

Abstract
Autophagy is now known to be an essential component of host innate and adaptive immunity. Several herpesviruses have developed various strategies to evade this antiviral host defense. Herpes simplex virus 1 (HSV-1) blocks autophagy in fibroblasts and in neurons, and the ICP34.5 protein is important for the resistance of HSV-1 to autophagy because of its interaction with the autophagy machinery protein Beclin 1. ICP34.5 also counteracts the shutoff of protein synthesis mediated by the double-stranded RNA (dsRNA)-dependent protein kinase PKR by inhibiting phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α) in the PKR/eIF2α signaling pathway. Us11 is a late gene product of HSV-1, which is also able to preclude the host shutoff by direct inhibition of PKR. In the present study, we unveil a previously uncharacterized function of Us11 by demonstrating its antiautophagic activity. We show that the expression of Us11 is able to block autophagy and autophagosome formation in both HeLa cells and fibroblasts. Furthermore, immediate-early expression of Us11 by an ICP34.5 deletion mutant virus is sufficient to render the cells resistant to PKR-induced and virus-induced autophagy. PKR expression and the PKR binding domain of Us11 are required for the antiautophagic activity of Us11. However, unlike ICP34.5, Us11 did not interact with Beclin 1. We suggest that the inhibition of autophagy observed in cells infected with HSV-1 results from the activity of not only ICP34.5 on Beclin 1 but also Us11 by direct interaction with PKR.
INTRODUCTION
Macroautophagy (here referred to as autophagy) is an evolutionarily conserved self-eating mechanism (1). The process starts with the formation of a vacuole, known as the autophagosome, that sequesters cytoplasmic components and subsequently fuses with a lysosome. Autophagosome formation is dependent on the hierarchical activity of ATG (autophagy)-related proteins (2). Autophagy is now known to be essential for tissue homeostasis and development, and defective autophagy is associated with a number of diseases (3, 4). During the course of an infection, viruses often interact with proteins that execute autophagy. Autophagy and/or autophagy genes probably have both antiviral and proviral effects in the life cycles and pathogenesis of many different virus families (5–7). With respect to their antiviral effects, the autophagy proteins target viral components or virions for lysosomal degradation in a process termed xenophagy (8), and they also play a role in initiating innate and adaptive immune system responses to viral infections (5, 9). In response to the antiviral effect of autophagy, some viruses encode virulence factors that interact with the host's autophagy machinery and block the execution of autophagy (6, 10). In contrast, other viruses appear to utilize components of the autophagic machinery to foster their own intracellular growth or nonlytic cellular egress (6).
Viruses of the Herpesviridae family have developed strategies to downregulate autophagy although the varicella-zoster virus does not seem to encode any autophagy inhibitors (11, 12). The herpes simplex virus 1 (HSV-1) ICP34.5 protein (13), the viral homologs of Bcl-2 of Kaposi's sarcoma herpesvirus and murine gammaherpesvirus 68 (γHV-68) (14, 15), and the human cytomegalovirus (HCMV) TRS1 protein (16) have all been shown to block the formation of autophagosomes through their interactions with the autophagy protein Beclin 1. Beclin 1 is a critical component of several highly regulated complexes that control the formation and maturation of autophagosomes. These viral proteins mimic the inhibitory effect of the cellular form of the Bcl-2 protein family (14). Another way that viruses could control autophagy would be to manipulate the host's protein synthesis machinery (17). Indeed, it is interesting that some signaling pathways that regulate autophagy are also known to control protein synthesis (18). For example, the mTOR kinase included in the mTOR complex 1 (mTORC1) and the eukaryotic translation initiation factor 2α (eIF2α) kinases, which control protein synthesis, are also known to be modulators of autophagy (18). Activation of mTORC1, by amino acids and growth factors, favors protein synthesis and represses autophagy, whereas activation of eIF2α kinases turns off protein translation and stimulates autophagy. All four members of the eIF2α kinase family block the initiation of translation by phosphorylating the eukaryotic translation initiation factor eIF2α in response to various stress situations (19). GCN2 is sensitive to amino acid starvation, the PKR-like endoplasmic reticulum kinase (PERK) responds to endoplasmic reticulum stress induced by the accumulation of unfolded proteins, the heme-regulated inhibitor (HRI) is activated in response to heme deficiency, and the interferon (IFN)-induced PKR kinase is activated by double-stranded RNA (dsRNA). PKR is known to be activated by many viruses because dsRNA is a frequent by-product of viral replication or a product of overlapping transcription from the compact genomes of DNA viruses. Moreover, PKR activation blocks viral protein synthesis and, consequently, stifles viral production.
Control of the host's protein machinery is essential for viral replication to occur, and herpesviruses are able to manipulate the mTORC1 and the PKR-eIF2α signaling pathways so as to seize control of the protein synthesis machinery (17). The HSV-1 protein ICP34.5 interacts with the cellular phosphatase PP1α to mediate the dephosphorylation of eIF2α and thus antagonizes the PKR signaling pathway (Fig. 1) (20). ICP34.5 is important in resisting the interferon (IFN)-induced inhibition of protein synthesis. However, HSV-1 encodes a second gene product, the Us11 protein, which is required for translation regulation late in the viral life cycle (21). Rather than performing redundant functions, it seems that ICP34.5 and Us11 fulfill unique roles at discrete points in the productive replication cycle (22). Us11 is an abundant viral protein produced late in the viral life cycle (it is a true-late or γ2 protein) which binds to dsRNA and physically associates with PKR (23, 24). Us11 can prevent PKR activation in response to either dsRNA or the PKR activator PACT and thereby preclude activation of the PKR/eIF2α signaling pathway (Fig. 1) (23, 25). Like ICP34.5, Us11 is a multifunctional protein that has been shown to protect HeLa cells from induced apoptosis, to downregulate the IFN-β pathway, and to inhibit 2′-5′ oligoadenylate synthetase, another IFN-induced antiviral enzyme (26–28). In addition, immediate-early expression of Us11 is able to compensate for ICP34.5 and so to counteract the shutoff of protein synthesis in a mutant virus with a deletion of 34.5 genes (Δ34.5 virus) (29). HSV-1 infection stimulates autophagy by activating the PKR/eIF2α signaling pathway (30); we therefore wondered whether Us11, which inhibits PKR, could also be an antiautophagic protein.
Fig 1.
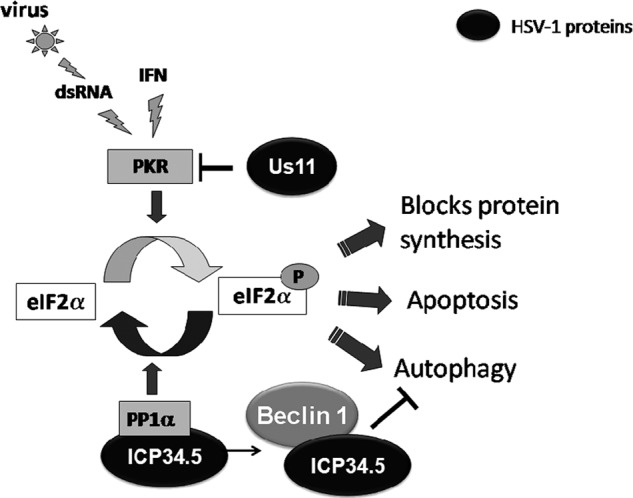
Schematic representation of PKR/eIF2α pathway. dsRNA, double-stranded RNA; IFN, interferon; PACT, PKR-activating protein; PKR, protein kinase RNA activated; PP1α, protein phosphatase 1α.
