acetate buffer, pH 4.0, 4.5, 5.0, 5.5, and 6.0; phosphate buffer, pH 6.0, 6.5, 7.0, and 8.0) on the biosynthesis of AA-2G by the wild-type and mutant CGTases were also investigated.
The kinetic analysis of the wild-type and mutant CGTases for AA-2G biosynthesis (using l-AA and maltodextrin as substrates) was performed by measuring the amount of AA-2G with different concentrations of maltodextrin (0.23, 0.46, 1.16, 2.3, 11.6, and 23.2 mM) while the concentration of the other substrate, l-AA, was fixed (2.83, 5.67, 28.3, 56.7, and 141.5 mM), and the results were subjected to kinetic analysis using SigmaPlot (Jandel Scientific). The following equations were used to fit the experimental data to determine which kinetic mechanism applies to the transglycosylation reactions catalyzed by CGTase.
The ping-pong mechanism is represented by equation 1:
The substrate inhibition mechanism is represented by equation 2:
where v is the reaction rate (the amount of AA-2G formed by 1 mg enzyme per hour (mM · mg−1 · h−1), Vmax is the maximal reaction rate (mM · mg−1 · h−1), a and b are the donor (maltodextrin) and acceptor (l-AA) concentrations (mM), respectively, KmA and KmB are the affinity constants for the substrates maltodextrin and l-AA, respectively, and KiB is the inhibitor constant for the substrate l-AA.
The thermostability of the wild-type and mutant CGTases was determined by incubating the purified enzyme in acetic acid-sodium acetate buffer (pH 5.5) at 40°C. At regular intervals of 2 h, samples were taken to catalyze the formation of AA-2G. The half-life at pH 5.5 of the wild-type and mutant CGTases was determined by incubating the purified enzyme at 4°C. At regular intervals of 5 h, samples were taken to catalyze the formation of AA-2G.
Analysis of CGTase activity.
The α-cyclodextrin-forming activity was determined using the methyl orange method described by Li et al. (23). Namely, 0.1 ml of the purified wild-type or mutant CGTase diluted with 50 mM phosphate buffer to 1 mg/ml was added to 0.9 ml of 3% (wt/vol) soluble starch in 50 mM phosphate buffer (pH 6.0). After incubation at 40°C for 10 min, the reaction was terminated by the addition of 1.0 M HCl (1.0 ml). Finally, 1.0 ml of 0.1 mM methyl orange in 50 mM phosphate buffer (pH 6.0) was added, and the absorbance at 505 nm was measured after incubation at 16°C for 20 min. One unit of α-cyclodextrin-forming activity was defined as the amount of enzyme that was able to produce 1 μmol of α-cyclodextrin per minute.
The hydrolyzing activity was analyzed by the starch-degrading method (20). The purified CGTase (1 mg/ml) was incubated with 1% (wt/vol) soluble starch solution in 50 mM phosphate buffer at pH 6.5 and 50°C for 10 min. One unit of hydrolyzing activity was defined as the amount of enzyme producing 1 μmol of reducing sugar (determined by the 3,5-dinitrosalicylic acid method) per minute.
The disproportionation activity was determined as described previously (20), and the reaction mixture contained 6 mM 4-nitrophenyl-α-d-maltoheptaoside-4-6-O-ethylidene (EPS; Megazyme, County Wicklow, Ireland) as a donor substrate, 10 mM maltose as an acceptor substrate, and 1 mg/ml purified CGTase in 10 mM citrate buffer (pH 6.0). The mixtures were incubated for at 50°C for 10 min. One unit of activity was defined as the amount of enzyme converting 1 μmol of EPS per minute.
Protein concentrations were determined by the Bradford method with a Bradford protein assay kit and bovine serum albumin as a standard (Beyotime, Jiangsu, China).
Structure modeling of the wild-type and mutant CGTases.
