

Abstract
Sir2 is an evolutionarily conserved NAD+-dependent deacetylase which has been shown to play a critical role in glucose and fat metabolism. In this study, we have perturbed Drosophila Sir2 (dSir2) expression, bidirectionally, in muscles and the fat body. We report that dSir2 plays a critical role in insulin signaling, glucose homeostasis, and mitochondrial functions. Importantly, we establish the nonautonomous functions of fat body dSir2 in regulating mitochondrial physiology and insulin signaling in muscles. We have identified a novel interplay between dSir2 and dFOXO at an organismal level, which involves Drosophila insulin-like peptide (dILP)-dependent insulin signaling. By genetic perturbations and metabolic rescue, we provide evidence to illustrate that fat body dSir2 mediates its effects on the muscles via free fatty acids (FFA) and dILPs (from the insulin-producing cells [IPCs]). In summary, we show that fat body dSir2 is a master regulator of organismal energy homeostasis and is required for maintaining the metabolic regulatory network across tissues.
INTRODUCTION
Metabolic and energy homeostasis are tightly linked to insulin signaling and fat metabolism, which together become crucial determinants of organismal physiology (1, 2). From a clinical perspective, dysfunctions in insulin signaling and metabolic defects affect each other, as seen in obesity and type 2 diabetes (2, 3). Comorbidity of these conditions arises because abrogated fat metabolism has been implicated in insulin resistance and because alterations in insulin signaling are known to regulate the expression of fat metabolism genes. However, the interplay between insulin signaling, fat metabolism, and mitochondrial functions in the etiology of metabolic diseases is still unclear (4).
Recent reports in mammals and flies clearly show that SIRT1/Sir2 plays an important role in fat metabolism (5–12) and affects starvation survival (5). Additionally, ablation of SIRT1 in liver and muscles has been shown to result in an insulin resistance-like phenotype (12, 13). However, it is not clear if fat metabolism and systemic insulin signaling are regulated independently by Sir2/SIRT1. An important link between insulin signaling and fat metabolism is the FOXO family of transcription factors. FOXOs are typically associated with the transcription of genes downstream to insulin/insulin-like growth factor (IGF) signaling, which include lipogenic and lipolytic genes (14, 15). Although SIRT1 regulates FOXO-dependent transcription (16), the importance of the SIRT1-FOXO cross talk in mediating the effects of insulin signaling at the organismal level is poorly addressed (17–19). In this respect, it is important to delineate the Sir2/SIRT1-dependent fat phenotype from insulin resistance in the liver and in peripheral tissues (muscles), in addition to investigating the role of FOXO in regulating these effects.
Impaired insulin signaling has been associated with mitochondrial dysfunctions and defects in energy homeostasis. Several reports highlight the role of SIRT1 in affecting the transcription of mitochondrial genes via PGC1α (20–22). Although SIRT1-mediated transcriptional regulation is expected to affect mitochondrial functions and energy homeostasis, the physiological relevance, specifically at the organismal level, is still unclear. Given the pivotal functions of SIRT1 in the liver, investigating its ability to affect metabolic parameters in peripheral tissues becomes important.
The ability of an organism to maintain metabolic and energy homeostasis has been implicated as a major determinant of survival in response to acute and chronic dietary alterations. Although longevity is regulated by homeostatic mechanisms, the robustness of such metabolic adaptations (specifically, energy metabolism) is often measured as a function of starvation survival or resistance.
Using Drosophila as a model system, we have addressed the role of dSir2 in maintaining tissue-specific as well as organismal energy homeostasis. Although previous reports have highlighted the role of Sir2/SIRT1 in muscles (13, 23–25), we show that its overexpression in this tissue is sufficient to regulate glucose homeostasis. We also show that an absence of dSir2 in the fat body leads to abrogated insulin signaling and impaired energy homeostasis in the muscles. Moreover, dSir2 in the fat body mimics the effects of dSir2 in the muscles, highlighting the similarity in the functions of dSir2 in these two tissues. We report an increase in insulin signaling and, hence, reduced nuclear localization of dFOXO within the fat body of fat body-specific dSir2 knockdown flies. Further, by simultaneous overexpression of a constitutively nuclear dFOXO (dFOXO-TM) and knockdown of dSir2 in the fat body, we delineate the effects of fat body dSir2 on fat metabolism from systemic insulin signaling. We have found that ablation of dSir2 in the fat body leads to an imbalance in energy homeostasis and causes a “nutrient-dependent mitochondrial stress” condition in the organism. This is evident from the rescue of the signaling defects in the muscles of fat body-specific dSir2 knockdown flies by the administration of l-carnitine. Finally, we report that although there are similarities in the metabolic functions of dSir2 in the muscles and fat body, the ability to adapt to an acute metabolic stress like starvation is differentially regulated. In conclusion, we highlight the interaction between two key metabolic sensors in the fat body in establishing communication across tissues for maintaining energy homeostasis, and we identify a physiological mechanism underlying the nonautonomous effects of fat body dSir2 on muscles.
MATERIALS AND METHODS
Fly strains.
P{Switch1}106-Gal4 (26–28), Sir2EP2300, and Sir2EP2384 flies were obtained from Bloomington Stock Center (Indiana University). The P{Switch tub-Gal4 } strain was a kind gift from Stephen Helfand. yw+; UAS-dFOXO-TM (dFOXOAAA) and yw+; UAS-dFOXO-WT flies were kind gifts from Marc Tatar. The UAS-dFOXO-GFP strain was provided to us by Gaiti Hasan, National Centre for Biological Sciences (NCBS), Bangalore, India. The rp298; P{Switch-MHC}-Gal4 (27) strain was obtained from NCBS, Bangalore, India. dSir2RNAi (CG5216:23201/GD and 23199/GD) and chicoRNAi (chRNAi) (CG5685:1402/GD) flies were obtained from the Vienna Drosophila RNAi Center (VDRC). mCherryRNAi flies were provided by Richa RIkhy from the Indian Institute of Science Education and Research (IISER), Pune, India. Flies were grown on normal food under noncrowding conditions at 25°C with a 12/12-h light/dark cycle. Age-matched virgin female flies 3 to 5 days old were used for all analyses.
Activation of inducible Gal4.
The inducible Gal4 (P{Switch} lines) was activated by rearing flies on medium containing 200 μM RU486 (mifepristone) (in ethanol). Flies reared on a diet containing only ethanol were used as controls. When two different upstream activation sequence (UAS) genes were activated simultaneously by the same Gal4, as in the case of dSir2-dFOXO and dSir2-chico stocks, 500 μM RU486 (mifepristone) was used to activate the Gal4.
Mitochondrial DNA estimation.