In this study, we investigated the role of Us11 in autophagy regulation by HSV-1. We found that expression of Us11 during viral infection or after ectopic expression is able to counteract the stimulation of autophagy by various different inducers, such as the transfection of dsRNA, in a manner independent of Beclin 1 and mTOR. The inhibition of autophagy by Us11 is dependent on its interaction with PKR. Moreover, the PKR binding domain and the N-terminal region of Us11 are both necessary for its antiautophagic activity.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were cultured at 37°C under 5% CO2 in RPMI medium supplemented with 10% fetal calf serum (FCS). Green fluorescent protein (GFP)-LC3 stably transfected HeLa cells (31) were provided by Aviva Tolkovsky (Cambridge Centre for Brain Repair, Cambridge, United Kingdom) and were grown in RPMI medium–10% FCS with 500 μg/ml of G418. Wild-type (wt) and PKR-null (PKR+/+ and PKR−/−, respectively) mouse embryonic fibroblasts (MEFs), kindly provided by B. R. G. Williams (Monash University, Victoria, Australia), were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS. The wild-type viruses used in this study were the KOS strain and the F strain of HSV-1 and were propagated in Vero cells. The recombinant viruses R3616 and R5104, which are described elsewhere (21), were a kind gift from Bernard Roizman (University of Chicago, Chicago, IL). R3616 lacks 1,000 bp in the domain of both copies of the HSV-1 γ134.5 gene. R5104 derives from R5103 and carries a chimeric DNA fragment containing the Us10 gene and the promoter of the α47 gene fused to the coding domain of the Us11 gene. R5103 lacks the γ134.5, Us8, -9, -10, and -11 and α47 (Us12) genes.
Plasmids.
The GFP-LC3 expression vector and LC3 tagged with GFP and monomeric red fluorescent protein (RFP) in tandem (mRFP-GFP-LC3) were kindly provided by Tamotsu Yoshimori (Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) (32). The GFP-LC3ΔG plasmid was a kind gift from Isei Tanida (Juntendo University School of Medicine, Tokyo, Japan). The Flag-ICP34.5 plasmid was a gift from Bin He (University of Illinois, Chicago, IL) (33). The Us11 plasmid was constructed by inserting the Us11 open reading frame, amplified from plasmid pRB4766 (29) provided by B. Roizman, into a pCDNA3 expression vector (Invitrogen). Hemagglutinin (HA)-tagged Us11 (HA-Us11) full-length and truncation constructs were made by amplification from the Us11 plasmid with different PCR primers (Table 1). The PCR products were inserted into a pSG5-HA plasmid using EcoRI and BglII restriction enzymes. Transfections were performed using FuGENE HD transfection reagent (Roche), as previously described (16).
Table 1.
Vectors and primers used to construct plasmids containing various different truncated forms of Us11
| Plasmid | Vector | Forward primerb | Reverse primerb | Restriction enzymes |
|---|---|---|---|---|
| Us11 | pCDNA3 | CATAAGCTTATGAGCCAGACCCAACCC | CAGAATTCCTATACAGACCCGCGAGCC | HindIII-EcoRI |
| HA-Us11_1–152 (FL)a | pSG5-HA | CAGAATTCGCATGAGCCAGACCCAACCC | ATAAGATCTCTATACAGACCCGCGAGCC | EcoRI-BglII |
| HA-Us11_1–91 | pSG5-HA | CAGAATTCGCATGAGCCAGACCCAACCC | ATAAGATCTCTAGGGAACACGCGGTGTCCT | EcoRI-BglII |
| Us11_1–121 | pSG5-HA | CAGAATTCGCATGAGCCAGACCCAACCC | ATAAGATCTCTAGGGGTCACGCGGTACCCT | EcoRI-BglII |
| Us11_40–152 | pSG5-HA | CAGAATTCGCCGCATGATCTCCGGACCC | ATAAGATCTCTATACAGACCCGCGAGCC | EcoRI-BglII |
| Us11_66–152 | pSG5-HA | CAGAATTCGCGTCGGTGCGGACACTACG | ATAAGATCTCTATACAGACCCGCGAGCC | EcoRI-BglII |
| Us11_90–152 | pSG5-HA | CAGAATTCGCGTTCCCCGGGAGCCCCGG | ATAAGATCTCTATACAGACCCGCGAGCC | EcoRI-BglII |
FL, full-length.
Restriction sites are underlined.
Antibodies.
To detect HSV-1-infected cells, we used a murine monoclonal antibody directed against the viral protein ICP0, purchased from Santa Cruz. The murine monoclonal antibody against Us11 was provided by B. Roizman (34). To detect Flag-tagged ICP34.5 and HA-tagged Us11 constructions, we used rabbit antibodies directed against Flag or HA (Cell Signaling Technology). Antibodies against eIF2α, phosphorylated eIF2α (Ser51), PKR (all, Cell Signaling Technology), phosphorylated PKR (Thr 451) (Millipore), Beclin 1 and Bcl-2 (both, Santa Cruz), and actin and LC3B (both, Sigma) were used in this study. For immunofluorescence studies, an antibody against LC3 was purchased from Cell Signaling (reference 2775). Phosphorylated 4E-BP1 (Thr37 and Thr46), 4E-BP1, phosphorylated p70S6K (Thr389), and p70S6K were purchased from Cell Signaling. For immunoprecipitation of Beclin-1, we used a goat antibody provided by Santa Cruz. Tetramethyl rhodamine isothiocyanate (TRITC)-conjugated or horseradish peroxidase-labeled goat anti-mouse or anti-rabbit secondary antibodies were purchased from Jackson. Alexa Fluor 350 donkey anti-mouse secondary antibody was obtained from Invitrogen.
PKR stimulation by poly(I · C).
Poly(I · C), an artificial dsRNA [poly(I)-poly(C)] purchased from Sigma, was transfected into cells using a liposome-mediated procedure to mimic the intracytoplasmic dsRNA generated during viral replication and to stimulate the PKR/eIF2α pathway. Oligofectamine (Invitrogen)-poly(I · C) mixes were prepared according to the manufacturer's instructions and added to cells for 4 h. After poly(I · C) treatment, the cells were fixed or lysed.
HSV-1 infection of HeLa cells.
HeLa or GFP-LC3 HeLa cells were grown in RPMI medium supplemented with 10% FCS (plus G418 for the GFP-LC3 cells). HSV-1 in serum-free RPMI medium was added to the cells at a multiplicity of infection (MOI) of 1 for adsorption for 1 h at 37°C. For mock-infected cells, serum-free RPMI medium alone was added to the cells for the same length of time. After the inoculum was removed, the cells were maintained in RPMI medium with 10% FCS, and autophagy was induced for the last 4 h of infection. Starvation-induced autophagy was carried out by culturing the cells in Earle's balanced salt solution (EBSS; Gibco) for 4 h before fixation.
Coimmunoprecipitation assays.
