

Abstract
The Vibrio cholerae BreR protein is a transcriptional repressor of the breAB efflux system operon, which encodes proteins involved in bile resistance. In a previous study (F. A. Cerda-Maira, C. S. Ringelberg, and R. K. Taylor, J. Bacteriol. 190:7441–7452, 2008), we used gel mobility shift assays to determine that BreR binds at two independent binding sites at the breAB promoter and a single site at its own promoter. Here it is shown, by DNase I footprinting and site-directed mutagenesis, that BreR is able to bind at a distal and a proximal site in the breAB promoter. However, only one of these sites, the proximal 29-bp site, is necessary for BreR-mediated transcriptional repression of breAB expression. In addition, it was determined that BreR represses its own expression by recognizing a 28-bp site at the breR promoter. These sites comprise regions of dyad symmetry within which residues critical for BreR function could be identified. The BreR consensus sequence AANGTANAC-N6-GTNTACNTT overlaps the −35 region at both promoters, implying that the repression of gene expression is achieved by interfering with RNA polymerase binding at these promoters.
INTRODUCTION
Vibrio cholerae is a Gram-negative bacterium that causes the severe diarrheal disease cholera and is acquired by oral ingestion of contaminated water or food. Upon infection, the bacteria colonize the intestinal epithelium via the toxin coregulated pilus (TCP) and produce cholera toxin (CT), which induces severe loss of fluid and ions. A virulence regulatory system coordinately controls the production of a number of virulence factors, including CT and TCP. ToxT, a member of the AraC family of transcriptional regulators, is the direct activator of the genes encoding CT and TCP (1, 2). Expression of toxT is dependent upon a complex transcriptional regulatory cascade (3, 4).
During the course of infection, enteric pathogens encounter different environments within the stomach and the intestinal lumen, including changes in pH, CO2, and osmolarity and the presence of bile (5). Bile is produced by the liver and stored in the gallbladder. It is secreted into the proximal portion of the duodenum upon ingestion of food. The main components of bile are bile salts, which are detergents that aid in the emulsification of dietary fats and in their absorption (6). Additionally, the detergent property of the bile salts affects the cell membrane of bacteria, acting in a bactericidal manner (5, 6).
Enteric bacteria, including V. cholerae, are able to employ bile as a host signal to modulate gene expression and overcome the bactericidal effect of bile (5, 7). V. cholerae resistance to bile depends on (i) increased motility in the presence of bile, which is hypothesized to be essential for evading the high concentrations of bile in the lumen, penetrating the mucus layer, and achieving access to the underlying epithelial cells for colonization (8, 9); (ii) biofilm formation, where cells inside the biofilm are resistant to the bactericidal effect of bile (10); (iii) selectively excluding bile from entering the cell by differentially expressing genes encoding the OmpU and OmpT porins (11–13); and (iv) extruding bile out of the cell by inducing the expression of genes encoding proteins involved in efflux, such as acrA (14), tolC (15), vceB (16), vexB (11), and breB (11, 17).
We have recently demonstrated that the expression of breB, encoding an efflux pump belonging to the RND family, is induced by bile and bile salts (cholate, deoxycholate, and chenodeoxycholate) (17). The expression of genes encoding multidrug transporters is usually tightly controlled by transcriptional regulators to prevent nonspecific transport and loss of membrane potential that could result in cell death (18, 19). We have established that BreR, a member of the TetR family of transcriptional regulators, is a direct repressor of breAB operon expression, and that it binds at two independent sites at this promoter (17). Typically, genes encoding TetR repressors that control the expression of genes encoding efflux pumps are located either in the same operon or adjacent and are divergently transcribed from the genes they regulate (20–25). However, the breR gene is located 8.99 kb upstream of the breAB operon, with several genes between them. Like other TetR regulators, BreR is able to regulate its own expression (negative regulation), and it binds to a single site at the breR promoter (17). Furthermore, breR expression is induced in the presence of bile, cholate, deoxycholate, and chenodeoxycholate, and it has been shown that deoxycholate prevents BreR binding to the breR promoter (17).
The present study examines BreR binding sites and their locations in the breR and breAB promoter regions to gain a better understanding of the mechanism of BreR-mediated repression. Electrophoretic mobility shift assays (EMSA) and DNase I footprinting demonstrate that BreR recognizes a single 28-bp binding site at its own promoter (−51 to −24) and two binding sites at the breAB promoter separated by 141 bp, a 29-bp proximal binding site (−54 to −26), and a 29-bp distal binding site (−224 to −196). Transcriptional assays showed that the mutations at the breR promoter resulted in PbreR-lacZ overexpression, and EMSAs confirmed that the mutations prevented BreR from binding. However, only mutations at the proximal site at the breAB promoter caused PbreAB-lacZ overexpression and loss of BreR binding by EMSA. The distal binding site did not exhibit any regulatory role in vivo. Altogether, these results indicate that the BreR binding sites overlap the −35 regions of the breAB and breR promoters, suggesting that BreR inhibits initiation of transcription by blocking RNA polymerase access to the promoter sequences.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and growth conditions.