For mitochondrial DNA estimation, total genomic DNA was isolated using a Bangalore Genei genomic DNA isolation kit (catalog number 118729). The relative mitochondrial content was quantified by real-time PCR using the primers for COX subunit I and nuclear Actin (for normalization).
Tissue isolation.
Indirect flight muscles and abdominal fat body were dissected in ice-cold phosphate-buffered saline (PBS) from anesthetized adult flies. Fresh tissue isolates were used for preparing protein lysates or for RNA extraction.
Hemolymph isolation.
Hemolymph was isolated from 12 adult flies by capillary action. The hemolymph was collected in capillaries after a small puncture in the head capsule. Twenty microliters of the hemolymph was diluted in 180 μl of PBS for further analyses.
RNA isolation and reverse transcription.
Total RNA was isolated from eight flies (pooled together) using TRIzol (catalog number 15596-026; Invitrogen,) according to the manufacturer's instructions. One microgram of RNA was used for cDNA synthesis using a SuperScript III reverse transcriptase kit (catalog number 18080-044; Invitrogen) per the manufacturer's instructions.
Real-time PCR analyses.
Quantitative PCR (qPCR) was performed using Quantifast SYBR green (catalog number 204054; Qiagen) and an Eppendorf Realplex instrument. The cycling conditions were as prescribed by the manufacturer. The primer pairs used for the assay are tabulated in Table S1 in the supplemental material. Various levels of actin (actin5c) and rp49 were used for normalization.
Lipid and glucose measurement.
Six (3- to 5-day-old) flies reared on a normal diet were snap-frozen in liquid nitrogen and homogenized in 600 μl of PBS–0.05% Tween 20 (PBST). The homogenate was heated to 70°C for 10 min. Following centrifugation at 12,000 rpm for 15 min, the supernatant was transferred to fresh tubes, which were assayed at a pathology laboratory (Shahbazker's Diagnostic Center, Mumbai, India).
FFA level measurement.
Hemolymph samples isolated from eight (3- to 5-day-old) flies were used to estimate free fatty acid (FFA) levels. Free fatty acid level measurement was done using a free fatty acid quantification kit from Abcam (catalog number ab65341) according to the manufacturer's instructions.
oGTT.
An oral glucose tolerance test (oGTT) was performed according to Haselton et al. (29) with modifications. Briefly, 3- to 5-day-old flies were starved for 16 h, and an oGTT was performed by shifting them to vials containing 10% glucose and 2% agar for 30 min before transferring them back to 2% agar. Following this, glucose levels were measured in the hemolymph at various time points.
Mitochondrial membrane potential.
Mitochondrial membrane potential was detected using 1,1′,3,3′-tetraethylbenzamidazolocarbocyanin iodide (JC-1) (Molecular Probes). A stock solution at 5 mg/ml was made in dimethyl sulfoxide (DMSO), as per the manufacturer's instruction. The final staining solution having 5 μM JC-1 was made in Schneider's incomplete medium (GIBCO). Flies were anesthetized using ether, and indirect flight muscles were dissected into Schneider's medium. Muscles were incubated in JC-1 solution for 20 min on a shaker. Samples were then washed with medium and finally with PBS. They were fixed with 4% paraformaldehyde (PFA) and mounted on slides for imaging on an LSM510 confocal microscope (Carl Zeiss) under a 63× objective. For quantitation, mean integrated densities from 30 sections from three flies per treatment or genotype were obtained using ImageJ software. The ratios of red and green intensities were used for depicting the change in membrane potential.
Western blotting.
Total fly lysates were incubated on ice for 15 to 20 min in a buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 6 mM EGTA, 20 mM NaF, 1% Triton X-100, and protease inhibitors (catalog number 05-056-489001; Roche). The lysates were then centrifuged at 12,000 rpm for 15 min at 4°C to pellet the debris. Supernatants were used for protein estimations using a bicinchoninic acid (BCA) kit (catalog number BCA9643; Sigma) and resolved by 10% SDS-PAGE. Following electrotransfer onto polyvinylidene difluoride (PVDF) membranes, they were probed with appropriate antibodies according to standard procedures. Anti-Akt, anti-phospho-AKT (anti-p-Akt), and antiactin antibodies were purchased from Cell Signaling (catalog numbers 9273 and 4054) and Sigma-Aldrich (catalog number A1978), respectively. Chemiluminescence detection (catalog number 12-015-196001; Roche) was used to visualize the bands. For quantitation, mean integrated densities of Western blots from three independent lysates (each obtained from multiple flies, as indicated in the figure legends) per treatment or genotype were obtained using ImageJ software. The ratios of p-Akt/Akt and actin intensities were used for depicting the change in phosphorylation status of AKT.
ATP assay.
Adult flies were snap-frozen and boiled in water for 15 min. The debris was pelleted by centrifugation at 12,000 rpm at 4°C for 6 min. The supernatant was used to determine total ATP levels using an ATP bioluminescent assay kit (FLAA-1KT; Sigma-Aldrich) and Berthold luminometer. ATP levels were normalized to total protein in the supernatant (determined by using Bradford's reagent) (catalog number 500-0205; Bio-Rad). Normalized ATP levels indicate total ATP per unit of optical density (OD).
l-Carnitine and etomoxir treatments.
For l-carnitine treatment, flies were reared for 24 h on a normal diet containing l-carnitine (catalog number C0283-5G; Sigma-Aldrich) at a concentration of 25 mg/ml in water. For etomoxir treatment, flies were reared for 12 h on a normal diet containing etomoxir (catalog number E1905; Sigma-Aldrich) at a concentration of 25 μM in water.
Statistical analyses.
SigmaPlot, version 12.0, was used for all statistical analyses. Student's t test and analysis of variance (ANOVA) were used to analyze statistical significance of the data. A log rank Mantel-Cox test was used for calculating significance for survival assays.
RESULTS
Muscle dSir2 regulates mitochondrial functions and energy homeostasis autonomously and mimics the nonautonomous effects of fat body dSir2.
