

Abstract
The vast majority of cellular mRNAs initiate their translations through a well-defined mechanism of ribosome recruitment that occurs at the 5′-terminal 7-methylguanosine cap with the help of several canonical protein factors. A subset of cellular and viral mRNAs contain regulatory motifs in their 5′ untranslated regions (UTRs), termed internal ribosome entry sites (IRES), that sidestep this canonical mode of initiation. On cellular mRNAs, this mechanism requires IRES trans-acting protein factors (ITAFs) that facilitate ribosome recruitment downstream of the cap. While several ITAFs and their target mRNAs have been empirically identified, the in silico prediction of targets has proved difficult. Here, we report that a high AU content (>60%) of the IRES-containing 5′ UTRs serves as an excellent predictor of dependence on NF45, a recently identified ITAF. Moreover, we provide evidence that cells deficient in NF45 ITAF activity exhibit reduced IRES-mediated translation of X-linked inhibitor of apoptosis protein (XIAP) and cellular inhibitor of apoptosis protein 1 (cIAP1) mRNAs that, in turn, leads to dysregulated expression of their respective targets, survivin and cyclin E. This specific defect in IRES translation explains in part the cytokinesis impairment and senescence-like phenotype observed in HeLa cells expressing NF45 RNA interference (RNAi). This study uncovers a novel role for NF45 in regulating ploidy and highlights the importance of IRES-mediated translation in cellular homeostasis.
INTRODUCTION
Translation is a process that is tightly regulated to ensure rapid and specific protein expression in response to different stimuli (1). Translation regulation occurs primarily at the initiation step through a canonical mechanism in which the ribosome is recruited at the 5′-terminal 7-methylguanosine cap with the help of several protein factors (2). However, a subset of cellular and viral mRNAs contains regulatory motifs in their 5′ untranslated regions (UTRs), termed internal ribosome entry sites (IRES), that sidestep this canonical mode of translation initiation. First discovered in picornaviruses (3), IRES are specific RNA elements that permit the recruitment of the ribosome to the translation initiation start, independently of the cap (1, 3). Unlike some viral IRES, which can directly recruit the ribosome, eukaryotic IRES appear to require IRES trans-acting protein factors (ITAFs) that facilitate ribosome recruitment by acting as either scaffold proteins or RNA chaperones (reviewed in references 4 and 5). While several ITAFs and their target mRNAs have been empirically identified, in silico prediction of targets has proved difficult due to the lack of consensus regarding IRES binding motifs and an overall poor understanding of the biology of cellular IRES (6).
We previously characterized NF45 as an ITAF that regulates the cellular inhibitor of apoptosis protein 1 (cIAP1) IRES during the unfolded protein response (7). Indeed, NF45 was shown to interact directly with the cIAP1 IRES and positively affect its activity, resulting in the increased translation of its mRNA during thapsigargin-induced endoplasmic reticulum (ER) stress. This was the first report of NF45 having a bona fide function in IRES-mediated translation, although there have been reports of the involvement of NF45 and its binding partner NF90 in transcription (8, 9), viral replication (10–12), and microRNA processing (13, 14).
More recently, NF45 has been implicated in the mitotic control of HeLa cells. Indeed, the depletion of NF45-NF90 complexes by RNA interference leads to the generation of large multinucleated cells, a result of impaired cytokinesis and cell growth (15, 16). Multinucleation can arise from defects in DNA replication, cell proliferation, or mitosis and from the aberrant expression of proteins regulating these key cellular events. One such protein that is essential to mitosis and whose aberrant expression has been linked to multinucleation is survivin (17, 18). Survivin (BIRC5), a member of the inhibitor of apoptosis protein (IAP) family, has been shown to play a dual role in cell division by regulating microtubule dynamics (19) and by being a member of the chromosomal passenger complex (reviewed in reference 20). Cyclin E is another protein that is crucial for cell cycle progression because of its interaction with cyclin-dependent kinase 2 (Cdk2), which leads to retinoblastoma protein (Rb) phosphorylation, the release of E2F transcriptional activity, and the expression of genes that drive the G1-to-S transition (21–23).
In this study, we developed a screen to identify new targets of NF45 regulation and to show that a high AU content in IRES-containing 5′ UTRs serves as an excellent predictor of NF45 dependence. Moreover, we provide evidence that cells deficient in NF45 ITAF activity exhibit reduced IRES-mediated translation of X-linked IAP (XIAP) and cIAP1 mRNAs. This in turn leads to dysregulated expression of their respective targets, survivin and cyclin E, thus explaining in part the multinucleated phenotype of NF45-deficient cells. Hence, we report the identification of an NF45-regulated operon that impinges on the regulation of cell division and cell cycle progression.
MATERIALS AND METHODS
Cell cultures, expression constructs, and transfection.
HeLa cells expressing a control (c) or NF45 (d5) small hairpin RNA (shRNA) were as characterized previously (15) and maintained under selective pressure conditions with 400 μg/ml of G418 (Invitrogen, Burlington, ON, Canada) in serum-, antibiotic-, and sodium pyruvate-supplemented Dulbecco's modified Eagle's medium (DMEM). HEK293 and HeLa cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and were routinely cultured in DMEM supplemented with serum, antibiotic, and l-glutamine. Transient DNA transfections were performed using Lipofectamine Plus reagent (Invitrogen) or jetPRIME (Polyplus Transfection, Illkirch, France) per the manufacturers' protocols. For Western blot analyses, the cells were transfected with 50 nM NF45 or control (nontargeting) small interfering RNAs (siRNAs) using RNAiMAX reagent (Invitrogen) for 72 h or 96 h. Unless otherwise stated, all assays were performed 24 h following transfection, except for the rescue experiments, which were performed at 48 h posttransfection. Green fluorescent protein (GFP) fusion expression plasmids were constructed by first ligating Turbo GFP (from pTurboGFP-dest1; Evrogen, Moscow, Russia) into a pcDNA3 backbone (Invitrogen), followed by ligation of the appropriate open reading frame (ORF) (NF45 or XIAP). The GFP-NF45R construct was obtained by introducing a silent mutation that is resistant to the d5 shRNA to the GFP-NF45 plasmid by site-directed mutagenesis. All plasmid constructs were verified by nucleotide sequencing.
Cloning of IRES and bicistronic assays.
Two databases, one with nonredundant human 5′ UTR sequences and one containing known IRES sequences, were used to calculate the AU content of known human IRES and 5′ UTRs (6). The aquaporin 4 (AQP4), cIAP1, ELG, nuclear respiratory factor (NRF), and XIAP IRES have been cloned into the pβgal/CAT bicistronic vector and characterized (7, 24). (As an aside, ELG1 is another name for the gene ATAD5 that is involved in DNA repair; ELG is the alias for the C17ORF85 ORF. In this study, “ELG” will be used to designate the C17ORF85-related IRES.) In addition to these IRES, we cloned the previously characterized NRF (25), SNM1 (26), MYT2 (27), UNR (28), TEK (29), and sphingolipid Ca2+ release-mediating protein from endoplasmic reticulum (Scamper) (30) IRES into the pβgal/CAT bicistronic vector (31). The Scamper and MYT2 IRES were synthesized with flanking NheI-XhoI sites and cloned into a pUC57 vector (Bio Basic, Inc., Toronto, ON, Canada), followed by subcloning into the pβgal/CAT bicistronic vector. A portion of the SNM1 (GenBank accession no. BI770136) 5′ UTR containing IRES activity described by Zhang et al. (26) (nucleotides −669 to −1, CTC TTC CT GCT AGC GGG ATT GTT CAT TGC TGC [forward primer] and CTC TTC CT CTC GAG GGC AAA ATG ATT TTA TCA [reverse primer]), the UNR (GenBank accession no. BI546285) 5′ UTR containing IRES activity described by Cornelis et al. (28) (nucleotides −462 to −1, CTC TTC CT GCT AGC TGC TGC TTA TGG CGG CGC [forward primer] and CTC TTC CT CTC GAG CGC AGT GAT ACT CAA ATA [reverse primer]), and the TEK (GenBank accession no. BI546285) 5′ UTR containing IRES activity described by Park et al. (29) (nucleotides −354 to −1, CTC TTC CT GCT AGC GCA GCA GCA AAA GCA GCA [forward primer] and CTC TTC CT CTC GAG GCT TCC CCA AAT CTC TCC [reverse primer]) were amplified by PCR from mammalian gene collection clones (all purchased from Thermo Fisher Scientific, Inc., Ottawa, ON, Canada) using primers with NheI-XhoI sites. The NRF 5′ UTR (nucleotides −653 to −1, CTC GAG CAG AGT AAT GAC ATG GTT CC [forward primer] and CTC TTC CAA GCG TGG GCT GTA CC [reverse primer]) was PCR amplified from a liver cDNA library using primers with NheI-XhoI sites. The amplified UTRs were then subcloned into the pβgal/CAT bicistronic vector. All constructs were verified by sequencing, and basal IRES activities in c cells (relative to cells transfected with empty bicistronic vector) were confirmed. β-Gal and CAT assays were performed as described previously (31).