The V. cholerae strains, plasmids, and primers used in this study are listed in Tables 1 and 2. The strains were grown in Luria-Bertani (LB) medium (26). Antibiotics (Sigma) were used at the following concentrations: kanamycin, 45 μg/ml; polymyxin B, 50 U/ml; and streptomycin, 100 μg/ml or 1 mg/ml (allelic exchange experiments). For PbreR-lacZ and PbreAB-lacZ induction experiments, strains were grown in subinhibitory concentrations of sodium cholate (crude bile; Sigma) as noted in the β-galactosidase assay methods. Bile stocks were freshly prepared in LB medium and filter sterilized. For allelic exchange experiments, LB agar contained 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; Gold Biotechnology Inc.).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype | Source or reference |
|---|---|---|
| Vibrio cholerae strains | ||
| C6706 str2 | E1 Tor Smr | Laboratory collection |
| KSK262 | C6706 str2 ΔlacZ3 | 3 |
| FCM274 | KSK262 ΔbreR1 | This work |
| FCM158 | KSK262 PbreR-lacZ1 | 17 |
| FCM191 | FCM158 ΔbreR1 | 17 |
| FCM313 | KSK262 PbreR-lacZ1 A-50G/A-44G | This work |
| FCM315 | FCM274 PbreR-lacZ1 A-50G/A-44G | This work |
| FCM345 | KSK262 PbreAB-lacZ1 | This work |
| FCM321 | FCM274 PbreAB-lacZ1 | This work |
| FCM291 | KSK262 PbreAB-lacZ1 A-223G/T-220C/A-217G | This work |
| FCM293 | FCM274 PbreAB-lacZ1 A-223G/T-220C/A-217G | This work |
| FCM295 | KSK262 PbreAB-lacZ1 A-52G/T-49C/A-46G | This work |
| FCM297 | FCM274 PbreAB-lacZ1 A-52G/T-49C/A-46G | This work |
| FCM301 | KSK262 PbreAB-lacZ1 A-223G/T-220C/A-217G A-52G/T-49C/A-46G | This work |
| FCM303 | FCM274 PbreAB-lacZ1 A-223G/T-220C/A-217G A-52G/T-49C/A-46G | This work |
| Plasmids | ||
| pKAS154 | pKAS32 derivative, Kanr | 27 |
| pGKK346 | lacZ in pGKK344 | 17 |
| pFC3 | pKAS154, ΔbraR1 | This work |
| pFC31 | pKAS154, 235 bp harboring the R site | This work |
| pFC29 | pKAS154, 230 bp harboring the ABd site | This work |
| pFC30 | pKAS154, 246 bp harboring the ABp site | This work |
| pFC27 | PbreR-lacZ1 in pGKK346 | This work |
| pFC42 | PbreR-lacZ1 A-50G/A-44G in pGKK346 | This work |
| pFC43 | PbreAB-lacZ1 in pGKK346 | This work |
| pFC38 | PbreAB-lacZ1 A-223G/T-220C/A-217G in pGKK346 | This work |
| pFC39 | PbreAB-lacZ1 A-52G/T-49C/A-46G in pGKK346 | This work |
| pFC40 | PbreAB-lacZ1 A-223G/T-220C/A-217G A-52G/T-49C/A-46G in pGKK346 pFC42 | This work |
Table 2.
Primers used in this study
| Name | Nucleotide sequence (5′ to 3′) |
|---|---|
| FC13F | GATCGGGATCCCGTAAGCAATCTCGCTACTG |
| FC18F | GATCGGAATTCACCATGAAACTCAGTGAGCAAAA |
| FC24 | AGCAGACACTCAGATTATCG |
| FC25 | CGATAATCTGAGTGTCTGCT |
| FC26 | TGTACGAATCCCCATGCCTT |
| FC27 | GTGGTTAACTTGTCGCGCAT |
| FC30 | GATTTAATGGTGTCTACACG |
| FC31 | AGGTGCCGCTCAAAGATACG |
| FC35 | GCACAAAGTAAACTCGTTGG |
| FC36 | GCTTTCTCCTTTTGGGTTGAGCAG |
| FC37 | CTTGAGTAATGATAAAAAGTAAAC |
| FC39 | GGCGATAATACCTTTATTTTTAG |
| FC40 | CGTTGGTGTACTTTTTTGTGCGTCG |
| FC41 | TTTTGCTCACTGAGTTTCAT |
| FC42F | GATCGGGATCCAGCACTGGAAACTACAGGTAAG |
| FC42R | GATCGGAATTCAGCGAGTTACCAATTGGGTTTCG |
| FC43F | GATCGGGATCCTCTGGTTGAAGCACTCTCTG |
| FC43R | GATCGGAATTCGTACGAATCCCCATGCCTTTG |
| FC44F | GATCGGGATCCGCTTCAATCAGCGCCAACCG |
| FC44R | GATCGGAATTCATACTTGGGGAGCAATGAATCTG |
| FC45 | TCTTGGTTTAACATGCTTTCTCC |
| FC46 | CGTATCTTTGAGCGGCACCTG |
| FC47 | CTTTCGGGTGTACATTTGTG |
| FC48 | GAACTGCTCAACCCAAAAGG |
| FC68 | GATCGTCTAGACGATTGAATCGACGTTGATCCCTTG |
| FC69 | GATCGTCTAGAGATCCATATTCGCTGCATGG |
| FC70 | GATCGTCTAGACGTAAGCAATCTCGCTACTG |
| FC71 | GATCGTCTAGAGAAGGATTCATAGTGTGTTG |
| FC1DIG | /5DigN/CGTAAGCAATCTCGCTACTGGCCTGCACCTTTG |
| FC2DIG | /5DigN/CGATAATCTGAGTGTCTGCTCAGCGAGTTA |
| FC3DIG | /5DigN/AGCAGACACTCAGATTATCGATAGAATAAA |
| C4DIG | /5DigN/GAAGGATTCATAGTGTGTTGCTCCTCAATT |
| FC7DIG | /5DigN/TTTCTGATCCCTGAATGCCATTTTGAGGCG |
| FC8DIG | /5DigN/GATCCATATTCGCTGCATGGAATCCAAACTG |
| FCmut2-Sap | GATCGGCTCTTCGCTCGTTGGTGTACTTTTTTG |
| FCmut3-Sap | GATCGGCTCTTCGGGTCTAGCACGCTTTCTCCTTTTGGGTTGAG |
| FCmut4-Sap | GATCGGCTCTTCGACCAAGAGTTTAGTTTTCAGG |
| FCmut5-Sap | GATCGGCTCTTCGGGGCGTGCACTTGTGTTGGGCTTTTTATTCTATCG |
| FCmut6-Sap | GATCGGCTCTTCGCCCGAAAGTTTACTTTTTATCATTACTCAAG |
| FCmut11-Sap | GATCGGCTCTTCGGAGCTTACTCTGTGCTAAAAATAAAGGTATTATCG |
| TR3B | GATCGGGATCCTAATCGCGGCAACCCAGCCAA |
| TR3E | GATCGGAATTCCGATTGAATCGACGTTGATCC |
| TR3N1 | GATCGGCGGCCGCGATCCATATTCGCTGCATGGA |
| TR3N2 | GATCGGCGGCCGCAAAGCCTTAGAGGCTAACGGAT |
Construction of in-frame deletion strains.
The deletions were obtained by PCR amplifying, from C6706 str2, ∼500-bp DNA fragments flanking the gene of interest while retaining several codons from the 5′ and 3′ ends of the gene fused in frame. The fragments were ligated into pKAS154 (27), and the different genes were deleted from the V. cholerae chromosome by allelic exchange (28). The deletion of breR was obtained using TR3B with TR3N2 and TR3N1 with TR3E. The constructs were confirmed by DNA sequencing.