To understand the link between dSir2 and energy homeostasis in the muscles, we wanted to determine whether perturbing the expression of dSir2 would affect mitochondrial functions. Muscle-specific dSir2 overexpression (mdSir2OE) (see Fig. S1 in the supplemental material) using two overexpression lines increased ATP levels and mitochondrial DNA (mtDNA) content autonomously (Fig. 1A and B). Muscle-specific dSir2 knockdown (mdSir2KD) (see Fig. S1) using two RNA interference (RNAi) lines led to a reduction in these parameters (Fig. 1A and B). In addition to muscles, fat body (equivalent to liver and adipocytes) is important for metabolic and energy homeostasis in flies. Fat body-specific dSir2 knockdown (fbdSir2KD) (see Fig. S1) and dSir2 overexpression (fbdSir2OE) (see Fig. S1) flies exhibited bidirectional changes in total ATP levels in the whole body (Fig. 2A). We checked if this was associated with changes in mitochondrial DNA content in fbdSir2KD and fbdSir2OE flies. Consistent with the ATP results, dSir2KD and dSir2OE in the fat body led to similar alterations in mtDNA content in the muscles (Fig. 2B). The electrochemical gradient across the inner mitochondrial membrane is the major driver for mitochondrial ATP synthesis and is an indicator of mitochondrial activity. We measured mitochondrial membrane potential in the muscles of fbdSir2OE and fbdSir2KD flies. fbdSir2OE flies showed a significant increase in mitochondrial membrane potential in the muscles while fbdSir2KD flies showed a decrease (Fig. 2C and D).
Fig 1.

Muscle dSir2 regulates mitochondrial functions and energy homeostasis. Total ATP and mitochondrial DNA content were measured in muscle-specific dSir2 overexpression, mdSir2OE (pSw-MHC-Gal4/Sir2EP2300 and pSw-MHC-Gal4/Sir2EP2384), and knockdown, mdSir2KD [pSw-MHC-Gal4; Sir2RNAi(23201) and pSw-MHC-Gal4; Sir2RNAi(23199)], flies. (A) Normalized total ATP levels in mdSir2OE and mdSir2KD flies. (B) Mitochondrial DNA content (normalized to nuclear DNA content) in mdSir2OE and mdSir2KD flies. All the assays were done on muscle samples. Sample sizes were 36 flies for the ATP assay and 24 flies for the measurement of mtDNA. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01.
Fig 2.

Fat body dSir2 nonautonomously regulates muscle mitochondrial functions and energy homeostasis. Total ATP, mitochondrial DNA content, and mitochondrial membrane potential were measured in fat body-specific dSir2 overexpression, fbdSir2OE (pSw-S1106-Gal4/Sir2EP2300 and pSw-S1106-Gal4/Sir2EP2384), and knockdown, fbdSir2KD [pSw-S1106-Gal4; UAS-Sir2RNAi(23201) and pSw-S1106-Gal4; UAS-Sir2RNAi(23199)], flies. (A) Normalized total ATP levels in fbdSir2OE and fbdSir2KD flies. (B) Mitochondrial DNA content (normalized to nuclear DNA content) in fbdSir2OE and fbdSir2KD flies. (C) Mitochondrial membrane potential as measured using JC-1 staining and confocal microscopy as detailed in Materials and Methods in fbdSir2OE and fbdSir2KD flies. All the assays were done on muscle samples. Sample sizes were 36 flies for the ATP assay and 24 flies for the measurement of mtDNA. For measurement of mitochondrial membrane potential and microscopy, four flies were used, and a total of 90 stacks were imaged and quantified using ImageJ. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01.
We wanted to investigate if the changes in mitochondrial parameters (Fig. 1 and 2) were also associated with changes in the expression of mitochondrial proteins due to changes in the nuclear genes encoding them. As shown in Fig. 3A to E, mdSir2OE flies showed a significant increase in dPGC1, dCyt.C-p, dCOX-IV, TFAM, and Delg (NRF2) in the muscles, while there was downregulation in the expression of these genes in mdSir2KD flies (Fig. 3; see also Fig. S2 and S5 in the supplemental material). The transcript levels in the muscles of fbdSir2OE and fbdSir2KD flies mimicked those of mdSir2OE and mdSir2KD flies (Fig. 3; see also Fig. S3 and S5 in the supplemental material). Overall, these results clearly demonstrated that both mdSir2 and fbdSir2 regulate mitochondrial functions and energy homeostasis in the muscles in an autonomous and nonautonomous manner, respectively. Additionally, these results also highlight the evolutionary conservation of the role of Sir2 and SIRT1 in the muscles to regulate energy homeostasis (30).
Fig 3.

Muscle and fat body dSir2 regulate the expression of nuclear genes for mitochondrial proteins in the muscles in an autonomous and nonautonomous manner, respectively. As indicated, relative mRNA expression levels of dPGC1, dCyt.C-p, dCOX-IV, TFAM, and Delg in the muscle samples of mdSir2OE (pSw-MHC-Gal4/Sir2EP2300 and pSw-MHC-Gal4/Sir2EP2384) and mdSir2KD [pSw-MHC-Gal4; UAS-Sir2RNAi(23201) and pSw-MHC-Gal4; UAS-Sir2RNAi(23199)] flies (A to E) and of fbdSir2OE (pSw-S1106-Gal4/Sir2EP2300 and pSw-S1106-Gal4/Sir2EP2384) and fbdSir2KD [pSw-S1106-Gal4; UAS-Sir2RNAi(23201) and pSw-S1106-Gal4; UAS-Sir2RNAi(23199)] (F to J) flies. Sample size, 60 flies. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Insulin signaling in the muscles is regulated autonomously as well as nonautonomously by dSir2.
Aberrations in mitochondrial functions are often associated with impaired insulin signaling. Our previous work shows that dSir2 affects insulin signaling by regulating the expression of the Drosophila insulin-like peptides (dILP2 and -5 and not the other dILPs) in a diet-dependent manner (5). To investigate if the altered mitochondrial functions observed in the results shown in Fig. 1 to 3 were also associated with changes in insulin signaling, we measured phospho-AKT levels and the expression of dilp-2 and dilp-5 in both muscles and fat body dSir2OE and dSir2KD flies. While perturbing dSir2 in the muscles affected phospho-AKT levels (Fig. 4A and D; and see also Fig. S4 and S5 in the supplemental material), the expression of both dilp-2 and -5 remained unaltered (Fig. 4B and F; see also Fig. S4 and S5). Interestingly, fbdSir2OE led to a significant downregulation in the expression of both dilp-2 and -5, while fbdSir2KD flies showed an increase in their expression (Fig. 4I and L; see also Fig. S4 and S5). The phospho-AKT levels increased in the muscles of fbdSir2OE flies while there was a reduction in phospho-AKT levels in the muscles of fbdSir2KD flies (Fig. 4G, H, K, and L; see also Fig. S4 and S5). These results suggested that dSir2 is a critical regulator of insulin signaling in the muscles.
Fig 4.