The homology models of the wild-type and mutant CGTases from P. macerans were constructed using the crystallographic structure of B. circulans strain 251 CGTase (PDB accession code 1CXK) (22) as the template by the program suite MODELLER 9v2 (25). The models had 67.9% identity with the template, and the sequence alignment showed only one gap, at position 249. All graphical molecular representations were generated using Accelrys Discovery Studio Client 2.5. Structural alignment was done according to the combinatorial extension method by using the server http://cl.sdsc.edu/ (26). The stereochemical quality of the model was examined with PROCHECK (27), Verify3D (28), and ProQ (29), having 95% of the residues in the most favored and 0.2% in disallowed regions. The root mean square deviation (RMSD) between the template and model alpha carbon backbones was calculated with the combinatorial extension method (26). The overall structures of model and template were very similar (RMSD of 0.5 Å for 687 superimposed Cα). A maltononaose substrate was transferred from the active site of PDB 1CXK CGTase to the active site of the model. Finally, an energy minimization of the enzyme-substrate interaction was carried out with the Amber-based energy minimization method provided by the Accelrys Discovery Studio Client 2.5.
RESULTS AND DISCUSSION
Site-directed mutagenesis.
All of the mutants were successfully constructed by site-directed mutagenesis and verified by DNA sequencing. When the wild-type and mutant CGTases were expressed in the host E. coli BL21(DE3), it was found that there was no difference in the expression levels and molecular masses of the enzymes (data not shown). The crude CGTase solution was purified by one-step nickel affinity chromatography on Ni-NTA resin. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), it was found that there was no difference in the molecular mass (about 75 kDa) between the wild-type and mutant CGTases, and this result was similar to what was previously reported (23).
Synthesis of AA-2G by the wild-type and mutant CGTases.
The yield of AA-2G produced by the wild-type CGTase with maltodextrin as the glycosyl donor was 1.01 g/liter. The single-site saturation mutagenesis results showed that the Y260R (tyrosine → arginine), Q265K (glutamine → lysine), and Y195S (tyrosine → serine) mutants produced the highest AA-2G yields (1.44, 1.40, and 1.23 g/liter, respectively) among all mutants. Based on this result, the double and triple Y260R/Q265K, Y260R/195S, Q265K/Y195S, and Y260R/Q265K/Y195S mutants were constructed. As shown in Table 1, the AA-2G yields produced by the Y260R, Q265K, Y195S, Y260R/Q265K, Y260R/195S, Q265K/Y195S, and Y260R/Q265K/Y195S mutants were increased by 44, 40, 23, 55, 57, 49, and 59%, respectively, over that of the wild-type CGTase.
Table 1.
Comparison of the reaction activities, AA-2G yields, and stability of the wild-type and seven mutant CGTasesa
| CGTase | Relative activity (%)b |
AA-2G yield (g/liter)c |
t1/2 (h) at: |
|||
|---|---|---|---|---|---|---|
| Cyclization (α-cyclodextrin-forming activity) | Hydrolysis (starch-degrading activity) | Disproportionation | 40°C | pH 5.5 | ||
| Wild type | 100 | 100 | 100 | 1.01 ± 0.05 | 7.6 ± 0.4 | 29.8 ± 0.9 |
| Y260R | ND | 226 ± 10 | 138.0 ± 1.0 | 1.44 ± 0.03 | 8.0 ± 0.8 | 29.3 ± 0.7 |
| Q265K | 15 ± 0.5 | 236 ± 6 | 130.7 ± 0.8 | 1.40 ± 0.07 | 7.8 ± 0.7 | 33.6 ± 1.1 |
| Y195S | ND | 200 ± 8 | 102.6 ± 0.5 | 1.23 ± 0.04 | 8.8 ± 0.3 | 31.3 ± 0.8 |
| Y260R/Q265K | 8 ± 0.7 | 213 ± 8 | 129.2 ± 0.8 | 1.55 ± 0.05 | 8.5 ± 0.6 | 30.2 ± 0.9 |
| Y260R/195S | ND | 492 ± 7 | 142.3 ± 0.8 | 1.57 ± 0.07 | 7.9 ± 0.5 | 28.9 ± 0.6 |
| Q265K/Y195S | ND | 498 ± 9 | 140.1 ± 0.7 | 1.49 ± 0.08 | 7.7 ± 0.7 | 30.4 ± 1.0 |
| Y260R/Q265K/195S | 12 ± 0.9 | 557 ± 5 | 148.0 ± 0.6 | 1.59 ± 0.05 | 7.9 ± 0.4 | 31.7 ± 0.5 |
Each value represents the mean of triple independent measurements, and the deviation from the mean was below 5%. t1/2, half-life.