Construction of the PbreAB-lacZ fusion.
The pGKK346 plasmid was linearized with XbaI between the chromate homology fragment and the promoterless lacZ gene (17). Approximately 500 bp of the breAB promoter region was amplified by PCR using FC70 with FC71. The resulting fragment was digested with XbaI and ligated into the linearized pGKK346 plasmid, generating pFC43. The lacZ fusions were transferred into the chromosome of a V. cholerae ΔlacZ strain by allelic exchange (28) between the chr and gal loci. All of the constructs were confirmed by DNA sequencing.
Introduction of base pair changes into the PbreR-lacZ and PbreAB-lacZ fusions.
The mutations were introduced into the R, the ABproximal (ABp), and the ABdistal (ABd) binding sites by PCR amplifying fragments from C6706 str2 using overlapping primers containing the site for the SapI enzyme. The A-50G/A-44G changes in the R binding site were obtained using FC68 with FCmut11-Sap and FC69 with FCmut2-Sap. The A-52G/T-49C/A-46G changes in the ABp binding site were obtained using FC70 with FCmut5-Sap and FC71 with FCmut6-Sap, and the A-223G/T-220C/A-217G changes in the ABd binding site were obtained using FC70 with FCmut3-Sap and FC71 with FCmut4-Sap. The resulting fragments were then digested with XbaI and SapI and ligated into a linearized (XbaI) pGKK346 plasmid, generating pFC38 (ABd mutations), pFC39 (ABp mutations), and pFC42 (R mutations). The double ABd and ABp mutations were obtained by PCR amplifying fragments from pFC38 using FC71 with FCmut6-Sap and FC70 with FCmut5-Sap. The fragments were digested with XbaI and SapI and ligated into a linearized (XbaI) pGKK346, generating pFC40. After screening for the correct orientation of the fragment, the lacZ fusions were transferred into the chromosome of a V. cholerae ΔlacZ strain by allelic exchange between the chr and gal loci. All of the constructs were confirmed by DNA sequencing.
β-Galactosidase assays.
Different V. cholerae strains harboring the wild-type or mutated PbreR-lacZ or PbreAB-lacZ transcriptional fusion were grown for 15 h in LB medium at 37°C with aeration. The cultures were then diluted 100-fold into LB medium with or without crude bile (0.4%) and were grown at 37°C with aeration until the OD600 of the culture had reached 0.8 to 1.0. β-Galactosidase assays were carried out as previously described (26).
EMSA.
The R3, R4, R5, and R6 fragments were amplified from C6706 str2 using primers pairs FC35 and TR3N1, FC18F and TR3N1, FC39 and FC41, and FC40 and FC41, respectively. The AB3, AB4, AB5, AB6, AB7, AB8, AB9, AB10, AB11, and AB12 fragments were amplified from C6706 str2 using primer pairs FC30 and FC25, FC31 and FC25, FC13F and FC46, FC13F and FC45, FC13F and FC36, FC24 and FC26, FC24 and FC27, FC31 and FC37, FC31 and FC47, and FC48 and FC37, respectively. Following amplification, these fragments were 3′ end labeled with digoxigenin (DIG) as previously described (29). The wild-type R1, AB1, and AB2 fragments were amplified from C6706 str2 using the DIG-labeled primer pairs FC7DIG and FC8DIG, FC3DIG and FC4DIG, and FC1DIG and FC2DIG, respectively. The mutated R1, AB1, and AB2 fragments were amplified from pFC42, pFC39, and pFC38, respectively, using FC7DIG and FC8DIG, FC3DIG and FC4DIG, and FC1DIG and FC2DIG, respectively. All of the fragments, including those amplified with DIG-labeled primers, were gel purified as previously described (29). Binding reactions were performed using formerly purified BreR-His6 (17) with different fragments in the presence of the nonspecific competitor poly(dI-dC), followed by electrophoresis in 6% polyacrylamide gels as previously described (22). The DNA was transferred, probed, and detected as previously described (30).
DNase I footprinting.
DNA fragments for footprinting assays were amplified from C6706 str2 by PCR. A 230-bp fragment carrying the ABd binding site was amplified using FC42F with FC42R, a 246-bp fragment harboring ABp was amplified using FC43F with FC43R, and a 235-bp fragment containing the R binding site was amplified using FC44F with FC44R. These fragments were ligated into pBluescript (Stratagene), generating pFC29, pFC30, and pFC31, respectively. For coding strand labeling, the inserts were excised with XbaI and EcoRI. For noncoding strand labeling, the inserts were excised with BamHI and HindIII. The fragments were gel purified (Qiagen), treated with shrimp alkaline phosphatase (NEB), and then end labeled with [γ-33P]ATP (3,000 Ci/mmol; NEN) using T4 polynucleotide kinase (Amersham Biosciences). Single end-labeled fragments were obtained by digestion with BamHI (coding strands) and EcoRI (noncoding strands). Binding reactions of BreR to the radiolabeled fragments were performed using the binding conditions defined above for EMSA. After incubation, the fragments were treated with various DNase I (Ambion) concentrations and loaded onto 6% acrylamide sequencing gels as previously described (29). The gels were dried and visualized by autoradiography.
Colonization assays.
The infant mouse competition assays were performed essentially as previously described (31). Suckling CD-1 mice (3 to 5 days old; Charles River) were inoculated orally, and the total CFU were obtained from the small intestine of four to six mice after 24 h by plating intestinal homogenates on streptomycin X-Gal plates.
RESULTS
BreR binds to a single site between −55 and −24 in the breR promoter.
To learn more about BreR regulation at the breR promoter, we sought to identify the DNA elements involved in BreR binding. Regulators belonging to the TetR family usually bind as homodimers to regions of complete or partial dyad symmetry (32). BreR was previously shown to bind to the breR promoter fragment R1 (−102 to 131) (17). In order to determine which region(s) of the breR promoter is critical for BreR binding, the R1 fragment was examined for the presence of regions of dyad symmetry using the Vienna RNA Secondary Structure Prediction program (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). Figure 1A shows, with arrows, putative regions of dyad symmetry in the R1 fragment (ds1, ds2, and ds3) as predicted by the Vienna program and in a size range typical of regulatory protein binding sites. Based on these predictions, we generated various fragments to delineate the minimal segments containing regions of dyad symmetry that are essential for BreR binding at the breR promoter (Fig. 1A).