Insulin signaling in the muscles is regulated autonomously as well as nonautonomously by dSir2. (A to C) Muscle-specific dSir2 overexpression (pSw-MHC-Gal4/Sir2EP2300, or mdSir2OE): phospho-AKT levels in muscle samples (A), ratio of p-Akt/Akt in these samples (B), and relative mRNA expression levels of dilp-2 and -5 in head samples (C). (D to F) Muscle-specific dSir2 knockdown (pSw-MHC-Gal4; Sir2RNAi, or mdSir2KD): phospho-AKT levels in muscle samples (D), ratio of p-Akt/Akt in these samples (E), and relative mRNA expression levels of dilp-2 and -5 in head samples (F). (G to I) Fat body-specific dSir2 overexpression (pSw-S1106-Gal4/Sir2EP2300, or fbdSir2OE): phospho-AKT levels in muscle samples (G), ratio of p-Akt/Akt in these samples (H), and relative mRNA expression levels of dilp-2 and -5 in head samples (I). (J to L) Fat body-specific dSir2 knockdown (S1106-Gal4; UAS-Sir2RNAi, or fbdSir2KD): phospho-AKT levels in muscle samples (J), ratio of p-Akt/Akt in these samples (K), and relative mRNA expression levels of dilp-2 and -5 in head samples (L). Sample sizes were 16 flies for phospho-AKT, Akt, and actin levels per loading and 24 flies for measurement of dilp mRNA expression. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. **, P < 0.01; ***, P < 0.001.
Insulin signaling governs the efficiency of glucose utilization in organisms. We performed an oral glucose tolerance test (oGTT) (29) in muscle and fat body dSir2OE and dSir2KD flies to check the physiological output of the effect of dSir2 on insulin signaling in the muscles. Overexpression of dSir2 in the muscles improved glucose clearance, while a muscle-specific knockdown of dSir2 hampered it (Fig. 5A and B). Similar to the results of overexpression in muscles, overexpression of dSir2 in the fat body improved the oGTT response while knocking it down worsened the oGTT response (Fig. 5C and D). These results clearly highlight that the role of Sir2 and SIRT1 in maintaining glucose homeostasis is evolutionarily conserved in both invertebrates and vertebrates (23, 31).
Fig 5.
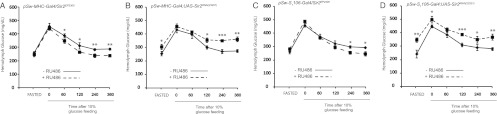
dSir2 in the fat body and muscles regulates glucose homeostasis. An oral glucose tolerance test (oGTT) was performed using hemolymph isolated from pSw-MHC-Gal4/Sir2EP2300 (mdSir2OE) (A), pSw-MHC-Gal4; UAS-Sir2RNAi (mdSir2KD) (B), pSw-S1106-Gal4/Sir2EP2300 (fbdSir2OE) (C), and pSw-S1106-Gal4; UAS-Sir2RNAi (fbdSir2KD) (D) flies. For every time point, 5 samples from hemolymph isolated from 12 flies each were used. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fat body dSir2 mediates its effects on dFOXO-dependent fat metabolism via dILPs and insulin/IGF signaling (IIS).
We have earlier shown that fat body dSir2 regulates systemic insulin signaling and affects fat metabolism (5). To determine if the aberrations in insulin signaling arose due to defects in fat metabolism, we looked at triglyceride levels, phospho-AKT levels, and expression of insulin signaling components in the fat body of fbdSir2KD flies. Knockdown of dSir2 in the fat body led to an increase in total triglyceride levels within the fat body (see Fig. S6 in the supplemental material). Unlike in the muscles, knocking down dSir2 in the fat body led to an increase in the phospho-AKT levels in the fat body (Fig. 6A and B), indicating an increase in insulin signaling. Increased insulin signaling reduces nuclear localization of FOXO and transcription of lipolytic genes (32, 33). To check if the increase in insulin signaling in the fat body of fbdSir2KD flies led to a reduction in the nuclear localization of dFOXO, we monitored the localization of dFOXO-green fluorescent protein (GFP) in the fat body of control and fbdSir2KD flies. As shown in Fig. 6C, knockdown of dSir2 in the fat body significantly reduces the nuclear localization of dFOXO-GFP (Fig. 6C). This in addition to an increase in dilp-2 and dilp-5 (Fig. 4) was interpreted as an increase in insulin signaling within the fat body in fbdSir2KD flies.
Fig 6.

Fat body dSir2 regulates dFOXO localization and activity in an insulin-dependent manner. Phospho-AKT levels in fat body samples of pSw-S1106-Gal4; UAS-Sir2RNAi (fbdSir2KD) flies (A) and the ratio of p-Akt/Akt in these samples are shown (B). (C) dFOXO-GFP localization in the fat bodies of control and fbdSir2KD flies. (D and E) Relative mRNA expression levels of brummer (bmm), dGADD45, dPEPCK, Cathepsin-L (dCPl), and d4eBP in the fat bodies of pSw-S1106-Gal4; UAS-Sir2RNAi, pSw-S1106-Gal4/UAS-ChRNAi, and pSw-S1106-Gal4/UAS-chRNAi; UAS-Sir2RNAi flies with relevant controls (n = 24 flies for measurement of mRNA expression). Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. Images were captured using a Zeiss 510 metaconfocal microscope at a magnification of ×63. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Decreased nuclear localization of FOXO is associated with a reduction in its transcriptional activity and a consequential downregulation of its target genes (14). In addition to a reduction in the nuclear localization of dFOXO-GFP (Fig. 6C), increased insulin signaling in the fat body of fbdSir2KD flies led to a downregulation of the expression of FOXO target genes (Fig. 6D). chico, the Drosophila homolog of mammalian IRS-1 is a critical component of insulin signaling, and abrogating chico expression leads to reduced insulin signaling (34). We wanted to check if reducing insulin signaling, using chicoRNAi, in fbdSir2KD flies restores FOXO-dependent transcription in the fat body. We compared the expression of FOXO target genes in the fat bodies of pSw-S1106-Gal4/chRNAi and pSw-S1106/UAS-chRNAi; UAS-Sir2RNAi flies. It is interesting that knocking down chico in the fat bodies of fbdSir2KD flies (pSw-S1106/UAS-chRNAi; UAS-Sir2RNAi) rescued the expression of the dFOXO target brummer lipase (bmm) (35, 36) but not that of the other FOXO target genes compared to fbdSir2KD (Fig. 6E). The rescue in the expression of bmm alone indicated that FOXO-dependent regulation of fat metabolism is probably independent of a direct interplay between dSir2 and FOXO in the fat body.
