

Abstract
The entry of Kaposi's sarcoma-associated herpesvirus (KSHV) into human dermal microvascular endothelial cells (HMVEC-d), natural in vivo target cells, via macropinocytosis is initiated through a multistep process involving the binding of KSHV envelope glycoproteins with cell surface α3β1, αVβ3, and αVβ5 integrin molecules and tyrosine kinase ephrin-A2 receptor, followed by the activation of preexisting integrin-associated signaling molecules such as focal adhesion kinase (FAK), Src, c-Cbl, phosphoinositide 3-kinase (PI-3K), and Rho-GTPases. Many viruses, including KSHV, utilize cellular reactive oxygen species (ROS) for viral genomic replication and survival within host cells; however, the role of ROS in early events of viral entry and the induction of signaling has not been elucidated. Here we show that KSHV induced ROS production very early during the infection of HMVEC-d cells and that ROS production was sustained over the observation period (24 h postinfection). ROS induction was dependent on the binding of KSHV to the target cells, since pretreatment of the virus with heparin abolished ROS induction. Pretreatment of HMVEC-d cells with the antioxidant N-acetylcysteine (NAC) significantly inhibited KSHV entry, and consequently gene expression, without affecting virus binding. In contrast, H2O2 treatment increased the levels of KSHV entry and infection. In addition, NAC inhibited KSHV infection-induced translocation of αVβ3 integrin into lipid rafts, actin-dependent membrane perturbations, such as blebs, observed during macropinocytosis, and activation of the signal molecules ephrin-A2 receptor, FAK, Src, and Rac1. In contrast, H2O2 treatment increased the activation of ephrin-A2, FAK, Src, and Rac1. These studies demonstrate that KSHV infection induces ROS very early during infection to amplify the signaling pathways necessary for its efficient entry into HMVEC-d cells via macropinocytosis.
INTRODUCTION
Reactive oxygen species (ROS)—hydrogen peroxide (H2O2), superoxide anion (·O2−), peroxide (·O2−2), hydroxyl radical (·OH), and hydroxyl anion (OH−)—are highly reactive molecules due to the presence of unpaired valence shell electrons and mediate important roles in cell signaling and homeostasis. They are ideally suited as signaling molecules, since they are small, can diffuse over short distances, can be induced by several mechanisms that are rapid and controllable, and have numerous mechanisms for their rapid removal. Reversible protein phosphorylation is the key biochemical event in most cell signaling pathways, and signal transduction involving ROS is no exception. As signaling molecules, hydrogen peroxide and other ROS posttranslationally modify target proteins by oxidizing thiol groups, thus forming disulfide bonds that reversibly alter protein structure and function. Specificity is achieved by localized production; targeted secondary oxidation occurs via glutaredoxins or thioredoxins. Target proteins containing reduction-oxidation (redox)-sensitive thiol groups include signal transduction pathway proteins such as phosphatases and mitogen-activated protein kinases, many transcription factors, RNA-binding proteins that direct DNA methylation, and proteins involved in histone acetylation, deacetylation, or methylation.
Kaposi's sarcoma-associated herpesvirus, also called human herpesvirus 8 (KSHV/HHV-8), is etiologically associated with the pathogenesis of Kaposi's sarcoma (KS), primary effusion B-cell lymphoma (PEL), and B-cell angiolymphoproliferative multicentric Castleman's disease (MCD) (1, 2). Like that of other members of the herpesvirus family, the KSHV life cycle includes latent and lytic cycles. Human B-cell lines from PEL, such as BCBL-1 and BC-3 cells, carry multiple copies of the KSHV genome and the latency-associated genes open reading frame 73 (ORF 73) (latency-associated nuclear antigen 1 [LANA-1]), ORF 72 (vCyclin), ORF 71 (vFLIP), K12 (kaposin), and ORF 10.5 (LANA-2), as well as 12 microRNAs that are expressed in these cells. The lytic cycle can be induced from the latently infected cells by the expression of KSHV lytic-cycle switch protein RTA (replication and transcriptional activator) (ORF 50) or by stimulation with 12-O-tetradecanoyl phorbol-12-acetate (TPA) or by the histone deacetylase inhibitor n-butyrate. KSHV particles induced from these cells serve as the source of virus for various studies, such as the infection of in vitro target human microvascular dermal endothelial (HMVEC-d) cells and human foreskin fibroblasts (HFF). In contrast to infection by alpha- or betaherpesviruses, in vitro infection of adherent HMVEC-d and HFF target cells by the gamma-2 herpesvirus KSHV does not result in a productive lytic cycle but instead is followed by the establishment of latency. Another novel feature of this in vitro latency in HMVEC-d cells and HFF is that as early as 2 h postinfection (p.i.), KSHV expresses its latent genes concurrently, as well as a limited set of lytic-cycle genes with antiapoptotic and immunomodulation functions, including the ORF 50 (RTA) gene (3). While the expression of latent genes such as ORF 73, ORF 72, and ORF 71 continues, the expression of nearly all lytic genes declines by 24 h p.i. (3).