To study the interaction between Us11 and Beclin 1, HeLa cells cultured in six-well tissue culture plates were transfected to coexpress Beclin 1 and the various different molecular partners tested (empty vector, HA-Us11, Bcl-2, or Flag-ICP34.5). Forty-eight hours after transfection, the cells were washed with cold, sterile phosphate-buffered saline (PBS) and then lysed at 4°C for 2 h in lysis buffer [20 mM Tris HCl, pH 7.5, 140 mM NaCl, 1 mM EDTA, 2% CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate), 10% glycerol]. Lysates were centrifuged at 15,000 rpm at 4°C for 30 min. Supernatants were incubated overnight at 4°C with 20 μg/ml of anti-Beclin 1 (BECN1) (sc-10086; Santa Cruz). The immune complexes were then incubated for 2 h at 4°C with 30 μl of protein A-Sepharose 4B (GE Healthcare). The immune complexes were successively washed twice with washing buffer 1 (20 mM Tris HCl, pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.5% CHAPS, 10% glycerol) followed by washing buffer 2 (20 mM Tris HCl, pH 7.5, 280 mM NaCl, 1 mM EDTA, 0.5% CHAPS, 10% glycerol). The immune complexes were finally boiled for 5 min in loading buffer (62.5 mM Tris, pH 6.8, 10% glycerol, 1.5% SDS, 0.025% bromophenol blue, 8% β-mercaptoethanol) before being analyzed by SDS-PAGE.
Immunoblot analysis.
HeLa cells were lysed in 65 mM Tris, pH 6.8, 4% SDS, and 1.5% β-mercaptoethanol and incubated at 100°C for 5 min. After SDS-PAGE, the proteins were electrotransferred onto a polyvinylidene difluoride membrane. After incubation in blocking buffer (PBS, 0.1% Tween 20, and 5% bovine serum albumin or nonfat dry milk), the blots were probed overnight with specific antibodies and then incubated with horseradish peroxidase-linked secondary antibodies (Jackson Immunology), followed by chemiluminescent detection, according to the manufacturer's instructions (Immobilon; Millipore). Scanning for quantification was monitored using ImageJ software.
Immunofluorescence analysis.
For indirect immunofluorescence, cells were cultured on glass coverslips. Cell monolayers were washed three times with PBS and then fixed with 3.5% paraformaldehyde (PFA) in PBS. The cells were treated with 50 mM NH4Cl in PBS for 10 min, permeabilized using 0.2% Triton X-100 in PBS for 4 min, washed twice with PBS, and then incubated for 1 h in PBS containing 0.2% gelatin and then with appropriate primary antibodies diluted in PBS–0.2% gelatin for 1 h. The cells were washed three times and then incubated with appropriate secondary antibodies diluted in PBS–0.2% gelatin. To detect Us11 or Flag-tagged ICP34.5 expression, cells were stained with mouse anti-Us11 or rabbit anti-HA and anti-Flag antibodies followed by TRITC-conjugated goat or Alexa Fluor 350 donkey anti-mouse or anti-rabbit secondary antibodies. After cells had been washed three times with PBS, coverslips were mounted in Glycergel (Dako). Specimens were examined using a Nikon Eclipse 80i epifluorescence microscope. Digitized images were stored and overlaid to evaluate two-color experiments.
Statistics.
Data are expressed as a means ± standard errors of the means (SEM) of at least three experiments. The statistical significance was assessed by Student's t test.
RESULTS
PKR activation induces autophagy.
It had previously been reported that the induction of autophagy by HSV-1 involves activation of the dsRNA-dependent protein kinase PKR via phosphorylation of eIF2α (30). Our hypothesis was that the viral protein Us11 would be able to block autophagy because it inhibits PKR. We therefore wanted to stimulate specifically autophagy via PKR to observe the effect of Us11. However, it has never been reported that stimulation of PKR by its classical inducer, the artificial dsRNA poly(I · C), induces autophagy. To date, three other antiviral cellular proteins have been identified that are able to recognize dsRNA: the two RNA helicases, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (mda-5), and Toll-like receptor 3 (TLR3), which is present at the cell surface or within endocytic vesicles (35). Therefore, to find out whether PKR activation had any effect on autophagy, poly(I · C)was transfected into HeLa cells and wt and PKR-null MEF cells using Oligofectamine (Fig. 2). We confirmed that transfection of poly(I · C) for 4 h in our system led to phosphorylation of eIF2α and activation of PKR in HeLa cells (Fig. 2A).
Fig 2.

Transfection of poly(I · C) induces autophagy. (A) Immunoblot analysis of phosphorylated eIF2α (P-eIF2α) and PKR (P-PKR) proteins in HeLa cells. Cells were transfected with 0, 5, or 10 μg/ml poly(I · C) for 4 h. Total eIF2α and PKR blotting was used as a loading control. (B) Representative images of HeLa GFP-LC3 cells (scale bar, 10 μm) transfected for 4 h with poly(I · C) and quantification of GFP-LC3-positive cells with GFP-LC3 dots. As a control, HeLa cells were transfected with a GFP-LC3ΔG. The results are the means of three independent experiments, and 50 cells were analyzed per assay. *, P < 0.05; **, P < 0.01 (t test). (C) Immunoblot analysis of LC3-II protein in HeLa cells. Cells were transfected with poly(I · C) and either left untreated or treated with bafilomycin A1 (Baf) for 4 h. Actin blotting was used as a loading control. (D) HeLa cells were transfected with the mRFP-GFP-LC3 plasmid and treated with poly(I · C) for 4 h. Autolysosomes were quantified by counting the number of GFP− RFP+ puncta per cell. The results are the means of three independent experiments. Twenty cells were analyzed per assay. *, P < 0.05 (t test). Amino-acid-starved cells were used as a positive control.
To assess the impact of poly(I · C) on the autophagic pathway, different assays monitoring autophagy were first performed in HeLa cells. As shown in Fig. 2B, HeLa cells stably expressing GFP-LC3 that were transfected with poly(I · C) (5 or 10 μg/ml for 4 h) displayed increased numbers of GFP-LC3-positive cells with GFP-LC3 dots. Variable sizes of GFP-LC3 dots have been occasionally observed after poly(I · C) treatment. To confirm that these LC3 dots were not LC3-nonspecific aggregates, HeLa cells were transfected with a construct expressing a mutant GFP-LC3ΔG protein, which is unable to conjugate with phosphatidyl-ethanolamine (PE) and therefore unable to associate with the autophagosomal membrane (36). Cells transfected with GFP-LC3ΔG did not display any punctate staining in response to poly(I · C) treatment (Fig. 2B), suggesting that the dots formed in the poly(I · C)-transfected cells were not merely the result of aggregation of GFP-LC3.
Monitoring static levels of autophagosomes is not sufficient to elucidate the effects of poly(I · C) on autophagy because an accumulation of autophagosomes in cells can result from either an increase in the rate of their formation or a decrease in their fusion with lysosomes (37). We therefore analyzed the accumulation of the PE-conjugated form of LC3 (LC3 form II [LC3-II]) by immunoblotting in cells which had or had not been treated with bafilomycin A1, which blocks the maturation of autophagosome and the turnover of LC3-II. We observed similar levels of LC3-II in HeLa cells in the presence and absence of poly(I · C) transfection. However, bafilomycin treatment revealed that the accumulation of LC3-II was higher in the poly(I · C)-transfected cells, which was consistent with conclusion that poly(I · C) stimulates autophagic flux (Fig. 2C). We confirmed this finding using the tandem mRFP-GFP-LC3 probe (Fig. 2D). Based on the fact that the GFP signal is quenched in acidic compartments, such as autolysosomes, this probe makes it possible to differentiate between autophagosomes (GFP-positive and RFP-positive [GFP+ RFP+] or yellow puncta) and autolysosomes (GFP-negative [GFP−] and RFP+ or red puncta) (38, 39). We observed that transfection with poly(I · C) increased the number of autolysosomes (GFP− RFP+ puncta). These findings clearly indicate that poly(I · C) stimulates the biosynthesis of autophagosomes and the autophagy flux in HeLa cells. To confirm that poly(I · C) stimulates autophagy by activating PKR, we assayed the ability of poly(I · C) to induce autophagy in murine fibroblasts that lack PKR (Fig. 3A). The number of autophagosomes was increased after poly(I · C) transfection in wt MEFs, but poly(I · C) was not able to stimulate autophagy in PKR-deficient cells. To quantify the autophagic flux, we monitored the accumulation of LC3-II by immunoblotting, with or without bafilomycin. Accordingly, poly(I · C) treatment induces an increase in the autophagic flux in wt MEFs but not in PKR−/− cells (Fig. 3B). The decrease of LC3-II in poly(I · C)-treated wt cells corresponds to a robust autophagic flux. In contrast, the decline of LC3-II in PKR−/− cells corresponds to a decrease in autophagosome synthesis because we observed a weak accumulation of LC3-II in the presence of bafilomycin (Fig. 3B).
