

Abstract
The Epstein-Barr virus (EBV) is an important human pathogen that is associated with multiple cancers. The major oncoprotein of the virus, latent membrane protein 1 (LMP1), is essential for EBV B-cell immortalization and is sufficient to transform rodent fibroblasts. This viral transmembrane protein activates multiple cellular signaling pathways by engaging critical effector molecules and thus acts as a ligand-independent growth factor receptor. LMP1 is thought to signal from internal lipid raft containing membranes; however, the mechanisms through which these events occur remain largely unknown. Lipid rafts are microdomains within membranes that are rich in cholesterol and sphingolipids. Lipid rafts act as organization centers for biological processes, including signal transduction, protein trafficking, and pathogen entry and egress. In this study, the recruitment of key signaling components to lipid raft microdomains by LMP1 was analyzed. LMP1 increased the localization of phosphatidylinositol 3-kinase (PI3K) and its activated downstream target, Akt, to lipid rafts. In addition, mass spectrometry analyses identified elevated vimentin in rafts isolated from LMP1 expressing NPC cells. Disruption of lipid rafts through cholesterol depletion inhibited PI3K localization to membranes and decreased both Akt and ERK activation. Reduction of vimentin levels or disruption of its organization also decreased LMP1-mediated Akt and ERK activation and inhibited transformation of rodent fibroblasts. These findings indicate that LMP1 reorganizes membrane and cytoskeleton microdomains to modulate signal transduction.
INTRODUCTION
More than 90% of the world's population is persistently infected with the Epstein-Barr virus (EBV), a member of the human gammaherpesvirus family (1). Latent EBV infection is associated with multiple lymphoid and epithelial cancers, including Burkitt lymphoma, Hodgkin disease, nasopharyngeal carcinoma (NPC), and gastric carcinoma. The malignant cells contain the viral genome as an episome and express only a subset of viral genes (2). One viral gene product that is expressed in many EBV-associated malignancies is latent membrane protein 1 (LMP1). LMP1 is considered the major oncogene of EBV because it is essential for B-lymphocyte immortalization, and the expression of LMP1 alone is sufficient to transform rodent fibroblasts cells in vitro (3, 4). Decreased expression of LMP1 levels or inhibition of LMP1-activated signaling pathways impairs growth and inhibits transformation, suggesting that targeting LMP1 signaling pathways may be a specific therapy (5, 6).
The oncogenic potential of LMP1 requires its ability to self-aggregate in the absence of ligand and function as a constitutively active tumor necrosis factor receptor through the recruitment of downstream signaling effector molecules including tumor necrosis factor associated factors (TRAFs) (7, 8). Multiple signaling pathways are activated by LMP1 in both B cells and epithelial cells, including mitogen-activated protein kinase (MAPK/ERK), phosphatidylinositol 3-kinase (PI3K)/Akt, NF-κB, and c-Jun N-terminal kinase (JNK) (9–13). It is important to further define how LMP1 specifically activates multiple signaling pathways. The localization of LMP1 to lipid raft microdomains is thought to contribute to its ability to signal effectively as mutants of LMP1 that are incapable of trafficking to lipid rafts are unable to activate NF-κB in B cells (14). In addition, LMP1 has been shown to interact with TRAF3 in lipid rafts, while cytosolic LMP1 associated with TRAF2. The effects of LMP1 on lipid rafts may well be cell type specific since simvastatin, a cholesterol synthesis inhibitor, depleted LMP1 from B-cell lipid rafts but did not affect LMP1 raft localization in NPC cells (15, 16).
Lipid rafts are membrane microdomains that are enriched in cholesterol and sphingolipids (17). Lipid rafts have been well characterized based on their biochemical properties of detergent insolubility and buoyancy in isopycnic density gradients and are often referred to as detergent-resistant membranes (DRMs) (18). These microdomains act as organization centers in membranes to mediate cellular processes, including signal transduction, virus entry and egress, endocytosis, and protein trafficking (17).
LMP1 alters the transcription of many host genes that are important for apoptosis, cell cycle progression, cell proliferation, and migration (19–21). One target of LMP1 transcriptional upregulation is vimentin. Vimentin levels are consistently increased by LMP1 expression in B cells and epithelial cells; however, its functions in LMP1-mediated signaling or transformation have not been evaluated (19, 22). Vimentin is an intermediate filament that is a major constituent of mesenchymal cells and contributes to cellular processes, including attachment, migration, protein trafficking, and signal transduction by organizing protein complexes (23). These protein networks can contain integrins, growth factor receptors or receptor-binding proteins, and kinases. Vimentin localizes to lipid rafts and is critical for the recruitment of specific proteins to these microdomains (24, 25). LMP1 was initially shown to colocalize with vimentin in B cells, and chemical disruption of vimentin localization induced relocalization of LMP1 to sites of vimentin redistribution (26). These findings suggest that vimentin and LMP1 may form a complex within cells in lipid rafts.
In this study, the recruitment of PI3K to lipid rafts in LMP1 expressing cells and the requirement of raft integrity in LMP1-mediated signal transduction was analyzed. It was found that in NPC cells and Rat-1 fibroblasts that express LMP1, PI3K, and the activated downstream target (p-Akt) are redistributed to DRMs. Importantly, disruption of lipid rafts through cholesterol depletion decreased PI3K membrane recruitment. Raft disruption impaired activation of the PI3K target, Akt, and activation of the ERK kinase in epithelial cells, suggesting that lipid rafts are essential for LMP1-mediated activation of these pathways and therefore transformation. Identification of additional molecules recruited to lipid rafts in LMP1-expressing cells using proteomic analysis revealed elevated levels of vimentin in DRMs. The importance of vimentin in LMP1 signaling and transformation was evaluated by short hairpin RNA (shRNA) knockdown, expression of dominant-negative vimentin protein, and chemical inhibitor experiments. These data indicated that vimentin is required for both activation of the PI3K and extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathways and transformation of Rat-1 cells by LMP1. This study provides the first evidence for the recruitment of PI3K to lipid rafts by LMP1 and reveals a critical role for vimentin in LMP1 signal transduction and transformation.
MATERIALS AND METHODS
Cells and plasmids.
Rat-1, 293T, and NPC cells (C666.1) were cultured in Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Thermo), penicillin-streptomycin (Gibco), and antibiotic-antimycotic (Gibco) at 37°C with 5% CO2. C666-1 cells were established from an NPC sample. These cells retain the viral genome but express little to no LMP1. Generation of stable Rat-1 and C666.1 cells expressing pBabe-HA-LMP1 or pBabe (empty vector) have been described previously (10, 21). Stable cells expressing shRNAs against vimentin (shVIM) or pGIPZ (scrambled control) were generated by FuGENE6 transfection according to the manufacturer's instructions (Roche) and selected in DMEM containing 8 μg of puromycin/ml. Plasmids expressing green fluorescent protein (GFP)-vimentin, GFP-vimentin-DN, or GFP alone were kindly provided by Stuart Martin (University of Maryland) and were used in transient transfection of Rat-1 cells using FuGENE6 (27). pBabe.V5-LMP1-DsRed was constructed by PCR amplification of LMP1 from the pBabe.HA-LMP1 vector with primers containing the V5 tag sequence and restriction sites (EcoRI and SacII). The resulting PCR product was digested and cloned in frame into the pDsRed-N1 vector (Clontech) using the EcoRI and SacII sites. The entire sequence containing V5-LMP1-DsRed was excised by digestion with EcoRI and SalI prior to ligation into the pBabe (puromycin) vector cut with the same enzymes.
Retrovirus production and focus formation assay.