The cyclization, hydrolysis, and disproportionation reaction activities for wild-type CGTase was 165 ± 5, 8.3 ± 0.1, and 806 ± 8 U/mg, respectively, and were defined as 100% for the relative activity. ND, not detectable.
Maltodextrin was the glycosyl donor.
Influence of reaction temperature and pH on the enzymatic synthesis of AA-2G.
The optimal temperatures of the wild-type and mutant CGTases for AA-2G synthesis were 36°C, which was the same as that for α-CGTase-catalyzed AA-2G synthesis with β-cyclodextrin as the glycosyl donor (14) but different from that of the recombinant α-CGTase for the cyclization activity (45°C) (23). The AA-2G titer decreased significantly when the temperature was lower than 20°C, and more than 60% of the highest titer was retained within the range from 28 to 52°C.
Figure 1 shows the influence of pH on AA-2G synthesis by the wild-type and mutant CGTases. It could be seen that the optimal pH of the Y260R, Q265K, Y195S, Y260R/Y195S, and Q265K/Y195S mutants exhibited a shift to a certain extent compared to that of the wild-type CGTase. The highest AA-2G yield was obtained with the Y260R and Q265K mutants at pH 5.0, with the Y195S and Y260R/Y195S mutants at pH 5.5, and with the Q265K/Y195S mutant at pH 7.0. The optimal pH for the Y260R/Q265K and Y260R/Q265K/Y195S mutants for AA-2G synthesis was 6.5, which was the same as that of the wild-type CGTase. The previous work indicated that the optimal pH of this recombinant α-CGTase for cyclization and AA-2G biosynthesis with β-cyclodextrin as the glycosyl donor was 5.5 (14, 23).
Fig 1.
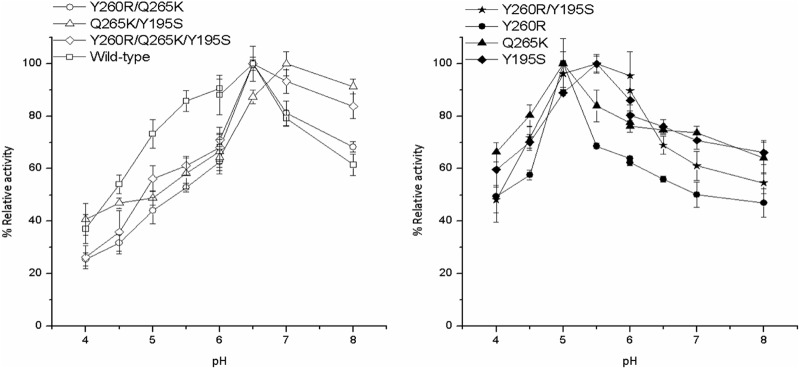
Influence of reaction pH on AA-2G synthesis by the wild-type and mutant CGTases with maltodextrin as the glycosyl donor. The pH 4.0 was 6.0 with acetic acid-sodium acetate buffer and 6.0 to 8.0 with phosphate buffer. The maximal AA-2G yield for the wild type and each mutant CGTase was defined as 100%.