Polysome profiling and quantitative reverse transcription-PCR analyses.
c and d5 cells were grown to 70 to 80% confluence on 15-cm plates in DMEM supplemented with sodium pyruvate. For rescue experiments, cells were transfected with 10 μg of GFP or 20 μg of GFP-NF45R for 24 h prior to harvesting. The cells were then incubated with 0.1 mg/ml cycloheximide for 3 min, washed with cold phosphate-buffered saline (PBS)-cycloheximide, and lysed in cold polysome lysis buffer (15 mM Tris-HCl [pH 7.4], 15 mM MgCl2, 300 mM NaCl, 1% [vol/vol] Triton X-100, 0.1 mg/ml cycloheximide, 100 U/ml RNasin). Equal OD254 (optical density at 254 nm) units were loaded onto 10-to-50% linear sucrose gradients (10 ml) and centrifuged at 39,000 rpm for 90 min at 4°C. The gradients were fractionated from the top into 1-ml fractions using an Isco gradient fractionation system (Teledyne Isco Inc., Lincoln, NE), and the RNA was monitored at 254 nm. One hundred nanograms of in vitro-synthesized CAT RNA was added to each fraction as an internal control, and the total RNA was isolated from individual fractions by proteinase K digestion followed by phenol-chloroform extraction. Equal volumes of RNA from each fraction were used to generate cDNA from the qScript cDNA SuperMix reverse transcription kit (Quanta Biosciences, Gaithersburg, MD). PCR primers specific for XIAP (AGGGCACATGTATGTCATGG [forward] and TAGAGGGTGGCTCAGGAAAA [reverse]), cIAP1 (TCTGGAGATGATCCATGGGTAGA [forward] and TGGCCTTTCATTCGTATCAAGA [reverse]), NRF (AAATCTGGTGAGGGCATACG [forward] and TCAAATCTGTGTGGCTCTCG [reverse]), SNM1 (TTCCTAGCCATTGCTGATGTT [forward] and TTGCATCATTGGGAGAAGGT [reverse]), ELG (AAACCCATCACCCCTAAATG [forward] and GGGCGGGGAATATATAAAGG [reverse]), apoptotic protease activating factor 1 (APAF1) (CTTGAGCCCTGGAGTTTGAG [forward] and TGCATGAACTGCCATGAAAT [reverse]), and CAT (GCGTGTTACGGTGAAAACCT [forward] and GGGCGAAGAAGTTGTCCATA [reverse]) were used to quantify mRNAs by quantitative PCR (qPCR) using PerfeCTa SYBR green supermix (Quanta Biosciences). The mRNA distribution profiles were plotted for each fraction (normalized to CAT RNA), and the ratios of heavy polysomes (HPs) (fractions 5 to 10) to light polysomes (LPs) (fractions 2 to 4) were calculated for each transcript. For steady-state mRNA levels, RNA was extracted from cells using the Absolutely RNA miniprep kit (Agilent Technologies, Cedar Creek, TX), and cDNA and qPCR analyses were performed as described above.
RNA immunoprecipitation.
HeLa c cells were plated on 10-cm plates at 50% confluence and were transfected the next day with 10 μg of pcDNA3-FLAG or pcDNA3-FLAG-NF45 plasmid using jetPRIME transfection reagent (Polyplus-Transfection). Twenty-four hours later, 1% formaldehyde was added to the cells to cross-link RNA-protein complexes for 30 min at room temperature. Glycine (0.2 M) was added to quench the cross-linking, and cells were lysed for 30 min at 4°C in RNA immunoprecipitation (RIP) buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA [pH 8.0], 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate) supplemented with 1 μg/ml each of aprotinin, leupeptin, pepstatin, and phenylmethylsulfonyl fluoride (PMSF) and 40 U/ml of RNase inhibitor (Promega). Cell lysates were sonicated and treated with DNase I for 30 min. Immunoprecipitation was carried out using 20 μl of anti-FLAG-M2 affinity gel (Sigma) per sample for 2 h at 4°C. The beads were washed 3 times in PBS, treated with 20 μg of proteinase K at 55°C for 1 h, and the cross-linking was reversed at 70°C for 45 min. RNA was extracted by phenol-chloroform purification, and cDNA was generated using qScript cDNA supermix (Quanta Biosciences). RNA immunoprecipitates were detected using primers specific to XIAP, cIAP1 (QuantiTect primer assay; Qiagen), NRF, ELG, and APAF1.
Western blot analyses.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer for 30 min at 4°C, followed by centrifugation at 10,000 × g to remove debris. Equal amounts of proteins were resolved by 10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes using a wet-transfer protocol, and probed with antibodies to NF45 (7), XIAP (anti-RIAP3 [24]), survivin (6EA; Cell Signaling Technology, Danvers, MA), cyclin E (clone C-19; Santa Cruz Biotechnologies, Santa Cruz, CA), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (BD Biosciences, Mississauga, ON, Canada). Membranes were incubated with Alexa 680-conjugated (Invitrogen, Burlington, ON, Canada) or IR800-conjugated (Li-Cor Biosciences, Lincoln, NE) secondary antibodies, followed by detection using the Li-Cor Odyssey infrared scanner (Li-Cor Biosciences, Lincoln, NE). Densitometry analyses were performed using the Li-Cor Odyssey software.
Metabolic labeling and immunoprecipitation.
Cells were plated on 10-cm dishes and transfected with 50 nM nontargeting siRNA or NF45 siRNA. After 96 h of knockdown, cells were metabolically labeled with [35S]methionine for 45 min at 37°C and lysed as described previously (7). Coimmunoprecipitations of cIAP1, XIAP, and GAPDH proteins were performed at 4°C overnight using protein G/protein A-agarose beads (EMD Chemicals) coated with, respectively, the antibodies anti-cIAP1 (R&D Systems, MN) at a 1:150 dilution, anti-GST-XIAP (32) (Aegera) at a 1:150 dilution, and anti-GAPDH (clone 6C5; Advanced ImmunoChemical, CA) at a 1:250 dilution. The immunoprecipitated beads were then washed with cold wash buffer (50 mM Tris [pH 7.4], 300 mM NaCl, 0.1% Triton X-100), resuspended in Laemmli buffer, and boiled to elute bound proteins. The immunoprecipitated proteins were separated on 10% SDS-PAGE gels along with 5% of the input proteins. The gel was stained with Coomassie blue, incubated with Amplify fluorogenic reagent (GE Biosciences, Baie d'Urfé, QC, Canada) for 30 min, and dried before exposure to film. The input proteins were also analyzed by Western blotting to verify the knockdown of NF45.
RNA-streptomycin affinity chromatography.