Fig 1.

Mapping of the BreR binding site within the breR promoter region using EMSA and dyad symmetry prediction. (A) Schematic representation of the R1 fragment showing the regions of dyad symmetry predicted by the Vienna program. Putative regions of dyad symmetry (ds1, ds2, and ds3) are indicated by gray or black arrows. Various DNA fragments (R3, R4, and R5) used for EMSA are shown by gray boxes. The transcriptional start site is indicated by a black arrow. The ATG start codon and putative −35 and −10 regions are also shown. (B) EMSA showing BreR binding to breR promoter region fragments R3, R4, and R5. In each panel, 10 ng DIG-dUTP-labeled DNA was incubated with 0 (first lane), 50 (second lane) or 250 ng (third lane) of purified BreR-His6 prior to electrophoresis.
To localize the BreR binding site at the breR promoter, EMSAs were performed using the R3 (−55 to +131), R4 (−77 to +60), and R5 (−41 to +60) fragments containing various regions of dyad symmetry (Fig. 1B). Increasing amounts of BreR caused a shift of the R3 and R4 fragments but not of the R5 fragment (Fig. 1B), indicating that the region from −55 to −41 containing the upstream halves of regions of dyad symmetry ds2 and ds3 is critical for BreR binding (Fig. 1).
DNase I footprinting analyses were performed to further localize the BreR binding site(s) in the breR promoter. BreR was incubated with a 235-bp radioactively labeled fragment extending from positions −158 to +81. This fragment showed a single region of strong protection, indicating that a single site is necessary for BreR protection at the breR promoter (Fig. 2A). On the coding strand, this site extended from positions −51 to −24 (Fig. 2A). On the noncoding strand, the protected region extended from −28 to −55 (Fig. 2A). Further examination of the protected region revealed the presence of a region of partial dyad symmetry (containing 2 mismatches) that consists of 9-bp half sites (Fig. 2B, indicated by gray arrows) separated by a 6-bp spacer. These results show that BreR DNase I protection overlaps both ds2 and ds3 regions of dyad symmetry and indicate that BreR directly represses breR expression by interacting with a binding site that overlaps the breR −35 region. The BreR binding site at the breR promoter will be referred to here as the R binding site.
Fig 2.

DNase I protection of the breR promoter by BreR. (A) The coding and noncoding strands of the 33P-end-labeled fragment from positions −158 to +81 were incubated with a 1:200 dilution of DNase I and no protein (NP; lanes 1 and 5), ∼150 ng BreR and a 1:200 DNase I dilution (lane 2) or a 1:400 DNase I dilution (lane 3), ∼300 ng BreR and a 1:200 dilution of DNase I (lane 4), and a G+A sequencing reaction (lane 6). The region of protection by BreR is indicated with a black bar. (B) Double-stranded DNA sequence showing the BreR-protected region at the breR promoter. The protected region for both strands is shaded in gray, and the endpoints of the protection above and below the shaded regions are labeled (−55 to −24). A region with partial dyad symmetry, involved in DNA binding, is indicated by gray arrows. The transcriptional start site (+1) (17) and putative −35 and −10 regions are highlighted in boldface and are underlined.
BreR binds to a proximal (−56 and −26) and a distal (−228 to −196) site in the breAB promoter.
In a previous study, a breAB promoter region (−382 to +131) utilized for the lacZ transcriptional fusions was divided into two fragments, an ∼230-bp fragment named AB1 (−95 to +132) and an ∼300-bp fragment named AB2 (−382 to −76) (17). It was shown by EMSA that BreR binds to the AB1 and AB2 fragments, indicating that BreR binds at two independent binding sites at the breAB promoter (17). To further define the binding sites for each of the individual fragments, a series of breAB promoter fragments (distal fragments included AB3 [−297 to −76], AB4 [−198 to −76], AB5 [−382 to −179], AB6 [−382 to −210], and AB7 [−382 to −224]; proximal fragments included AB8 [−95 to +79], AB9 [−95 to +10], AB10 [−198 to −15], and AB11 [−198 to −38]) were used in gel mobility shift assays with increasing amounts of purified BreR (Fig. 3A and B). For the distal region, BreR bound to AB3 and AB5 but not AB4, AB6, or AB7 (Fig. 3B). This suggests that a region from −210 to −179 is critical for binding. For the proximal region, we observed that BreR bound to AB8, AB9, and AB10 but not AB11 (Fig. 3B). This result indicates that a region from −38 to −15 is critical for binding. As a whole, these binding patterns suggest that the regions from −95 to −15, containing ds11 and ds12, and from −297 to −179, containing ds5, ds6, and ds7, are critical for BreR binding to the proximal and distal sites, respectively (Fig. 3A and B). These data confirm our previous findings and establish that in EMSAs BreR binds the breAB promoter at both a proximal (named ABp) and a distal (named ABd) site with respect to the +1 position. Additionally, we generated the AB12 fragment containing both the ABp and the ABd binding sites. When the AB12 fragment was incubated with BreR, a double shift was observed at a low BreR concentration (50 ng), with the lower band having a much greater intensity than the upper band (Fig. 3B, second lane). At a higher BreR concentration (250 ng), the intensity of the upper band was significantly increased (Fig. 3B, third lane), supporting the hypothesis that BreR recognizes two independent sites present at the breAB promoter.
Fig 3.

Mapping of the distal and proximal BreR binding sites at the breAB promoter region using a series of DNA fragments. (A) Schematic representation of the ABd (−382 to −76) and ABp (−95 to +132) fragments. Putative regions of dyad symmetry (ds4, ds5, ds6, ds7, ds8, ds9, ds10, ds11, ds12, ds13, ds14, and ds15) predicted by the Vienna program are indicated by gray or black arrows. Various DNA fragments (AB3, AB4, AB5, AB6, AB7, AB8, AB9, AB10, AB11, and AB12) used for EMSA are shown by gray boxes. The transcriptional start site is indicated by a black arrow. The ATG start codon and putative −35 and −10 regions are also shown. (B) EMSA showing BreR binding to promoter region fragments. In each panel, 10 ng DIG-dUTP-labeled DNA was incubated with 0 (first lane), 50 (second lane), or 250 ng (third lane) of BreR-His6 prior to electrophoresis.