However, it was not clear if the defects in fat metabolism resulted in abrogated signaling or if increased insulin signaling led to fat accumulation in the absence of fat body dSir2. To dissect the interplay between insulin signaling and fat metabolism in fbdSir2KD flies, we overexpressed both wild-type and constitutively nuclear dFOXO (dFOXO-TM) (37). Overexpression of phosphorylation-defective FOXO has been shown to act independent of alterations in insulin signaling in both mammals and flies (33, 37). Interestingly, while overexpression of dFOXO-TM alone in the fat body did not alter total triglycerides (TAGs), it brought down the TAGs to basal levels in fat body dSir2 knockdown flies, thereby rescuing the fat phenotype (Fig. 7A). This rescue was associated with a 4-fold increase in the expression of bmm in fbdSir2KD+FOXO-TMOE compared to fbdSir2KD flies (Fig. 7B). In contrast, flies overexpressing only wild-type dFOXO (fbdFOXO-WTOE) and in the background of fat body dSir2KD (fbdSir2KD; FOXO-WTOE) showed TAGs and bmm mRNA levels similar to those of fat body dSir2KD flies (see Fig. S7 and S8 in the supplemental material).
Fig 7.

Fat body dFOXO-TM overexpression in the absence of dSir2 rescues triglyceride levels but not muscle insulin signaling defects. As indicated on the figure, data are for fat body-specific dSir2 knockdown (pSw-S1106-Gal4; UAS-Sir2RNAi), fat body-specific overexpression of dFOXO-TM (pSw-S1106-Gal4/UAS-dFOXO-TM), and simultaneous fat body-specific knockdown and overexpression of dSir2 and dFOXO-TM (pSw-S1106-Gal4/UAS-FOXO-TM; UAS-Sir2RNAi). (A) Total triglyceride levels. (B) Expression of brummer lipase. (C) Relative expression of dilp-5 in head sample. (D) p-Akt levels in muscle samples. Relative mRNA expression levels of dPGC1 (E), dCyt.C-p (F), and dCOX-IV (G) in muscle samples are shown. (H and J) Circulating free fatty acid levels. (I) Relative mRNA expression of fatty acid synthase in the fat body. Sample sizes were 16 flies for phospho-AKT levels and 60 flies for analysis of the expression levels of genes. For free fatty acid measurement eight sets with five flies were used. Control, −RU486; overexpression/knockdown, +RU486 (200 μM or 400 μM, for driving single or double UAS genes). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Hyperlipidemia in mammals is associated with severe inflammatory responses (38, 39). We investigated if the hyperlipidemia in the fbdSir2KD flies led to an increase in the inflammatory response. As shown in Fig. S9 in the supplemental material, fbdSir2KD flies showed a significant increase in 18w (interleukin-2 [IL-2]) and egr (tumor necrosis factor alpha [TNF-α]) in the fat body, which indicated the evolutionary conservation of the functions of Sir2 and SIRT1 (40, 41).
Nonautonomous effect of fat body dSir2 on insulin signaling in the muscles is mediated by free fatty acids.
Based on the results presented above, we wanted to assess the contributions of hyperlipidemic and hyperinsulinemic effects on insulin signaling in the muscles of fbSir2KD flies. There was a reduction in the p-Akt levels in the muscles of fbdSir2KD; FOXO-TMOE flies similar to that in the fbSir2KD flies (Fig. 7D). Additionally, the decreased p-Akt levels in the muscles of these flies were associated with increased dilp-2 and dilp-5 expression (Fig. 7C; see also Fig. S10 in the supplemental material). There was also a significant reduction in the expression of dPGC1, dCyt.C-p, and dCOX-IV (Fig. 7E to G) in the muscles of fbdSir2KD; FOXO-TMOE flies which mimicked that in fbdSir2KD flies.
In addition to TAGs, circulating free fatty acids have been shown to affect insulin signaling and lead to insulin resistance (42, 43). To examine the effect of fbdSir2 on free fatty acids, we measured circulating free fatty acid levels in the hemolymph. As shown in Fig. 7H, fbdSir2KD led to a significant increase in circulating free fatty acid levels. Additionally, these flies also exhibited a marked increase in the expression of fatty acid synthase (FAS) gene (Fig. 7I; see Fig. S5 in the supplemental material). These results indicate that elevated free fatty acid levels, which are associated with abrogated fat metabolism in the fat body, might be contributing to the reduced insulin signaling in the muscles of these flies. Intriguingly, free fatty acid levels remained elevated in fbdSir2KD with FOXO-TMOE and fbFOXO-TMOE flies (Fig. 7J).
To address the role of free fatty acids in the manifestation of the metabolic defects associated with fbdSir2KD, we tried to rescue the phenotype by administering l-carnitine. As shown in Fig. 8A, administering l-carnitine not only reduced circulating free fatty acid levels in control flies but also decreased the elevated free fatty acid levels that were observed in fbdSir2KD flies. l-Carnitine increases beta-oxidation by enhancing the mitochondrial uptake of fatty acids via carnitine palmitoyl transferase 1 (CPT1). Inhibition of CPT1 activity using etomoxir abrogated the l-carnitine-mediated decrease in circulating free fatty acids in both control and fbdSir2KD flies (Fig. 8A).
Fig 8.

l-Carnitine rescues the defects in muscle physiology associated with fat body-specific knockdown of dSir2. Fat body-specific dSir2 knockdown (pSw-S1106-Gal4; UAS-Sir2RNAi or fbdSir2KD) flies were treated or not treated with 25 mg/ml l-carnitine for 24 h and with 25 μM etomoxir for 12 h. Circulating free fatty acid levels (A), phospho-AKT levels (B), total ATP levels (C), mtDNA content (D), and mitochondrial membrane potential (E) were determined. (F to H) Relative mRNA expression levels of dPGC1, dCOX-IV, and dCyt.C-p, as indicated. Sample size, eight sets each with 5 flies for free fatty acid measurement, 24 flies for measurement of phospho-AKT levels, and 36 flies for total ATP levels. Mitochondrial membrane potential was measured using JC-1 staining using four flies. In total, 90 stacks were imaged and quantified by ImageJ. Control, −RU486; overexpression/knockdown, +RU486 (200 μM). All data are shown as means ± standard errors of the means. *, P < 0.05; **, P < 0.01.
Next, we wanted to check if the l-carnitine-mediated reduction in free fatty acid levels also rescued reduced insulin signaling in the muscles of fbSir2KD flies. As expected, we found that l-carnitine administration rescued the reduced phospho-AKT levels in the muscles of fbSir2KD flies, and levels were comparable to those of control flies (Fig. 8B).
l-Carnitine rescues the defects in muscle mitochondrial physiology associated with fat body-specific knockdown of dSir2.
Results shown in Fig. 2 and 3 demonstrate that fbdSir2KD flies showed severe defects in mitochondrial functions. To investigate the contribution of free fatty acids in the mitochondrial dysfunctions associated with fbdSir2KD flies, we tried to rescue the mitochondrial defects by l-carnitine. In response to l-carnitine treatment, fbdSir2KD flies showed a rescue in ATP levels (Fig. 8C), mitochondrial DNA content (Fig. 8D), and mitochondrial activity (Fig. 8E) as well as the expression of nuclear genes dPGC1, dCOX IV, and dCyt.C-p encoding mitochondrial proteins (Fig. 8F to H).