Previous studies have indicated a role for ROS in KSHV lytic-cycle induction. Oxidative stress has been shown to reactivate KSHV from latency in PEL and endothelial cells (4–6). Several inducers of KSHV reactivation, such as TPA, cytokines, and hypoxia, induce KSHV lytic replication through an increase in intracellular H2O2 production and the activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2), Jun N-terminal protein kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathways (4–6). In addition to their role in KSHV lytic induction, ROS are also involved in KSHV pathogenesis. In the KSHV-infected human umbilical vein endothelial cell (KSHV-HUVEC) latency model, endothelial junction dysregulation and increased vascular permeability have been observed (7). That study demonstrated that latent KSHV infection leads to the activation of the Rac1/NADPH oxidase/ROS production pathway to regulate the phosphorylation of junctional proteins such as VE-cadherin and β-catenin, and this activation was hypothesized to be participating in viral spread to other cell types.
Inhibition of ROS by the antioxidant N-acetylcysteine (NAC) has also been shown to prevent KSHV tumorigenesis (6, 8). In a PEL mouse model, NAC-treated animals had an extended life span relative to the control group (6). In addition, NAC reduced the tumor size in an endothelial KS-mouse model by inhibiting the expression of both viral and cellular genes involved in proliferation and angiogenesis (8). In all these studies, the role of ROS has been elucidated only in endothelial or B-cell latency models. In contrast, nothing is known about whether primary infection of target cells with KSHV induces ROS and whether ROS play roles in primary infection.
Primary in vitro infection of HMVEC-d cells by KSHV is considered the closest model mimicking in vivo infection of endothelial cells. Our earlier detailed studies have highlighted the different stages of in vitro endothelial cell infection (9). We and others have shown that KSHV initiates its infection by binding to heparan sulfate (HS) on the cell surface via its envelope glycoproteins, followed by interactions with integrins αVβ5, α3β1, and αVβ3, the transporter molecule xCT (CD98), and tyrosine kinase ephrin-A2 (EphA2) receptor (10–14). We have also shown that KSHV hijacks several integrin-associated signaling pathways to effectively enter the target cell and to create an intracellular environment that is conducive to the establishment of infection. Similarly to the integrin interactions with its natural ligands, KSHV binding to the target cells induces several integrin-dependent signaling events, such as the phosphorylation of focal adhesion kinase (FAK), a nonreceptor tyrosine kinase, that is followed by the activation of Src, phosphoinositide 3-kinase (PI-3K), Rho-GTPases (Rac1, RhoA, and Cdc42), and the adaptor molecule c-Cbl, as well as their downstream effector molecules, such as AKT, ezrin, protein kinase Cζ (PKC-ζ), MEK, ERK1/2, and p38 MAPK (15–23). We have shown previously that these KSHV binding-induced signal molecules play key roles in virus entry via bleb-mediated macropinocytosis, actin remodeling, microtubule acetylation, transport toward the nucleus, and initiation of viral and host gene expression (9, 24).
Several studies demonstrate that ROS are important mediators that transduce the signals associated with integrin activation as well as modulating integrin functions (25–27). Studies have shown that integrin engagement triggers a transient and localized increase in ROS. Among the proposed mechanisms, integrin engagement with extracellular matrix ligand proteins or with antibodies has been shown to modify mitochondrial function and to activate oxidases such as NADPH oxidase (28, 29). The small GTP-binding protein Rac1 is an essential protein directly interacting with integrin molecules involved in ROS production (25, 28–31). ROS production induced by integrin engagement signals has been to shown to induce the reversible oxidation of target proteins such as protein tyrosine phosphatases (32, 33).
Although KSHV has been shown to interact with integrins and to activate several integrin-associated signaling events, the role(s) of ROS in the modulation of KSHV-induced signaling during viral entry has not been deciphered (9, 24). The studies presented here demonstrate that ROS are induced very early during primary infection of HMVEC-d cells with KSHV. Treatment with nontoxic doses of the antioxidant NAC reduced KSHV infection by blocking virus entry, the recruitment of integrin to the lipid rafts (LRs), the membrane bleb formation observed during macropinocytosis, phosphorylation of the ephrin-A2 receptor, and integrin-associated signaling, such as FAK, Src, and Rac1 activation. These studies collectively demonstrate for the first time that KSHV infection-induced ROS production promotes KSHV entry.