DNase I footprinting was used to further map the BreR binding sites within the breAB promoter using a 230-bp (−327 to −97) and a 246-bp (−167 to +78) radioactively labeled fragment containing the individual ABd and ABp sites, respectively. BreR protected a single region from DNase I digestion on each fragment (Fig. 4A and B). The ABd binding site extends from −224 to −196 and from −201 to −228 on the coding and noncoding strands, respectively (Fig. 4A). DNase I hypersensitivity in the presence of BreR was observed at position −195 in the noncoding strand (Fig. 4A). The ABd site contains a region of dyad symmetry that has a total of 10 mismatches and consists of 9-bp half sites separated by 6 bp (Fig. 4C, indicated by gray arrows).
Fig 4.

DNase I footprinting analyses for BreR at the breAB promoter. The coding and noncoding strands of two radioactively 33P-end-labeled fragments of 230 bp (−327 to −97) (A) and 246 bp (−167 to +78) (B) were incubated with a 1:200 dilution of DNase I and no protein (NP; lanes 1 and 5), ∼150 ng BreR and a 1:200 DNase I dilution (lane 2) or a 1:400 DNase I dilution (lane 3), ∼300 ng BreR and a 1:200 dilution of DNase I (lane 4), and a G+A sequencing reaction (lane 6). The region of protection by BreR and a region of hypersensitivity induced by BreR are indicated by thick black lines and a black arrow, respectively. (C) Double-stranded DNA sequence of the breAB promoter. BreR-protected regions are shaded in gray for both strands. Regions of partial dyad symmetry, involved in DNA binding, are shown for the ABd and ABp binding sites with gray arrows. The transcriptional start site (+1) (17), ATG start codon, and putative −35 and −10 regions are highlighted in boldface and are underlined.
The ABp binding site was located 141 bp downstream of the ABd site, and it extends from positions −54 to −26 and from −30 to −56 on the coding and noncoding strands, respectively (Fig. 4B). The ABp site overlaps the −35 region, similar to the R site at the breR promoter (Fig. 2 and 4), and contains a region of dyad symmetry that has a total of 4 mismatches and consists of 9-bp half sites separated by 6 bp (Fig. 4C, indicated by gray arrows). These results show that the ABp site contains the region of dyad symmetry ds11 predicted by the Vienna program (Fig. 3A), and the ABd site contains the regions ds6 and ds7. Additionally, they suggest that BreR represses breAB expression by interacting with the ABp site that overlaps the −35 region and the ABd site located 141 bp upstream of the ABp site.
Identification of conserved base pairs within the R, ABp, and ABd binding sites.
To determine if a BreR recognition consensus sequence could be discerned, we compared the BreR binding sites at the breR and breAB promoters. Sequence alignment of the protected nucleotides of the coding and noncoding strands of the R, ABp, and ABd sites was performed using DNASTAR software. Using a cutoff of 5/6 residue identity in any given position as consensus (Fig. 5A, shaded in gray tones), an 18-bp region of dyad symmetry separated by 6 bp (AANGTANAC-N6-GTNTACNTT) was found within the protected regions (Fig. 5A). Of these 18 bp, 6 are 100% conserved in the three binding sites (Fig. 5A, shaded in dark gray).
Fig 5.
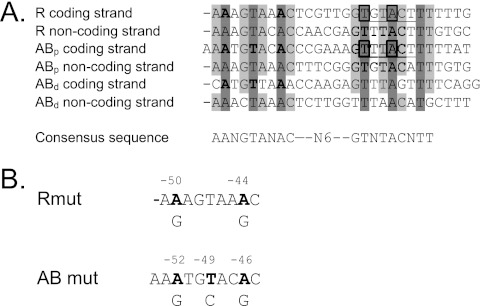
BreR binding consensus sequence. (A) Sequence alignment of the coding and noncoding strands of the R, ABp, and ABd binding sites. Conserved base pairs are shaded in gray tones. Base pairs selected for substitution are shaded in dark gray and are highlighted in boldface. The −35 regions at the R and ABp sites are underlined, and the consensus region is also shown. Boxes indicate conserved nucleotides within the −35 region of the R and AB promoters. (B) Schematic representation of the bases selected for substitutions (A-50 and A-44 in the R site, A-52, T-49, and A-46 in the ABp site, and A-223, T-220, and A-217 in the ABd site). Each mutated base is in boldface. The position number is shown above, and the base to which it was changed is shown below.
Mutations at the R, ABp, and ABd binding sites prevent BreR binding in vitro.
The nucleotides identified in the binding site alignment that were 100% conserved (Fig. 5A, shaded in dark gray) were then mutated to investigate their importance for BreR binding at the R, ABp, and ABd binding sites. Two of the six conserved nucleotides were within the −35 consensus region in the breR and breAB promoters (Fig. 5A, boxed nucleotides). In order to maintain the integrity of the −35 region, mutations were only made in the conserved base pairs located in the half site upstream of the −35 site (Fig. 5A, shaded in dark gray and highlighted in boldface). The A residues at positions −50 and −44 were changed to G residues (A-50G/A-44G; named Rmut) within the breR promoter (Fig. 5A, shaded in dark gray and highlighted in boldface, and B, highlighted in boldface). Base pair substitutions within the breAB promoter were performed in the ABp (A-52G/T-49C/A-46G; named ABpmut) and ABd (A-223G/T-220C/A-217G; named ABdmut) binding sites (Fig. 5A, shaded in dark gray and highlighted in boldface, and B, highlighted in boldface). The mutations were incorporated into the wild-type R1 (−102 to +131), AB1 (−95 to +132), and AB2 (−382 to −76) fragments that BreR is capable of binding as previously determined by EMSA (17). The A-50G/A-44G changes in the R1 fragment (breR promoter) prevented BreR binding at both small (50 ng) and large (250 ng) amounts of BreR (Fig. 6A, lanes 5 and 6), whereas the same amounts of protein were capable of shifting the wild-type fragment (Fig. 6A, lanes 2 and 3). These results establish that the base pair substitutions at the R site are important for BreR binding at this site in vitro. At the breAB promoter, the A-52G/T-49C/A-46G mutations in the ABp site prevented BreR from binding to the AB1 fragment at small and large protein amounts (Fig. 7A, lanes 5 and 6) that were able to shift the wild-type AB1 fragment (Fig. 7A, lanes 2 and 3). The A-223G/T-220C/A-217G changes within the ABd site also prevented BreR from binding to the AB2 fragment at small and large protein amounts (Fig. 7A, lanes 11 and 12), while the same amounts of BreR could bind the wild-type AB2 fragment (Fig. 7A, lanes 8 and 9). These data suggest that the mutations at the ABp and ABd sites are important for BreR binding in vitro. Further analysis of BreR binding at the wild-type AB1 and AB2 fragments indicates that 250 ng of BreR fully shifted the AB1 fragment, containing the ABp site, compared to a partial shift of the AB2 fragment, containing the ABd site (Fig. 7A). This is also observed with the wild-type AB3 and AB6 fragments (Fig. 2C). These results suggest that, in vitro, BreR affinity for the ABd site is lower than the affinity for the ABp site.