S10 cytoplasmic lysate from HeLa cells was prepared as described previously (7). Briefly, HeLa-S cells (2-ml packed cell volume [PCV]) (BioVest International, Tampa, FL) were resuspended in 2 ml of hypotonic buffer (10 mM Tris-HCl [pH 7.6], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT]) containing EDTA-free protease inhibitor cocktail (Roche) and lysed using a Dounce homogenizer (30 strokes, pestle B). A mixture of 4 ml of HeLa S10 cytoplasmic lysate and 12 ml binding buffer (20 mM Tris [pH 7.6], 10 mM MgCl2, 120 mM KCl, 8% sucrose, 2 mM DTT) containing EDTA-free protease inhibitor cocktail (Roche) and RNase inhibitor (Promega) was incubated at 37°C for 10 min. An in vitro-transcribed, poly(A)-tailed and strepto-tagged XIAP IRES or β-hemoglobin 5′ UTR RNAs were added to the mixture and incubated further for 10 min at 37°C. RNA-dihydrostreptomycin affinity chromatography was performed as described previously (33), and the RNA-associated proteins were analyzed using Western blot analysis.
Fluorescence microscopy.
d5 and c cells (1.0 × 105) were seeded onto 12-well flat-bottom plates. For HeLa cell immunofluorescence, 1.0 × 105 cells were seeded on coverslips in 6-well plates and transfected with 50 nM nontargeting or NF45-targeting siRNA for 72 h. Twenty-four hours later, the cells were fixed in 3.7% formaldehyde, permeabilized with 0.1% Triton X-100, and simultaneously stained for nuclei (5 μg/ml Hoechst 3342) and filamentous actin (F-actin) filaments (0.2 U/ml Alexa Fluor 568-conjugated phalloidin) for 30 min at room temperature. The staining for d5 and c cells was captured using a Cellomics ArrayScan VTI automated fluorescence imaging system (Thermo Fisher Scientific, Inc., Ottawa, ON, Canada), and staining for HeLa cells was captured using an Olympus Fluoview FV1000 confocal microscope (Richmond Hill, ON, Canada).
Propidium iodide staining and flow cytometry.
d5 and c cells (3.0 × 105) were seeded onto 6-well flat-bottom plates. Twenty-four hours posttransfection, the cells were scraped from the plates, resuspended in 500 μl of PBS, and fixed in 500 μl of chilled ethanol for 30 min at 4°C. The cells were then spun down at 300 × g for 5 min at 4°C and resuspended in 500 μl of PBS. DNase-free RNase (Qiagen, Toronto, ON, Canada) was added to the suspension at 9 μg/ml, and the samples were briefly vortexed and incubated for 1 h at 37°C. Propidium iodide (Sigma, Oakville, ON, Canada) was added to a final concentration of 13 μg/ml, and the samples were incubated for 30 min at room temperature. Flow cytometry was performed using a BD FACSCanto flow cytometer (BD Biosciences, Mississauga, ON, Canada).
Statistical analyses.
All data are expressed as the mean ± standard error of the mean (SEM). Unless otherwise stated, all results were obtained through a minimum of three independent experimental replications. A t test analysis was performed to determine the significance of the data using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA).
RESULTS
AU content of 5′ UTRs correlates with NF45-dependent IRES activity.
We initially identified NF45 as an ITAF that positively regulates the cIAP1 IRES during the unfolded protein response (7). In that study, NF45 was found to interact with an AU-rich stem-loop within the cIAP1 IRES. We noticed that the entire cIAP1 IRES region, and indeed its entire 5′ UTR, was unusually AU rich (68%). This was in stark contrast to several GC-rich IRES, whose activities were found to be unaffected by changes in NF45 expression (Bcl-xL, DAP5 APAF1, and VCIP) (Fig. 1A, square data points). It is important to note that the nucleotide composition of cellular IRES identified to date does not deviate significantly from those of their 5′ UTRs (6). Therefore, on its own, this constraint cannot be used to predict novel IRES. We hypothesized, however, that we could predict whether known IRES with a high AU content would be dependent on NF45 for their activities.
Fig 1.
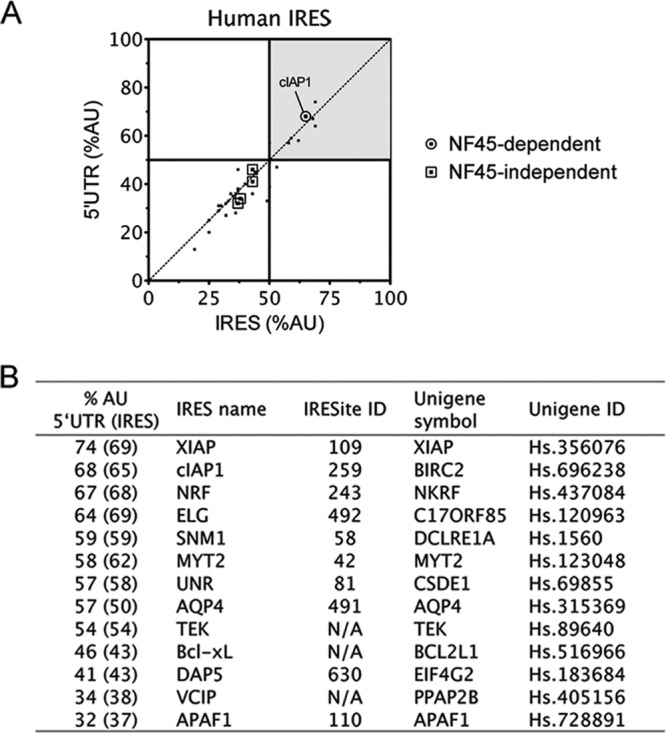
The AU content of 5′ UTRs correlates with NF45-dependent IRES activity. (A) Diagonal plot of the AU content of human 5′ UTRs and IRES. We showed previously (7) that NF45 expression does not affect Bcl-xL, DAP5, APAF1, or VCIP IRES activities, but it does impact the cIAP1 IRES. (B) List of IRES-containing human mRNAs that have a >50% AU content within their 5′ UTRs. The percent AU content of the minimal IRES is indicated in brackets (if unknown, the content is equivalent to that of the 5′ UTR).
To address this hypothesis, we surveyed the set of known IRES-containing RNAs (6) and calculated the AU content of their 5′ UTRs and, if known, their minimal IRES regions (Fig. 1A). We ranked a list of 58 known eukaryotic IRES and 38 viral IRES based on their AU content and further considered only human IRES with AU contents of >50% (Fig. 1A). This resulted in a list of 9 IRES (including cIAP1) that we would anticipate to be dependent on NF45, and we decided to test them in the wet laboratory, along with the GC-rich IRES of Bcl-xL, DAP5, VCIP, and APAF1 (Fig. 1B).
AU-rich 5′ UTRs harboring IRES are regulated by NF45.