Overexpression of dSir2 in the fat body but not muscles increases starvation survival.
Based on our earlier report and the results presented above, we wanted to determine if overexpression of dSir2 in the fat body and muscles influenced starvation survival. As shown in Fig. 9A, fbdSir2OE led to a significant increase in starvation survival, and fbdSir2KD reduced starvation resistance, consistent with our earlier findings (5). Corroborating our previous report and highlighting the importance of fat body dSir2, overexpression and knockdown of dSir2 in the muscles did not have any effect on starvation survival (Fig. 9B).
Fig 9.
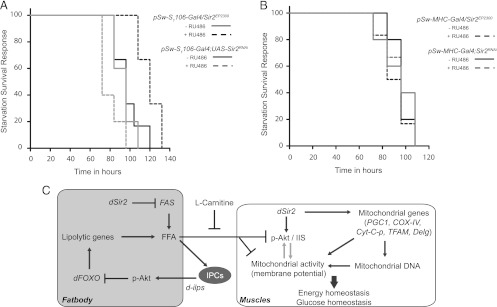
dSir2 in the fat body is a key factor in maintaining metabolic regulatory network in the organism with consequences on survival. (A and B) Starvation survival of fat body-specific dSir2 overexpression, fbdSir2OE (pSw-S1106-Gal4/Sir2EP2300), and knockdown, fbdSir2KD (pSw-S1106-Gal4; UAS-Sir2RNAi), flies (A) and muscle-specific dSir2 overexpression, mdSir2OE (pSw-MHC-Gal4/Sir2EP2300), and knockdown, mdSir2KD (pSw-MHC-Gal4; UAS-Sir2RNAi), flies (B). Control, −RU486; overexpression/knockdown, +RU486 (200 μM). For survival analyses a Mantel-Cox log rank test was used, and the statistical significance was as follows: P = 0.0039 for fbdSir2OE and the control, P = 0.049 for fbdSir2KD and the control, and P was nonsignificant for both mdSir2OE and the control and for mdSir2KD and the control. (C) Schematic indicating the functions of dSir2 on organismal physiology. dSir2 in the muscle is sufficient to regulate mitochondrial functions and glucose homeostasis. The ability of dSir2 to maintain organismal physiology is elicited by its functions in the fat body (liver-equivalent tissue). Fat body dSir2-dependent changes in free fatty acids (FFA) might influence dilp production in the IPCs, which impinges on insulin signaling. dilp/IIS-dependent regulation of dFOXO activity affects lipid homeostasis in the fat body. We hypothesize that an absence of dSir2 in the fat body leads to elevated FFA, thus resulting in a metabolic stress condition in muscles. This manifests as a prediabetic state, which is associated with reduced insulin signaling and mitochondrial dysfunctions in the muscle. Metabolic intervention using l-carnitine rescues this phenotype and supports the mechanistic insights into dSir2-dependent alterations in energy homeostasis.
DISCUSSION
An inability to synchronize metabolic adaptation across tissues leads to physiological changes that are detrimental to the organism. Factors that mediate cellular responses to altered dietary inputs have also been implicated in establishing regulatory networks between metabolic tissues. In this study, we report the central role that dSir2 plays in mediating organismal physiology. We also describe its metabolic functions in the fat body, which impinge upon muscle mitochondrial physiology and insulin signaling in a nonautonomous manner.
The importance of SIRT1 in insulin signaling has been reported by studies that have overexpressed or knocked down SIRT1 (12, 13, 31, 44, 45). Using multiple lines to knock down and overexpress dSir2 in the fat body (liver and adipocyte equivalent) and muscles, we show the evolutionary conservation of the role of Sir2 and SIRT1.
dSir2 enhancer trap lines (EP2300 and EP2384), which contain an enhancer inserted in the upstream regulatory region of the dSir2 gene, have been extensively used to overexpress the native protein (46–48). Due to the proximity of this P-element to the adjacent DNAJ-H gene, there are concerns about a simultaneous overexpression of DNAJ-H and dSir2 in a Gal4-dependent manner. However, similar to an earlier report (46), we found that inducible overexpression of dSir2 using various concentrations of RU486 did not alter the expression of DNA J-H (see Fig. S11 in the supplemental material).
In this report, we show for the first time that overexpressing Sir2 (dSir2) in the muscles improves insulin signaling and glucose homeostasis. Our results clearly point out the functional similarities between dSir2 in the fat body and muscles in maintaining glucose homeostasis (Fig. 5). We show that muscle dSir2 affects insulin signaling in an autonomous manner (Fig. 4), which is associated with alterations in dInR expression (see Fig. S12 in the supplemental material). It remains to be seen if muscle dSir2 regulates other components of insulin signaling as in the case of mammalian SIRT1 (13, 31, 49). Interestingly, the nonautonomous effects of fat body dSir2 on muscle insulin signaling involved dilp-2 and dilp-5 and free fatty acids (Fig. 4 and 7). The increase in p-Akt levels in the muscles of fbdSir2OE (Fig. 4) indicated enhanced insulin signaling.
We have earlier shown that ablating dSir2 in the fat body results in defective fat metabolism (5). In this study, we have found that fat body dSir2-mediated changes affect lipid homeostasis, influence dilp production in the IPCs, and lead to an increase in insulin signaling in the fat body (Fig. 4, 6, and 7). In order to elucidate the complex interactions between abrogated fat metabolism and insulin signaling in the fat body, we have addressed the role of dFOXO. Alterations in dFOXO-GFP localization and changes in the expression of dFOXO target genes in fbdSir2KD flies (Fig. 6) indicated that dSir2 influenced dFOXO activity in an insulin-signaling-dependent manner. Simultaneous knockdown of dSir2 and chico clearly demonstrated the involvement of insulin signaling in dSir2-dependent changes in dFOXO activity (Fig. 6). However, it was interesting that the expression of brummer lipase was independent of a direct interplay between dSir2-dFOXO in the fat body, unlike the other dFOXO target genes such as GADD45, PEPCK, dCP1, and d4eBP (Fig. 6). These results indicated a novel interplay between dSir2 and dFOXO, which is mediated by insulin signaling.