Recombinant retrovirus particles were generated as previously described (13). Briefly, sub confluent 293T cells grown in 100-mm dishes were transfected with pBabe, pBabe.HA-LMP1, or pBabe.LMP1-DsRed and pVSVG and pGag/Pol plasmids. After 24 h of incubation at 37°C, the medium was replaced with fresh DMEM supplemented with 10% FBS and antibiotics and incubated for 48 h at 33°C. The cell supernatant containing retrovirus particles was clarified by centrifugation 1,000 × g for 10 min and frozen at −80°C until use. The cells were transduced with medium containing retrovirus particles and 4 μg of Polybrene/ml for 24 h at 37°C. After transduction, the medium was changed to fresh DMEM supplemented with 10% FBS and antibiotics. For the focus formation assay, the medium was changed every other day for 10 to 14 days. The cells were stained with 1% crystal violet in 50% methanol, and the average number of foci per field was determined from 10 independent fields. In experiments using a retrovirus expressing a LMP1-DsRed construct foci were visualized using phase-contrast and fluorescence microscopy. Fluorescent foci were measured with a ruler within the optical unit, and the average foci diameter was determined from 30 independent foci.
Generation of cell lysates and immunoblot analysis.
Cells were harvested at full confluence by washing with cold phosphate-buffered saline (PBS), scraping into cold PBS, pelleted at 1,000 × g for 5 min, and lysed with radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate [SDS], 0.1% deoxycholic acid) supplemented with 0.5 mM phenylmethylsulfonyl fluoride, protease, and phosphatase inhibitor cocktails (Sigma) and 1 mM sodium orthovanadate for 30 min on ice. Insoluble material was removed by centrifugation for 5 min at 16,100 × g, and the protein concentration of the soluble lysate fraction was determined by using the Bio-Rad DC protein assay system. Equal amounts of protein (25 μg) were mixed with 5× Laemmli sample buffer (10% [wt/vol] SDS, 250 mM Tris [pH 6.8], 1 mg of bromophenol blue/ml, 0.5 M dithiothreitol [DTT], 5% β-mercaptoethanol, 50% [wt/vol] glycerol) to make a 1× final solution, boiled for 5 min, separated by SDS-PAGE, and transferred to Optitran (Schleicher & Schuell) for immunoblot analysis. Membranes were blocked for 1 h at room temperature in a Tris-buffered saline solution containing 0.1% Tween 20 and 5% nonfat dry milk. Primary antibodies included anti-p85 (PI3K; Upstate), anti-phospho-Akt (S473), anti-Akt (Cell Signaling), anti-LMP1 (CS1-4 and S12), anti-HSC70, anti-vimentin, anti-phospho-ERK, anti-ERK2, anti-transferrin receptor (Santa Cruz), anti-vimentin (Abcam), and anti-Flotillin-2 (Flot-2; BD Biosciences). Secondary antibodies include horseradish peroxidase-conjugated anti-mouse and anti-rabbit (Amersham Pharmacia). After incubation with secondary antibodies, blots were soaked in Pierce SuperSignal West Pico or Femto chemiluminescence system and exposed to X-ray film (ISCBioexpress).
Immunofluorescence.
Semiconfluent Rat-1 or C666 cells grown on glass-bottom dishes (MatTek) were washed two times with PBS and fixed with a 1:1 solution of 95% ethanol and acetone for 20 min at −20°C. The cells were then washed with 0.3% Triton X-100 in PBS (wash solution) and blocked for 1 h at room temperature with 5% goat serum in 0.3% Triton X-100 PBS solution (blocking buffer). After blocking, the cells were incubated overnight with primary antibodies at a 1:100 dilution in blocking buffer, washed three times with 0.3% Triton X-100 in PBS, and incubated for 45 min with secondary antibodies (1:200 dilution in blocking buffer) conjugated to fluorescent molecules specific for primary antibodies (goat anti-rabbit Alexa Fluor 647 [Molecular Probes] or goat anti-mouse Cy3 [Jackson]). Primary antibodies included anti-LMP1 (CS1-4; Dako); anti-p85 (PI3K), anti-flotillin-2 (Flot-2), and anti-caveolin-1 (Cav-1; Cell Signaling); and anti-vimentin (Santa Cruz). The cells were then washed four times, covered with 1 ml of mounting medium (10 mM Tris-HCl [pH 8.5], 90% glycerol, 2%, [wt/vol] n-propyl gallate, 0.25% [wt/vol] 1,4-diazabicyclo[2.2.2]octane [DABCO]), and visualized using an Olympus FV500 inverted laser scanning confocal microscope.
Lipid raft isolation.
Lipid rafts were purified from confluent monolayers of cells as described previously (28). Briefly, five 150-mm culture plates were washed twice with PBS, scraped into PBS, and centrifuged at 1,000 × g for 10 min, and the remaining cell pellet (equivalent to 300 μl of packed cells) was frozen at −80°C prior to lysis. The cells were lysed in 700 μl of 1% Triton X-100 in MNE buffer (25 mM morpholineethanesulfonic acid, 150 mM NaCl, 5 mM EDTA [pH 8.5]), homogenized with a tight-fit Dounce homogenizer (30 strokes), and incubated on ice for 30 min. After incubation, the lysate was then mixed with the same volume (1 ml) of 80% sucrose (wt/vol) in MNE buffer and transferred into a 12-ml polyallomer SW41 ultracentrifuge tube (Beckman). Then, 7 ml of 30% sucrose followed by 3 ml of 5% sucrose, both in MNE, were layered on top of the sample-sucrose solution and subjected to ultracentrifugation at 187,813 × g at 4°C using an SW41 rotor (Beckman). Fractions (1 ml) were collected from the top of the gradient, and proteins in each fraction were concentrated using Amicon Ultra 0.5-ml 10k filters (Millipore) prior to separation by SDS-PAGE.
In some experiments, the floating opaque band corresponding to the detergent-resistant membrane (DRM) fraction present between the 30 and 5% interface was removed (2 ml), diluted in 9 ml of PBS, and pelleted by ultracentrifugation at 111,132 × g for 1 h in a SW41 rotor. The remaining pellet containing the DRMs and associated proteins was dissolved in RIPA buffer, and equal amounts of total protein were mixed with Laemmli sample buffer, separated by SDS-PAGE, and analyzed by immunoblotting. Methyl-β-cyclodextrin (MβCD) is routinely used to remove cholesterol from membrane preparations. To analyze the requirement of cholesterol for DRM localization, the cells were serum starved for 1 h and then treated with 10 mM MβCD for 30 min prior to DRM isolation.
Cytoskeletal disruption.
To determine the importance of intact cytoskeletal networks on LMP1 signaling, the cells were grown to confluence, serum starved for 1 h, and then treated with a final concentration of 5 μg of nocodazole/ml (tubulin disruption), 1 μg of cytochalasin D/ml (actin disruption), 1% 3,3′-iminodipropionitrile (IDNP; vimentin disruption), or mock treated (DMSO) for 1 h. The cells were then washed twice with PBS, scraped into PBS, pelleted, and lysed in RIPA buffer. The soluble protein lysates were then analyzed by SDS-PAGE and immunoblot analysis.
In-gel trypsin digestion and matrix-assisted laser desorption ionization (MALDI) mass spectrometry.
