

Abstract
The efficacy of the innate immune system depends on its ability to mount an appropriate response to diverse infections and damaging agents. Key components of this system are pattern recognition receptors that detect pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPs). Nlrp1b is a pattern recognition receptor that forms a caspase-1 activation platform, known as an inflammasome, upon sensing the proteolytic activity of anthrax lethal toxin. The activation of caspase-1 leads to the release of proinflammatory cytokines that aid in the clearance of the anthrax infection. Here, we demonstrate that Nlrp1b also becomes activated in cells that are subjected to energy stress caused by metabolic inhibitors or by nutrient deprivation. Glucose starvation and hypoxia were used to correlate the level of cytosolic ATP to the degree of inflammasome activation. Because lowering the ratio of cytosolic ATP to AMP activates the main cellular energy sensor, AMP-activated protein kinase (AMPK), we assessed whether AMPK promoted inflammasome activity by using a combination of small interfering RNA (siRNA) and transfection of a dominant negative AMPK subunit. We found that AMPK promoted inflammasome activity, but activation of AMPK in the absence of ATP depletion was not sufficient for caspase-1-mediated pro-interleukin 1β (pro-IL-1β) processing. Finally, we found that mutation of the ATP-binding motif of Nlrp1b caused constitutive activation, suggesting that ATP might inhibit the Nlrp1b inflammasome instead of being required for its assembly.
INTRODUCTION
Immune cells that respond to infection initiate energy-demanding processes, often in inflamed tissues that are low in oxygen and glucose. These conditions cause energetic stress that results in a metabolic switch from oxidative phosphorylation to glycolysis (1). Although glycolysis is a less efficient means to generate ATP, it is rapid and capable of meeting the needs of activated cells. The glycolytic pathway is also upregulated by Toll-like receptor signaling (2), indicating that immune cells that detect pathogens prepare to fight infection by altering their metabolism.
Studies on the NLRP3 inflammasome have revealed additional links between metabolism and innate immunity (3). Inflammasomes are protein complexes that activate the proinflammatory caspase-1 in response to pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) (4). NLRP3 becomes activated when cells are exposed to certain microbe-derived or endogenous stimuli that presumably generate a common signal or signals that trigger inflammasome assembly. Among the stimuli that activate NLRP3 are fatty acids and the islet amyloid polypeptide (5, 6), which are associated with obesity and type 2 diabetes. These metabolic disorders are exacerbated by the activation of the NLRP3 inflammasome, as this interferes with insulin signaling and thereby decreases insulin sensitivity.
Comparatively, few studies have focused on NLRP1 and its murine homolog Nlrp1b. Nlrp1b was discovered to be activated by anthrax lethal toxin (LeTx) using a genetic approach to determine why macrophages from some strains of mice are rapidly killed by the toxin while others are not (7). The macrophages that were killed were found to express a sensitive allele of Nlrp1b (allele 1 or 5) and to undergo a caspase-1-dependent form of cell death known as pyroptosis, whereas those that were not killed expressed a resistant allele (allele 2, 3, or 4). Although it was originally believed that the induction of pyroptosis by LeTx was a virulence strategy used by Bacillus anthracis to subvert the immune response, subsequent studies have shown that mice that express a sensitive allele are more resistant to an anthrax infection because of the beneficial release of interleukin 1β (IL-1β) (8, 9).
The enzymatic component of LeTx is a metalloprotease that cleaves mitogen-activated protein kinase kinases (MAPKKs) (10), and this disruption of MAPKK signaling has been suggested to interfere with several processes involved in the immune response (11). It was recently discovered that LeTx also cleaves Nlrp1b and that this proteolysis is required for inflammasome activity (12, 13). It remains to be determined, however, if this cleavage event is sufficient or whether LeTx also causes a form of cellular dysfunction that is detected by Nlrp1b. Monitoring for cellular dysfunction would enable Nlrp1b to detect more than just anthrax infections, which is in line with how pattern recognition receptors are thought to be activated by diverse pathogens or injury (14).
Given the energy requirements of an immune response and the connection between innate immune receptors and metabolism, we speculated that energy stress might be a danger signal that activates Nlrp1b. Here, we have used a reconstituted system in human fibroblasts to demonstrate that reduction of cytosolic ATP levels induces murine Nlrp1b inflammasome activity. Reducing the expression of the AMP-activated protein kinase (AMPK), the major energy sensor in cells, decreased the level of Nlrp1b activation, but signaling by AMPK was not sufficient to induce inflammasome activity. LeTx did not activate AMPK or cause a global reduction in ATP levels, suggesting that ATP depletion and LeTx activity represent distinct activation signals. Finally, we assessed the requirement for a functional ATPase domain in Nlrp1b. Surprisingly, mutation of the Nlrp1b Walker A motif, which is involved in ATP-binding, caused constitutive activation. This result contrasted with the abolition of activity observed when the same mutation was introduced into NLRP3. These results suggest that Nlrp1b does not require energy from ATP to activate caspase-1, which would allow it to function under conditions of severe energy stress.
MATERIALS AND METHODS
Cell culture and reagents.
HT1080 cells (ATCC) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. J774A.1 cells that express control short hairpin RNA (shRNA) or shRNA directed against Nlrp1b were described previously (15). Protective antigen (PA) and lethal factor (LF) were purified as described previously, and each were applied to cells at a final concentration of 10−8 M (16). The proteasome inhibitor, MG-132 (Calbiochem), was used at 10 μM. Sodium azide, 2-deoxyglucose (2DG), N-acetyl-cysteine, and staurosporine were purchased from Sigma-Aldrich and used at the indicated concentrations.
Plasmid construction and site-directed mutagenesis.
Plasmids encoding NTAP-Nlrp1b allele 1, NTAP-Nlrp1b allele 3, procaspase-1-T7, and pro-IL-1β–hemagglutinin (HA) have been described previously (17). QuikChange site-directed mutagenesis (Stratagene) was performed according to the manufacturer's instructions to generate the pNTAP-Nlrp1b-WA mutant, containing mutations G137A, K138A, and S139A. These three alanine mutations were introduced to pNTAP-Nlrp1b by using the oligonucleotide 5′-GAA GGG GCT GCT GGG ATT GCG GCG GCA ACA CTG GCC AGG CTG GTG AAG-3′ and its complement. The WA mutation was introduced into Nlrp1b allele 3 using the oligonucleotide 5′-GAA GGG GCT GCT GGG ATT GCG GCG GCA ACA CTG GCC AGG CTG-3′ and its complement.