Fig 6.

Mutational analysis of the R binding site at the breR promoter. (A) BreR binding to the wild-type and mutated (Rmut) R1 fragment. EMSA was performed using DIG-dUTP-labeled DNA (10 ng) incubated with 0 (lanes 1 and 4), 50 (lanes 2 and 5), or 250 ng (lanes 3 and 6) of BreR-His6 prior to electrophoresis. (B) Expression of the PbreR-lacZ fusion carrying the wild-type or mutated promoter in different strain backgrounds. β-Galactosidase activity was measured by growing the strains in the absence or presence of 0.4% bile in LB at 37°C until the optical density at 600 nm of the cultures had reached 0.8 to 1.0. Results are from three independent experiments. Error bars indicate standard deviations.
Fig 7.

Mutational analysis of the ABp and ABd binding sites at the breAB promoter. (A) Binding of BreR to the wild-type and mutated (ABpmut and ABdmut) ABp and ABd fragments. EMSA was performed using DIG-dUTP-labeled DNA (10 ng) incubated with 0 (lanes 1, 4, 7, and 10), 50 (lanes 2, 5, 8, and 11), or 250 ng (lanes 3, 6, 9, and 12) of BreR-His6 prior to electrophoresis. (B) Influence of base pair mutations in PbreAB-lacZ expression in various strain backgrounds. β-Galactosidase activity was measured as described for Fig. 6.
Mutations within the R binding site affect BreR repression of the breR promoter in vivo.
To further investigate if the A-50G/A-44G changes that prevent BreR binding to the R1 fragment in vitro affected autoregulation in vivo, these mutations were introduced into a PbreR-lacZ transcriptional fusion. BreR repression of the breR promoter was assessed in wild-type and ΔbreR strains harboring a PbreR-lacZ fusion at the lacZ locus carrying the wild-type or mutated promoters. It has previously been reported that expression of breR is induced in response to bile and that deletion of BreR increases PbreR-lacZ expression in the absence and presence of bile compared to the response of the wild type, suggesting that expression of breR is obtained by BreR derepression (17). Similarly, the mutations that disrupt BreR binding also caused derepression of PbreR-lacZ in the absence and presence of bile. The A-50G/A-44G mutations increased PbreR-lacZ expression 9.5- and 2-fold in the absence and presence of bile, respectively, compared to the wild type (Fig. 6B). Deletion of breR in the presence of these mutations did not further derepress PbreR-lacZ expression, suggesting that these mutations cause a loss of BreR repression. This expression was not as high as that of the breR mutation, suggesting that these mutations alter basal expression by RNA polymerase. These data establish that repression of breR expression results from BreR binding to the R binding site (region from −50 to −24), and that conserved nucleotides A-50, A-44, or both are essential for BreR repression.
Mutations only in the ABp binding site affect BreR binding to the breAB promoter in vivo.
To determine if the mutations affecting BreR binding to the ABp and ABd sites in vitro alter BreR repression of breAB in vivo, the A-52G/T-49C/A-46G and -223G/T-220C/A-217G mutations were introduced into a PbreAB-lacZ transcriptional fusion. The influence of the mutations on BreR-mediated repression of breAB expression was determined using wild-type and ΔbreR strains harboring PbreAB-lacZ fusions carrying the wild-type or mutated promoters. As shown previously (17), Fig. 7B confirms that PbreAB-lacZ expression is higher in the presence of bile and that deletion of breR causes increased PbreAB-lacZ expression in the absence and presence of bile. Expression from the fusion containing the substitutions at the ABp region (A-52G/T-49C/A-46G [ABpmut]) showed a 19.7-fold increase in the absence of bile and a 1.8-fold increase in the presence of bile compared to the wild type, indicating a critical role for the ABp binding site in vivo (Fig. 7B). Introduction of the breR mutation into this background did not further derepress PbreAB-lacZ expression, suggesting that the mutations in the ABp site cause a loss of BreR repression (Fig. 7B). The mutations at the ABp site did not increase PbreAB-lacZ expression to the levels observed in the ΔbreR strain, suggesting that these mutations alter basal expression. Expression from the PbreAB-lacZ fusion containing the mutations in the ABd region (A-223G/T-220C/A-217G [ABdmut]) in the wild-type and ΔbreR backgrounds was similar to that of the wild-type promoter lacZ fusion grown with or without bile in the wild-type and ΔbreR strains (Fig. 7B). These results suggest that disruption of BreR binding at this site does not influence the repression of PbreAB-lacZ expression by BreR in vivo. A PbreAB-lacZ fusion containing mutations at both the ABp and ABd regions was also defective for binding, and lacZ expression was similar to that obtained with the promoter lacZ fusion carrying the mutations in the ABp site (Fig. 7B). These results indicate that BreR represses breAB expression in vivo by binding exclusively to the ABp binding site (region from −54 to −26), and that binding at this region can be disrupted by a 3-bp change at positions −52, −49, and −46.
Deletion of breR does not affect colonization in the infant mouse model.
It has previously been shown that in the absence of BreR, the breAB genes are overexpressed (17) and there is more resistance to cholate when this strain is grown in liquid media compared to that of the wild type (data not shown). To determine if BreR plays a role in an in vivo infection model, the ΔbreR mutant was competed against a wild-type strain in the infant mouse colonization model. The ΔbreR mutant was not reduced in colonization compared to the wild type (data not shown). These results indicate that BreR does not play a role in intestinal colonization in this model, and this may be attributed to the fact that the infant mouse does not produce bile.
DISCUSSION
The TetR regulator, BreR, and the efflux system BreAB have been identified as being important in the response of V. cholerae to bile (17). The work presented here begins to define the regulatory mechanisms employed by BreR to regulate the expression of the breR gene and the breAB operon.