Purified lipid rafts and associated proteins were dissolved in Laemmli sample buffer, separated in SDS–4 to 20% polyacrylamide gels, and fixed and stained with Imperial protein stain (Thermo) according to the manufacturer's instructions. After destaining in ultrapure water, protein bands were excised from the gel with a clean scalpel, placed in a clean microcentrifuge tube (Eppendorf), and cut into small pieces. Gel slices were washed with ultrapure water at room temperature with shaking for 10 min. To destain the gel pieces, the water was removed and replaced with 25 mM ammonium bicarbonate in 50% acetonitrile (ACN) and incubated an additional 10 min. The ammonium bicarbonate-ACN solution was removed and replaced with fresh solution. This was repeated until the gel pieces were completely destained (i.e., small hard and opaque). The final ammonium bicarbonate-ACN solution was removed, and the samples were dehydrated with 100% ACN and dried in a SpeedVac for 5 min after removal of the ACN with a pipette. Proteins within the gel plugs were reduced with 10 mM DTT at room temperature for 30 min. The DTT solution was then removed, replaced with 55 mM iodoacetamide, and incubated for 45 min at room temperature in the dark to alkylate the proteins in the gel. After reduction and alkylation, the gel slices were washed with 25 mM ammonium bicarbonate for 10 min, dehydrated again with 100% ACN, and dried in a SpeedVac as described above. Gels were rehydrated with 20 μg of trypsin (sequencing grade; Promega)/ml in 25 mM ammonium bicarbonate for 30 min on ice. The excess trypsin solution was removed, and then the gel slices were covered with 30 μl of 25 mM ammonium bicarbonate, followed by incubation overnight at 36°C.
To extract tryptic peptides, 50 μl of ACN was added to the digests and incubated for 10 min with shaking at room temperature. After incubation, the solution was removed, and the gel was rehydrated with 30 μl of high-pressure liquid chromatography-grade water for 10 min with shaking. ACN was then added as before for a second extraction, which was combined with the first, frozen at −80°C, and dried in a SpeedVac. Peptides were then reconstituted in 5 μl of 50% ACN–0.1% trifluoroacetic acid (TFA) prior to spotting on MALDI plates with Matrix solution (50% ACN, 0.1% TFA, 5 mM ammonium citrate, 10 mg of α-cyano-4-hydroxy-cinnamic acid methyl ester/ml). The spots were analyzed using an AB Sciex 4800 plus MALDI-time of flight (TOF)/TOF mass spectrometer. The most intense peptides (up to 25) were selected for tandem mass spectrometry (MS/MS) analysis, and the combined MS and MS/MS data were analyzed by using ProteinPilot (AB Sciex) interfaced with the Mascot 2.3 search engine (Matrix Science, London, United Kingdom). The NCBInr database was used with the Homo sapiens taxonomy restriction applied. The data was searched with tolerances of 100 ppm for the precursor ions and 0.5 Da for the fragment ions, trypsin as the cleavage enzyme (up to two missed cleavages), carbamidomethyl modification of cysteines as a fixed modification, and methionine oxidation selected as a variable modification. Protein identifications were considered significant (P ≤ 0.05) with at least two unique peptides and a Mascot score of ≥66.
RESULTS
Previous studies using density gradients have shown that LMP1 can be detected in cytosolic, cytoskeletal, and lipid raft fractions within cells and that a significant proportion of the LMP1 containing rafts are within intracellular fractions (29, 30). LMP1 specifically recruits TRAF3 to these microdomains and mutations within LMP1 that affect its ability to target to lipid rafts also inhibit LMP1-mediated NF-κB activation (14, 31). These data indicate that proper localization or trafficking of LMP1 is critical for its ability to signal effectively. In addition to NF-κB, LMP1 also activates the PI3K/Akt and ERK pathways which are required for the transformation of rodent fibroblasts by LMP1 (10).
LMP1 colocalizes with PI3K within internal lipid raft containing membranes.
Visualization of LMP1 using confocal microscopy identified the majority of LMP1 localized to internal perinuclear membranes. Interestingly, PI3K was also predominantly located in these internal membranes (Fig. 1, top panels). The lipid raft markers flotillin-2 (Flot-2) and caveolin-1 (CAV-1) also colocalized with LMP1 in perinuclear regions but were not detected as extensively as LMP1 throughout the cytoplasm (Fig. 1, bottom panels). These data support previous findings that a subpopulation of LMP1 localizes to lipid raft microdomains and suggests that these domains also contain PI3K (14, 31).
Fig 1.
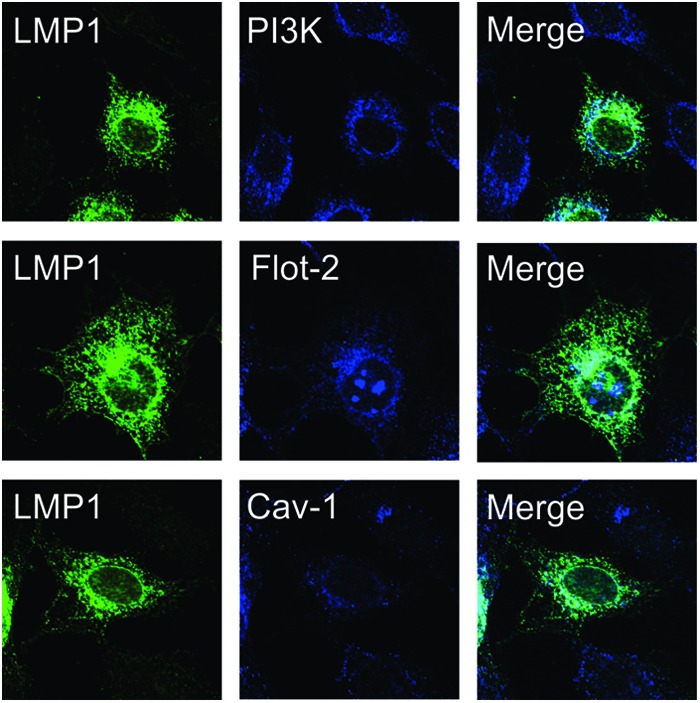
LMP1 colocalizes with PI3K in internal membranes containing lipid raft resident proteins. Rat-1 cells stably expressing LMP1 were fixed and stained with primary antibodies specific for LMP1 (CS1-4), p85 (PI3K), flotillin-2 (Flot-2), or caveolin-1 (Cav-1). Cells were visualized by confocal microscopy with a ×60 oil objective lens.
LMP1 recruits PI3K to lipid raft microdomains.
To determine whether LMP1 affects the localization of PI3K to lipid rafts, DRMs were purified from lysates prepared using a nonionic detergent. The lysates were layered at the bottom of a discontinuous sucrose gradient (28). Based on their buoyant density, lipid rafts and associated proteins float to the top of the gradient after ultracentrifugation (Fig. 2A). Fractions were collected and analyzed for specific proteins. Lipid raft resident protein flotillin-2 was found in the DRM fraction where the non-lipid-raft-resident transmembrane protein transferrin receptor remained at the bottom of the gradient (Fig. 2A). Immunoblotting analysis of the lipid raft fractions purified from NPC cells (C666) stably expressing LMP1 or vector control (pBabe) detected increased levels of PI3K and its phosphorylated, activated target (pAkt) in LMP1 DRMs (Fig. 2B). Quantitation of four independent experiments revealed an 8-fold increase of PI3K in DRMs isolated from LMP1 expressing cells compared to vector control DRMs.
Fig 2.

LMP1 induces the lipid raft localization of PI3K. (A) Schematic diagram of lipid raft isolation protocol. Detergent-resistant membranes (DRMs) or lipid rafts were isolated from cell lysates separated on a discontinuous sucrose gradient by ultracentrifugation. Due to their buoyant density, lipid rafts and associated proteins migrate to the top of the gradient. Fractions were collected from the gradient, separated by SDS-PAGE, and analyzed by immunoblot for flotillin-2 and transferrin receptor as controls. The lipid raft fraction or whole-cell lysates of C666 cells (B) or Rat-1 cells (C) expressing LMP1 or vector control (pBabe) were separated by SDS-PAGE and analyzed by immunoblot analysis for the indicated proteins. The band intensities of PI3K were determined from four independent experiments using ImageJ software, normalized to flotillin-2 levels, and represented relative to the pBabe control level. (D) Rat-1 cells were grown in the absence of serum for 24 h prior to the isolation of lipid rafts and analysis of PI3K levels.