Fig 3.

Stimulation of autophagy by poly(I · C) is PKR dependent. (A) Representative images of GFP-LC3 in PKR+/+ and PKR−/− MEFs and the quantification of GFP-LC3 dots per cell. Cells were transfected with GFP-LC3 for 48 h and then grown in complete or starvation medium (EBSS) or treated with poly(I · C) for 4 h before fixation. The results are the means of three independent experiments; 50 to 100 cells were analyzed per assay. *, P < 0.05; **, P < 0.01 (t test). (B) Immunoblot analysis of LC3 levels in PKR+/+ and PKR−/− cells after poly(I · C) treatment for 4 h. Bafilomycin A1 was added to all the samples in order to block the fusion with the lysosome.
During the course of these experiments, we used starvation as a control to induce autophagy in MEF cells (Fig. 3). To study the regulation of autophagy by starvation in PKR−/− cells more accurately, autophagic flux was evaluated by monitoring the number of endogenous LC3 dots in the presence and absence of bafilomycin (Fig. 4). We observed that the autophagic flux induced by starvation was weaker in PKR−/− cells than in wt cells. Bafilomycin treatment, by blocking maturation of autophagosomes, allows us to visualize the total number of autophagosomes synthesized by the cell in response to a stress. We observed that there were fewer LC3 dots after starvation in bafilomycin-treated PKR−/− cells than in wt MEFs. These findings suggest that PKR−/− cells produce fewer autophagosomes during starvation-induced autophagy and that PKR may play a role in the regulation of autophagy in response to nutrient deprivation.
Fig 4.
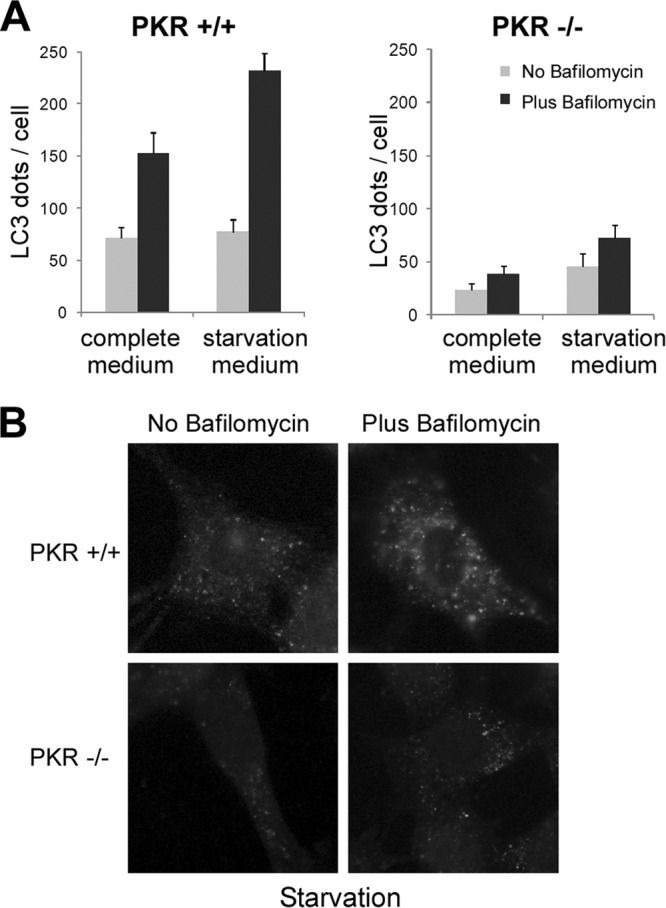
Stimulation of autophagy by starvation in PKR-deficient and wt cells. PKR+/+ and PKR−/− MEFs were grown in complete or starvation medium (EBSS) for 4 h before fixation. Cells were either left untreated or treated with bafilomycin A1 for 4 h in order to block the fusion with the lysosome. (A) Quantification of LC3 dots per cell by ImageJ software. The results are the means of three independent experiments; 50 to 100 cells were analyzed per assay. (B) Representative images of endogenous LC3 after starvation-induced autophagy in cells treated or not with bafilomycin A1.
The viral protein Us11 inhibits autophagy.
To study the impact of Us11 on autophagy, we transiently transfected HeLa cells that stably express GFP-LC3 either with a Us11 expression vector or with an empty vector. We then induced autophagy either with poly(I · C) transfection or by starvation for 4 h prior to fixation (Fig. 5A). Since HSV-1 ICP34.5 has been reported to block autophagy, we used a Flag-tagged ICP34.5 plasmid as a positive control (13). Us11- and ICP34.5-expressing cells were detected by immunofluorescence using anti-Us11 and anti-FLAG antibodies, respectively, and autophagy was quantified in these cells. As expected, the number of puncta per cell was lower in HeLa cells expressing ICP34.5 than in the control cells, regardless of the autophagy inducers used (Fig. 5A). Similarly, Us11 blocked autophagy whether this had been induced by poly(I · C) transfection or by nutrient starvation. To find out whether the inhibition of autophagy caused by Us11 led to inhibition of the autophagic flux, we used the tandem mRFP-GFP-LC3 probe (Fig. 5B). Autophagy was induced in HeLa cells by poly(I · C) or by starvation for 4 h prior fixation. The number of GFP− RFP+ puncta per cell, corresponding to autolysosomes, was clearly lower in cells expressing either ICP34.5 or Us11. These results suggest that Us11 blocked the autophagic flux to the same extent as ICP34.5. To confirm these findings, we analyzed the accumulation of endogenous LC3 dots in cells transfected with the Us11 expression vector by immunofluorescence (Fig. 5C). There were fewer LC3 dots in cells expressing Us11 than in the control cells, after both poly(I · C) and EBSS treatment. We concluded from these experiments that Us11 blocks both the formation of autophagosomes and the autophagic flux stimulated by starvation or poly(I · C).
Fig 5.