The AMPKα1 gene was amplified by using the forward primer 5′-CGC GCG GCC GCA ATG GCG ACA GCC GAG AAG CAG-3′ and the reverse primer 5′-CGC CGC GAG TTG TGC AAG AAT TTT AAT TAG-3′ from an Open Biosystems plasmid, clone ID LIFESEQ90091361. The PCR product was digested with NotI and XhoI and then ligated into pcDNA3-His-FLAG. The pcDNA3-His-FLAG vector was generated by amplifying a triple FLAG tag with the forward primer 5′-CGC GGG CCC GAC TAC AAA GAC CAT GAC GGT G-3′ and the reverse primer 5′-CGC GCT AGC CTA CTT GTC ATC GTC ATC CTT GTA GTC G-3′ from pMZI3F (18) and ligating the PCR product into the ApaI and NheI sites of the pcDNA3-His-HA vector (17). The pcDNA3-His-AMPKα1-T174A-FLAG was made by using the mutagenic oligonucleotide 5′-GGT GAA TTT TTA AGA GCA AGT TGT GGC TCA CC-3′ and its complement.
The murine NLRP3 gene was amplified by using the forward primer 5′-CGC AAG CTT ATG ACG AGT GTC CGT TGC AAG-3′ and the reverse primer 5′-CGC CTC GAG TCA CCA GGA AAT CTC GAA GAC TAT AG-3′ from an Open Biosystems plasmid, clone ID 40048724. The PCR product was digested with HindIII and XhoI and then ligated into pNTAP-B (Stratagene 240101-52). The pNTAP-NLRP3-WA mutant, containing G227A, K228A, and T229A, was made by using the oligonucleotide 5′-GTG TTC CAG GGA GCA GCA GGC ATC GCG GCA GCC ATC CTA GCC AGG AAG ATT ATG-3′ and its complement. Additionally, an R258W mutation was introduced into pNTAP-NLRP3 by using the oligonucleotide 5′-CTA TTT GTT CTT TAT CCA CTG CTG GGA GGT GAG CCT CAG GAC G-3′ and its complement.
The murine NLRP6 gene was amplified by using the forward primer 5′-CGC GAA TTC GAT GCT GAA GTC TGC AGG CAC C-3′ and the reverse primer 5′-CGC CTC GAG TCA TTT TGA ATA TAT GAT GGA CAG-3′ from an Open Biosystems plasmid, clone ID 4972485. The PCR product was digested with EcoRI and XhoI and then ligated into pNTAP-A (Stratagene 240101-51).
The ASC (apoptosis-associated speck-like protein containing a CARD) gene was amplified from an Open Biosystems plasmid, clone ID 2648391, by using the forward primer 5′-CGC GGA TCC ATG GGG CGG GCA CGA GAT GCC-3′ and the reverse primer 5′-CGC GCG GCC GCT CAG CTC TGC TCC AGG TCC ATC AC-3′. The PCR product was digested with BamHI and NotI and then ligated into pcDNA3-FLAG.
IL-1β assay and ATP level assay.
One million HT1080 cells were seeded on a 10-cm dish the day before transfection. On the day of transfection, 1 μg each of pNTAP-Nlrp1b, pcDNA3–procaspase-1-T7, and pcDNA3–pro-IL-1β–HA were transfected using 9 μl of 1 mg/ml polyethyleneimine, pH 7.2. Approximately 24 h after transfection, cells were treated with LF (10−8 M) and PA (10−8 M) or 50 mM 2DG and 10 mM NaN3 in 5 ml medium for 3 h. The cell supernatant was mixed with 1 μl of α-HA antibody (Sigma-Aldrich H9658) overnight, followed by the addition of 100 μl of protein A Sepharose (GE Healthcare) and 2 h of incubation. Proteins were eluted from the protein A Sepharose beads with SDS loading dye and subjected to immunoblotting using a polyclonal α-HA antibody (Santa Cruz sc805).
Cell pellets were harvested and then lysed with 300 μl of EBC buffer (0.5% NP-40, 20 mM Tris [pH 8], 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 60 min. Equivalent amounts of cell lysate protein (∼30 μg) were subjected to SDS-PAGE and immunoblotted with α-HA (Santa Cruz sc805) and α-β-actin (Sigma-Aldrich A5441) antibodies.
Intracellular ATP levels were measured by a CellTiter-Glo luminescent cell viability assay (Promega G7571) in accordance with the manufacturer's instructions. Approximately 24 h after transfection in a 10-cm dish, cells were trypsinized, and 1 × 105 cells were seeded in each well in a 96-well plate. After 2 h, these transfected cells were treated with LeTx, 2DG, and NaN3 as indicated for 3 h, and cell lysates were assayed for intracellular ATP.
Knockdown of endogenous AMPK.
Half a million HT1080 cells were seeded on a 10-cm dish the day before transfection. On the day of transfection, 15 μl of 10 μM control siRNA (Santa Cruz sc-37007) or 10 μM AMPK siRNA (Santa Cruz sc-45312) were transfected using 15 μl Lipofectamine RNAiMAX transfection reagent (Invitrogen 13778-150). Approximately 48 h after transfection, cell lysates were harvested and cytosolic proteins were extracted by using the NE-PER nuclear and cytoplasmic extraction reagent kit (Fisher Scientific PI78833) in accordance with the manufacturer's instructions. Equivalent amounts of protein (∼60 μg) were subjected to SDS-PAGE and immunoblotted with α-phospho-AMPK (Santa Cruz sc-101630), α-total-AMPK (Santa Cruz sc-74461), and α-β-actin (Sigma-Aldrich A5441) antibodies.
Detection of TAP-tagged proteins.
One 10-cm dish of HT1080 cells was transfected with 1 μg plasmid encoding TAP-tagged proteins, 1 μg plasmid encoding procaspase-1-T7, and 1 μg plasmid encoding pro-IL-1β-HA. Approximately 24 h after transfection, cell pellets from each plate were lysed with 300 μl EBC buffer at 4°C for 1 h. Cell lysates from 3 plates were incubated with 25 μl streptavidin agarose resin (Thermo Scientific 20349) for ∼2 h. Beads were washed 3 times with 1 ml EBC buffer. Proteins were eluted with SDS and analyzed by immunoblotting with α-calmodulin binding peptide antibody (Upstate 07-482).
RESULTS
ATP depletion activates Nlrp1b.
To determine whether reduction of cytosolic ATP can activate the Nlrp1b inflammasome, we used a reconstituted system in which HT1080 human fibroblasts were transfected with plasmids encoding Nlrp1b, procaspase-1, and pro-IL-1β. This system allows for the study of Nlrp1b in isolation of other inflammasomes, and we have shown previously that LeTx induces Nlrp1b inflammasome assembly in the transfected cells as monitored by the cleavage of pro-IL-1β by caspase-1 and the subsequent release of processed IL-1β from the cells (17). To inhibit the production of ATP, we treated cells with the glycolysis inhibitor 2-deoxyglucose (2DG) and an inhibitor of the mitochondrial electron transport chain, sodium azide. Treatment of cells with these two compounds greatly reduced cytosolic ATP and caused the release of IL-1β (Fig. 1A and B). Two forms of IL-1β were secreted, which correspond in molecular weight to cleavage events at the canonical site, yielding a 17-kDa form, and at an amino-terminal site, yielding an ∼25-kDa form. Secretion of IL-1β was observed from cells expressing Nlrp1b allele 1 but not Nlrp1b allele 3 or from cells expressing the related control proteins NLRP3 or NLRP6 (Fig. 1C to E; see also Fig. S1 in the supplemental material). The Nlrp1b allele specificity for inflammasome activation by ATP depletion mirrors that observed in LeTx-mediated inflammasome activation in HT1080 cells (Fig. 1A) and in murine macrophages (7).