Similar analysis of Rat-1 cells grown in the presence of serum readily detected PI3K in rafts and LMP1 expression slightly increased PI3K in these microdomains (Fig. 2C). The constitutive presence of PI3K in rafts likely reflects the activation and recruitment of PI3K to lipid rafts induced by growth factors present in serum. Importantly, when grown in the absence of serum, PI3K was only detected in LMP1-containing DRMs (Fig. 2D). These findings reveal that PI3K is constitutively recruited to lipid raft microdomains by LMP1 and substantiates the model that LMP1 is able to act as a constitutively active growth factor receptor in the absence of external ligands.
To further evaluate the contribution of lipids to LMP1 raft localization and signaling, cells were treated with MβCD which removes cholesterol from membranes and disrupts lipid rafts (32). Rat-1 (Fig. 3A) or C666 (Fig. 3B) cells expressing LMP1 were treated with MβCD or mock treated for 30 min, and the DRMs were isolated from these cells and analyzed by immunoblotting. MβCD treatment decreased the localization of PI3K in microdomains in both cell types. The localization of Flot-2 to the raft fractions was also decreased by MβCD treatment confirming raft disruption by this drug. Unexpectedly, the levels of LMP1 in DRMs increased with MβCD treatment. Since LMP1 is known to self-aggregate and cholesterol depletion increases membrane fluidity, it is possible that MβCD treatment increases the formation of LMP1-LMP1 containing protein complexes that also float in these experiments (14, 33). These findings indicate that LMP1 induces the recruitment of PI3K to cholesterol-rich lipid rafts.
Fig 3.
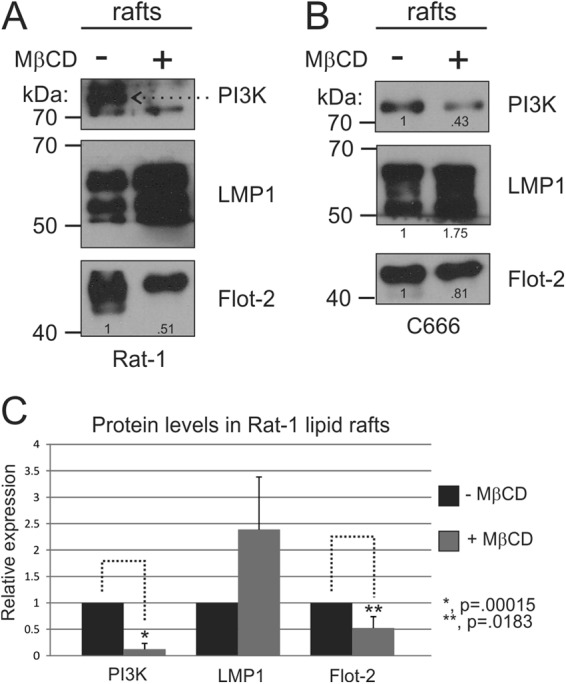
Lipid raft disruption through cholesterol depletion inhibits PI3K DRM localization. Rat-1 (A) or C666 (B) cells expressing LMP1 were serum starved for 1 h and then treated with MβCD for 30 min at 37°C. DRMs were then isolated, and equivalent amounts by volume were analyzed by immunoblotting for PI3K, LMP1, and flotillin-2 (Flot-2) levels. The band intensities of PI3K, LMP1, and flotillin-2 were determined using ImageJ software and are represented relative to the untreated control level. (C) The results of three independent experiments were graphed as mean averages with standard errors of the mean. Statistical significance was determined by using the paired two-tailed Student t test.
Disruption of lipid rafts through cholesterol depletion inhibits LMP1 Akt and ERK activation.
To determine the requirement for PI3K lipid raft localization for activation of Akt, C666-pBabe or C666-LMP1 cells were mock treated or treated with MβCD for 10 min or 30 min, and the lysates were analyzed by immunoblotting. LMP1 expression in these cells greatly increased Akt activation as determined using a phospho-specific antibody for pS473-Akt antibody and treatment with MβCD rapidly inhibited this activation (Fig. 4A). Longer treatment decreased total Akt levels possibly due to degradation of the protein. Similarly, treatment with MβCD also inhibited LMP1-induced activation of ERK and decreased pERK levels in C666-LMP1 cells (Fig. 4A). Treatment with MβCD did not affect the levels of LMP1. These data indicate that raft structure is an important factor in LMP1-mediated activation of both Akt and ERK.
Fig 4.
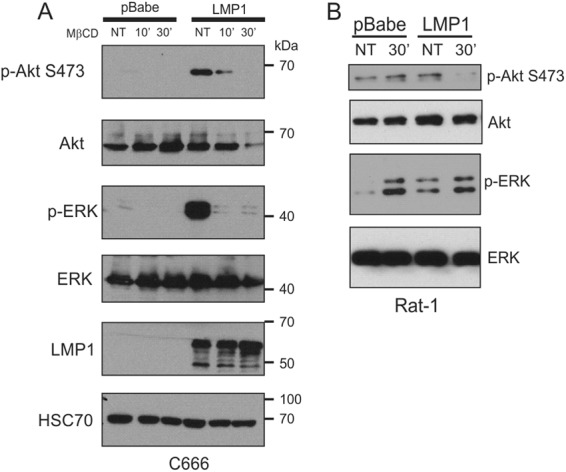
Lipid raft disruption inhibits LMP1-mediated Akt and ERK activation. C666 (A) or Rat-1 (B) cells expressing pBabe or LMP1 were serum starved for 1 h and then treated with vehicle (NT) or MβCD for the indicated times. After treatment, cell lysates were prepared and separated in SDS–10% polyacrylamide gels, transferred to nitrocellulose, and subjected to immunoblot analysis with the indicated primary antibodies.
Rat-1 cells have higher basal levels of Akt activation, which was slightly increased by LMP1. Treatment with MβCD completely blocked LMP1-induced Akt activation but did not affect the basal levels of Akt activation in the pBabe control cells. These findings indicate that PI3K/Akt activation by LMP1 requires the recruitment of PI3K to cholesterol-rich rafts, while the activation of Akt in control cells is independent of raft localization (Fig. 4B). In contrast, treatment with MβCD greatly increased levels of p-ERK in both control and LMP1-expressing cells. These findings suggest that in the presence or absence of LMP1, disruption of lipid rafts results in high levels of ERK activity. Raft disruption by treatment with MβCD has been previously shown in Rat-1 cells to increase ERK activation, EGFR signaling, and basal alpha(1A)-adrenergic receptor signaling (34, 35). This may indicate that in rapidly growing Rat-1 fibroblasts, raft disruption releases upstream activators of ERK that are sequestered in rafts, while in the presence of LMP1 ERK is activated possibly through increased proximity of ERK to specific activators in rafts.
LMP1 induces vimentin protein expression and lipid raft localization.
Previous studies and the data presented here indicate that LMP1 alters the constituents within lipid raft microdomains to activate signaling pathways. To identify other potential proteins recruited to lipid rafts by LMP1 expression, DRMs were isolated from C666-pBabe and C666-LMP1 cells, separated by SDS-PAGE, and the total protein profile was visualized following Coomassie blue staining. The relative abundances of multiple proteins were changed between pBabe and LMP1 lipid rafts (Fig. 5A). Proteins that were specifically detected in LMP1 rafts but not in the pBabe rafts were excised, digested with trypsin, and analyzed by MALDI TOF/TOF MS. Using Protein Pilot and Mascot database searches of the MS and MS/MS spectra, myosin-9, cytoskeletal-associated protein 4 (CKAP4), vimentin, and actin were found to be significant and major components of the excised bands (Fig. 5A, arrows). These data support previous findings that LMP1 associates with cytoskeleton components including vimentin, the first protein identified to form a complex with LMP1. The increased level of vimentin in LMP1 lipid rafts was confirmed by immunoblot analysis in comparison to HSC70 and Flot-2 (Fig. 5B). These findings reveal that in epithelial cells, similarly to EBV-infected B cells, LMP1 increased expression of the intermediate filament protein vimentin and induced its localization to the lipid raft membrane fraction.