Figure 2 shows the time profiles of AA-2G synthesis by the wild-type and mutant CGTases under the obtained optimal conditions. At the initial stage of the reaction, the amount of AA-2G increased dramatically. The Y260R mutant produced the highest AA-2G titer of 1.75 g/liter at 16 h. The yields of AA-2G by the Y260R/Q265K/Y195S, Q265K, Y260R/Q265K, and Q265K/Y195S mutants were highest, i.e., 1.92, 1.70, 1.85 and 1.80 g/liter, respectively, at 24 h. The yields of AA-2G produced by the wild-type, Y260R/Y195S and Y195S were highest, i.e., 1.20, 1.88 and 1.48 g/liter, respectively, at 20 h. The AA-2G yield produced by Y260R/Q265K/Y195S was 1.6-fold that produced by the wild-type CGTase.
Fig 2.
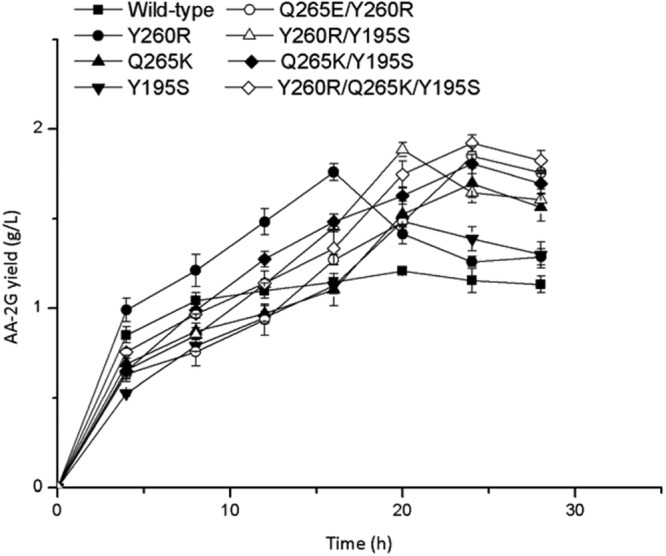
Time profiles of AA-2G synthesis by the wild-type and mutant CGTases with l-AA and maltodextrin as the substrates.
Influence of mutation on the reaction kinetics of CGTase.
The Lineweaver-Burk plots of the kinetics for the wild-type CGTase and Y260R are shown in Fig. S1 in the supplemental material. As shown in Fig. S1A, the parallel lines in the Lineweaver-Burk plot for the wild-type CGTase indicate a normal ping-pong type of kinetics, and the linear regression of the experimental data corresponds well to the values calculated by equation 1. However, for the Y260R mutant, the experimental data correspond well to the values calculated by equation 2 (see Fig. S1B in the supplemental material), which represents a substrate inhibition mechanism, indicating that too high a concentration of l-AA would inhibit the CGTase activity for AA-2G synthesis. In addition, the experimental data obtained with the other mutant CGTases were also best fitted by equation 2 (data not shown), and the detailed kinetic parameters are listed in Table 2. The maximal reaction rate (Vmax) of the mutant CGTases was higher than that of the wild-type CGTase. Meanwhile, compared to the wild-type CGTase, the Km of the Y260R, Q265K, Y195S, Y260R/Q265K, Y260R/195S, Q265K/Y195S, and Y260R/Q265K/Y195S mutants with maltodextrin as the substrate decreased by 20, 18, 3, 25, 26, 22, and 31%, respectively, while the Kcat/Km increased by 53, 43, 15, 77, 89, 63, and 126%, respectively. The kinetics results indicate that the affinity and catalytic efficiency of these mutants for maltodextrin increased compared to those of the wild-type CGTase. However, the Km values of the mutants for l-AA were somewhat increased compared to that of the wild-type CGTase, suggesting that the affinity of the mutants for l-AA decreased. Meanwhile, the catalytic efficiency of the mutants for l-AA slightly decreased, as indicated by the lower Kcat/Km(l-AA) values relative to the wild-type CGTase. The inhibitor constant Ki(l-AA) values indicated that the substrate inhibition by l-AA was most pronounced for the mutant Q265K, although this mutant did not show the highest affinity for l-AA.