MATERIALS AND METHODS
Cells.
HMVEC-d cells (CC-2543; Lonza Walkersville, Walkersville, MD) were cultured in endothelial basal medium 2 (EBM-2) with growth factors (Lonza Walkersville). BCBL-1 cells (KSHV-carrying human B cells) were propagated and maintained by procedures described previously (10, 16, 34).
Reagents.
Antibodies against total Src and phospho-Src (dilutions, 1:1,000 for Western blotting [WB] and 1:50 for the immunofluorescence assay [IFA]) and against total EphA2 and phospho-EphA2 (1:1,000 for WB and 1:50 for the IFA) were from Cell Signaling Technology, Danvers, MA. Antibodies against total FAK, phospho-FAK, and Rac1 (1:1,000 for WB and 1:100 for the IFA) were from BD Biosciences, San Jose, CA. The anti-tubulin antibody, heparin, N-acetylcysteine, and H2O2 were from Sigma-Aldrich, St. Louis, MO. The antibody against Rac1-GTP (1:200 for the IFA) was from NewEast Biosciences, King of Prussia, PA. Mouse monoclonal antibody 4A4 against glycoprotein K8.1A (gpK8.1A) (1:100 for the IFA), rabbit polyclonal antibody UK-218 against glycoprotein B (gB) (1:100 for the IFA), and a rabbit polyclonal antibody against LANA-1 (1:50 for the IFA) were generated in our laboratory (35–37). Alexa Fluor 488 (1:500)- and Alexa Fluor 594 (1:1,000)-conjugated anti-mouse and anti-rabbit secondary antibodies were from Molecular Probes, Invitrogen, Grand Island, NY. Anti-rabbit and anti-mouse antibodies linked to horseradish peroxidase were from KPL Inc., Gaithersburg, MD.
Virus.
Induction of the KSHV lytic cycle in BCBL-1 cells, supernatant collection, and virus purification procedures have been described previously (3). KSHV DNA was extracted from the virus, and the copy numbers were quantitated by real-time DNA PCR using primers amplifying the KSHV ORF 73 gene as described previously (3, 38).
ROS measurement.
HMVEC-d cells were cultured in a 12-well plate until they were confluent and were incubated with EBM-2 without growth factors for 2 h. Cells were loaded with dye by replacing the medium with fresh EBM-2 containing 10 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA [C6827]; Invitrogen, Grand Island, NY) for 1 h at 37°C under 5% CO2. After loading, the medium was removed and was replaced with fresh EBM-2 with or without KSHV (40 DNA copies/cell). Fluorescence was measured using a Synergy HT microplate reader (BioTek Instruments) with a 485/20 excitation, 528/20 emission filter pair and a photomultiplier tube (PMT) sensitivity setting of 55. Readings were made from the bottom at the times indicated in Fig. 1. Between each two time points, the cells were kept in the culture incubator. For ROS measurement after 24 h of infection, HMVEC-d cells were infected with KSHV (40 DNA copies/cell) for 24 h before dye loading. Fluorescence was measured 1 h after the medium was exchanged with fresh EBM-2.
Fig 1.

(A) ROS production upon primary infection of endothelial cells with KSHV. Confluent HMVEC-d cells in 12-well plates were serum starved for 2 h, incubated with EBM-2 containing 10 μM CM-H2DCFDA for 1 h at 37°C, and either mock infected or infected with KSHV (40 DNA copies/cell). Fluorescence was measured with a Synergy HT microplate reader (BioTek Instruments) using a 485/20 excitation and 528/20 emission filter pair and a PMT sensitivity setting of 55. Each reading was done in triplicate, and the data are means for three independent experiments ± standard deviations. Statistical analysis was conducted using a two-tailed Student test. **, P < 0.01. (a) ΔDCFDA fluorescence between infected and uninfected cells. A.U., arbitrary units. (b) Fold induction of ROS production in infected cells relative to uninfected cells. (B) Binding of KSHV to endothelial cells induces ROS production. HMVEC-d cells were starved for 2 h, loaded with CM-H2DCFDA (10 μM) for 1 h, and infected either with untreated KSHV (40 DNA copies/cell) or with KSHV preincubated with 100 μg/ml heparin for 1 h at 37°C (40 DNA copies/cell). The respective control cells were mock infected either with medium alone or with medium containing 100 μg/ml heparin. (a) Fluorescence was measured at the indicated time points, and ROS production is expressed as fold induction relative to the respective controls. Each reading was done in triplicate, and the data are means for two independent experiments ± standard deviations. Statistical analysis was performed using a two-tailed Student test. *, P < 0.05. (b) The inhibition of ROS production in cells infected with heparin-treated virus was compared with ROS production in cells infected with untreated KSHV, and the percentage of inhibition is shown. (C) Elevated ROS production during KSHV latency in endothelial cells. Serum-starved (2 h) HMVEC-d cells were either mock infected or infected with KSHV (40 DNA copies/cell); the virus was removed; the cells were loaded with CM-H2DCFDA for 1 h at 12 h, 24 h, and 48 h p.i.; and fluorescence was measured. **, P < 0.01.