Fig 5.
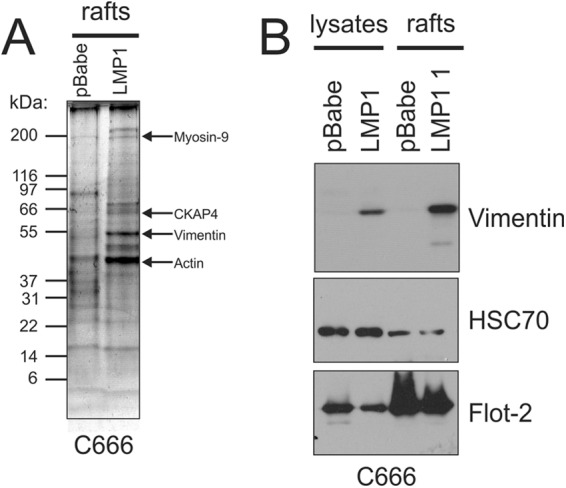
LMP1 increases vimentin levels within DRMs isolated from NPC cells. DRMs were isolated from C666 cells expressing pBabe or LMP1 and separated in an SDS–4 to 20% gradient polyacrylamide gel. The gel was fixed and stained with Coomassie blue to visualize proteins. The predominant bands in the LMP1 rafts not detected in the corresponding region within pBabe rafts were excised, and proteins within that band were digested with trypsin and analyzed by MALDI MS. The major protein components of these bands based on significant Mascot ion sores were determined to be myosin-9, CKAP4, vimentin, and actin. (B) The increased levels of vimentin within lipid rafts were confirmed by immunoblot analysis of pBabe and LMP1 lipid rafts, with HSC70 and Flot-2 serving as loading controls.
LMP1 expression in B cells results in vimentin reorganization to perinuclear regions of the cell that colocalize with LMP1 (26). Vimentin relocalization to the perinuclear region was also observed when LMP1 was expressed in Rat-1 cells (Fig. 6A). The majority of LMP1 colocalized with vimentin within internal structures (Fig. 6B). Vimentin localization was also reorganized in C666-LMP1 cells compared to C666-pBabe cells (data not shown), providing further evidence that LMP1 and vimentin may form a complex within cells that is linked to lipid raft microdomains.
Fig 6.
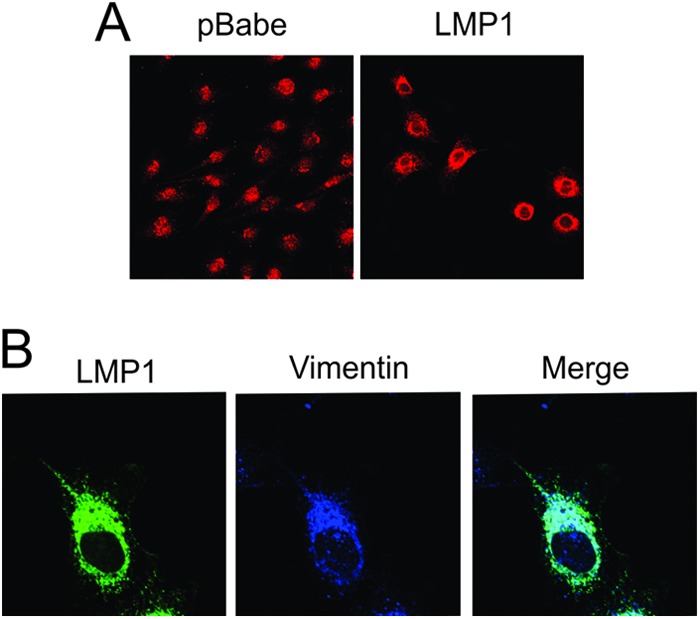
LMP1 colocalizes with and reorganizes vimentin within cells. Rat-1 cells expressing pBabe or LMP1 were fixed and subjected to immunofluorescence using primary antibodies against vimentin (A) or LMP1 (CS1-4) and vimentin (B). Cells were visualized by confocal microscopy after incubation with fluorescently labeled secondary antibodies.
Vimentin is important for LMP1 MAPK/ERK.
Vimentin has been identified in lipid rafts and is thought to function as a scaffold for the organization of cellular processes such as signal transduction (24, 25). Recent findings implicate vimentin as a critical mediator of ERK activation (36, 37). To evaluate the contribution of vimentin to LMP1 effects on raft components and signaling, IDNP, a chemical inhibitor that disrupts vimentin filament organization, was tested for its effects on LMP1 activation of ERK signaling (36). Tubulin (Noco; nocodazole) and actin (Cyto; cytochalasin D) depolymerizing agents were also used in these experiments. IDNP treatment of Rat1-LMP1 cells consistently inhibited ERK activation but had little effect on the vector control cells (Fig. 7A). In contrast, IDNP treatment did not consistently decrease Akt activation (Fig. 7A).
Fig 7.

Vimentin organization is essential for LMP1-mediated ERK activation. (A) pBabe or LMP1 stable Rat-1 cells were serum starved for 1 h, treated for 1 h with the indicated compounds, and then analyzed by immunoblot for the activation of ERK and Akt with phospho-specific antibodies. HSC70 and total ERK and Akt antibodies were used as loading controls. (B) pBabe or LMP1 Rat-1 cells were transfected with GFP, VIM-GFP, or VIM.DN-GFP using FugeneHD. At 24 h posttransfection, the cells were harvested, and protein lysates were analyzed for p-Akt, p-ERK, and total protein levels by immunoblotting. The band intensities of p-Akt and p-ERK from were determined using ImageJ software, normalized to their corresponding total protein levels, and represented relative to DMSO-treated cells. The results of three independent experiments were graphed as mean averages with the standard errors of the mean. Statistical significance was determined using the paired two-tailed Student t test.
To further evaluate the contribution of vimentin in LMP1-mediated signaling, LMP1 cells were transfected with a dominant-negative vimentin (VIM-DN-GFP), GFP-tagged vimentin (VIM-GFP), or GFP alone, and the lysates were analyzed for ERK and Akt activation. Expression of the VIM-DN-GFP construct significantly inhibited LMP1-mediated ERK and Akt activation (Fig. 7B) with little effect on p-Akt or the low levels of p-ERK in control cells detected on long exposures. VIM-DN-GFP expression did not affect LMP1 levels. In addition, expression of the GFP-vimentin slightly impaired LMP1-mediated ERK activation and had variable effects on pAkt levels. Taken together, these data suggest that proper organization of vimentin is essential for the activation of Akt and ERK by LMP1.
Vimentin is important for LMP1-mediated transformation.
Activation of the ERK pathway in rodent fibroblasts by LMP1 is required for transformation as monitored by loss of contact inhibition in a typical focus formation assay (13). Therefore, it was predicted that vimentin would be important for LMP1-mediated focus formation in Rat-1 cells. To test this, stable cell lines were generated that express a shRNA against vimentin (shVIM) or a scrambled control (pGIVZ). These cell lines were transduced with equivalent retrovirus particles that expresses LMP1, and 10 to 14 days posttransduction the cells were stained with crystal violet and visualized by microscopy for the formation of foci. Although vimentin was only partially decreased (∼65%) (Fig. 8B, vimentin panel), there was a marked reduction (∼10-fold) in the number of foci induced by LMP1 expression (Fig. 8A). Importantly in the shVIM cells, LMP1-mediated activation of both Akt and ERK was greatly reduced (Fig. 8B). This experiment was repeated with a retrovirus that expresses a fluorescently tagged LMP1 construct. Transduction of Rat-1 cells with this LMP1 construct produced large fluorescent foci and shRNA reduction of vimentin resulted in significantly smaller and fewer fluorescent foci when measured by microscopy. Expression of the fluorescent LMP1 was detected throughout an example of a large LMP1-induced focus with multiple LMP1+ cells and compared to the smaller focus in the presence of the vimentin shRNA with fewer LMP1+ cells (Fig. 8C). In the presence of the shRNA to vimentin, the cells in the small focus had levels of LMP1 apparently equivalent to the cells in the large focus formed in the presence of the scrambled shRNA. Thus, the impaired cellular growth, as evidenced by smaller foci, is not a result of effects on LMP1 expression. Expression of the shRNA constructs was evaluated using GFP expression and revealed expression in all cells (data not shown). These data indicate that vimentin is a critical contributor to LMP1-mediated signaling, ERK activation, and transformation.