Table 2.
Kinetic parameters of the wild-type and mutant CGTasesa
| Enzyme | Vmax (mM/mg/h) | Kcat (h−1) | Km (l-AA) (mM) | Kcat/Km (l-AA) (mM−1 h−1) | Km (maltodextrinb) (mM) | Kcat/Km (maltodextrin) (mM−1 h−1) | Ki (l-AA) (mM) |
|---|---|---|---|---|---|---|---|
| Wild type | 0.18 ± 0.03 | 13.50 | 38.32 ± 0.12 | 0.35 | 0.64 ± 0.03 | 21.09 | ND |
| Y260R | 0.22 ± 0.05 | 16.50 | 43.44 ± 0.25 | 0.38 | 0.51 ± 0.02 | 32.35 | 200 |
| Q265K | 0.21 ± 0.06 | 15.75 | 44.32 ± 0.43 | 0.36 | 0.52 ± 0.04 | 30.28 | 196 |
| Y195S | 0.20 ± 0.02 | 15.00 | 48.47 ± 0.38 | 0.31 | 0.62 ± 0.05 | 24.19 | 320 |
| Y260R/Q265K | 0.24 ± 0.03 | 18.00 | 44.52 ± 0.64 | 0.40 | 0.48 ± 0.07 | 37.50 | 221 |
| Y260R/Y195S | 0.25 ± 0.04 | 18.75 | 48.84 ± 0.35 | 0.38 | 0.53 ± 0.05 | 35.38 | 402 |
| Q265K/Y195S | 0.23 ± 0.05 | 17.25 | 47.21 ± 0.52 | 0.36 | 0.50 ± 0.02 | 34.5 | 286 |
| Y260R/Q265K/Y195S | 0.28 ± 0.03 | 21.00 | 51.07 ± 0.18 | 0.41 | 0.47 ± 0.08 | 44.68 | 394 |
Each value represents the mean of three independent measurements, and the deviation from the mean was below 5%.
The average molecular mass of maltodextrin was 2,150.68 Da.
The influence of mutations on the cyclization, hydrolysis, and disproportionation activities of the CGTase was investigated. As shown in Table 1, the wild-type CGTase had a much higher relative cyclization (α-cyclodextrin-forming) activity than all the mutants, and the Y260R, Y195S, Y260R/Y195S, and Q265K/Y195S mutants nearly lost the ability to cyclize. Compared with the wild-type CGTase, the Y260R, Q265K, Y260R/Q265K, Y260R/195S, Q265K/Y195S, and Y260R/Q265K/Y195S mutants showed increases in hydrolysis (starch-degrading) activity of 1.26-, 1.36-, 1.00-, 1.13-, 3.92-, 3.98-, and 4.57-fold, respectively. Except for the Y195S mutant, which had the same disproportionation activity as the wild-type CGTase, the mutants (Y260R, Q265K, Y260R/Q265K, Y260R/195S, Q265K/Y195S, and Y260R/Q265K/Y195S) showed increases in disproportionation activity of 38, 30.7, 29.2, 42.3, 40.1, and 48%, respectively. The change trend for the disproportionation activity of the mutants was in accordance with the increase in AA-2G yield, indicating that the disproportionation of CGTase was the main reaction for AA-2G biosynthesis with linear oligosaccharides as glycosyl donors.