Fig 1.

Metabolic inhibitors activate the Nlrp1b inflammasome. (A) Plasmids pcDNA3–procaspase-1-T7 and pcDNA3–pro-IL-1β–HA were cotransfected with either pNTAP-Nlrp1b allele 1 or allele 3 into HT1080 cells. Approximately 24 h after transfection, cells were treated with LeTx or 50 mM 2DG and 10 mM NaN3 for 3 h. The cell lysates were probed for HA-tagged pro-IL-1β and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β and pro-IL-1β by immunoblotting. The asterisk indicates an ∼25 kDa HA-tagged pro-IL-1β-cleaved product. (B) Cell lysates from panel A were assayed for ATP. Error bars represent standard deviation for at least three independent experiments. (C) Plasmids encoding procaspase-1-T7 and pro-IL-1β–HA were cotransfected with pNTAP-Nlrp1b (1 μg), pNTAP-NLRP3 (1 μg), or pNTAP-NLRP6 (1 μg). pcDNA3-FLAG-ASC (50 ng) was cotransfected with the NLRP3 and NLRP6 constructs. Approximately 24 h after transfection, cells were treated with 50 mM 2DG and 10 mM NaN3 for 3 h. The cell lysates were probed for HA-tagged pro-IL-1β and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β and pro-IL-1β by immunoblotting. (D) Cell lysates from panel C were assayed for ATP. Error bars represent standard deviations for at least three independent experiments. (E) To establish relative expression levels of NLRP proteins, plasmids pcDNA3–procaspase-1-T7 and pcDNA3–pro-IL-1β–HA were cotransfected into HT1080 cells with pNTAP-Nlrp1b, pNTAP-NLRP3, and pcDNA3-FLAG-ASC or pNTAP-NLRP6 and pcDNA3-FLAG-ASC. Approximately 24 h after transfection, cells were lysed and TAP-tagged proteins were precipitated using streptavidin resin and immunoblotted using antibody directed against the calmodulin binding peptide segment of the TAP tag. Blots shown represent at least three independent experiments.
We demonstrated previously that autoproteolysis of Nlrp1b allele 1 is important for inflammasome assembly and that Nlrp1b allele 3 does not undergo autoproteolysis because of an amino acid difference at position 927 (15). Restoration of autoproteolysis in Nlrp1b allele 3 by the D927V mutation did not, however, restore inflammasome function in response to either LeTx or ATP depletion (see Fig. S2 in the supplemental material), indicating that additional amino acid differences impair allele 3 function (15).
To determine the threshold level of cytosolic ATP that triggers Nlrp1b inflammasome activation, we grew cells in minimal medium (containing no serum or glucose) with various concentrations of either glucose or 2DG (Fig. 2A and B). IL-1β was released from cells grown in 1 mM glucose but not from cells grown in 10 mM glucose. This reduction of glucose in the medium caused a drop in cytosolic ATP levels from ∼90% to ∼80% of that observed in control cells grown in complete medium. Further decreasing the concentration of glucose in the medium or adding 2DG caused a greater reduction in ATP levels and increased the release of IL-1β. LeTx, and depletion of ATP to a lesser extent, caused release of pro-IL-1β into the medium, which was likely through passive release, because the release of pro-IL-1β, but not processed IL-1β, was inhibited by the addition of the osmotic stabilizer PEG-6000 (data not shown). These results suggest that LeTx is a more effective inducer of pyroptosis than ATP depletion is, possibly because the toxin is a stronger activator of the inflammasome.
Fig 2.

Glucose starvation and hypoxia deplete cytosolic ATP and activate the inflammasome. (A) Cells were transfected with plasmids containing pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b allele 1. These transfected cells were trypsinized, and 1 × 105 cells were seeded per well in a 96-well plate. After 2 h, the cells were left in minimal DMEM containing 20 mM HEPES, pH 7.5, and the indicated concentrations of 2DG or glucose for 3 h. The cell lysates were then assayed for ATP. (B) Cells were transfected as described for panel A. Approximately 24 h after transfection, cells were incubated in minimal DMEM containing 20 mM HEPES, pH 7.5, and the indicated concentrations of 2DG or glucose. After 3 h of treatment, HA-tagged pro-IL-1β and IL-1β were detected by immunoblotting. (C) Cells were transfected with plasmids containing pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b allele 1 or 3. These transfected cells were trypsinized, and 1 × 105 cells were seeded per well in a 96-well plate. After 2 h, these cells were left in minimal DMEM containing 20 mM HEPES, pH 7.5, and incubated under normoxic or hypoxic (1% O2) conditions for 3 h. The cell lysates were then assayed for intracellular ATP. (D) Plasmids encoding pNTAP-Nlrp1b allele 1 or 3, procaspase-1-T7, and pro-IL-1β–HA were transfected into HT1080 cells. Approximately 24 h after transfection, cells were incubated in minimal DMEM containing 20 mM HEPES, pH 7.5, in normoxic or hypoxic conditions. After 3 h of treatment, HA-tagged pro-IL-1β and IL-1β were detected as described above. Blots shown represent at least three independent experiments. The asterisk indicates an ∼25 kDa HA-tagged pro-IL-1β-cleaved product. Error bars represent standard deviations for at least three independent experiments.
We next assessed whether the Nlrp1b inflammasome functions under hypoxia by growing cells in minimal medium with 1% oxygen. These conditions caused intracellular ATP levels to drop to ∼50% of control levels and stimulated the release of IL-1β into the cell supernatants (Fig. 2C and D; see also Fig. S3 in the supplemental material). Hypoxia did not reduce the ATP levels in cells grown in complete medium to an extent that triggered inflammasome activation (data not shown). These results suggest that various conditions that lead to the reduction of cytosolic ATP below a threshold level activate Nlrp1b.
AMPK promotes inflammasome activation.
Depletion of cellular ATP is sensed by the master regulator of metabolism, AMPK (19). AMPK is phosphorylated when a high AMP/ATP ratio exists in cells and it in turn phosphorylates numerous substrates to increase catabolic and decrease anabolic processes. To determine whether AMPK signaling is involved in the activation of Nlrp1b by ATP depletion or by LeTx, we used siRNA to reduce AMPK levels (Fig. 3A). A combination of 2DG and sodium azide, but not LeTx, caused an increase in phosphorylation of AMPK in control cells. In cells treated with AMPK siRNA, there was a reduced level of total and phosphorylated AMPK upon ATP depletion and less IL-1β was secreted compared to that in control cells, whereas similar amounts of IL-1β were secreted by control and AMPK knockdown cells in response to LeTx (Fig. 3A; see also Fig. S4A and B in the supplemental material).