Since most of the TetR family regulators bind to their binding sites (complete or partial regions of dyad symmetry) as dimers (32), we initially searched for regions of dyad symmetry present at the breR and breAB promoters using a program that predicts RNA secondary structures. EMSA studies using these predictions followed by DNase I footprinting showed that BreR binds at the breR promoter (R binding site) at a unique region from position −55 to −24 and binds to the breAB promoter at two independent sites, a proximal site (ABp) located at −56 to −26 and a distal site (ABd) located at −228 to −196. A DNase I hypersensitive site was induced by BreR at the ABd binding site (Fig. 4A), indicating a change in DNA conformation upon BreR binding. When the coding and noncoding sequences of these binding sites were aligned, the analysis revealed that BreR recognizes the consensus sequence AANGTANAC-N6-GTNTACNTT. This sequence contains 18 conserved base pairs that are symmetrical, suggesting that BreR binds as a homodimer, as has been shown for other TetR family repressors (32).
In order to genetically determine if the consensus site defined requirements for BreR binding, substitutions were performed on consensus base pairs that were 100% conserved. Introduction of the A-50G/A-44G base pair mutations into the breR promoter completely abolished BreR binding to the breR promoter and prevented transcriptional repression by BreR. These data established that BreR binds the breR promoter region at a position that overlaps the −35 consensus site. Mutations at the ABp site (A-52G/T-49C/A-46G) inhibited BreR binding to this site and eliminated repression at the breAB promoter. The base pair changes at the ABd site (A-223G/T-220C/A-217G) prevented BreR binding to ABd; however, they did not prevent breAB transcriptional repression by BreR, suggesting that the ABd site does not play a key role in BreR repression in vivo. A possible explanation for why the ABd site did not show a role in vivo is based on the position of this site. The ABd site is located 158 bp upstream of the −35 consensus region, suggesting that although BreR can bind to the ABd site, it is excessively distant in order to interfere with RNA polymerase binding. Another explanation could be based on the sequence of the ABd binding site. While the R and ABp sites display a high degree of sequence similarity to the BreR consensus sequence (Fig. 5), the ABd site exhibits four mismatches (Fig. 5). This observation suggests that even though BreR can bind to the ABd site in vitro, this interaction is of low affinity compared to those of the other BreR binding sites. This is supported by the fact that 250 ng of BreR protein completely shifted the wild-type AB1 fragment (−95 to +132) compared to a lower percentage of shift for the wild-type AB2 fragment (−382 to −76) (Fig. 7A) (17). Overall, it appears that BreR represses breAB expression by binding at a single site (ABp) in the breAB promoter that overlaps the −35 region. Moreover, the mutations generated at the R and ABp sites abolish BreR binding (Fig. 2B and 4C), suggesting that these base pairs play an important role in BreR binding.
Double or triple mutations in the dyad symmetry of the R and ABp binding sites in the wild-type and ΔbreR backgrounds significantly reduced the level of BreR binding, resulting in overexpression of the mutated PbreR-lacZ and PbreAB-lacZ fusions in vivo. However, overexpression from these promoters was not as high as the overexpression from the ΔbreR strain harboring either PbreR-lacZ or PbreAB-lacZ wild-type lacZ transcriptional fusions (Fig. 6B and 7B). A partial overexpression could be attributed to the fact that the mutations were localized 9 to 15 bp upstream of the −35 region, and they could affect expression by influencing RNA polymerase interaction with the DNA or alter the binding site of an unknown activator. Another possible explanation arises when comparing the shift of the Rmut and ABpmut fragments induced by addition of large amounts of BreR (250 ng) (Fig. 6A and 7A). The comparison shows that there is a slight but detectable shift with both mutated probes in the presence of 250 ng of BreR, suggesting that the mutations do not abolish BreR binding completely. This also could take place in vivo, explaining the expression levels of the mutated PbreR-lacZ and PbreAB-lacZ (Fig. 6A and 7A).
A number of TetR regulators, such as VceR (33, 34), CmeR (22), MexL (35), MexZ (36), TtgV (37), LmrA (25), TtgR (38), QacR (39), MtrR (40), and TcmR (41), also repress the expression of genes encoding efflux pumps by binding to complete or partial regions of dyad symmetry within the promoter region of their target genes (18, 32). Some of the operator sequences overlap both the −35 and −10 consensus sites (35–38, 40, 41), whereas others overlap only the −10 (25, 33, 39) or the −35 region (22), indicating that they most probably block RNA polymerase binding. The location of the R and ABp binding sites, overlapping the −35 region at the breR and breAB promoters, respectively, is consistent with the role of BreR as a negative regulator and shows a binding pattern shared by other TetR repressors. It is most likely that BreR inhibits initiation of transcription by physically preventing RNA polymerase access to the breR and breAB promoter region.
Since the expression of breR and breAB is controlled by bile, we propose the following regulation model. In the absence of bile, BreR binds to the consensus sequence AANGTANAC-N6-GTNTACNTT overlapping the −35 region of the breR and breAB promoters. As a result, BreR inhibits initiation of transcription by blocking RNA polymerase binding and maybe binding of an activator. In the presence of bile, V. cholerae has to be able to resist its bactericidal effect, therefore it utilizes bile as a signal to turn on the breAB resistance genes. In addition, breR expression is also turned on to tightly control breAB expression to prevent extrusion of vital cellular metabolites and loss of membrane potential. We have previously proposed that certain bile salts (cholate, deoxycholate, and/or glycodeoxycholate) bind BreR, inducing a conformational change that disrupts its interaction with DNA (17), identified here as the R and ABp binding sites. As a consequence, expression of breAB takes place and the toxic compounds are extruded out of the cell, upon which BreR is available to rapidly repress breAB gene expression and prevent further extrusion of critical metabolites or loss of membrane potential.
ACKNOWLEDGMENTS
We thank Emily Stonehouse for helpful discussions and critical reading of the manuscript.
This work was supported by NIH grant AI039654 and NSF grant OCN-0120677.