Fig 8.

Vimentin is important for the ability of LMP1 to transform cells and activate ERK and Akt. Rat-1 cells stably expressing an shRNA against vimentin (shVIM) or scrambled control (pGIVZ) were transduced with retrovirus particles expressing HA-LMP1 (A) or LMP1-DsRed (C) and incubated 10 to 14 days with medium being replaced every 2 days. After incubation, the cells were visualized for focus formation by crystal violet staining (A) or fluorescence microscopy (C). (A) The average numbers of foci per field were counted and are represented as average means of 10 fields with the standard errors of the mean. (B) Cell lysates were prepared from pBabe or LMP1 Rat-1 cells expressing pGIVZ or shVIM constructs and analyzed by immunoblot for vimentin, p-Akt, p-ERK, ERK, and Akt. The band intensities of p-ERK and p-Akt were determined using ImageJ software, normalized to total Akt and ERK protein levels, and represented relative to the shRNA scrambled cells. The results of three independent experiments were graphed as mean averages with standard errors of the mean. The statistical significance was determined using the paired two-tailed Student t test. (C) The foci formed using the fluorescent DsRed LMP1 construct were measured to determine the relative sizes of each focus by measuring the diameter of the fluorescent focus. The graph represents the mean focus diameters of 25 foci with standard errors of the mean. An individual fluorescent focus formed in the presence of the scrambled or vimentin-specific shRNA is shown to indicate the greater number of LMP1 expressing cells in the larger focus formed in the presence of the scrambled shRNA. (D) The levels of expression of LMP1, PI3K, and vimentin in total lysates or purified rafts in the presence of the scrambled or vimentin-specific shRNAs were determined by immunoblotting.
Assessment of the effect of decreased vimentin on raft components revealed that, similarly to the effects of depletion of cholesterol on LMP levels in rafts, decreased vimentin also increased LMP1 in rafts and slightly increased the levels of PI3K in rafts (Fig. 8D). These findings suggest that in the absence of vimentin- or cholesterol-containing lipid rafts, LMP1 aggregation and flotation occurs and PI3K is also affected possibly due to its indirect or direct link to LMP1. Overall, these data demonstrate that raft structure and the components of rafts that are modulated by LMP1 are essential for LMP1 signaling.
DISCUSSION
In this study it was found that LMP1 expression results in the reorganization of lipid rafts and that these microdomains are important for LMP1-mediated signal transduction and transformation capabilities. Specifically, in LMP1-expressing cells increased levels of PI3K, pAkt, and vimentin were found in lipid rafts. Lipid raft disruption through cholesterol depletion resulted in a significant reduction of PI3K localization to rafts and a reduction in the activation of the downstream target Akt, although the amount of LMP1 in rafts was increased by cholesterol depletion. Cholesterol depletion also inhibited LMP1-mediated activation of ERK in epithelial cells. In contrast, in Rat-1 fibroblast cells, cholesterol depletion increased the high basal levels of activated Akt in the control cells but effectively inhibited LMP1-mediated activation of Akt. MβCD treatment also greatly activated ERK in both the control and the LMP1-expressing cells. The significant activation of ERK in the control cells by cholesterol depletion may mask any specific inhibitor effects on LMP1-mediated ERK activation. However, the levels of activated ERK are equivalent in the MβCD-treated control and LMP1-expressing cells, suggesting that MβCD treatment indeed blocked LMP1-mediated ERK activation.
Recent studies using keratinocyte cell lines demonstrated that membrane cholesterol is critical for PI3K association with lipid raft microdomains and Akt activity (38). Similar results have been observed in lymphocyte, breast, prostate, and melanoma cell lines (39–41). The data presented here reveal that a viral oncoprotein similarly activates Akt through relocalization of PI3kinase to cholesterol rich lipid rafts. The recruitment of PI3K to lipid rafts is likely the result of interaction with LMP1 since LMP1 localizes to these microdomains and forms a complex with PI3K (42). It is currently unknown whether the two proteins interact directly or are potentially linked through an adaptor protein possibly the TRAF molecules. TRAF3 represents a potential bridging protein since it has previously been shown to bind PI3K and interact with CTAR1, is recruited to lipid rafts by LMP1, and is important for LMP1-mediated transformation (43, 44). Further studies will help clarify the mechanism of LMP1-mediated recruitment of PI3K to lipid rafts. This effect on lipid raft components may represent a potential target to inhibit LMP1 transformation effects.
This study also revealed that LMP1 increases vimentin association with lipid raft microdomains and that this effect was required for LMP1-mediated signaling since decreased vimentin expression by shRNA and expression of a dominant-negative vimentin inhibited LMP1-induced Akt and ERK activation. Chemical disruption of vimentin filaments consistently impaired ERK activation but had variable effects on the activation of Akt. Vimentin was also found to be important for transformation of Rat-1 fibroblasts by LMP1. The findings of the present study indicate that LMP1 modulates protein trafficking through lipid raft microdomains to activate signal transduction and provides compelling evidence that LMP1 interaction with the vimentin cytoskeleton is a major contributing factor for the activation of Akt and ERK and for transformation (Fig. 9). We have previously shown that PKCδ is a key mediator of LMP1 ERK activation and transformation (13). Interestingly, PKCδ has been shown to bind to vimentin and also localizes to detergent-insoluble fractions (45, 46). Therefore, it may be that vimentin forms a cytoskeletal scaffold for the recruitment of a signaling complex containing LMP1, PKCδ, MEK, and ERK (Fig. 9). The importance of the vimentin network has recently been shown in β-adrenergic receptor activation of ERK signaling (36). Additionally, in neurons, vimentin has been shown to bind activated pERK1 and pERK2 and transport them from the site of axonal lesion to the nerve cell body where they activate substrates necessary for lesion repair (37). These observations indicate that vimentin is likely a key mediator of the MAPK/ERK signaling pathway through the organization and transport of signal transduction molecules. This property may be particularly important for LMP1 transformation since LMP1-mediated focus formation was blocked by reducing vimentin levels within the cell.
Fig 9.
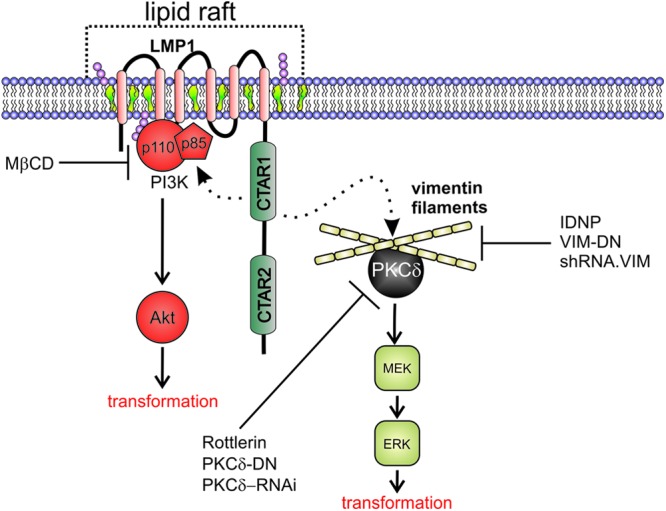
LMP1 modulation of membrane rafts and the vimentin cytoskeleton to activate Akt and ERK signal transduction pathways. LMP1 self-aggregates in lipid raft microdomains rich in cholesterol and sphingolipids. LMP1 expression leads to the recruitment of PI3K to lipid rafts and the activation of downstream target Akt that is essential for transformation. These effects can be disrupted through cholesterol depletion from cellular membranes with MβCD. LMP1 also induces vimentin expression and raft localization. Disruption of vimentin organization or depletion of vimentin expression blocks LMP-mediated ERK and Akt activation and transformation. PKCδ binds vimentin filaments and is also required for LMP1-mediated ERK activation. These results suggest a two component model that requires the localization of LMP1 and associated molecules into lipid rafts and that the interaction of LMP1 with vimentin may facilitate signaling complexes containing PKCδ and downstream kinases important for ERK signaling.