Fig 3.

AMPK facilitates Nlrp1b inflammasome activation. (A) HT1080 cells were first transfected with either control siRNA or AMPK siRNA. After 24 h, these cells were then transfected with pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b. Approximately 24 h after the second transfection, cells were treated with LeTx or 50 mM 2DG and 10 mM NaN3 for 3 h. Cell lysates were probed for phospho-AMPK, total AMPK, HA-tagged IL-1β, and β-actin by immunoblotting. Supernatants were immunoprecipitated with anti-HA antibodies and probed for HA-tagged IL-1β and pro-IL-1β by immunoblotting. (B) Plasmids pNTAP-Nlrp1b, pcDNA3–procaspase-1-T7, and pcDNA3–pro-IL-1β–HA were cotransfected with pcDNA3-His-FLAG vector, pcDNA3-His-AMPKα1-FLAG, or pcDNA3-His-AMPKα1-T174A-FLAG into HT1080 cells. Approximately 24 h after transfection, cells were treated with LeTx or 50 mM 2DG and 10 mM NaN3 for 3 h. Cell lysates were probed for phospho-AMPK, total-AMPK, HA-tagged IL-1β, and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β and pro-IL-1β by immunoblotting. (C) Plasmids encoding procaspase-1-T7, pro-IL-1β–HA, and pNTAP-Nlrp1b were transfected into HT1080 cells. These transfected cells were treated with LeTx, 50 mM 2DG, and 10 mM NaN3 or with 2 μM staurosporine (STS) as indicated. After 3 h, cell lysates were harvested and probed for phospho-AMPK, total-AMPK, HA-tagged IL-1β, and β-actin by immunoblotting. HA-tagged pro-IL-1β and IL-1β in the supernatants were detected as described above. (D) Cell lysates from panel C were assayed for ATP. (E) Plasmids pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b were cotransfected into HT1080 cells. Approximately 24 h after transfection, cells were treated with LeTx or 50 mM 2DG and 10 mM NaN3 in the presence or absence of 10 μM cycloheximide (CHX) for 3 h. The cell lysates were probed for HA-tagged pro-IL-1β and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β and pro-IL-1β by immunoblotting. Blots shown represent at least three independent experiments. The asterisk indicates an ∼25-kDa HA-tagged pro-IL-1β-cleaved product. Error bars represent standard deviations for at least three independent experiments.
AMPK activity is dependent on phosphorylation of the alpha subunit at a conserved threonine residue (19). In order to confirm that AMPK facilitated Nlrp1b activation by ATP depletion, the mutant AMPKα1-T174A subunit was overexpressed, which had a dominant negative effect, as indicated by the diminished level of AMPK phosphorylation in ATP-depleted cells (Fig. 3B). Consistent with the knockdown experiment, overexpression of AMPKα1-T174A, but not wild-type AMPKα1, reduced the amount of IL-1β secreted upon ATP depletion (Fig. 3B; see also Fig. S4C in the supplemental material). AMPKα1-T174A expression had only a minor effect on inflammasome activation by LeTx (Fig. 3B; see also Fig. S4D in the supplemental material).
We next assessed whether signaling by AMPK is sufficient to activate Nlrp1b by treating cells with staurosporine. Staurosporine is a kinase inhibitor that causes activation of AMPK in the absence of ATP depletion (Fig. 3C and D). Staurosporine did not cause the release of IL-1β from cells (Fig. 3C), indicating that Nlrp1b activation is not controlled solely by AMPK.
Because a major function of AMPK is to inhibit translation, we speculated that inhibition of ribosomal function may explain how AMPK facilitates inflammasome activation. Treatment of cells with cycloheximide did not, however, stimulate the release of IL-1β from cells treated with sodium azide and 2DG or with LeTx (Fig. 3E; see Fig. S4E and F in the supplemental material).
Effects of MG-132 and N-acetylcysteine on inflammasome activation.
Several studies have suggested that reactive oxygen species (ROS) generated from the mitochondrial electron transport chain are required to trigger NLRP3 assembly downstream of diverse stimuli such as extracellular ATP, particulates, and palmitate (5, 20). The ROS required to activate NLRP3 may be derived from damaged mitochondria or from excessive oxidative phosphorylation. Recently, however, the requirement for ROS has been suggested to be at the priming stage—the expression of NLRP3—rather than the activation stage (21). To address whether activation of the Nlrp1b inflammasome required ROS, we pretreated transfected HT1080 cells with the nonspecific ROS scavenger N-acetylcysteine (NAC). We found that NAC did not inhibit IL-1β secretion from cells treated with LeTx (Fig. 4A; see also Fig. S5A in the supplemental material) but did diminish the amount of IL-1β secreted from cells treated with the metabolic inhibitors (Fig. 4A; see Fig. S5B and C in the supplemental material). NAC did not affect cytosolic ATP levels (Fig. 4B), however, which suggests that cellular redux potential influences inflammasome assembly.
Fig 4.