To determine whether NF45 acts as an ITAF for AU-rich IRES, we measured IRES activities using an experimental system consisting of HeLa cells that stably express shRNA targeting NF45 d5 cells) and cells expressing a nontargeting shRNA (c cells). These previously characterized cells exhibit significantly reduced expression of NF45 (7, 15). We transiently transfected these cell lines with bicistronic pβgal/CAT vectors harboring selected cellular or viral IRES and determined IRES activity levels in the cells deficient in NF45 relative to the cells expressing normal levels of NF45. Using this system, we showed in our previously published assessment of six cellular IRES that decreased expression of NF45 considerably affects the activity levels of the AU-rich cIAP1 IRES (>2-fold decrease in activity) but spares the activity of GC-rich IRES, such as APAF1, Bcl-xL, DAP5, and VCIP (7). In the present study, we extended the screen to include the AU-rich IRES that are predicted to be NF45 dependent (Fig. 1B). We observed that the ratios of IRES activities in d5 versus c cells were significantly less (<50% of that in c cells) for cIAP1, XIAP, NRF, and ELG IRES (Fig. 2A) and were indeed significantly affected compared to that of the encephalomyocarditis virus (EMCV) IRES that was used as a control. The EMCV IRES (60% GC content) does not require most of the known cellular ITAFs for its optimal activity, except for the polypyrimidine tract binding (PTB) and La proteins (34, 35); therefore, it is not affected by the lack of NF45 in d5 cells (Fig. 2A, black bar). IRES with an AU content of <60% (MYT2, UNR, AQP4, TEK, Bcl-xL, DAP5, VCIP, and APAF1) were largely unaffected by the decreased expression of NF45 in d5 cells (Fig. 2A). Importantly, the decrease in IRES activity levels observed for XIAP, NRF, and ELG in d5 cells was not due to an increase in β-Gal expression but to a decrease in the CAT protein level (data not shown). We also controlled for possible splicing of the bicistronic pβgal/CAT plasmids used and found that the ratios of β-Gal to CAT transcripts were equal in all cases (data not shown); this shows that NF45 regulation of these AU-rich IRES is specific and not due to spurious events associated with the use of reporter constructs. Although we did see a measurable decrease in SNM1 activity (59% AU content, 24% decrease), it was not significant compared to the EMCV IRES; therefore, we did not consider it further. Altogether, these data strongly argue that NF45 is required for the optimal activity of AU-rich IRES.
Fig 2.

AU-rich 5′ UTRs harboring IRES are regulated by NF45. (A) IRES activity was tested in d5 (NF45 shRNA) stable cells relative to c (nontargeting shRNA) cells using a pβgal/IRES/CAT-based bicistronic assay (schematic) with the IRES listed in Fig. 1B, as described in Materials and Methods. Note that a ratio of 1.0 is indicative of no change in IRES activities between the two cell lines. The IRES are ranked in order of decreasing IRES activities and 5′ UTR AU content from left to right (bottom panel). P values are shown for the identified NF45-dependent IRES compared to the EMCV IRES used as a control. (B) Steady-state mRNA levels of the indicated mRNAs were determined by qPCR in c and d5 cells, as well as in d5 cells transfected with GFP (d5-GFP) or a GFP-NF45R (d5-GFP-NF45R). Their expression was normalized to that of β-actin and expressed as a ratio of d5 to c or d5-NF45R to d5-GFP for each transcript. (C) Polyribosome-associated mRNAs from the indicated cell lines were determined as described in Materials and Methods. General polysome profiles, as well as heavy-to-light polysome ratios (HP/LP), are shown for d5 relative to c (d5-to-c ratio) and for d5-GFP-NF45R relative to d5-GFP (d5-NF45R–to–d5 ratio). (D) Detection and enrichment of cIAP1, XIAP, and NRF mRNAs from FLAG-NF45 RNA immunoprecipitation compared to a FLAG control, showing that NF45 specifically associates with these transcripts in vivo.
Next, we analyzed the steady-state mRNA levels of the affected IRES-containing mRNAs by quantitative reverse transcription-PCR (qRT-PCR) and found that they were not significantly decreased in d5 relative to c cells or in d5 cells overexpressing a GFP-NF45 construct harboring a silent mutation resistant to the d5 shRNA (GFP-NF45R) (Fig. 2B, d5-NF45R). These results further confirm that the decreases in IRES activities observed for cIAP1, XIAP, NRF, and ELG in d5 cells are not due to changes at the mRNA level (transcription or mRNA stability) but likely occur through a specific effect on protein translation. To ensure that the NF45-dependent changes in IRES activity assays reflect the translational efficiencies of endogenous mRNAs, polysome profiling of select IRES-containing mRNAs was performed. Cycloheximide-treated d5 and c cell extracts were fractionated on a 10- to -50% linear sucrose gradient by ultracentrifugation. In vitro-transcribed CAT RNA was introduced into each fraction to serve as an internal control, and total RNA was extracted. The distributions of specific transcripts were analyzed by quantitative RT-PCR and normalized to CAT RNA. NF45 knockdown in d5 cells resulted in a decrease in XIAP, cIAP1, NRF, and ELG polysomal loading, as shown by a decrease in the ratio of heavy polysomes (HPs) to light polysomes (LPs) (Fig. 2C, right, white bars); this is reflective of a shift in their mRNA distribution towards less charged polysomes. Importantly, this decrease in polysome loading could be rescued for all target mRNAs by overexpression of the GFP-NF45R complex (Fig. 2C, right, black bars). Unexpectedly, we found that polysome loading of the GC-rich IRES, APAF1, was also decreased in d5 cells. However, this reduction could not be rescued by NF45 overexpression and possibly reflects an indirect effect of NF45-NF90 transcriptional regulation of a wide array of genes. These results confirm that the translation of AU-rich IRES-containing mRNAs of XIAP, cIAP1, NRF, and ELG is enhanced specifically by NF45. Moreover, NF45 knockdown or overexpression did not affect the general translation rates, as indicated by polysome profiles (Fig. 2C, left) or by [35S]methionine incorporation (see Fig. 4C). These observations further demonstrate that NF45 is not a general modulator of translation but is a regulator of IRES-mediated translation of specific transcripts.
Fig 4.

NF45 regulates XIAP translation through interaction with its IRES. (A) Western blots of endogenous XIAP and cIAP1 protein expression in d5 cells relative to that in c cells. The blots and densitometry analyses are representative of at least three experiments. (B) NF45 reexpression in NF45-deficient HeLa cells rescues XIAP protein expression. The HeLa cells were transfected with 50 nM control nontargeting siRNA or NF45 siRNA (d5 siRNA) for 48 h, followed by the overexpression of GFP or GFP-NF45R for an additional 48 h. The cell extracts were analyzed by Western blotting for NF45, XIAP, cIAP1, and GAPDH expression, and protein expression was quantified. (C) De novo protein expression of cIAP1 and XIAP in NF45-deficient cells. c cells were transfected with 50 nM control or NF45 siRNA and were pulse-labeled with [35S]methionine. 35S-labeled and Coomassie blue-stained total protein, as well as specific cIAP1, XIAP, and GAPDH immunoprecipitates, are shown. (D) NF45 interacts specifically with the XIAP IRES. NF45 and PABP Western blotting of an RNA affinity chromatography preparation was performed using the XIAP IRES or hemoglobin RNA.
To demonstrate that NF45 interacts specifically with the cIAP1, XIAP, ELG, and NRF IRES in cells, we set out to identify interactions between NF45 and these mRNAs by RNA immunoprecipitation. HeLa c cells were transfected with a pCDNA3-FLAG-NF45 plasmid or a control pcDNA3-FLAG plasmid, and RNA immunoprecipitation was carried out using anti-FLAG-M2 affinity beads. Immunoprecipitates were analyzed by qPCR for the presence of the target mRNAs using specific primers. Indeed, we observed an enrichment of the cIAP1, XIAP, and NRF mRNAs in FLAG-NF45 compared to the FLAG immunoprecipitates, indicating specific interactions between NF45 and these transcripts in cells (Fig. 2D). However, we could not detect the ELG mRNA in any of the precipitates (Fig. 2D). ELG has two different transcripts, of which the one containing IRES, ELGa (24), is not very abundant in c and d5 HeLa cells and is therefore likely below the PCR detection limit in our assay. Importantly, the GC-rich IRES mRNA APAF1, whose polysome distribution was unaffected by NF45 rescue (Fig. 2C), was not present in FLAG-NF45 immunoprecipitates, further suggesting that NF45 specifically interacts with a select group of AU-rich IRES mRNAs.
Taken together, these data demonstrate that NF45 is an ITAF that specifically regulates IRES activities and translation of the human AU-rich IRES-containing mRNAs of cIAP1, XIAP, ELG, and NRF.
Prediction of NF45-dependent IRES.