Inhibition of autophagy by ectopic expression of Us11. (A) Representative images of GFP-LC3 HeLa cells transfected with a pCDNA3 empty vector (vector), Us11, or Flag-ICP34.5 plasmid for a period of 48 h and then fixed after 4 h of starvation (EBSS) or poly(I · C) treatment (scale bar, 10 μm). Us11-transfected cells were visualized by an anti-Us11 antibody, and ICP34.5-transfected cells were visualized by an anti-Flag antibody. Insets show the viral protein staining. Autophagy was quantified by counting the number of GFP-LC3 dots per transfected cell; 50 to 100 cells were analyzed per assay. (B) Representative images of HeLa cells cotransfected with mRFP-GFP-LC3 and vector, Us11, or Flag-ICP34.5 plasmid for 24 h and cultured in starvation medium (EBSS) or treated with poly(I · C) (scale bar, 10 μm). Quantification of LC3 dots was performed in cells expressing Us11 or ICP34.5, as described in the Materials and Methods section. Red and yellow dots indicate GFP− RFP+ and GFP+ RFP+ puncta, respectively. Insets show part of the cytoplasm at a higher magnification. The number of GFP− RFP+ LC3 puncta per cell was quantified; 50 cells were analyzed per assay. The results are the means of three independent experiments. *, P < 0.05; **, P < 0.01 (t test). (C) Representative images of endogenous LC3 protein in HeLa cells. Cells were transfected with empty vector or Us11 vector for 48 h. Autophagy was quantified by counting the number of LC3 dots per cell, using ImageJ software. The results are the means of three independent experiments; 50 to 100 cells were analyzed per assay.
Us11 inhibits autophagy during viral infection.
Wild-type HSV-1 is able to control autophagy, whereas a Δ34.5 mutant virus clearly induces autophagy in several cell types in a PKR-dependent manner (30). In addition to stimulating autophagy, the Δ34.5 mutant virus is known to be neuro-attenuated in mice, to stimulate the TANK binding kinase 1-mediated signaling pathway, and to block host and viral protein synthesis (30, 33, 40). In a first series of experiments, we examined the expression of Us11 after infection of HeLa cells with the Δ34.5 mutant virus R3616 (Fig. 6A). We did not detect Us11 expression at 18 h postinfection (p.i.) although the Us11 gene is present in the genome of this virus. The failure to translate Us11 mRNA has previously been attributed to the cessation of cellular and viral synthesis (22). Thus, it is possible that the failure of HSV-1 R3616 to express Us11 contributes to the observed phenotype regarding autophagy.
Fig 6.

Immediate-early expression of Us11 inhibits autophagy during infection with a recombinant virus lacking ICP34.5. (A) Immunoblot analysis of the Us11 protein in HeLa cells at 18 h p.i. (B) Immunoblot analysis of phosphorylated eIF2α protein in HeLa cells. Cells were infected with HSV-1, R3616, or R5104 at an MOI of 1 and transfected with poly(I · C) (5 μg/ml) for the last 4 h of infection. Total eIF2α blotting was used as a loading control. (C) Immunoblot analysis of LC3 protein in HeLa cells infected with HSV-1, R3616, or R5104 at an MOI of 1 at indicated times of infection. (D) Representative images of infected GFP-LC3 HeLa cells at 18 h p.i. Infected cells were visualized by an anti-ICP0 antibody, and GFP-LC3-positive cells with GFP-LC3 dots were quantified over the time course of infection. (E) Representative images of infected GFP-LC3 HeLa cells at 18 h p.i. when treated with poly(I · C) to induce autophagy. GFP-LC3-positive cells with GFP-LC3 dots were quantified in ICP0-positive cells. The results are the means of three independent experiments; 100 cells were analyzed per assay. **, P < 0.01 (t test). Scale bar, 10 μm.
In order to investigate the role of Us11 in blocking autophagy in the context of viral infection, we used another previously described HSV-1 Δ34.5 mutant virus (R5104) in which the Us11 gene, which is normally expressed with late kinetics, is expressed under the control of the α47 promoter (29). As a result, Us11 is expressed in this mutant as an early protein and so can compensate for the lack of ICP34.5 with regard to protein synthesis shutoff (21, 29). We wanted to find out whether Us11 can also replace the antiautophagic activity of ICP34.5 in this context of viral infection. We first verified the pattern of expression of Us11 (Fig. 6A) and the inhibition of eIF2α phosphorylation (Fig. 6B) after infection of HeLa cells with R5104. We then analyzed the levels of LC3-II by Western blotting after infection with these three different viruses, wt HSV-1, R3616, and R5104 (Fig. 6C). LC3-II was notably increased after infection with the Δ34.5 mutant virus (R3616), whereas the level of LC3-II in cells infected with R5104, in which Us11 is expressed early in infection, was similar to that in wt HSV-1-infected cells (Fig. 6C). We also performed a kinetic analysis of autophagy in GFP-LC3 HeLa cells infected with these three viruses (Fig. 6D). Infected cells were identified by their expression of the viral protein ICP0. The number of GFP-LC3-positive cells with GFP-LC3 dots remained low in mock-infected cells and in cells infected with wt HSV-1 during the course of infection, which was consistent with the ability of wt virus to control autophagy. In contrast, and as expected from a previous study (30), GFP-LC3-positive cells increased significantly after R3616 infection. Finally, the number of positive cells was low in R5104-infected cells. In fact, this virus is as effective as wild-type HSV-1 in blocking autophagy throughout the course of infection, even in the absence of ICP34.5 protein expression (Fig. 6D). We also evaluated autophagy in HSV-1-infected cells after autophagy had been induced by poly(I · C) transfection (Fig. 6E). Autophagy was induced after poly(I · C) treatment in mock-infected cells and after infection with the mutant virus R3616 but was blocked by wild-type HSV-1 infection. Once again, R5104 was able to control autophagy as well as the wt HSV-1 virus, even after PKR-induced autophagy and even in the absence of ICP34.5. We observed the same results after starvation-induced autophagy (data not shown). Taken together, these results showed that Us11 is able to inhibit autophagy when induced by viral infection, by PKR activation, or by starvation.
Us11 inhibits autophagy in a PKR-dependent manner.
To investigate the impact of PKR on the ability of Us11 to inhibit autophagy, assays were performed in murine fibroblasts lacking PKR. HSV-1 ICP34.5 was used as a control. PKR+/+ and PKR−/− cells were transiently cotransfected with GFP-LC3 and either Us11 or a Flag-tagged ICP34.5 expression vector, and autophagy was induced by starvation. Us11- and ICP34.5-expressing cells were identified using the anti-Us11 or anti-Flag antibody, respectively, and GFP-LC3 dots were quantified in those cells (Fig. 7A). As previously reported, ICP34.5 blocked autophagy independently of the expression of PKR (13, 16). In contrast, Us11 was unable to inhibit autophagy in PKR-deficient cells (Fig. 7A). These results suggest that Us11 inhibits autophagy in a PKR-dependent manner. Interestingly, starvation induced a greater accumulation of autophagosomes and a greater autophagic flux in wt PKR cells than in PKR−/− cells (Fig. 4). These results suggest that two signaling pathways emanating from PKR and mTORC1, respectively, may contribute to the regulation of starvation-induced autophagy. Us11 probably acts only on the PKR-dependent component of starvation-induced autophagy.
Fig 7.

The PKR binding domain of Us11 is required for inhibition of autophagy. (A) PKR+/+ and PKR−/− MEFs were cotransfected with a GFP-LC3 plasmid and an empty vector, Us11, or Flag-ICP34.5 plasmid for 48 h and starved for 4 h. Insets show the viral protein staining obtained. Autophagy was quantified by counting the number of GFP-LC3 dots per cell. The results are the means of three independent experiments. (B) Schematic representation of Us11 showing the positions of the PKR binding domain and the RNA binding domain. PKR phosphorylates Us11 on threonine 71, serine 76, and threonine 83. Different Us11 constructions with an HA tag at the amino terminus were analyzed. (C) Expression of each HA-tagged transgene in HeLa cells after 48 h of transfection. Actin blotting was used as a loading control. (D) Quantification of GFP-LC3 dots in cells transfected with different Us11 constructs after poly(I · C)-induced autophagy. The results are the means of three independent experiments. *, P < 0.05; **, P < 0.01 (t test).