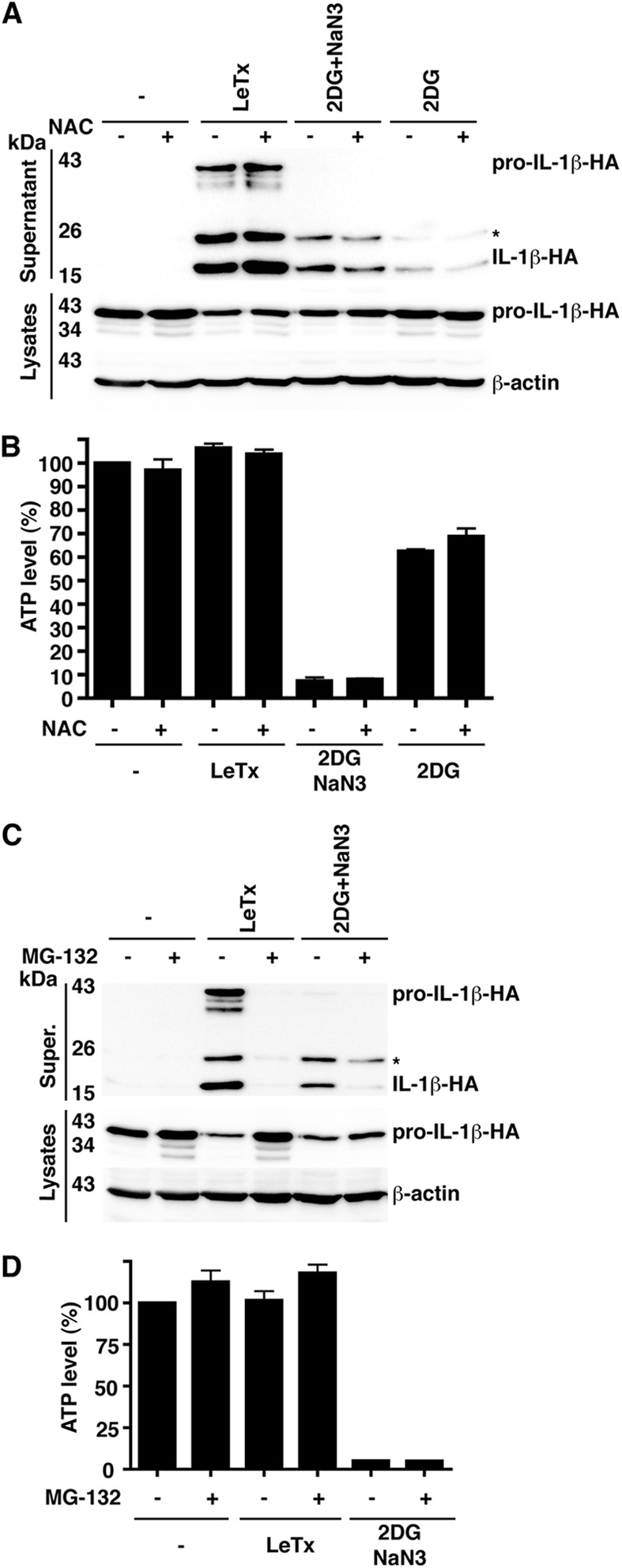
Roles of reactive oxygen species and proteasome activity in Nlrp1b activation. (A) Plasmids pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b were transfected into HT1080 cells. Approximately 24 h after transfection, cells were left untreated or were treated with LeTx, 50 mM 2DG, and 10 mM NaN3 or 50 mM 2DG in the absence or presence of 25 mM N-acetyl cysteine (NAC). After 3 h, cell lysates were collected and probed for HA-tagged pro-IL-1β and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and probed for HA-tagged pro-IL-1β and IL-1β by immunoblotting. (B) Cell lysates from panel A were assayed for ATP. (C) Cells were transfected with pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b. Approximately 24 h after transfection, cells were treated with LeTx or 50 mM 2DG and 10 mM NaN3 in the presence or absence of 10 μM MG-132. After 3 h, cell lysates were collected and probed for HA-tagged pro-IL-1β; HA-tagged pro-IL-1β and IL-1β in supernatants were detected as described above. (D) Cell lysates from panel C were assayed for ATP. Blots shown represent at least three independent experiments. The asterisk indicates an ∼25-kDa HA-tagged pro-IL-1β-cleaved product. Error bars represent standard deviations for at least three independent experiments.
It has been established that proteasome inhibitors block the activation of Nlrp1b by LeTx (22, 23), so we sought to determine whether this was also the case for activation by ATP depletion. The proteasome inhibitor MG-132 blocked the secretion of IL-1β in response to both LeTx and 2DG-sodium azide (Fig. 4C; see also Fig. S5D and E in the supplemental material). MG-132 did not, however, prevent the depletion of cytosolic ATP by 2DG-sodium azide (Fig. 4D), suggesting that proteasome inhibition diminishes Nlrp1b inflammasome activity triggered by both LeTx and ATP depletion.
Mutation of the Nlrp1b Walker A motif causes constitutive activation.
The NACHT domains of NLRPs are thought to mediate homo-oligomerization by using energy derived from ATP (24). Because this notion seemed at odds with Nlrp1b sensing low ATP levels, we mutated the Walker A motif of the Nlrp1b NACHT domain to assess its involvement in inflammasome activation. In contrast to wild-type Nlrp1b, the Walker A mutant was constitutively active, suggesting that ATP hydrolysis is not required to form a functional inflammasome (Fig. 5A and B). The lower level of the Walker A mutant may result from its constitutive secretion, as it has been demonstrated previously that inflammasome components are secreted upon activation (25). The Walker A mutation did not, however, activate allele 3 of Nlrp1b (Fig. 5C and D). This was not surprising, as we have shown previously that the inability of Nlrp1b allele 3 to undergo autocatalyic proteolysis prevents the assembly of a functional inflammasome (15).
Fig 5.
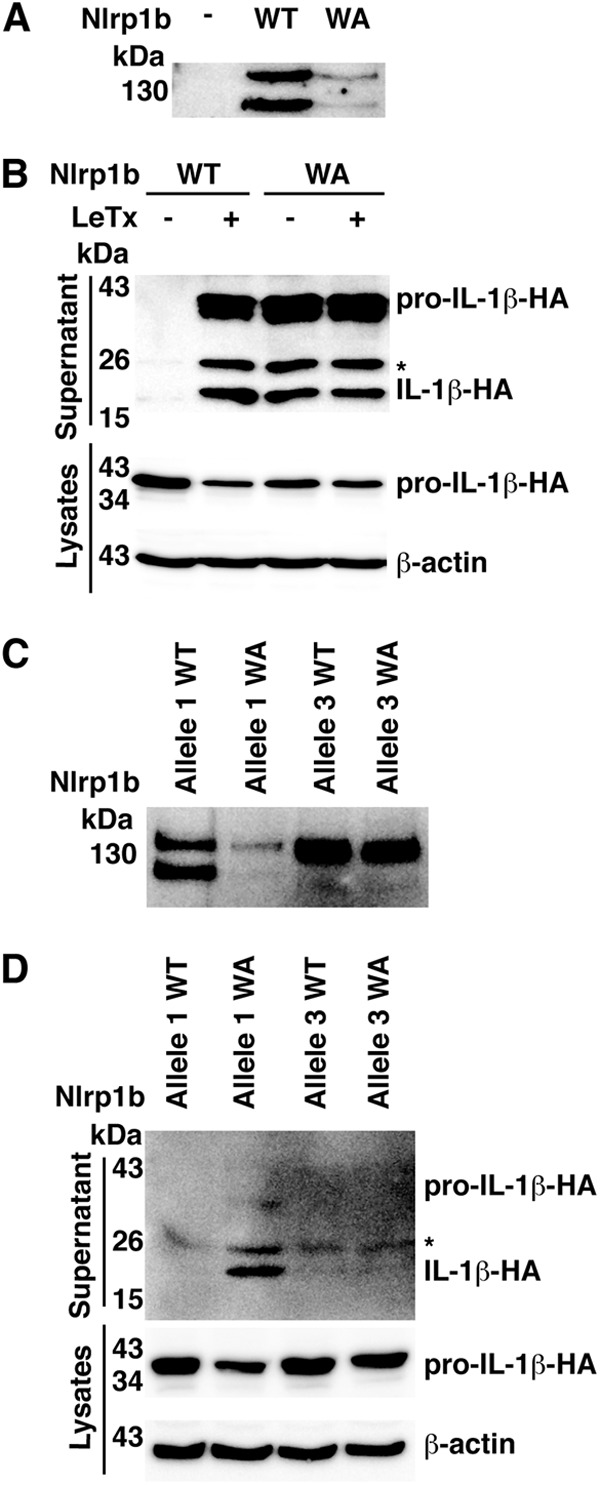
Walker A motif mutant of Nlrp1b is constitutively active. (A) HT1080 cells were transfected with pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-Nlrp1b wild-type (WT) or Walker A (WA) mutant. Approximately 24 h after transfection, cells were lysed and TAP-tagged proteins were precipitated using streptavidin resin and immunoblotted using antibody directed against the calmodulin binding peptide segment of the TAP tag. (B) Plasmids encoding procaspase-1-T7 and pro-IL-1β–HA were cotransfected into cells with either pNTAP-Nlrp1b-WT or pNTAP-Nlrp1b-WA. Approximately 24 h after transfection, cells were treated with LeTx, and then cell lysates were collected and probed for HA-tagged IL-1β and β-actin by immunoblotting; supernatants were immunoprecipitated with anti-HA antibodies and probed for HA-tagged pro-IL-1β and IL-1β by immunoblotting. (C) As described for panel A, except that both Nlrp1b allele 1 and allele 3 were used. (D) As described for panel B, except that both Nlrp1b allele 1 and allele 3 were used. Blots shown represent at least three independent experiments. The asterisk indicates an ∼25-kDa HA-tagged pro-IL-1β-cleaved product.