We have further explored the interplay between dFOXO and dSir2 by overexpressing a constitutively nuclear form of dFOXO (dFOXO-TM). Interestingly, while TAG levels are brought back to control levels (Fig. 7), fbdSir2KD; dFOXO-TMOE flies have high circulating free fatty acids and elevated dilp levels and reduced p-Akt in the muscles (Fig. 7). Importantly, by comparing the phenotypes of fbdSir2KD; dFOXO-TMOE and fbdSir2KD; dFOXO-WTOE flies, it becomes obvious that in the absence of fbdSir2, a dilp-mediated increase in insulin signaling in the fat body impairs dFOXO-dependent transcription of lipolytic genes. It should also be noted that there is an increase in fatty acid synthase expression in the fat body of fbdSir2KD flies (Fig. 7). A very recent report by Koh et al. has shown that an autonomous dSir2-dFOXO interaction is important for modulating mitochondrial functions in the background of a PINK mutation (47). However, our report is one of the first studies which describe the significance of dSir2-dFOXO interactions at the organismal level, specifically from a metabolic perspective. Moreover, our findings also point out that dSir2 has a nonautonomous top-down control on dFOXO (and lipolytic effects) via dilp-IIS.
It is important that abrogating dSir2 in the fat body leads to hyperlipidemic and hyperinsulinemic conditions, which are associated with insulin resistance in the muscles. Since these flies mimicked a prediabetic state, we wanted to investigate if fbdSir2KD led to compromised mitochondrial functions and energy deficiency in the muscles. Interestingly, we found that both overexpression and knockdown of dSir2 in the fat body resulted in an increase and decrease, respectively, of ATP in the muscles (Fig. 2). This was associated with changes in muscle mitochondrial DNA content and activity, which were positively correlated with dSir2 expression in the fat body (Fig. 2). Further, the nonautonomous effects of dSir2 on muscle mitochondrial physiology were phenocopied by flies that had dSir2 perturbations (overexpression and knockdown) in the muscle (Fig. 1).
Obesity and diabetes are intrinsically characterized by elevated TAG/free fatty acid levels, impaired insulin signaling, and mitochondrial dysfunctions. Although several hypotheses have been proposed to explain the causal relationship between fat metabolism, insulin signaling, and mitochondrial functions, a clear mechanistic insight is still lacking (4). We have examined whether defects in insulin signaling and mitochondrial functions originate due to the hyperlipidemic condition of fbdSir2KD flies. We show that absence of fat body dSir2 results in aberrant energy metabolism, which leads to an “energy-excess condition” with excessive accumulation of triglycerides (Fig. 7). This energy-excess condition leads to nutrient-induced mitochondrial stress, which is a major cause of insulin signaling defects. More importantly, nutrient-induced stress is associated with improper transport of fatty acids and impaired fatty acid oxidation in the mitochondria. A considerable body of work shows that l-carnitine improves the transport of fatty acids into the mitochondria and activates fatty acid oxidation (50–52). Additionally, l-carnitine has been previously used to rescue fat-dependent metabolic defects in diabetic and obese conditions (53, 54). In order to identify the primary cause of the phenotype associated with fbdSir2KD, we attempted a metabolic rescue using l-carnitine. l-Carnitine administration led to a significant decrease in the fatty acids in fbdSir2KD flies (Fig. 8). However, in the presence of etomoxir, which is an inhibitor of CPT1 (55, 56), l-carnitine administration failed to reduce the circulating free fatty acid levels (Fig. 8). This indicates that the ability of l-carnitine to reduce free fatty acids involves CPT1-dependent fatty acid oxidation in the mitochondria. Moreover, the l-carnitine-mediated rescue in free fatty acid levels was associated with the restoration of insulin signaling defects and mitochondrial functions in the muscles of these flies (Fig. 8). The metabolic rescue observed in these flies highlights the importance of elevated free fatty acids in manifestation of the metabolic defects associated with fbdSir2KD flies (Fig. 8).
A recent report in mammals indicated that the ability of SIRT1 in the liver to regulate insulin signaling in the muscles and adipose tissue arises due to alterations in gluconeogenesis and glycemia (12). However, our findings in fbdSir2KD flies indicate that aberrant fat metabolism and a systemic accumulation of free fatty acids are the primary cause of abrogated insulin signaling in the whole organism.
Our previous report and the results presented above highlight the importance of fat body dSir2 in affecting inflammatory, metabolic, and energy parameters that affect organismal survival. Our results show that overexpression of dSir2 in the fat body, but not muscle, increases starvation survival (Fig. 9). Although the functions of fbdSir2 and mdSir2 in regulating muscle-specific metabolic and glucose homeostasis are similar, the differential effects on organismal survival exemplify the importance of fbdSir2 on organismal physiology.
In summary, we establish the central role that dSir2 plays in maintaining organismal physiology. By comparing and contrasting its functions in the muscles and the fat body, we delineate the role of dSir2 on fat metabolism from insulin signaling (Fig. 7). Our results also point out the top-down control that fat body dSir2 exerts on insulin signaling, mitochondrial functions, and energy homeostasis in peripheral tissues, such as muscles (Fig. 9). In addition to describing the significance of the dSir2-dFOXO interactions at the organismal level, we provide mechanistic insights into dSir2-dependent cross talk across metabolic tissues, which is vital for physiological homeostasis (Fig. 9). Importantly, we show that overexpression of dSir2 in fat body and muscles leads to better metabolic and energy homeostasis, which is phenocopied by flies that have been treated with l-carnitine (Fig. 8 and 9). By demonstrating a metabolic rescue of the phenotype exhibited by fat body dSir2 knockdown flies, we not only highlight its role in fat metabolism but also emphasize the clinical implications of Sir2 orthologs in diseases such as type 2 diabetes and obesity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gaiti Hasan from NCBS and Stephen Helfand and Marc Tatar from Brown University for providing us the fly stocks.
We acknowledge the funding from TIFR/DAE, Government of India.
Footnotes
Published ahead of print 5 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00976-12.