Figure S2A to C in the supplemental material shows the replacement of tyrosine 260 and glutamine 265 by arginine and lysine, respectively. There were great differences in side chain length between the wild-type and mutant CGTases. Thus, the position of the hydrogen bond and polarity between the amino acid residues at positions 260 and 265 and the substrate sugars might be different for the Y260R and Q265K mutants. The possible increased hydrogen bond might make the substrate or intermediate more bent toward the 260 position, thereby blocking the formation of cyclodextrin from a linear substrate. This might explain why the cyclization of the Y260R and Q264K mutants was sharply reduced. Similarly, due to the possible presence of hydrogen-bonding interactions, the affinity between the enzyme and the linear substrate (such as maltodextrin) increased, and thus the AA-2G yield formed by the Y260R and Q265K mutants was higher than that of the wild-type CGTase. The increases in Km and Kcat/K[r]m verified this point. The Y260R/Q265K mutant produced more AA-2G because its ability to bind a linear substrate (such as maltodextrin) was higher than that of the Y260R and Q265K mutants. In addition, the introduction of hydrophilic residues near the catalytic site would reduce the hydrophobic character of the active site, which might favor the presence of water molecules, which could react with the intermediate (30). Therefore, the hydrolyzing activities of the mutants (Y260R and Q265K) increased significantly compared to that of the wild-type CGTase.
Figure S2D in the supplemental material shows a close-up view of the Y195S mutant with the substrate maltononaose. When the tyrosine at 195 is replaced by serine, two hydrogen-bonding interactions with the sugar substrate might be removed due to the short side chain. Therefore, this mutant could not form an inclusion complex with a spiral substrate and hindered the formation of α-cyclodextrin (31). Meanwhile, due to the possible decrease in hydrogen bonding with the substrate sugars, the ability of the mutant Y195S to bind cyclodextrin decreased, while the effect of linear oligosaccharide was unapparent (31). However, when the Ser195 participated in action with either Arg260 or Lys265 or both of them, as shown in Fig. S3 in the supplemental material, the removal of the hydrogen-bonding interactions between residue 195 and the substrate sugars resulted in a decrease in cyclodextrin binding specificity (see Fig. S3B), and therefore, more linear substrates were easily able to bind with Arg260 and Lys265 via hydrogen-bonding interactions (see Fig. S3A). This might explain why, for the Y260R/Y195S, Q265K/Y195S, and Y260R/Q265K/Y195S mutants, the substrate specificity for maltodextrin clearly increased, although the affinity of the Y195S mutant for maltodextrin did not observably change (Table 2).
The thermal stabilities of the wild-type and mutant CGTases are shown in Table 1. The half-life at 40°C of all the mutants was almost the same as that of the wild-type CGTase, indicating that the mutation did not affect the thermal stability of CGTase significantly. Table 1 also shows that the half-lives of the wild-type and mutant CGTases at pH 5.5 were about 30 h, suggesting that these enzymes had a strong acid resistance, which favored AA-2G biosynthesis, because l-AA is easily oxidized in neutral and alkaline environments (7).
In summary, seven CGTase mutants with enhanced maltodextrin specificity were obtained by site-saturation mutagenesis strategies, and the AA-2G yield was significantly improved when these mutant CGTases were used as biocatalysts. The enhanced specificity for maltodextrin was confirmed by reactions kinetics, and the inherent mechanism was revealed by structure modeling. The results produced a deeper understanding of the interaction of +2 and +3 subsites of CGTase with the linear sugars. It should be noted that the AA-2G yield produced with maltodextrin as the glycosyl donor was much lower than that with α- and β-cyclodextrins as the glycosyl donors (14), and much work, including the systems engineering of the other substrate binding sites of CGTase, enzyme immobilization, and transformation optimization, should be conducted to further improve the AA-2G yield.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the Key Technologies R & D Program of Jiangsu Province, China (SBE201170459), Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (111-2-06), National High Technology Research and Development Program of China (863 Program, 2011AA100905), and 973 Program (2012CB720802 and 2012CB720806).
Footnotes
Published ahead of print 16 November 2012
Supplemental material for this article may be found at 10.1128/AEM.02883-12.