We intended to use our results from the IRES screen described above to establish specific constraints that could be used to predict NF45-dependent IRES in silico. From a plot of log2-transformed mean IRES activities versus the AU content of the IRES-containing 5′ UTRs, we can see that those IRES whose activities are reduced by >2-fold (log2 < −1) in NF45-deficient cells all lie within 5′ UTRs with an AU content of ≥60% (Fig. 3A). From these data, we propose that IRES-containing 5′ UTRs with AU content of >60% can be predicted to require NF45 for optimal IRES activity. To confirm this prediction on an unrelated AU-rich IRES that was not part of the original screen, we tested for the activity of the Canis lupus subsp. familiaris IRES, Scamper, in d5 cells. The AU content of the Scamper 5′ UTR and its respective IRES is 63% and therefore fits our constraint. As predicted, we found that Scamper IRES activity was decreased by 50% in d5 relative to c cells, confirming that Scamper expression is regulated by human NF45 (Fig. 3B). It is important to note that C. lupus subsp. familiaris NF45 is 99.7% identical to that of Homo sapiens, suggesting the presence of functional NF45-IRES interactions in C. lupus subsp. familiaris.
Fig 3.
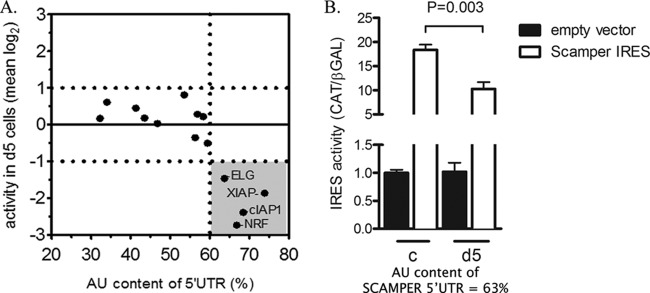
Prediction of NF45-dependent IRES. (A) Determination of threshold AU content for NF45-dependent IRES. IRES activity data from Fig. 2A were transformed into log2 space and plotted against the AU content of the 5′ UTRs tested. A constraint of >60% AU content was chosen based on the clustered activities of IRES that changed by more than 2-fold up or down in d5 versus c cells (shaded quadrant). (B) Conservation of NF45-IRES interactions allows prediction of NF45-dependent IRES in Canis lupus subsp. familiaris. The Scamper IRES (63% AU content) was cloned into a bicistronic vector as described in Materials and Methods and was tested for IRES activity as described for Fig. 2A (n = 3; mean ± SD).
NF45 regulates XIAP protein levels through interaction with its IRES.
We previously investigated the effect of NF45 on modulating cIAP1 IRES activity in the context of endoplasmic reticulum stress (7). To further characterize NF45 regulation of predicted IRES-containing mRNAs, we next focused our attention on the other inhibitor of apoptosis protein from our screen, XIAP, as its IRES (31, 36, 37), and the downstream cellular functions are well demarcated (reviewed in reference 38). Our initial screen of XIAP IRES activity in d5 versus c cells revealed a significant (75%) decrease in activity (Fig. 2A). Next, we assessed the steady-state levels of XIAP protein in c and d5 cells by Western blotting to determine whether they correlated with impaired IRES activity and translation. As was reported previously (7), we observed a significant decrease in cIAP1 protein levels in d5 cells compared to those in the control c cells (Fig. 4A). Similarly, there was a 50% decrease in XIAP protein levels in d5 cells relative to those in the control cell line (Fig. 4A). Next, we attempted to rescue XIAP protein expression in HeLa cells that were transiently knocked down for NF45 expression, since the chronic state of d5 cells makes them refractory to efficient steady-state protein rescue. Similarly to the stably transfected cells, the transient knockdown of NF45 in HeLa cells caused a comparable decrease (41%) in the XIAP protein level compared to that of control siRNA-transfected cells (Fig. 4B). In addition, the transient NF45 knockdown recapitulated the multinucleated phenotype observed in d5 cells (Fig. 5A, bottom right panel), showing that this system can be used to study the pathways associated with the d5 cell phenotype. Importantly, the reduction in XIAP protein expression caused by NF45 knockdown was significantly rescued by the overexpression of the siRNA-resistant GFP-NF45 construct (Fig. 4B, GFP-NF45R).
Fig 5.

NF45 regulates survivin and cyclin E expression downstream of XIAP and cIAP1. (A) Immunofluorescence images showing the multinucleation phenotype of the d5 HeLa cell line (arrow, bottom left panel) compared to the c HeLa cell line. The same phenotype can be reproduced by the transient siRNA knockdown of NF45 for 72 h (arrow, bottom right panel and inset). Cells were stained with Alexa Fluor 568 phalloidin for F-actin and with Hoechst stain for nuclei. (B) c and d5 cell phenotypes were quantified by propidium iodide staining and flow cytometry analysis. The number of multinucleated cells expressed as a percentage of the total number of viable cells was quantified and normalized to that of the control cell line (bottom). The number of cells in G2/M phase is also shown. (C) Western blot and densitometry analyses showing increased survivin expression in HeLa cells treated with 50 nM NF45 siRNA for 96 h compared to control siRNA. (D) survivin expression was blunted by XIAP overexpression in NF45 knocked-down HeLa cells. The HeLa cells were transfected with 50 nM NF45 or control siRNA and transfected 48 h later with GFP-XIAP for an additional 48 h. The protein extracts were analyzed by Western blotting and densitometry for survivin, XIAP, NF45, and GAPDH. (E) NF45 regulates cyclin E expression. HeLa cells were treated with 50 nM NF45 or control siRNA for 96 h, and expression of the indicated proteins was analyzed by Western blotting and densitometry. (F) NF45 controls cyclin E protein levels through its regulation of cIAP1 IRES-mediated translation. The HeLa cells were treated with 100 nM Smac mimetic (SM) or DMSO for 24 h and transfected with a pcDNA3-GFP or pcDNA3-GFP-NF45R plasmid for an additional 24 h. Protein extracts were analyzed by Western blotting for NF45, cIAP1, cyclin E, and GAPDH expression, and densitometry was performed.
To further confirm NF45 regulation of XIAP protein translation, we measured de novo protein synthesis. Control or NF45-targeting siRNA-transfected cells were pulse labeled with [35S]methionine, and the lysates were immunoprecipitated with XIAP, cIAP1, or GAPDH antibodies. NF45 knockdown did not alter general protein synthesis, as shown by the equal incorporation of [35S]methionine into newly synthesized proteins (Fig. 4C, left and middle panels). In contrast, cIAP1 and XIAP de novo protein synthesis was significantly reduced by NF45 knockdown, whereas GAPDH translation remained unchanged (Fig. 4C, right panel). This decrease in XIAP de novo protein synthesis in NF45-deficient cells, together with decreases in IRES activity, polysome distribution, and steady-state protein levels, confirms that NF45 specifically regulates the IRES-mediated translation of the inhibitor of apoptosis XIAP.
Previously published data from our laboratory showed that NF45 directly interacts with an AU-rich stem-loop structure present within the cIAP1 IRES (7). Hence, we wanted to determine if NF45 directly interacts with the XIAP IRES. To determine if NF45 interacts with the XIAP IRES, we used an RNA affinity chromatography approach to isolate XIAP IRES binding proteins from a HeLa cytoplasmic lysate using in vitro-transcribed XIAP IRES as bait. A Western blot of this affinity preparation revealed that, indeed, NF45 specifically interacts with the XIAP IRES but not with a control β-hemoglobin 5′ UTR RNA (Fig. 4D). The presence of poly(A) binding protein (PABP) shows that both the XIAP IRES and the β-hemoglobin 5′ UTR RNA were functional in this assay. Next, we attempted to an NF45 binding site(s) on the XIAP IRES by an in vitro UV-cross-linking RNA binding assay. However, we were unable to detect any complex formation between the purified recombinant NF45 and the XIAP IRES (data not shown), indicating that either NF45 needs to be posttranslationally modified in cells before binding to the XIAP IRES or it does not bind directly to the IRES in the absence of other proteins. Nevertheless, our results show that NF45 is a bona fide ITAF that interacts with the XIAP IRES and positively regulates its activity, thus driving XIAP protein expression.