Us11 contains a PKR-binding domain located in a 30-amino-acid stretch close to the C terminus (between amino acids 91 and 121) and a PKR substrate domain adjacent to this domain, with three identified phosphorylation sites (Fig. 7B) (23). The carboxyl-terminal half of Us11 contains an arginine/proline-rich RNA binding domain (Fig. 7B) (41). In order to delineate the domain of Us11 responsible for inhibiting autophagy, we made serial truncations of Us11 from the carboxyl and the amino termini (Fig. 7B and C). HeLa cells stably expressing GFP-LC3 were transiently transfected with the various different constructions of Us11 or an empty vector, and autophagy was monitored after activating PKR by poly(I · C) transfection (Fig. 7D). We observed that Us11 consisting of residues 1 to 91 [Us11(1–91)] was unable to counteract the stimulation of autophagy, showing that the carboxyl-terminal half of the Us11 protein is necessary for autophagy inhibition. However, the reduction in the number of GFP-LC3 dots after transfection with Us11(1–121) was the same as after transfection with full-length Us11. Thus, the region of amino acids 91 to 121 (which corresponds to the PKR-binding domain of Us11) seems to be required for autophagy inhibition to occur. Although this region is included in the RNA-binding domain (mapped to amino acids 91 to 152), it has previously been reported that a truncated form of Us11 protein at amino acid 123 (Δ123–152) is not able to bind RNA (41). These findings suggest that the PKR-binding domain of Us11 is necessary for autophagy inhibition to occur, whereas the binding of Us11 to RNA is not. Surprisingly, truncations of Us11 containing amino acids 40 to 152, 66 to 152, and 90 to 152 also failed to inhibit poly(I · C)-induced autophagy, suggesting that the inhibition of autophagy by Us11 also requires the N-terminal region (residues 1 to 40) of Us11. So far, no known cellular proteins have been reported to interact with the N-terminal region of Us11. Nevertheless, several herpesvirus proteins, such as HSV-1 ICP34.5 and human cytomegalovirus (HCMV) TRS1 (13, 16), are known to interact with Beclin 1, a protein involved in the autophagy machinery. Following on from these observations, we wondered whether Us11 interacts with Beclin 1.
Us11 does not block autophagy by inhibiting Beclin 1 or mTOR.
In order to find out whether Us11 interacts with Beclin 1, we performed a coimmunoprecipitation experiment involving HA-tagged Us11 and Beclin 1 plasmids (Fig. 8A). We did not observe any binding of Beclin 1 to Us11, whereas a clear interaction was observed with two known partners of Beclin 1: Bcl-2 and ICP34.5. Many herpesviruses, such as HSV-1 or HCMV, stimulate mTOR kinase, promoting 4E-BP1 phosphorylation, viral protein synthesis, and viral replication (17). Since activation of mTOR downregulates autophagy, we went on to investigate whether Us11 blocks autophagy via mTOR, which would mean that PKR might signal upstream of mTOR. Because starvation inhibits mTOR activity, we studied the modulation of mTOR after expression of Us11 and starvation by studying the phosphorylation of two of its substrates: 4E-BP1 and p70S6K. We observed that starvation induced rapid dephosphorylation of 4E-BP1 and p70S6K in both PKR+/+ and PKR−/− cells (Fig. 8B). Our results showed that the mTOR signaling pathway was inhibited by nutrient deprivation, independently of PKR. Expression of Us11 did not modify the level of phosphorylation of 4E-BP1 or of p70S6K in MEF PKR+/+ cells subjected to starvation even though Us11 blocked starvation-induced autophagy in these cells (Fig. 7A). These findings show that Us11 inhibits autophagy independently of mTOR.
Fig 8.
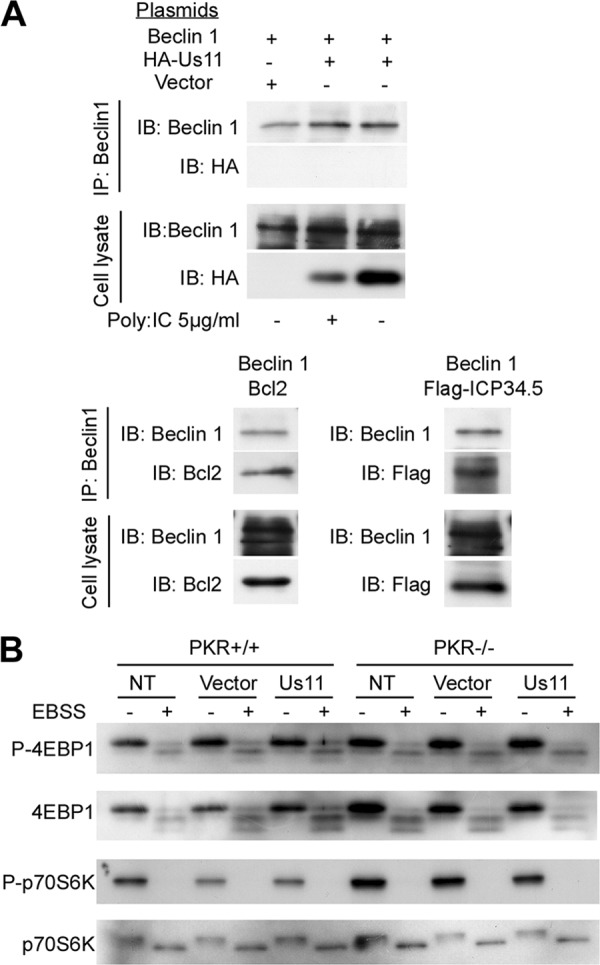
Us11 does not block autophagy by inhibiting Beclin 1 or mTOR. (A) HeLa cells were transiently transfected to express Beclin 1 and empty vector pSG5-HA, HA-Us11, Flag-ICP34.5, or Bcl-2. The Us11-Beclin 1 interaction was tested in complete medium or after stimulating autophagy by poly(I · C) (top panel). Beclin 1 was immunoprecipitated as described in Materials and Methods. Coimmunoprecipitations of Beclin 1 and Bcl-2 or of Beclin 1 and Flag-ICP34.5 in HeLa cells were performed as positive controls (bottom panels). Each immunoblot was representative of three independent experiments. IP, immunoprecipitation; IB, immunoblot. (B) PKR+/+ and PKR−/− MEFs were transfected with empty vector or Us11 for 24 h and grown in starvation medium (EBSS) for 4 h. Cells were analyzed for mTOR activity by immunoblotting for levels of phosphorylated 4E-BP1 (4E-BP1-P), total 4E-BP1, phosphorylated p70S6K (p70S6K-P), and total p70S6K. NT, not transfected.