REFERENCES
- 1. Cheng Z, Tseng Y, White MF. 2010. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21:589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marshall S. 2006. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Sci. STKE 2006:re7 doi:10.1126/stke.3462006re7 [DOI] [PubMed] [Google Scholar]
- 3. Taguchi A, White MF. 2008. Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 70:191–212 [DOI] [PubMed] [Google Scholar]
- 4. Muoio DM, Neufer PD. 2012. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banerjee KK, Ayyub C, Sengupta S, Kolthur-Seetharam U. 2012. dSir2 deficiency in the fat body, but not muscles, affects systemic insulin signaling, fat mobilization and starvation survival in flies. Aging 4:206–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chalkiadaki A, Guarente L. 2012. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 16:180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, Hsiao JJ, Iwasaki T, Stark R, Weismann D, Yu XX, Murray SF, Bhanot S, Monia BP, Horvath TL, Gao Q, Samuel VT, Shulman GI. 2009. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc. Natl. Acad. Sci. U. S. A. 106:11288–11293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haigis MC, Guarente LP. 2006. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20:2913–2921 [DOI] [PubMed] [Google Scholar]
- 9. Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, Kemper JK. 2010. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285:33959–33970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. 2009. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9:327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reis T, Van Gilst MR, Hariharan IK. 2010. A buoyancy-based screen of Drosophila larvae for fat-storage mutants reveals a role for Sir2 in coupling fat storage to nutrient availability. PLoS Genet. 6:e1001206 doi:10.1371/journal.pgen.1001206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. 2011. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J. Clin. Invest. 121:4477–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schenk S, McCurdy CE, Philp A, Chen MZ, Holliday MJ, Bandyopadhyay GK, Osborn O, Baar K, Olefsky JM. 2011. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Invest. 121:4281–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barthel A, Schmoll D, Unterman TG. 2005. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16:183–189 [DOI] [PubMed] [Google Scholar]
- 15. Chakrabarti P, Kandror KV. 2009. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J. Biol. Chem. 284:13296–13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015 [DOI] [PubMed] [Google Scholar]
- 17. Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, Luo Z, Farmer SR, Kandror KV. 2011. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 52:1693–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giannakou ME, Partridge L. 2004. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 14:408–412 [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, III, Olefsky J, Guarente L, Montminy M. 2008. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456:269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. 2007. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 282:15439–15450 [DOI] [PubMed] [Google Scholar]
- 21. Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. 2004. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U. S. A. 101:6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. 2003. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U. S. A. 100:8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. 2010. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. 2007. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 26:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pardo PS, Boriek AM. 2011. The physiological roles of Sirt1 in skeletal muscle. Aging 3:430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giannakou ME, Goss M, Jacobson J, Vinti G, Leevers SJ, Partridge L. 2007. Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 6:429–438 [DOI] [PubMed] [Google Scholar]
- 27. Osterwalder T, Yoon KS, White BH, Keshishian H. 2001. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. U. S. A. 98:12596–12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roman G, Endo K, Zong L, Davis RL. 2001. P{Switch}, a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 98:12602–12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haselton A, Sharmin E, Schrader J, Sah M, Poon P, Fridell YW. 2010. Partial ablation of adult Drosophila insulin-producing neurons modulates glucose homeostasis and extends life span without insulin resistance. Cell Cycle 9:3063–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA. 2012. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15:675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, Donmez G, Li J, Luo Z, Walsh K, Guarente L, Zang M. 2011. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 25:1664–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiAngelo JR, Birnbaum MJ. 2009. Regulation of fat cell mass by insulin in Drosophila melanogaster. Mol. Cell. Biol. 29:6341–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsumoto M, Han S, Kitamura T, Accili D. 2006. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Invest. 116:2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. 2001. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292:104–106 [DOI] [PubMed] [Google Scholar]
- 35. Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, Muller G, Jackle H, Kuhnlein RP. 2005. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1:323–330 [DOI] [PubMed] [Google Scholar]
- 36. Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, Yates JR, III, Fischer WH, Thomas JB, Montminy M. 2011. A hormone-dependent module regulating energy balance. Cell 145:596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Puig O, Marr MT, Ruhf ML, Tjian R. 2003. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 17:2006–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weber C, Soehnlein O. 2011. ApoE controls the interface linking lipids and inflammation in atherosclerosis. J. Clin. Invest. 121:3825–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wellen KE, Hotamisligil GS. 2005. Inflammation, stress, and diabetes. J. Clin. Invest. 115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S, Banks AS, Qiang L, Bhanot S, Olefsky JM, Sears DD, Caprio S, Shulman GI. 2011. SirT1 regulates adipose tissue inflammation. Diabetes 60:3235–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh DY, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. 2010. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 298:E419–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, Xiang X, Luo Z, Ruderman N. 2005. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-κB pathway in rat liver. Diabetes 54:3458–3465 [DOI] [PubMed] [Google Scholar]
- 43. Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. 1996. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 97:2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang F, Kume S, Koya D. 2009. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 5:367–373 [DOI] [PubMed] [Google Scholar]
- 45. Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. 2007. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 6:307–319 [DOI] [PubMed] [Google Scholar]
- 46. Bauer JH, Morris SN, Chang C, Flatt T, Wood JG, Helfand SL. 2009. dSir2 and Dmp53 interact to mediate aspects of CR-dependent lifespan extension in D. melanogaster. Aging 1:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koh H, Kim H, Kim MJ, Park J, Lee HJ, Chung J. 2012. Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant. J. Biol. Chem. 287:12750–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rogina B, Helfand SL. 2004. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. U. S. A. 101:15998–16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang J. 2007. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 282:34356–34364 [DOI] [PubMed] [Google Scholar]
- 50. Karlic H, Lohninger S, Koeck T, Lohninger A. 2002. Dietary l-carnitine stimulates carnitine acyltransferases in the liver of aged rats. J. Histochem. Cytochem. 50:205–212 [DOI] [PubMed] [Google Scholar]
- 51. Muller DM, Seim H, Kiess W, Loster H, Richter T. 2002. Effects of oral l-carnitine supplementation on in vivo long-chain fatty acid oxidation in healthy adults. Metabolism 51:1389–1391 [DOI] [PubMed] [Google Scholar]
- 52. Oyanagi E, Yano H, Uchida M, Utsumi K, Sasaki J. 2011. Protective action of l-carnitine on cardiac mitochondrial function and structure against fatty acid stress. Biochem. Biophys. Res. Commun. 412:61–67 [DOI] [PubMed] [Google Scholar]
- 53. Ferrari R, Merli E, Cicchitelli G, Mele D, Fucili A, Ceconi C. 2004. Therapeutic effects of l-carnitine and propionyl-l-carnitine on cardiovascular diseases: a review. Ann. N. Y. Acad. Sci. 1033:79–91 [DOI] [PubMed] [Google Scholar]
- 54. Mingorance C, Rodriguez-Rodriguez R, Justo ML, Herrera MD, de Sotomayor MA. 2011. Pharmacological effects and clinical applications of propionyl-l-carnitine. Nutr. Rev. 69:279–290 [DOI] [PubMed] [Google Scholar]
- 55. Rupp H, Elimban V, Dhalla NS. 1992. Modification of subcellular organelles in pressure-overloaded heart by etomoxir, a carnitine palmitoyltransferase I inhibitor. FASEB J. 6:2349–2353 [DOI] [PubMed] [Google Scholar]
- 56. Rupp H, Schulze W, Vetter R. 1995. Dietary medium-chain triglycerides can prevent changes in myosin and SR due to CPT-1 inhibition by etomoxir. Am. J. Physiol. 269:R630–640 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.