NF45 regulates downstream proteins involved in cell cycle progression and cytokinesis.
Next, we investigated the link between the impaired IRES-mediated translation observed in d5 cells and their overt cellular phenotype. The d5 HeLa stable cell line exhibits a striking senescence-like morphology (15) typified by an overall flat nonelliptical shape and increased F-actin expression, together with an overall increase in cell size (Fig. 5A, left panel). These cells also display multinucleation, suggesting either a cell fusion event or a defect in cytokinesis (Fig. 5A, arrow). Quantitative analyses of propidium iodide-labeled d5 cells by flow cytometry indicated a 2.4-fold increase in the multinucleated population compared to the control cells, but there was no change in the number of cells arrested in the G2/M phase (Fig. 5B), as reported previously (15). Importantly, this multinucleation phenotype could be reproduced by transient siRNA knockdown of NF45 for 72 h in HeLa cells (Fig. 5A, bottom right panel).
The slow-growth phenotype observed in d5 cells could be indicative of dysregulated NF-κB signaling, a key transcriptional program that affects cellular proliferation. Given that the IAPs have been well established as mediators of NF-κB signaling (reviewed in reference 39), we decided to assess NF-κB activity levels in the cells lacking NF45. However, we found no significant difference in activities between d5 and c cells, measured using a luciferase-based NF-κB reporter system (data not shown), although there was an increase in the basal NF-κB activity levels in both c and d5 cells when NF45 was overexpressed. Furthermore, the reduced cIAP1 expression in d5 cells might have been expected to trigger the transcriptional activation of cIAP2 expression by NF-κB activity (40). However, we did not observe any changes in cIAP2 mRNA expression between the two cell lines, consistent with our finding that NF-κB signaling was unaffected (data not shown).
Another pathway through which slow growth and multinucleation could arise is through the dysregulation of survivin function (17, 41). Survivin is a member of the IAP family that plays important roles in apoptosis inhibition and in microtubule spindle checkpoint regulation (17, 18). Survivin has been shown to play a dual role in cell division, first by regulating microtubule dynamics, through its association with polymerized tubulin (19), and also by being a member of the chromosomal passenger complex, in which it associates with the regulators of cytokinesis, such as inner centromere protein (INCENP), Aurora B kinase, and borealin (reviewed in reference 20). Interestingly, survivin protein stability is regulated by XIAP (42). Thus, we hypothesized that the loss of NF45 leading to the downregulation of XIAP IRES-mediated translation will result in altered survivin expression and the deregulation of mitosis. We used HeLa cells, in which we transiently knocked down NF45 using the d5 siRNA, and we set out to rescue survivin expression through the overexpression of GFP-XIAP. We observed that the NF45 knockdown caused a 2.5-fold increase in survivin protein levels (Fig. 5C). Moreover, GFP-XIAP overexpression in NF45 siRNA-treated cells significantly blunted survivin expression compared to the GFP control (Fig. 5D), while survivin steady-state mRNA levels remained unchanged in NF45 knocked-down cells (data not shown).
Although cIAP1 is primarily known as a regulator of the NF-κB signaling pathways, a recent report showed that nuclear cIAP1 transcriptionally upregulates cyclin E expression through its interaction with the E2F1 transcription factor (43). Given that NF45 is necessary for IRES-mediated cIAP1 expression (7), we wanted to test whether knockdown of NF45 alters cyclin E levels in HeLa cells and thus contributes to the distinct phenotype of the NF45-deficient cells. Indeed, NF45 transient knockdown caused a 50% decrease in cyclin E protein levels (Fig. 5E, left panel). Importantly, this decrease in cyclin E levels was almost entirely rescued by GFP-NF45R expression in NF45-depleted cells (Fig. 5E, right panel). To further confirm that cIAP1 is required for cyclin E regulation downstream of NF45, we overexpressed GFP-NF45R in the presence of a Smac mimetic compound that causes the rapid degradation of cIAP1 protein (44). As expected, NF45 overexpression increased cyclin E protein relative to the GFP control in dimethyl sulfoxide (DMSO)-treated cells (Fig. 5F). However, when the cells were treated with a Smac mimetic, which leads to the depletion of cIAP1, the NF45-driven upregulation of cyclin E was blunted.
These results identify a new pathway in which NF45 regulates cIAP1 and XIAP IRES-mediated translation, which in turn regulates survivin and cyclin E expression. The decrease in cyclin E expression, coupled with the increased expression of survivin protein, likely contributes to the senescence-like and multinucleated phenotype observed in NF45-deficient cells.
DISCUSSION
We have shown for the first time that NF45 regulates the translation of a cohort of IRES-containing transcripts that were initially predicted solely on the basis of the unusually high AU content (>60%) of their respective 5′ UTRs. We then confirmed that NF45 specifically regulates the IRES-mediated translation of XIAP and cIAP1 mRNAs and that the reduced expression of these proteins cause changes in the ploidy of HeLa cells through dysregulated expression of the XIAP and cIAP1 downstream targets, survivin and cyclin E, respectively.
IRES trans-acting factors are cellular proteins that help in the recruitment of the ribosome by acting as either scaffold proteins or RNA chaperones (4, 5). We previously identified and characterized NF45 as an ITAF that positively regulates the AU-rich cIAP1 IRES during the unfolded protein response (7). In this study, we show that the high AU content (>60%) of 5′ UTRs that contain IRES can serve as an excellent predictor of dependence on NF45. Indeed, we show that the IRES activity levels of the AU-rich IRES of cIAP1, XIAP, NRF, and ELG are significantly decreased in d5 cells lacking NF45; similarly, the loading of these mRNAs onto translating polysomes is reduced in NF45-deficient cells (Fig. 2). Importantly, this decrease in translation efficiency can be rescued by reexpression of NF45 (Fig. 2C), and all of these AU-rich IRES-bearing transcripts were detected in the NF45 immunocomplexes, with the exception of ELG (Fig. 2E). From these data, we propose that IRES-containing 5′ UTRs with an AU content of >60% can be predicted to require NF45 for optimal IRES activity. Further validation experiments, such as polysome profiling, de novo protein synthesis, and RNA binding assays, would be necessary to confirm that NF45 is a bona fide ITAF for a particular IRES, as we have done previously for the cIAP1 IRES (7) and as we have shown in this study for the XIAP IRES.
The non-AU-rich IRES APAF1 was also decreased in d5 cells (Fig. 2C). However, this reduction could not be rescued by NF45 overexpression, showing that it was not due to a direct effect of NF45 on translation. NF45 was first identified as a nuclear factor of activated T cells (NFAT)-related transcription factor that regulates interleukin-2 transcription, together with its binding partner NF90 (8). Therefore, a general reduction in the polysome loading of APAF1 could reflect an indirect transcriptional effect of NF45 and its binding partner, NF90, in a complex that is known to target a wide array of genes (9, 45). Moreover, we showed previously that the attenuation of cIAP1 IRES-dependent translation induction in d5 cells during ER stress can be rescued by reexpression of NF45 but not NF90 (7); this further argues that the NF45-NF90 transcriptional effects are independent of the function of NF45 as an ITAF.