Footnotes
Published ahead of print 9 November 2012
REFERENCES
- 1. Champion GA, Neely MN, Brennan MA, DiRita VJ. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23: 323– 331 [DOI] [PubMed] [Google Scholar]
- 2. Withey JH, DiRita VJ. 2006. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 59: 1779– 1789 [DOI] [PubMed] [Google Scholar]
- 3. Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181: 4250– 4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 75: 5542– 5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Begley M, Gahan CG, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol. Rev. 29: 625– 651 [DOI] [PubMed] [Google Scholar]
- 6. Hofmann AF. 1998. Bile secretion and the enterohepatic circulation of bile salts, 6th ed. W. B. Saunders, Philadelphia, PA: [Google Scholar]
- 7. Gunn JS. 2000. Mechanisms of bacterial resistance and response to bile. Microbes Infect. 2: 907– 913 [DOI] [PubMed] [Google Scholar]
- 8. Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 65: 1131– 1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schuhmacher DA, Klose KE. 1999. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 181: 1508– 1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. 2006. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol. Microbiol. 59: 193– 201 [DOI] [PubMed] [Google Scholar]
- 11. Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. 2006. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch. Microbiol. 186: 171– 181 [DOI] [PubMed] [Google Scholar]
- 12. Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc. Natl. Acad. Sci. U. S. A. 97: 10220– 10224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect. Immun. 68: 1491– 1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chatterjee A, Chaudhuri S, Saha G, Gupta S, Chowdhury R. 2004. Effect of bile on the cell surface permeability barrier and efflux system of Vibrio cholerae. J. Bacteriol. 186: 6809– 6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect. Immun. 69: 4681– 4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colmer JA, Fralick JA, Hamood AN. 1998. Isolation and characterization of a putative multidrug resistance pump from Vibrio cholerae. Mol. Microbiol. 27: 63– 72 [DOI] [PubMed] [Google Scholar]
- 17. Cerda-Maira FA, Ringelberg CS, Taylor RK. 2008. The bile response repressor BreR regulates expression of the Vibrio cholerae breAB efflux system operon. J. Bacteriol. 190: 7441– 7452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grkovic S, Brown MH, Skurray RA. 2002. Regulation of bacterial drug export systems. Microbiol. Mol. Biol. Rev. 66: 671– 701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li XZ, Nikaido H. 2004. Efflux-mediated drug resistance in bacteria. Drugs 64: 159– 204 [DOI] [PubMed] [Google Scholar]
- 20. Aires JR, Kohler T, Nikaido H, Plesiat P. 1999. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 43: 2624– 2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagman KE, Shafer WM. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J. Bacteriol. 177: 4162– 4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin J, Akiba M, Sahin O, Zhang Q. 2005. CmeR functions as a transcriptional repressor for the multidrug efflux pump CmeABC in Campylobacter jejuni. Antimicrob. Agents Chemother. 49: 1067– 1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. 1996. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 19: 101– 112 [DOI] [PubMed] [Google Scholar]
- 24. Namwat W, Lee CK, Kinoshita H, Yamada Y, Nihira T. 2001. Identification of the varR gene as a transcriptional regulator of virginiamycin S resistance in Streptomyces virginiae. J. Bacteriol. 183: 2025– 2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshida K, Ohki YH, Murata M, Kinehara M, Matsuoka H, Satomura T, Ohki R, Kumano M, Yamane K, Fujita Y. 2004. Bacillus subtilis LmrA is a repressor of the lmrAB and yxaGH operons: identification of its binding site and functional analysis of lmrB and yxaGH. J. Bacteriol. 186: 5640– 5648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. Kovacikova G, Skorupski K. 2002. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol. Microbiol. 46: 1135– 1147 [DOI] [PubMed] [Google Scholar]
- 28. Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169: 47– 52 [DOI] [PubMed] [Google Scholar]
- 29. Kovacikova G, Skorupski K. 2001. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41: 393– 407 [DOI] [PubMed] [Google Scholar]
- 30. Kovacikova G, Lin W, Skorupski K. 2005. Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol. Microbiol. 57: 420– 433 [DOI] [PubMed] [Google Scholar]
- 31. Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 84: 2833– 2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69: 326– 356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alatoom AA, Aburto R, Hamood AN, Colmer-Hamood JA. 2007. VceR negatively regulates the vceCAB MDR efflux operon and positively regulates its own synthesis in Vibrio cholerae 569B. Can. J. Microbiol. 53: 888– 900 [DOI] [PubMed] [Google Scholar]
- 34. Borges-Walmsley MI, Du D, McKeegan KS, Sharples GJ, Walmsley AR. 2005. VceR regulates the vceCAB drug efflux pump operon of Vibrio cholerae by alternating between mutually exclusive conformations that bind either drugs or promoter DNA. J. Mol. Biol. 349: 387– 400 [DOI] [PubMed] [Google Scholar]
- 35. Chuanchuen R, Gaynor JB, Karkhoff-Schweizer R, Schweizer HP. 2005. Molecular characterization of MexL, the transcriptional repressor of the mexJK multidrug efflux operon in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49: 1844– 1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsuo Y, Eda S, Gotoh N, Yoshihara E, Nakae T. 2004. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 238: 23– 28 [DOI] [PubMed] [Google Scholar]
- 37. Rojas A, Segura A, Guazzaroni ME, Teran W, Hurtado A, Gallegos MT, Ramos JL. 2003. In vivo and in vitro evidence that TtgV is the specific regulator of the TtgGHI multidrug and solvent efflux pump of Pseudomonas putida. J. Bacteriol. 185: 4755– 4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Teran W, Felipe A, Segura A, Rojas A, Ramos JL, Gallegos MT. 2003. Antibiotic-dependent induction of Pseudomonas putida DOT-T1E TtgABC efflux pump is mediated by the drug binding repressor TtgR. Antimicrob. Agents Chemother. 47: 3067– 3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grkovic S, Brown MH, Roberts NJ, Paulsen IT, Skurray RA. 1998. QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J. Biol. Chem. 273: 18665– 18673 [DOI] [PubMed] [Google Scholar]
- 40. Lucas CE, Balthazar JT, Hagman KE, Shafer WM. 1997. The MtrR repressor binds the DNA sequence between the mtrR and mtrC genes of Neisseria gonorrhoeae. J. Bacteriol. 179: 4123– 4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guilfoile PG, Hutchinson CR. 1992. The Streptomyces glaucescens TcmR protein represses transcription of the divergently oriented tcmR and tcmA genes by binding to an intergenic operator region. J. Bacteriol. 174: 3659– 3666 [DOI] [PMC free article] [PubMed] [Google Scholar]