In addition to its function in ERK signaling, vimentin also affects the localization and activity of surface molecules such as integrins that play important roles in cell attachment and migration (47, 48). Interestingly, LMP1 expression results in the induction of cell surface molecules and the activation of signaling pathways that contribute to cell attachment, migration, and invasion (49–51). The molecular mechanisms driving many of these changes are still unclear, but transcriptional upregulation by LMP1 clearly contributes. The ability of LMP1 to affect vimentin expression levels and localization may represent an additional mechanism contributing to these important cellular processes that is altered by LMP1.
Vimentin has not only been found in lipid rafts but is also present in exosomes, small endocytically derived vesicles secreted from cells that participate in cell-cell communication (52). Lipid rafts have been proposed as a potential mechanism of protein targeting to exosomes for secretion from the cell (53). We have recently shown that LMP1 alters the protein composition and function of exosomes, resulting in increased exosome secretion of EGFR and PI3K (54). LMP1 also enhanced the ability of NPC exosomes to bind to target cells and activate Akt and ERK in recipient cells. The ability of LMP1 to reorganize the components of lipid rafts and vimentin-containing microdomains would likely affect exosome content (55). This would allow virus-infected cells to secrete factors into exosomes that may be beneficial to the virus and could contribute to pathogenesis within the host.
Previous studies have shown that the ability of LMP1 to self-aggregate through its transmembrane domains is critical for its signaling properties. A heptad leucine repeat within the cytoplasmic amino terminus was identified and suggested to contribute to LMP1 membrane localization and a FWLY motif within the first transmembrane domain was shown to facilitate interactions with additional transmembrane domains and enhanced signaling (14, 56). The LMP1 cytoplasmic carboxy-terminal domain contains two major activation domains, CTAR1 and CTAR2, that can activate NF-κB. However, only the CTAR1 domain is uniquely required for transformation and for activation of PI3K and ERK (3, 13). Interestingly, the interaction of LMP1 with the cytoskeleton is also dependent upon CTAR1 (29).
The data presented here suggest that the localization of LMP1 to lipid rafts also induces the localization of signaling components, such as PI3K, to the lipid rafts and through the interaction of LMP1 with vimentin and the cytoskeleton, signaling pathways are activated to induce transformation. These findings suggest a two component model where LMP1 localizes to lipid rafts through its transmembrane domains and alters the raft components by increasing the abundance of LMP1 interacting proteins (Fig. 9). The direct interaction of LMP1 CTAR1 with the cytoskeleton through vimentin may provide a scaffolding function to activate and potentially modulate multiple cell signaling pathways.
ACKNOWLEDGMENTS
We thank Phillip Heaton for careful review of the manuscript, Kathy Shair for the DsRed LMP1 construct, and Stuart Martin for the vimentin constructs.
This study was funded by National Institutes of Health grants CA32979 and CA19014 (to N.R.-T.). D.G.M. was supported by training grant T32CA009156 and an American Cancer Society fellowship PF-11-158-01-MPC.
Footnotes
Published ahead of print 14 November 2012
REFERENCES
- 1. Raab-Traub N. 2012. Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol. 2:453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raab-Traub N. 2007. EBV-induced oncogenesis, p 986–1006 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 3. Kaye KM, Izumi KM, Kieff E. 1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. U. S. A. 90:9150–9154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang D, Liebowitz D, Kieff E. 1985. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 43:831–840 [DOI] [PubMed] [Google Scholar]
- 5. Mei Y-P, Zhou J-M, Wang Y, Huang H, Deng R, Feng G-K, Zeng Y-X, Zhu X-F. 2007. Silencing of LMP1 induces cell cycle arrest and enhances chemosensitivity through inhibition of AKT signaling pathway in EBV-positive nasopharyngeal carcinoma cells. Cell Cycle 6:1379–1385 [DOI] [PubMed] [Google Scholar]
- 6. Hannigan A, Wilson J. 2010. Evaluation of LMP1 of Epstein-Barr virus as a therapeutic target by its inhibition. Mol. Cancer 9:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller WE, Cheshire JL, Raab-Traub N. 1998. Interaction of tumor necrosis factor receptor-associated factor signaling proteins with the latent membrane protein 1 PXQXT motif is essential for induction of epidermal growth factor receptor expression. Mol. Cell. Biol. 18:2835–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mosialos G, Birkenbacht M, Yalamanchill R, Van Arsdale T, Ware C, Kleff E. 1995. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80:389–399 [DOI] [PubMed] [Google Scholar]
- 9. Eliopoulos AG, Young LS. 1998. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 16:1731–1742 [DOI] [PubMed] [Google Scholar]
- 10. Mainou BA, Everly DN, Raab-Traub N. 2005. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene 24:6917–6924 [DOI] [PubMed] [Google Scholar]
- 11. Paine E, Scheinman RI, Baldwin AS, Raab-Traub N. 1995. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-κB/Rel family proteins. J. Virol. 69:4572–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roberts ML, Cooper NR. 1998. Activation of a Ras/MAPK-dependent pathway by Epstein-Barr virus latent membrane protein 1 is essential for cellular transformation. Virology 240:93–99 [DOI] [PubMed] [Google Scholar]
- 13. Kung C-P, Meckes DG, Raab-Traub N. 2011. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCδ. J. Virol. 85:4399–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yasui T, Luftig M, Soni V, Kieff E. 2004. Latent infection membrane protein transmembrane FWLY is critical for intermolecular interaction, raft localization, and signaling. Proc. Natl. Acad. Sci. U. S. A. 101:278–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pioche-Durieu C, Keryer C, Souquère S, Bosq J, Faigle W, Loew D, Hirashima M, Nishi N, Middeldorp J, Busson P. 2005. In nasopharyngeal carcinoma cells, Epstein-Barr virus LMP1 interacts with galectin 9 in membrane raft elements resistant to simvastatin. J. Virol. 79:13326–13337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katano H, Pesnicak L, Cohen JI. 2004. Simvastatin induces apoptosis of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines and delays development of EBV lymphomas. Proc. Natl. Acad. Sci. U. S. A. 101:4960–4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50 [DOI] [PubMed] [Google Scholar]
- 18. Staubach S, Hanisch F-G. 2011. Lipid rafts: signaling and sorting platforms of cells and their roles in cancer. Expt. Rev. Proteomics 8:263–277 [DOI] [PubMed] [Google Scholar]
- 19. Everly DN, Mainou BA, Raab-Traub N. 2004. Induction of Id1 and Id3 by latent membrane protein 1 of Epstein-Barr virus and regulation of p27/Kip and cyclin-dependent kinase 2 in rodent fibroblast transformation. J. Virol. 78:13470–13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fries KL, Miller WE, Raab-Traub N. 1996. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 70:8653–8659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shair KHY, Schnegg CI, Raab-Traub N. 2008. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res. 68:6997–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Birkenbach M, Liebowitz D, Wang F, Sample J, Kieff E. 1989. Epstein-Barr virus latent infection membrane protein increases vimentin expression in human B-cell lines. J. Virol. 63:4079–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivaska J, Pallari Nevo H-MJ, Eriksson JE. 2007. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313:2050–2062 [DOI] [PubMed] [Google Scholar]