DISCUSSION
Previous studies have shown that wild-type HSV-1 is able to control autophagy in neurons and in fibroblasts, whereas a mutant virus lacking the genes encoding ICP34.5 stimulates autophagy in a PKR/eIF2α-dependent manner (30). It has been shown that the ICP34.5 protein, already known to form a complex with the protein phosphatase PP1α to dephosphorylate eIF2α, thus reversing the effect of PKR, is an antiautophagic protein. However, the main target of ICP34.5 in the autophagic machinery is Beclin 1 (13, 20). The ICP34.5 protein deleted of the Beclin 1 binding domain (ICP34.5 Δ68-87) interacts with PP1α and overcomes the inhibition of protein synthesis but does not block autophagy (13). Interestingly, HSV-1 encodes Us11, a protein that also counteracts the host translation shutoff (29). Us11 precludes the phosphorylation of eIF2α via a direct inhibition of PKR during viral infection (23). PKR-Us11 interaction is RNA dependent and requires a 30-amino-acid domain within the carboxyl-terminal RNA binding domain of the Us11 protein (23). However, the status of Us11 with regard to autophagy was unknown prior to this study. Our working hypothesis was that one of the functions of Us11 might be to inhibit the induction of autophagy by interacting with PKR. Interestingly, Us11 mRNA is not translated in cells infected with a Δ34.5 HSV-1 mutant because of the shutoff of protein synthesis (Fig. 6) (22). Our results demonstrate that the stimulation of autophagy observed in cells infected with Δ34.5 HSV-1, previously ascribed solely to the lack of ICP34.5, actually results from the combined loss of both ICP34.5 and Us11 functions.
In this study, we provide evidence that Us11 antagonizes the host's autophagy response. Ectopic expression of Us11 leads to a blockade of autophagy, characterized by significant decreases in the numbers of both autophagosomes and autolysosomes (Fig. 5). This inhibition at an early step of autophagy is similar to the inhibitory effect of the other antiautophagic protein of HSV-1, ICP34.5. In a viral context, immediate-early expression of Us11 in a mutant virus in which 34.5 genes have been deleted restores the control of autophagy to a level similar to that of wild-type HSV-1.
We next investigated the mechanism of action of Us11. Us11 did not interact with Beclin 1 and did not inhibit the mTORC1 signaling pathway. In line with previous results demonstrating that Us11 binds to PKR and prevents its activation in the context of viral infection (23, 42), we showed that interaction of Us11 with PKR is required to block autophagy. Moreover, the PKR binding domain of Us11, located at its carboxyl-terminal region, was necessary for the inhibition of autophagy. We thus demonstrated that interaction of Us11 with PKR is necessary for autophagy to be inhibited, probably by blocking PKR activity. One other hypothesis, which we would like to test in the future, is that Us11 needs PKR to be phosphorylated and to act as an antiautophagic protein. We were surprised to discover that the first 40 amino acids of the amino-terminal region also seemed to be essential for this activity. To date, no interaction between this region of Us11 and a cellular protein has been identified. Us11 shuttles between the nucleus and the cytoplasm (43), and infection by two herpesviruses, HCMV and murine cytomegalovirus, causes the sequestration of PKR in the nucleus (44, 45). We observed that a similar relocalization occurs in HSV-1-infected cells and that this depends on Us11 expression (data not shown). Redistribution of PKR from the cytoplasm to the nucleus by Us11 may prevent the interaction of activated PKR with cytoplasmic eIF2α. It would be interesting to test whether the N-terminal domain of Us11 could be involved in this relocalization since no classical nuclear localization signal (NLS) has so far been identified in Us11 (46). However, it has been reported that Us11 contains both a constitutive nucleolar retention signal (NoRS) and a nuclear export signal (NES), both of which are located between amino acids 88 and 125 (46).
PKR is an antiviral protein which acts via numerous effectors. The effects of PKR activation are known to alter cellular translation, to induce apoptosis, and to activate nuclear factor-κB (NF-κB) (47). For these reasons, PKR activation can be counteracted by numerous viral proteins from different families (48). For example, the vaccinia virus E3L protein is capable of inhibiting PKR by sequestrating dsRNA, and the influenza virus nonstructural protein NS1 blocks the kinase activity of PKR (48). As shown by Tallozcy et al. in 2002, activation of the PKR/eIF2α signaling pathway during Δ34.5 HSV-1 infection leads to stimulation of autophagy (30). We report here that activation of PKR by a classical inducer of PKR, the transfection of poly(I · C), clearly induces a stimulation of the autophagic flux in a PKR-dependent manner. A long-standing hypothesis proposes that viral proteins that block the PKR/eIF2α signaling also modulate autophagy (49). TRS1 and IRS1, which are both HCMV viral proteins, directly interact and block PKR (50). Nevertheless, we have demonstrated that TRS1 inhibits autophagy in a PKR-independent manner and via its interaction with Beclin 1 (16). In a similar manner, ICP34.5 binds to Beclin 1 and inhibits its autophagy function; its mechanism of action does not seem to rely on the dephosphorylation of eIF2α (13). However, it is tempting to speculate that other antiautophagic viral proteins may act through their effects on the PKR/eIF2α signaling pathway.
Recent data have shown that, among its numerous functions, Us11 binds to RIG-I and mda-5 helicases, two cellular antiviral proteins involved in viral dsRNA recognition, and inhibits their downstream signaling pathway (28). Moreover, it has recently been reported that stimulation of mda-5 by poly(I · C) induces autophagy in cancer cells (51). It would therefore be interesting to explore the relationships between autophagy, mda-5, and Us11. Indeed, another way that Us11 might block autophagy could be through its interaction with mda-5. However, the fact that Us11 does not inhibit autophagy in the absence of PKR (in PKR-deficient MEFs) suggests that interaction between Us11 and PKR is mandatory to counteract autophagy stimulation during viral infection.
We were both surprised and interested to observe that Us11 was also able to partially block autophagy induced by nutrient starvation (Fig. 5). Amino acid deprivation is known to stimulate autophagy via the inhibition of mTOR (52); however, we have shown here that Us11 does not modulate mTOR activity. Moreover, the eIF2α kinase GCN2 senses amino acid deficiency, but no interaction between Us11 and GCN2 has been reported. We note that PACT, a cellular activator of PKR that acts in an RNA-independent manner, is activated by growth factor deprivation and that Us11 can block PKR activation by PACT (25). In addition, it has recently been reported that PKR senses and responds to nutrients and to endoplasmic reticulum stress (53). These data suggest that the regulation of PKR by nutrients is underestimated and that, in addition to its function as a viral sensor, PKR is also able to sense various stresses. Finally, it is not very surprising to find that stimulation of autophagy by starvation might be blocked by Us11 because part of the starvation-induced autophagy is PKR dependent.
Why does HSV-1 need at least two different proteins, Us11 and ICP34.5, to control autophagy during viral infection? It is interesting that these two proteins also have similar functions with regard to the restoration of host protein synthesis during viral infection by interacting with PKR and PP1α, respectively (20, 23). The viral proteins Us11 and ICP34.5 are equally capable of inhibiting translational arrest by host defenses. Initially, it was proposed that these two proteins encode redundant functions, that Us11 represents an archaic version present in the genome, and that during evolution, HSV-1 acquired the γ134.5 gene, which may encode a more efficient inhibitor of the PKR/eIF2α pathway (29). More recently, it has been proposed that, since ICP34.5 is a γ1 protein (i.e., it does not require viral DNA replication to be expressed) and since Us11 is a γ2 protein (i.e., one which does require viral DNA replication to be expressed), these two proteins may act successively at discrete times during the replication cycle (22). It is only after viral replication has occurred that Us11 emerges as a major regulator of PKR during the late phase. We propose that, in a similar manner, these two proteins could successively block autophagy during the viral life cycle. A ΔUs11 mutant virus has a clear phenotype late in the infectious program (22). Lower translation rates at late times postinfection and decreased viral replication have been observed in cells infected with a ΔUs11 mutant virus compared to those infected with a wild-type virus. It has been suggested that the Us11 protein acts late in infection to antagonize PKR activation in response to the copious levels of dsRNA produced in virus-infected cells (22). It will be interesting to assess the capacity of Us11, expressed in its natural context as a true-late gene, to preclude virus-induced autophagy.