We next measured NLRP3 activity by using a constitutively active mutant identified in patients with the auto-inflammatory disorder Muckles-Wells syndrome (26). NLRP3-R258W caused secretion of IL-1β in the presence of the ASC adaptor but not in its absence (Fig. 6A and B). In contrast to what was observed for Nlrp1b, mutation of the Walker A motif in NLRP3 did not cause constitutive activation (Fig. 6C and D). Furthermore, the introduction of the Walker A mutation into NLRP3-R258W impaired its activity, indicating that a functional ATPase domain is required for NLRP3 function (Fig. 6D). These data suggest that the NACHT domains of Nlrp1b and NLRP3 have different requirements for ATP.
Fig 6.
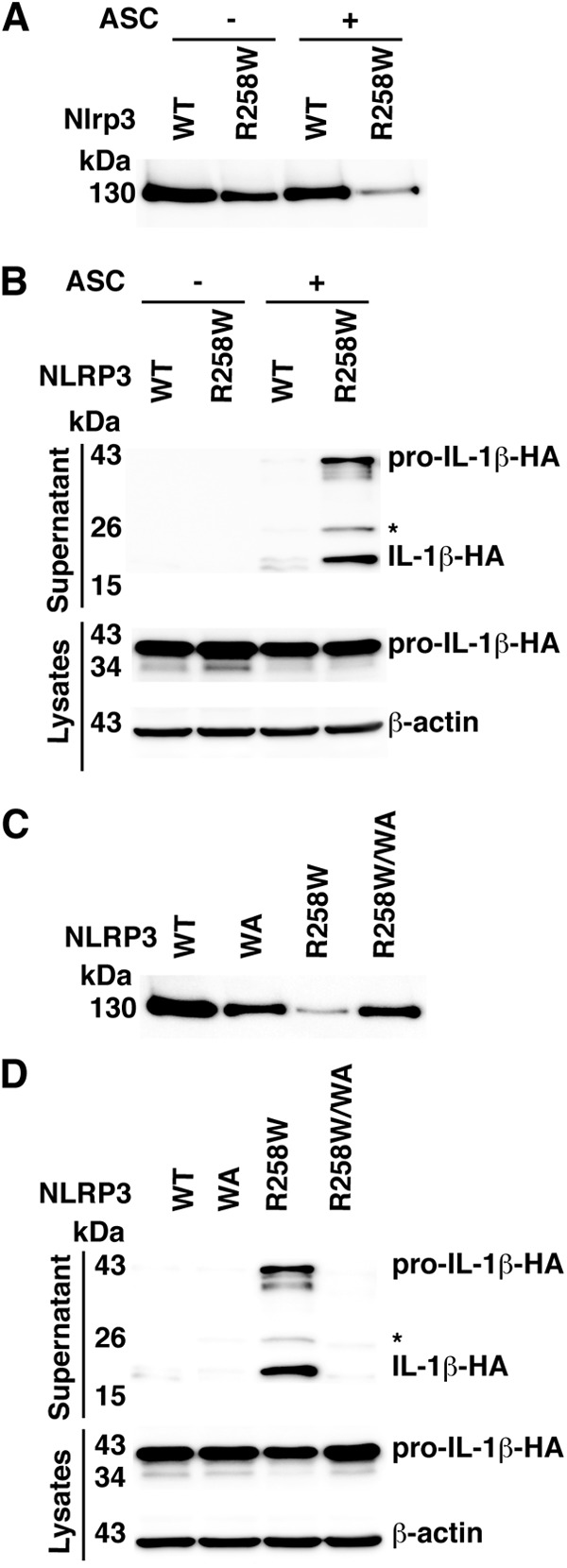
Mutation of the NLRP3 Walker A motif impairs activity. (A) Cells were transfected with pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and indicated pNTAP-NLRP3 plasmids along with 50 ng pcDNA3-FLAG-ASC or 50 ng empty vector. Approximately 24 h after transfection, TAP-tagged proteins were precipitated using streptavidin resin and immunoblotted using antibody directed against the calmodulin binding peptide segment of the TAP tag. (B) Plasmids encoding the indicated proteins were cotransfected into HT1080 cells with pcDNA3–procaspase-1-T7 and pcDNA3–pro-IL-1β–HA. Approximately 24 h after transfection, cell lysates were harvested and probed for HA-tagged IL-1β and β-actin; supernatants were probed for HA-tagged pro-IL-1β and IL-1β. (C) Plasmids pcDNA3-FLAG-ASC, pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and various pNTAP-NLRP3 constructs were transfected into HT1080 cells. TAP-tagged proteins were detected as described above. (D) Cells were transfected with pcDNA3-FLAG-ASC, pcDNA3–procaspase-1-T7, pcDNA3–pro-IL-1β–HA, and pNTAP-NLRP3. HA-tagged IL-1β and pro-IL-1β were detected as described above. The asterisk indicates an ∼25-kDa HA-tagged pro-IL-1β-cleaved product. Blots shown represent at least three independent experiments.
Depletion of ATP activates the Nlrp1b inflammasome in J774A macrophages.