REFERENCES
- 1. Englard S, Seifter S. 1986. The biochemical functions of ascorbic acid. Annu. Rev. Nutr. 6:365–406 [DOI] [PubMed] [Google Scholar]
- 2. Fujiwara S, Kakihara H, Sakaguchi K, Imanaka T. 1992. Analysis of mutations in cyclodextrin glucanotransferase from Bacillus stearothermophilus which affect cyclization characteristics and thermostability. J. Bacteriol. 174:7478–7481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacob RA, Sotoudeh G. 2002. Vitamin C function and status in chronic disease. Nutr. Clin. Care. 5:66–74 [DOI] [PubMed] [Google Scholar]
- 4. Naidu KA. 2003. Vitamin C in human health and disease is still a mystery? An overview. Nutr. J. 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li ZF, Zhang JY, Wang M, Gu ZB, Du GC, Li JK, Wu J, Chen J. 2009. Mutations at subsite-3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing alpha-cyclodextrin specificity. Appl. Microbiol. Biotechnol. 83:483–490 [DOI] [PubMed] [Google Scholar]
- 6. Penninga D, Strokopytov B, Rozeboom HJ, Lawson CL, Dijkstra BW, Bergsma J, Dijkhuizen L. 1995. Site-directed mutations in tyrosine 195 of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 affect activity and product specificity. Biochemistry 34:3368–3376 [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto I, Muto N, Murakami K, Suga S, Yamaguchi H. 1990. l-Ascorbic acid alpha-glucoside formed by regioselective transglucosylation with rat intestinal and rice seed alpha-glucosidases: its improved stability and structure determination. Chem. Pharm. Bull. (Tokyo) 38:3020–3023 [DOI] [PubMed] [Google Scholar]
- 8. Han R, Liu L, Li J, Du G, Chen J. 2012. Functions, applications and production of 2-O-d-glucopyranosyl-l-ascorbic acid. Appl. Microbiol. Biotechnol. 95:313–320 [DOI] [PubMed] [Google Scholar]
- 9. Sin KA, Nakamura A, Masaki H, Matsuura Y, Uozumi T. 1994. Replacement of an amino acid residue of cyclodextrin glucanotransferase of Bacillus ohbensis doubles the production of gamma-cyclodextrin. J. Biotechnol. 32:283–288 [DOI] [PubMed] [Google Scholar]
- 10. van der Veen BA, Uitdehaag JC, Penninga D, van Alebeek GJ, Smith LM, Dijkstra BW, Dijkhuizen L. 2000. Rational design of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 to increase alpha-cyclodextrin production. J. Mol. Biol. 296:1027–1038 [DOI] [PubMed] [Google Scholar]
- 11. Muto N, Suga S, Fujii K, Goto K, Yamamoto I. 1990. Formation of a stable ascorbic acid 2-glucoside by specific transglucosylation with rice seed α-glucosidase. Agric Biol. Chem. 54:1697–1703 [Google Scholar]
- 12. Aga H, Yoneyama M, Sakai S, Yamamoto I. 1991. Synthesis of 2-O-α-d-glucopyranosyl L-ascorbic acid by cyclomaltodextrin glucanotransferase from Bacillus stearothermophilus. Agric. Biol. Chem. 55:1751–1756 [Google Scholar]
- 13. Zhang ZC, Li JH, Liu L, Sun J, Hua ZZ, Du GC, Chen JA. 2011. Enzymatic transformation of 2-O-alpha-d-glucopyranosyl-l-ascorbic acid (AA-2G) by immobilized alpha-cyclodextrin glucanotransferase from recombinant Escherichia coli. J. Mol. Catal. B Enzym. 68:223–229 [Google Scholar]
- 14. Zhang ZC, Li JH, Liu L, Sun J, Hua ZZ, Du GC, Chen JA. 2011. Enzymatic transformation of 2-O-alpha-d-glucopyranosyl-l-ascorbic acid by alpha-cyclodextrin glucanotransferase from recombinant Escherichia coli. Biotechnol. Bioprocess Eng. 16:107–113 [Google Scholar]