- 24. Runembert I, Queffeulou G, Federici P, Vrtovsnik F, Colucci-Guyon E, Babinet C, Briand P, Trugnan G, Friedlander G, Terzi F. 2002. Vimentin affects localization and activity of sodium-glucose cotransporter SGLT1 in membrane rafts. J. Cell Sci. 115:713–724 [DOI] [PubMed] [Google Scholar]
- 25. Sprenger RR, Fontijn RD, van Marle J, Pannekoek H, Horrevoets AJ. 2006. Spatial segregation of transport and signalling functions between human endothelial caveolae and lipid raft proteomes. Biochem. J. 400:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liebowitz D, Kopan R, Fuchs E, Sample J, Kieff E. 1987. An Epstein-Barr virus transforming protein associates with vimentin in lymphocytes. Mol. Cell Biol. 7:2299–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whipple RA, Balzer EM, Cho EH, Matrone MA, Yoon JR, Martin SS. 2008. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res. 68:5678–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim KB, Lee JS, Ko YG. 2008. The isolation of detergent-resistant lipid rafts for two-dimensional electrophoresis. Methods Mol. Biol. 424:413–422 [DOI] [PubMed] [Google Scholar]
- 29. Higuchi M, Izumi KM, Kieff E. 2001. Epstein–Barr virus latent-infection membrane proteins are palmitoylated and raft-associated: protein 1 binds to the cytoskeleton through TNF receptor cytoplasmic factors. Proc. Natl. Acad. Sci. U. S. A. 98:4675–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lam N, Sugden B. 2003. LMP1, a viral relative of the TNF receptor family, signals principally from intracellular compartments. EMBO J. 22:3027–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ardila-Osorio H, Pioche-Durieu C, Puvion-Dutilleul F, Clausse B, Wiels J, Miller W, Raab-Traub N, Busson P. 2005. TRAF interactions with raft-like buoyant complexes, better than TRAF rates of degradation, differentiate signaling by CD40 and EBV latent membrane protein 1. Int. J. Cancer 113:267–275 [DOI] [PubMed] [Google Scholar]
- 32. Brown DA. 2001. Isolation and use of rafts, p 51:11.10.1–11.10.23 In Current protocols in immunology. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 33. Chabanel A, Flamm M, Sung KL, Lee MM, Schachter D, Chien S. 1983. Influence of cholesterol content on red cell membrane viscoelasticity and fluidity. Biophys. J. 44:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lei B, Morris DP, Smith MP, Schwinn DA. 2009. Lipid rafts constrain basal α1A-adrenergic receptor signaling by maintaining receptor in an inactive conformation. Cell Signal. 21:1532–1539 [DOI] [PubMed] [Google Scholar]
- 35. Furuchi T, Anderson RGW. 1998. Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J. Biol. Chem. 273:21099–21104 [DOI] [PubMed] [Google Scholar]
- 36. Kumar N, Robidoux J, Daniel KW, Guzman G, Floering LM, Collins S. 2007. Requirement of vimentin filament assembly for β3-adrenergic receptor activation of ERK MAP kinase and lipolysis. J. Biol. Chem. 282:9244–9250 [DOI] [PubMed] [Google Scholar]
- 37. Perlson E, Michaelevski I, Kowalsman N, Ben-Yaakov K, Shaked M, Seger R, Eisenstein M, Fainzilber M. 2006. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J. Mol. Biol. 364:938–944 [DOI] [PubMed] [Google Scholar]
- 38. Calay D, Vind-Kezunovic D, Frankart A, Lambert S, Poumay Y, Gniadecki R. 2010. Inhibition of Akt signaling by exclusion from lipid rafts in normal and transformed epidermal keratinocytes. J. Invest. Dermatol. 130:1136–1145 [DOI] [PubMed] [Google Scholar]
- 39. Li YC, Park MJ, Ye S-K, Kim C-W, Kim Y-N. 2006. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 168:1107–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lasserre R, Guo XJ, Conchonaud F, Hamon Y, Hawchar O, Bernard AM, Soudja SM, Lenne PF, Rigneault H, Olive D, Bismuth G, Nunes JA, Payrastre B, Marguet D, He HT. 2008. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat. Chem. Biol. 4:538–547 [DOI] [PubMed] [Google Scholar]
- 41. Fedida-Metula S, Elhyany S, Tsory S, Segal S, Hershfinkel M, Sekler I, Fishman D. 2008. Targeting lipid rafts inhibits protein kinase B by disrupting calcium homeostasis and attenuates malignant properties of melanoma cells. Carcinogenesis 29:1546–1554 [DOI] [PubMed] [Google Scholar]
- 42. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. 2003. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 278:3694–3704 [DOI] [PubMed] [Google Scholar]
- 43. Ha YJ, Lee JR. 2004. Role of TNF receptor-associated factor 3 in the CD40 signaling by production of reactive oxygen species through association with p40phox, a cytosolic subunit of nicotinamide adenine dinucleotide phosphate oxidase. J. Immunol. 172:231–239 [DOI] [PubMed] [Google Scholar]
- 44. Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, Kieff E, Mosialos G. 1996. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-κB activation. Mol. Cell. Biol. 16:7098–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Owen PJ, Johnson GD, Lord JM. 1996. Protein Kinase C-δ Associates with vimentin intermediate filaments in differentiated HL60 cells. Exp. Cell Res. 225:366–373 [DOI] [PubMed] [Google Scholar]
- 46. Chen Y-W, Lang ML, Wade WF. 2004. Protein kinase C-α and -δ are required for FcαR (CD89) trafficking to MHC class II compartments and FcαR-mediated antigen presentation. Traffic 5:577–594 [DOI] [PubMed] [Google Scholar]
- 47. Ivaska J, Whelan RDH, Watson R, Parker PJ. 2002. PKCε controls the traffic of β1 integrins in motile cells. EMBO J. 21:3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. 2005. PKCε-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 24:3834–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vockerodt M, Tesch H, Kube D. 2001. Epstein-Barr virus latent membrane protein-1 activates CD25 expression in lymphoma cells involving the NFκB pathway. Genes Immun. 2:433–441 [DOI] [PubMed] [Google Scholar]
- 50. Walter J, Schirrmacher V, Mosier D. 1995. Induction of CD44 expression by the Epstein-Barr virus latent membrane protein LMP1 is associated with lymphoma dissemination. Intern. J. Cancer 61:363–369 [DOI] [PubMed] [Google Scholar]
- 51. Shair KHY, Schnegg CI, Raab-Traub N. 2009. Epstein-Barr virus latent membrane protein-1 effects on junctional plakoglobin and induction of a cadherin switch. Cancer Res. 69:5734–5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buschow SI, van Balkom BWM, Aalberts M, Heck AJR, Wauben M, Stoorvogel W. 2010. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 88:851–856 [DOI] [PubMed] [Google Scholar]
- 53. Meckes DG, Raab-Traub N. 2011. Microvesicles and viral infection. J. Virol. 85:12844–12854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meckes DG, Shair KHY, Marquitz AR, Kung Edwards C-PRH, Raab-Traub N. 2010. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. U. S. A. 107:20370–20375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Verweij FJ, van Eijndhoven MAJ, Hopmans ES, Vendrig T, Wurdinger T, Cahir-McFarland E, Kieff E, Geerts D, van der Kant R, Neefjes J, Middeldorp JM, Pegtel DM. 2011. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-κB activation. EMBO J. 30:2115–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee J, Sugden B. 2007. A membrane leucine heptad contributes to trafficking, signaling, and transformation by latent membrane protein 1. J. Virol. 81:9121–9130 [DOI] [PMC free article] [PubMed] [Google Scholar]