To assess whether reduction of cytosolic ATP activates the Nlrp1b inflammasome in a macrophage cell line, we generated J774A cells that stably express either a control shRNA or an shRNA directed against Nlrp1b. Treatment of the control cells with either LeTx or 2DG led to the appearance of caspase-1 fragments of ∼43 kDa and ∼33 kDa; in addition to these fragments, several smaller fragments were observed in lysates of the LeTx-treated cells (Fig. 7A). Cells that express Nlrp1b shRNA exhibited reduced levels of caspase-1 fragments compared to the control cells upon treatment with either LeTx or 2DG (Fig. 7A; see also Fig. S7 in the supplemental material). As expected, the 2DG treatment reduced ATP levels in both control and Nrlp1b knockdown cells (Fig. 7B), indicating that ATP depletion activates the Nlrp1b inflammasome. In contrast, LeTx caused a large reduction of ATP in control cells but not in the Nlrp1b knockdown cells, which is likely a consequence of LeTx-induced pyroptosis causing loss of ATP.
Fig 7.
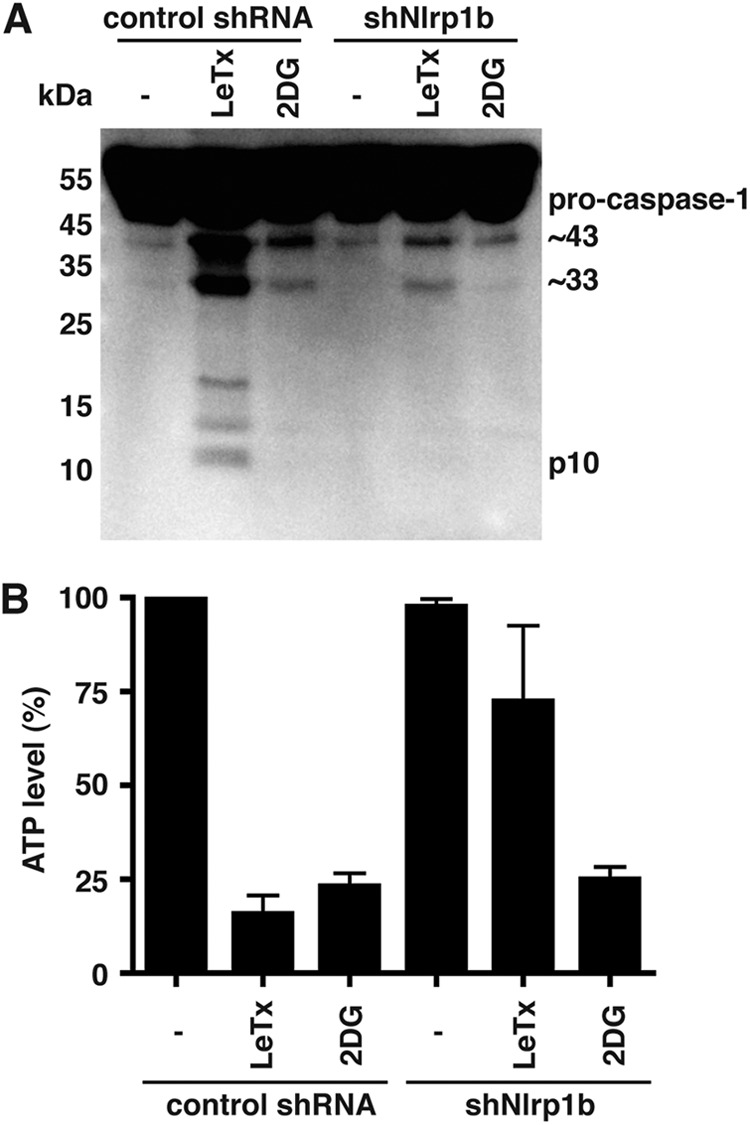
Depletion of ATP activates the Nlrp1b inflammasome in J774 macrophages. (A) J774A cells were grown in minimal medium and treated either with LeTx (5 × 10−10 M LF plus 10−8 M PA) or with 50 mM 2DG for 4 h. Supernatants were collected and probed for caspase-1 by immunoblotting. The blot represents three independent experiments. (B) Cell lysates from panel A were assayed for ATP. Error bars indicate standard deviations from three independent experiments.
DISCUSSION
The innate immune system is able to detect a diversity of infections. It does so by using pattern recognition receptors to sense conserved microbial structures and to discern between normal events and those that are associated with cellular damage (14). The damage signals that are detected directly by pattern recognition receptors are poorly characterized, although the events that elicit these signals are being uncovered. We have demonstrated here that Nlrp1b is activated by energy deprivation in a reconstituted system and in a macrophage cell line. That Nlrp1b can detect a danger signal in addition to LeTx is an attractive idea because the notion that a pattern recognition receptor has evolved to sense a virulence factor from a single pathogen is counterintuitive. Nlrp1b may therefore be able to detect viral replication, which can deplete cellular ATP, and a number of bacterial pathogens that secrete pore-forming toxins as sublytic concentrations of these toxins reduce ATP levels (27–30). Activation of Nlrp1b by energy stress and the subsequent release of IL-1β could benefit the host in at least two ways. First, the chemokine activity of IL-1β could recruit neutrophils to inflamed tissue to combat infection. Second, IL-1β might facilitate tissue repair and immune cell function by stimulating glucose uptake (31).
The molecular events that link the reduction of cytosolic ATP levels to the activation of the Nlrp1b inflammasome are not clear. The observation that a Walker A mutant is constitutively active raises the idea that Nlrp1b is a direct sensor of ATP levels—decreased concentrations of ATP might promote the formation of active ADP-bound or nucleotide-free Nlrp1b from inactive ATP-bound Nlrp1b. This model does not, however, explain the effects of AMPK, NAC, or MG-132 on inflammasome activity. It is conceivable that in addition to an activating signal, either particular cellular conditions must exist or a distinct inhibitory mechanism must be overcome for the inflammasome to assemble.
The amino-terminal region of Nlrp1b may be involved in its auto-inhibition, because cleavage of this region is required for LeTx-mediated inflammasome activation (12, 13). We did not, however, observe cleavage of Nlrp1b in response to ATP depletion (see Fig. S6 in the supplemental material). Nonetheless, there may be commonalities between the two activating signals. One possibility is that LeTx may inactivate an ATP-binding protein, which is likewise inactivated by ATP depletion, to initiate inflammasome activation. Alternatively, LeTx may cause a highly localized depletion of ATP that was not detected in our assays.
Lee and colleagues implicated AMP deaminase 3 (AMPD3), which coverts AMP to IMP, in LeTx-mediated pyroptosis of a murine macrophage cell line (32). These results are intriguing, but we do not know if they relate to the findings reported here. Lee and colleagues also demonstrated that a small-molecule activator of AMPK, AICAR, did not influence pyroptosis, which is in agreement with our work showing that activation of AMPK is not sufficient to trigger the Nlrp1b inflammasome. AMPK does, however, enhance inflammasome assembly caused by ATP depletion. Although the role of AMPK in this process is unclear, it does not appear to involve the downregulation of translation, as cycloheximide did not stimulate inflammasome activation.