- 15. Lee SB, Nam KC, Lee SJ, Lee JH, Inouye K, Park KH. 2004. Antioxidative effects of glycosyl-ascorbic acids synthesized by maltogenic amylase to reduce lipid oxidation and volatiles production in cooked chicken meat. Biosci. Biotechnol. Biochem. 68:36–43 [DOI] [PubMed] [Google Scholar]
- 16. Kwon T, Kim CT, Lee JH. 2007. Transglucosylation of ascorbic acid to ascorbic acid 2-glucoside by a recombinant sucrose phosphorylase from Bifidobacterium longum. Biotechnol. Lett. 29:611–615 [DOI] [PubMed] [Google Scholar]
- 17. Jun HK, Bae KM, Kim SK. 2001. Production of 2-O-alpha-d-glucopyranosyl L-ascorbic acid using cyclodextrin glucanotransferase from Paenibacillus sp. Biotechnol. Lett. 23:1793–1797 [Google Scholar]
- 18. Tanaka M, Muto N, Yamamoto I. 1991. Characterization of Bacillus stearothermophilus cyclodextrin glucanotransferase in ascorbic acid 2-O-alpha-glucoside formation. Biochim. Biophys. Acta 1078:127–132 [DOI] [PubMed] [Google Scholar]
- 19. Henrissat B. 1991. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280:309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L. 2000. The role of arginine 47 in the cyclization and coupling reactions of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 implications for product inhibition and product specificity. Eur. J. Biochem. 267:3432–3441 [DOI] [PubMed] [Google Scholar]
- 21. Uitdehaag JC, Kalk KH, van Der Veen BA, Dijkhuizen L, Dijkstra BW. 1999. The cyclization mechanism of cyclodextrin glycosyltransferase (CGTase) as revealed by a gamma-cyclodextrin-CGTase complex at 1.8-A resolution. J. Biol. Chem. 274:34868–34876 [DOI] [PubMed] [Google Scholar]
- 22. Uitdehaag JC, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW. 1999. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the alpha-amylase family. Nat. Struct. Biol. 6:432–436 [DOI] [PubMed] [Google Scholar]
- 23. Li Z, Li B, Gu Z, Du G, Wu J, Chen J. 2010. Extracellular expression and biochemical characterization of alpha-cyclodextrin glycosyltransferase from Paenibacillus macerans. Carbohydr. Res. 345:886–892 [DOI] [PubMed] [Google Scholar]
- 24. Li ZF, Zhang JY, Sun Q, Wang M, Gu ZB, Du GC, Wu J, Chen J. 2009. Mutations of Lysine 47 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhance beta-cyclodextrin specificity. J. Agric. Food Chem. 57:8386–8391 [DOI] [PubMed] [Google Scholar]
- 25. Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 15:5.6.1–5.6.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shindyalov IN, Bourne PE. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11:739–747 [DOI] [PubMed] [Google Scholar]
- 27. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291 [Google Scholar]
- 28. Bowie JU, Luthy R, Eisenberg D. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:164–170 [DOI] [PubMed] [Google Scholar]
- 29. Wallner B, Elofsson A. 2003. Can correct protein models be identified? Protein Sci. 12:1073–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van der Veen BA, Leemhuis H, Kralj S, Uitdehaag JC, Dijkstra BW, Dijkhuizen L. 2001. Hydrophobic amino acid residues in the acceptor binding site are main determinants for reaction mechanism and specificity of cyclodextrin-glycosyltransferase. J. Biol. Chem. 276:44557–44562 [DOI] [PubMed] [Google Scholar]
- 31. Nakamura A, Haga K, Yamane K. 1994. Four aromatic residues in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: effects of replacements on substrate binding and cyclization characteristics. Biochemistry 33:9929–9936 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.