NF45 protein structure is well conserved across mammalian species; thus, we predicted that its function also ought to be so and that the IRES dependency for NF45 could be held across species. To test this prediction, the IRES activity levels of the Canis lupus subsp. familiaris IRES, Scamper (63% AU content), was determined in d5 cells. Indeed, Scamper IRES activity was decreased by 50% in d5 relative to c cells (Fig. 3), confirming that AU content could be used to predict the NF45 regulation of IRES-containing mRNAs in other species. We have also calculated the AU content of 5′ UTRs and minimal IRES regions from known rodent, Drosophila, and virus IRES-containing mRNAs and identified several that could potentially be regulated by NF45 (Fig. 6). For instance, outside the class Mammalia, the CG5641 protein in Drosophila melanogaster is homologous to mammalian NF45 and may regulate IRES in this species. Of the few D. melanogaster IRES identified to date, grim, hid, ultrabithorax, reaper, hsp70, and hsp90 possess >60% AU content in their respective 5′ UTRs and would therefore be predicted to be regulated at the level of translation by CG5641. It would be interesting to test whether these AU-rich IRES transcripts are indeed regulated by NF45 in D. melanogaster. Interestingly, no NF45 orthologue exists in Saccharomyces cerevisiae, perhaps reflecting the apparent distinct mechanism of IRES regulation in this species that requires short stretches of adenosine nucleotides to recruit ribosomes through interaction with PABP (46). Collectively, these observations highlight the importance of NF45 ITAF activity in the regulation of AU-rich IRES across eukaryotic species.
Fig 6.

Diagonal plot of the AU content of 5′ UTRs and IRES from rodents, Drosophila, and viruses, showing that AU-rich IRES are present across species.
Next, we wanted to further characterize NF45 regulation of predicted IRES-containing mRNAs and the functional consequences of the loss of NF45. ELG is one of the IRES whose activity and polysomal loading were significantly reduced in NF45-depleted cells, although we were unable to verify its in vivo interaction with NF45 due to low transcript abundance (Fig. 2). Unfortunately, the ill-defined function of the ELG/C17ORF85 gene product and the lack of antibodies prevented us from further validating the effects of NF45 on ELG IRES-mediated translation. We were also unable to detect the NRF protein in either the d5 or c cells by Western blotting (data not shown). We therefore focused our attention on the XIAP IRES and the previously characterized cIAP1 IRES and their potential roles in the d5 cell phenotype. The d5 cell line exhibits a striking senescence-like and multinucleated phenotype compared to that of control c cells (Fig. 5A and B). Guan et al. (15) were the first to characterize the d5 cells and showed that their slow growth was due to a block in the cell cycle and defects in DNA synthesis, whereas the multinucleated cells were probably due to a defect in cytokinesis. Furthermore, recent studies using time-lapse microscopy of nuclear division were able to recapitulate the phenotype observed in d5 cells and indicated that multinucleated cells arose from the incomplete cytokinesis of daughter cells followed by the fusion of several binucleated cells (16, 47). These observations pointed to a role for NF45 in regulating important cellular functions, such as in the cell cycle and in cell division. Looking downstream of XIAP and cIAP1 for regulators of the cell cycle or mitosis, we identified the survivin and cyclin E proteins as potential targets. survivin is well characterized for its important roles in apoptosis inhibition and microtubule spindle checkpoint regulation (17, 18). Furthermore, survivin antiapoptotic effects are mediated by its interaction with XIAP, and the two regulate each other's protein stability through the proteasomal degradation pathway (42). In addition, the formation of a complex between XIAP and XIAP-associated factor 1 (XAF1) leads to a proteasomal degradation of survivin (48). We found that NF45 knockdown in HeLa cells leads to an increase in survivin protein due to a decrease in XIAP expression (Fig. 5C and D).
The reduction or loss of survivin in mammalian cells gives rise to a variety of cell division defects, including cytokinesis failure and multinucleated cells (41, 49, 50). These phenomena are due to survivin's role in regulating microtubule dynamics, through its association with polymerized tubulin (19), but also by being a member of the chromosomal passenger complex (reviewed in reference 20). However, the overexpression of survivin has also been linked to defects in cytokinesis and the generation of multinucleated cells. These defects are due to a reduction in microtubule dynamics, such as microtubule growth, the number of growing microtubules, and disorganized mitotic spindles (51–53).
On the other hand, our results show that NF45 knockdown in HeLa cells also leads to cyclin E downregulation, which can be rescued by the reexpression of NF45 (Fig. 5E) and is dependent on cIAP1 (Fig. 5F). Indeed, it was shown previously that a nuclear form of cIAP1, found in HeLa cells (54), can stimulate cyclin E expression at the transcriptional level through its direct interaction with E2F1 (43). Our results thus confirm that cyclin E regulation occurs by cIAP1, and they show an additional layer of regulation by NF45 through the control of cIAP1 IRES-mediated translation. Cyclin E downregulation in NF45-depleted cells could explain their senescence-like phenotype and, together with an increase in survivin, would lead to a block in the cell cycle, mitotic catastrophe, and defects in cytokinesis. Moreover, the significant decrease in XIAP and cIAP1 expression in these cells would also be expected to reduce their apoptotic threshold. Surprisingly, although aneuploidy is increased in d5 cells, the apoptotic index is unaffected (15, 47), probably due to the absence of the p53 protein in this HeLa cell line derivative (55). Recently, Shamanna et al. suggested that the d5 multinucleated phenotype is caused by a defect in DNA damage repair. They identified the NF90-NF45 complex to be a regulator of nonhomologous end-joining DNA damage repair mediated by DNA-activated protein kinase (DNA-PK) and suggested that structured RNA may modulate this process (16). These data, in combination with our observations, suggest that defects in DNA repair mechanisms, cell cycle progression, and cytokinesis form the basis of the multinucleated phenotype of NF45-deficient cells.
In conclusion, we show that NF45 regulates the translation of a set of IRES-containing transcripts that were predicted initially only on the basis of their high AU content. This regulation is important for maintaining normal levels of survivin and cyclin E and controlling normal ploidy during the HeLa cell cycle and mitosis.
ACKNOWLEDGMENTS
We thank members of the Apoptosis Research Centre for critical discussions.
This work was supported by an operating grant from the Canadian Institutes of Health Research (FRN 74740). M.D.F. was supported by the Vanier Canada Graduate Scholarship, and T.E.G. was supported by the Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award. M.H. is the CHEO Volunteer Association Endowed Scholar.
We have no competing financial interests in relation to the work described in this article.