The fact that HSV-1 encodes two antiautophagic proteins strongly suggests that autophagy has an antiviral effect against HSV-1. Autophagy is thought to protect the cell against intracellular pathogens by engulfing and degrading microorganisms using the autophagy machinery in a process termed xenophagy (11). HSV-1 may inhibit autophagy in order to evade xenophagy (54). In addition, a mutant virus which is unable to control autophagy because it lacks the Beclin 1 binding domain of ICP34.5 has a growth defect in the brains of mice (13). However, autophagy does not limit viral growth in vitro since HSV-1 replicates at the same rates in autophagy-deficient and wild-type cells (55). It has also been proposed that the inhibition of autophagy by HSV-1 contributes to its neurovirulence as a result of an interaction between the viral protein, ICP34.5, and Beclin 1 (13). Δ34.5 mutant viruses are incapable of efficient replication after being directly inoculated into the central nervous system and do not cause encephalitis; however, the neurovirulence and protein synthesis functions of ICP34.5 are discrete and separable (40). Moreover, a virus containing a mutation in ICP34.5 that abrogates binding to Beclin 1 is neuro-attenuated in mice (13). We report in this study that a Δ34.5 mutant virus that expresses Us11 as an immediate-early protein is able to control autophagy in vitro. It will be interesting to test this mutant virus for its neurovirulence in vivo. However, a spontaneous compensatory HSV-1 mutant obtained through serial passage selection, and equivalent to R5104 virus, does not restore the virulence phenotype of the wild-type virus in mice (56). It would therefore also be important to find out whether the expression of Us11 in these two compensatory Δ34.5 mutant viruses modulates autophagy in vivo.
ACKNOWLEDGMENTS
We thank Bernard Roizman for providing us the R3616 and R5104 mutant viruses. We are also very grateful to T. Yoshimori for providing us with the GFP-LC3 and mRFP-GFP-LC3 constructs. We thank Adam Geballe for his critical reading of the manuscript.
This work was supported by institutional funding from The Institut National de la Santé et de la Recherche Médicale (INSERM), from Paris Sud University, and grants from the Agence Nationale de la Recherche (ANR MIME 2007) to A.E.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Yang Z, Klionsky DJ. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizushima N, Yoshimori T, Ohsumi Y. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132 [DOI] [PubMed] [Google Scholar]
- 3. Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147:728–741 [DOI] [PubMed] [Google Scholar]
- 4. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kirkegaard K. 2009. Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 335:323–333 [DOI] [PubMed] [Google Scholar]
- 7. Virgin HW, Levine B. 2009. Autophagy genes in immunity. Nat. Immunol. 10:461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine B. 2005. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120:159–162 [DOI] [PubMed] [Google Scholar]
- 9. Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kudchodkar SB, Levine B. 2009. Viruses and autophagy. Rev. Med. Virol. 19:359–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavignac Y, Esclatine A. 2010. Herpesviruses and autophagy: catch me if you can! Viruses. 2:314–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takahashi MN, Jackson W, Laird DT, Culp TD, Grose C, Haynes JI, II, Benetti L. 2009. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J. Virol. 83:5466–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35 [DOI] [PubMed] [Google Scholar]
- 14. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939 [DOI] [PubMed] [Google Scholar]
- 15. Sinha S, Colbert CL, Becker N, Wei Y, Levine B. 2008. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4:989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 86:2571–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Esclatine A, Chaumorcel M, Codogno P. 2009. Macroautophagy signaling and regulation. Curr. Top. Microbiol. Immunol. 335:33–70 [DOI] [PubMed] [Google Scholar]
- 19. Dever TE. 1999. Translation initiation: adept at adapting. Trends Biochem. Sci. 24:398–403 [DOI] [PubMed] [Google Scholar]
- 20. He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cassady KA, Gross M, Roizman B. 1998. The second-site mutation in the herpes simplex virus recombinants lacking the γ134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2α. J. Virol. 72:7005–7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mulvey M, Poppers J, Sternberg D, Mohr I. 2003. Regulation of eIF2α phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 77:10917–10928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cassady KA, Gross M. 2002. The herpes simplex virus type 1 US11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J. Virol. 76:2029–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poppers J, Mulvey M, Khoo D, Mohr I. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 74:11215–11221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peters GA, Khoo D, Mohr I, Sen GC. 2002. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 76:11054–11064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Javouhey E, Gibert B, Arrigo AP, Diaz JJ, Diaz-Latoud C. 2008. Protection against heat and staurosporine mediated apoptosis by the HSV-1 US11 protein. Virology 376:31–41 [DOI] [PubMed] [Google Scholar]
- 27. Sanchez R, Mohr I. 2007. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 81:3455–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 86:3528–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Talloczy Z, Jiang W, Virgin HW, IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. 2005. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 1:23–36 [DOI] [PubMed] [Google Scholar]
- 32. Kimura S, Fujita N, Noda T, Yoshimori T. 2009. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 452:1–12 [DOI] [PubMed] [Google Scholar]
- 33. Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 66:3624–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sen GC, Peters GA. 2007. Viral stress-inducible genes. Adv. Virus Res. 70:233–263 [DOI] [PubMed] [Google Scholar]
- 36. Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K. 2008. Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 4:131–134 [DOI] [PubMed] [Google Scholar]
- 37. Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460 [DOI] [PubMed] [Google Scholar]
- 39. Klionsky DJ, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266 [DOI] [PubMed] [Google Scholar]
- 41. Roller RJ, Monk LL, Stuart D, Roizman B. 1996. Structure and function in the herpes simplex virus 1 RNA-binding protein US11: mapping of the domain required for ribosomal and nucleolar association and RNA binding in vitro. J. Virol. 70:2842–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mulvey M, Poppers J, Ladd A, Mohr I. 1999. A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J. Virol. 73:3375–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greco A, Arata L, Soler E, Gaume X, Coute Y, Hacot S, Calle A, Monier K, Epstein AL, Sanchez JC, Bouvet P, Diaz JJ. 2012. Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J. Virol. 86:1449–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Child SJ, Geballe AP. 2009. Binding and relocalization of protein kinase R by murine cytomegalovirus. J. Virol. 83:1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 80:11817–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Catez F, Erard M, Schaerer-Uthurralt N, Kindbeiter K, Madjar JJ, Diaz JJ. 2002. Unique motif for nucleolar retention and nuclear export regulated by phosphorylation. Mol. Cell. Biol. 22:1126–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams BR. 2001. Signal integration via PKR. Sci. STKE 2001:re2 doi:10.1126/stke.2001.89.re2 [DOI] [PubMed] [Google Scholar]
- 48. Garcia MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811 [DOI] [PubMed] [Google Scholar]
- 49. Kirkegaard K, Taylor MP, Jackson WT. 2004. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2:301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Child SJ, Hakki M, De Niro KL, Geballe AP. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78:197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tormo D, Checinska A, Alonso-Curbelo D, Perez-Guijarro E, Canon E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megias D, Mulero F, Piris MA, Dash R, Barral PM, Rodriguez-Peralto JL, Ortiz-Romero P, Tuting T, Fisher PB, Soengas MS. 2009. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 16:103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS. 2010. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140:338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Talloczy Z, Virgin HW, IV, Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29 [DOI] [PubMed] [Google Scholar]
- 55. Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. 2007. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81:12128–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mohr I, Sternberg D, Ward S, Leib D, Mulvey M, Gluzman Y. 2001. A herpes simplex virus type 1 γ34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J. Virol. 75:5189–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]