A recent study has shown that a strain of B. anthracis that produces LeTx activates caspase-1 in macrophages expressing a resistant allele of Nlrp1b (33). The authors demonstrated that both the bacterium and the toxin were required to activate caspase-1 and that the danger signal was mediated by extracellular ATP that had leaked from the macrophages. The authors did not directly test which inflammasome was responsible for caspase-1 activation by knocking down NLRP expression, but the data are consistent with NLRP3 activation. Extracellular ATP, a well-characterized trigger of NLRP3 (34), binds purinergic receptors to cause the opening of pannexin-1 channels. Membrane openings cause an efflux of potassium that is required for the assembly of the NLRP3 inflammasome.
A Walker A motif mutation in Nlrp1b caused constitutive activation, whereas the same mutation prevented activity of NLRP3 (Fig. 5 and 6). We speculate that the NACHT domain facilitates auto-inhibition of both proteins and that mutation of the Walker A motif abolishes auto-inhibition. The NLRP3 Walker A mutant is not active because its NACHT domain is also required for its oligomerization; oligomerization of Nlrp1b, however, is not dependent on a functional NACHT domain, so it is constitutively active. We showed previously that a truncation mutant of Nlrp1b consisting of the FIIND and CARD domains alone oligomerizes and activates procaspase-1 (17).
Our finding that Nlrp1b detects energy stress adds to a growing body of work that links metabolism and inflammation. This connection is likely a consequence of the requirement of immune cells to function in inflamed tissues that are deprived of oxygen and glucose. Hypoxia-inducible factor 1α (HIF-1α) is a transcription factor that controls the cellular response to low-oxygen conditions by upregulating genes involved in glycolysis, angiogenesis, and glucose uptake; in myeloid cells, HIF-1α also increases the expression of tumor necrosis factor alpha (TNF-α) and antimicrobial factors (35). The importance of HIF-1α for macrophage function was demonstrated by the observation that HIF-1α-null macrophages exhibit impaired inflammatory function and a metabolic defect that results in an ∼80% reduction in ATP levels (36). Thus, sensors of oxygen and ATP appear to coordinate to mount an effective immune response.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH grant RO1 AI067683.
J. M. holds the Canada Research Chair in Bacterial Pathogenesis.
Footnotes
Published ahead of print 10 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01003-12.
REFERENCES
- 1. Imtiyaz HZ, Simon MC. 2010. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 345:105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. 2010. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115:4742–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tannahill GM, O'Neill LA. 2011. The emerging role of metabolic regulation in the functioning of Toll-like receptors and the NOD-like receptor Nlrp3. FEBS Lett. 585:1568–1572 [DOI] [PubMed] [Google Scholar]
- 4. Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832 [DOI] [PubMed] [Google Scholar]
- 5. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. 2011. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12:408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O'Neill LA. 2010. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11:897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyden ED, Dietrich WF. 2006. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38:240–244 [DOI] [PubMed] [Google Scholar]
- 8. Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL, LeVine SM, Bradley KA. 2010. Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J. Immunol. 184:17–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, Liu S, Sastalla I, Leppla SH. 2010. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 6:e1001222 doi:10.1371/journal.ppat.1001222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280:734–737 [DOI] [PubMed] [Google Scholar]
- 11. Moayeri M, Leppla SH. 2009. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 30:439–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. 2012. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8:e1002638 doi:10.1371/journal.ppat.1002638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M. 2012. Anthrax lethal factor cleaves mouse Nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 7:e49741 doi:10.1371/journal.pone.0049741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4:a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frew BC, Joag VR, Mogridge J. 2012. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8:e1002659 doi:10.1371/journal.ppat.1002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kassam A, Der SD, Mogridge J. 2005. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol. 7:281–292 [DOI] [PubMed] [Google Scholar]
- 17. Liao KC, Mogridge J. 2009. Expression of Nlrp1b inflammasome components in human fibroblasts confers susceptibility to anthrax lethal toxin. Infect. Immun. 77:4455–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeghouf M, Li J, Butland G, Borkowska A, Canadien V, Richards D, Beattie B, Emili A, Greenblatt JF. 2004. Sequential peptide affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 3:463–468 [DOI] [PubMed] [Google Scholar]
- 19. Carling D, Mayer FV, Sanders MJ, Gamblin SJ. 2011. AMP-activated protein kinase: nature's energy sensor. Nat. Chem. Biol. 7:512–518 [DOI] [PubMed] [Google Scholar]
- 20. Zhou R, Yazdi AS, Menu P, Tschopp J. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225 [DOI] [PubMed] [Google Scholar]
- 21. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. 2011. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187:613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang G, Leppla SH. 1999. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect. Immun. 67:3055–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Squires RC, Muehlbauer SM, Brojatsch J. 2007. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J. Biol. Chem. 282:34260–34267 [DOI] [PubMed] [Google Scholar]
- 24. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, Ting JP. 2007. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U. S. A. 104:8041–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. 2007. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr. Biol. 17:1140–1145 [DOI] [PubMed] [Google Scholar]
- 26. Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, Lachmann HJ, Bybee A, Gaudet R, Woo P, Feighery C, Cotter FE, Thome M, Hitman GA, Tschopp J, McDermott MF. 2002. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 46:2445–2452 [DOI] [PubMed] [Google Scholar]
- 27. Ando T, Imamura H, Suzuki R, Aizaki H, Watanabe T, Wakita T, Suzuki T. 2012. Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 8:e1002561 doi:10.1371/journal.ppat.1002561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dickman KG, Hempson SJ, Anderson J, Lippe S, Zhao L, Burakoff R, Shaw RD. 2000. Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G757–G766 [DOI] [PubMed] [Google Scholar]
- 29. Dragneva Y, Anuradha CD, Valeva A, Hoffmann A, Bhakdi S, Husmann M. 2001. Subcytocidal attack by staphylococcal alpha-toxin activates NF-kappaB and induces interleukin-8 production. Infect. Immun. 69:2630–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lizak M, Yarovinsky TO. 2012. Phospholipid scramblase 1 mediates type I interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 11:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Del Rey A, Roggero E, Randolf A, Mahuad C, McCann S, Rettori V, Besedovsky HO. 2006. IL-1 resets glucose homeostasis at central levels. Proc. Natl. Acad. Sci. U. S. A. 103:16039–16044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee S, Wang Y, Kim SO, Han J. 2011. AMPD3 is involved in anthrax LeTx-induced macrophage cell death. Protein Cell 2:564–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ali SR, Timmer AM, Bilgrami S, Park EJ, Eckmann L, Nizet V, Karin M. 2011. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 35:34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232 [DOI] [PubMed] [Google Scholar]
- 35. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. 2005. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 115:1806–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.