Footnotes
Published ahead of print 5 November 2012
REFERENCES
- 1. Holcik M, Sonenberg N. 2005. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6:318–327 [DOI] [PubMed] [Google Scholar]
- 2. Pestova TV, Lorsch JR, Hellen CUT. 2007. Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3. Pelletier J, Sonenberg N. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320–325 [DOI] [PubMed] [Google Scholar]
- 4. Stoneley M, Willis AE. 2004. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23:3200–3207 [DOI] [PubMed] [Google Scholar]
- 5. King HA, Cobbold LC, Willis AE. 2010. The role of IRES trans-acting factors in regulating translation initiation. Biochem. Soc. Trans. 38:1581–1586 [DOI] [PubMed] [Google Scholar]
- 6. Baird SD, Turcotte M, Korneluk RG, Holcik M. 2006. Searching for IRES. RNA 12:1755–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graber TE, Baird SD, Kao PN, Mathews MB, Holcik M. 2010. NF45 functions as an IRES trans-acting factor that is required for translation of cIAP1 during the unfolded protein response. Cell Death Differ. 17:719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kao PN, Chen L, Brock G, Ng J, Kenny J, Smith AJ, Corthesy B. 1994. Cloning and expression of cyclosporin A- and FK506-sensitive nuclear factor of activated T-cells: NF45 and NF90. J. Biol. Chem. 269:20691–20699 [PubMed] [Google Scholar]
- 9. Kiesler P, Haynes PA, Shi L, Kao PN, Wysocki VH, Vercelli D. 2010. NF45 and NF90 regulate HS4-dependent interleukin-13 transcription in T cells. J. Biol. Chem. 285:8256–8267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Isken O, Grassmann CW, Sarisky RT, Kann M, Zhang S, Grosse F, Kao PN, Behrens SE. 2003. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 22:5655–5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merrill MK, Gromeier M. 2006. The double-stranded RNA binding protein 76:NF45 heterodimer inhibits translation initiation at the rhinovirus type 2 internal ribosome entry site. J. Virol. 80:6936–6942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y. 2009. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol. Cell. Biol. 29:3754–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Volk N, Shomron N. 2011. Versatility of microRNA biogenesis. PLoS One 6:e19391 doi:10.1371/journal.pone.0019391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guan D, Altan-Bonnet N, Parrott AM, Arrigo CJ, Li Q, Khaleduzzaman M, Li H, Lee CG, Pe'ery T, Mathews MB. 2008. Nuclear factor 45 (NF45) is a regulatory subunit of complexes with NF90/110 involved in mitotic control. Mol. Cell. Biol. 28:4629–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shamanna RA, Hoque M, Lewis-Antes A, Azzam EI, Lagunoff D, Pe'ery T, Mathews MB. 2011. The NF90/NF45 complex participates in DNA break repair via nonhomologous end joining. Mol. Cell. Biol. 31:4832–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. 1998. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396:580–584 [DOI] [PubMed] [Google Scholar]
- 18. Mita AC, Mita MM, Nawrocki ST, Giles FJ. 2008. survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 14:5000–5005 [DOI] [PubMed] [Google Scholar]
- 19. Altieri DC. 2006. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr. Opin. Cell Biol. 18:609–615 [DOI] [PubMed] [Google Scholar]
- 20. Lens SM, Vader G, Medema RH. 2006. The case for survivin as mitotic regulator. Curr. Opin. Cell Biol. 18:616–622 [DOI] [PubMed] [Google Scholar]
- 21. Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. 1991. The E2F transcription factor is a cellular target for the RB protein. Cell 65:1053–1061 [DOI] [PubMed] [Google Scholar]
- 22. DeGregori J, Kowalik T, Nevins JR. 1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15:4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. 1992. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70:993–1006 [DOI] [PubMed] [Google Scholar]
- 24. Baird SD, Lewis SM, Turcotte M, Holcik M. 2007. A search for structurally similar cellular internal ribosome entry sites. Nucleic Acids Res. 35:4664–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oumard A, Hennecke M, Hauser H, Nourbakhsh M. 2000. Translation of NRF mRNA is mediated by highly efficient internal ribosome entry. Mol. Cell. Biol. 20:2755–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X, Richie C, Legerski RJ. 2002. Translation of hSNM1 is mediated by an internal ribosome entry site that upregulates expression during mitosis. DNA Repair (Amst.) 1:379–390 [PubMed] [Google Scholar]
- 27. Kim JG, Armstrong RC, Berndt JA, Kim NW, Hudson LD. 1998. A secreted DNA-binding protein that is translated through an internal ribosome entry site (IRES) and distributed in a discrete pattern in the central nervous system. Mol. Cell Neurosci. 12:119–140 [DOI] [PubMed] [Google Scholar]
- 28. Cornelis S, Tinton SA, Schepens B, Bruynooghe Y, Beyaert R. 2005. UNR translation can be driven by an IRES element that is negatively regulated by polypyrimidine tract binding protein. Nucleic Acids Res. 33:3095–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park EH, Lee JM, Blais JD, Bell JC, Pelletier J. 2005. Internal translation initiation mediated by the angiogenic factor Tie2. J. Biol. Chem. 280:20945–20953 [DOI] [PubMed] [Google Scholar]
- 30. De Pietri Tonelli D, Mihailovich M, Schnurbus R, Pesole G, Grohovaz F, Zacchetti D. 2003. Translational control of Scamper expression via a cell-specific internal ribosome entry site. Nucleic Acids Res. 31:2508–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG. 1999. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol. 1:190–192 [DOI] [PubMed] [Google Scholar]
- 32. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. 2008. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 105:11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thakor N, Holcik M. 2012. IRES-mediated translation of cellular messenger RNA operates in eIF2alpha- independent manner during stress. Nucleic Acids Res. 40:541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim YK, Jang SK. 1999. La protein is required for efficient translation driven by encephalomyocarditis virus internal ribosomal entry site. J. Gen. Virol. 80:3159–3166 [DOI] [PubMed] [Google Scholar]
- 35. Pestova TV, Shatsky IN, Hellen CU. 1996. Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol. Cell. Biol. 16:6870–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holcik M, Korneluk RG. 2000. Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Mol. Cell. Biol. 20:4648–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewis SM, Holcik M. 2005. IRES in distress: translational regulation of the inhibitor of apoptosis proteins XIAP and HIAP2 during cell stress. Cell Death Differ. 12:547–553 [DOI] [PubMed] [Google Scholar]
- 38. Liston P, Fong WG, Korneluk RG. 2003. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene 22:8568–8580 [DOI] [PubMed] [Google Scholar]
- 39. Gyrd-Hansen M, Meier P. 2010. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 10:561–574 [DOI] [PubMed] [Google Scholar]
- 40. Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. 1997. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Natl. Acad. Sci. U. S. A. 94:10057–10062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC. 1999. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1:461–466 [DOI] [PubMed] [Google Scholar]
- 42. Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, Salvesen GS, Reed JC, Altieri DC. 2004. An IAP-IAP complex inhibits apoptosis. J. Biol. Chem. 279:34087–34090 [DOI] [PubMed] [Google Scholar]
- 43. Cartier J, Berthelet J, Marivin A, Gemble S, Edmond V, Plenchette S, Lagrange B, Hammann A, Dupoux A, Delva L, Eymin B, Solary E, Dubrez L. 2011. Cellular inhibitor of apoptosis protein-1 (cIAP1) can regulate E2F1 transcription factor-mediated control of cyclin transcription. J. Biol. Chem. 286:26406–26417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. 2006. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 1:525–533 [DOI] [PubMed] [Google Scholar]
- 45. Reichman TW, Muniz LC, Mathews MB. 2002. The RNA binding protein nuclear factor 90 functions as both a positive and negative regulator of gene expression in mammalian cells. Mol. Cell. Biol. 22:343–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gilbert WV, Zhou K, Butler TK, Doudna JA. 2007. Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317:1224–1227 [DOI] [PubMed] [Google Scholar]
- 47. Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, Cetin C, Sieckmann F, Pau G, Kabbe R, Wunsche A, Satagopam V, Schmitz MH, Chapuis C, Gerlich DW, Schneider R, Eils R, Huber W, Peters JM, Hyman AA, Durbin R, Pepperkok R, Ellenberg J. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464:721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arora V, Cheung HH, Plenchette S, Micali OC, Liston P, Korneluk RG. 2007. Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J. Biol. Chem. 282:26202–26209 [DOI] [PubMed] [Google Scholar]
- 49. Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, Cowsert LM, Bennett F, Krajewski S, Krajewska M, Welsh K, Reed JC, Ng SC. 2000. Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia 2:235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH. 2000. survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 10:1319–1328 [DOI] [PubMed] [Google Scholar]
- 51. Rosa J, Canovas P, Islam A, Altieri DC, Doxsey SJ. 2006. survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol. Biol. Cell 17:1483–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Speliotes EK, Uren A, Vaux D, Horvitz HR. 2000. The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol. Cell 6:211–223 [DOI] [PubMed] [Google Scholar]
- 53. Uren AG, Beilharz T, O'Connell MJ, Bugg SJ, van Driel R, Vaux DL, Lithgow T. 1999. Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc. Natl. Acad. Sci. U. S. A. 96:10170–10175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samuel T, Okada K, Hyer M, Welsh K, Zapata JM, Reed JC. 2005. cIAP1 localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 65:210–218 [PubMed] [Google Scholar]
- 55. Matlashewski G, Banks L, Pim D, Crawford L. 1986. Analysis of human p53 proteins and mRNA levels in normal and transformed cells. Eur. J. Biochem. 154:665–672 [DOI] [PubMed] [Google Scholar]