KSHV binding and entry by real-time DNA PCR.
For virus binding, HMVEC-d cells were infected for 1 h with KSHV (20 DNA copies/cell) at 4°C. Cells were washed twice with phosphate-buffered saline (PBS) to remove unbound virus, and total DNA was extracted by using a DNeasy kit (Qiagen, Inc., Valencia, CA) according to the manufacturer's instructions. For virus entry, HMVEC-d cells were infected with KSHV (20 DNA copies/cell) for 1 h at 37°C. Cells were washed twice with PBS to remove unbound virus, treated with trypsin-EDTA for 5 min at 37°C to remove the bound but noninternalized virus, and washed, and total DNA was isolated. A total of 100 ng of DNA from each sample was used in real-time DNA PCR using KSHV ORF 73 gene-specific primers anda TaqMan probe. The KSHV ORF 73 gene cloned into the pGEM-T vector (Promega) was used as the external standard. Known amounts of the ORF 73 plasmid were used in the amplification reactions along with the test samples. Cycle threshold values were used to generate the standard curve and to calculate the relative copy numbers of viral DNA in the samples.
Immunoblotting.
Cells were harvested in radioimmunoprecipitation assay (RIPA) lysis buffer (125 mM NaCl, 0.01 M sodium phosphate [pH 7.2], 0.1% sodium dodecyl sulfate [SDS], 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, and 50 mM sodium fluoride) with a protease inhibitor cocktail and a phosphatase inhibitor cocktail (Sigma). Cellular debris was removed by centrifugation at 13,000 × g for 5 min at 4°C, and equal amounts of protein samples were resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and were subjected to Western blotting with the antibodies indicated in the figures. To confirm equal protein loading, blots were also probed with antibodies against human tubulin or actin. Secondary antibodies conjugated to horseradish peroxidase were used for detection. Immunoreactive bands were visualized by enhanced chemiluminescence.
RNA extraction, reverse transcription, and real-time RT-PCR.
Total RNA was extracted by using TRIzol reagent (Invitrogen), quantified by densitometric analysis at 260 nm, and analyzed by real-time reverse transcription-PCR (RT-PCR) using ORF 73, ORF 50, and K8 primers as described previously (3, 20). PCR was performed using an ABI Prism 7500 real-time PCR system utilizing TaqMan EZ RT-PCR Core reagents (Applied Biosystems).
Immunofluorescence assay.
HMVEC-d cells seeded on 8-well chamber slides (Nalge Nunc International, Naperville, IL) were used for the IFA. Infected and uninfected cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 5 min, and blocked with Image-iT FX signal enhancer (Invitrogen) for 20 min. The cells were reacted with primary antibodies against the specific proteins, followed by fluorescent dye-conjugated secondary antibodies. Nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, Invitrogen), and stained cells were viewed with the appropriate filters under a fluorescence microscope with a 40× objective and the Nikon MetaMorph digital imaging system. All experiments were performed at least three times.
Rac1 activation analysis.
One hundred fifty micrograms of cell lysates was incubated with beads containing the glutathione S-transferase (GST)–Rac1-binding domain of PAK1 (PBD) for 2 h, and the resulting bound Rac1-GTP complexes were analyzed by Western blotting for Rac1.
RESULTS
ROS production is increased during primary infection of HMVEC-d cells with KSHV.
To determine whether ROS production is induced early during primary KSHV infection of HMVEC-d cells, we used purified virus prepared from a latently infected PEL cell line (BCBL-1) and quantitated the virus by methods described previously (3, 10). HMVEC-d cells loaded with CM-H2DCFDA, a fluorescent ROS indicator dye, were infected with KSHV (40 DNA copies/cell), and fluorescence was measured at the time points indicated in Fig. 1A up to 6 h p.i. The CM-H2DCFDA dye is nonfluorescent when reduced but becomes fluorescent after cellular oxidation and the removal of acetate groups. The differences in fluorescence (ΔDCFDA fluorescence) between uninfected and KSHV-infected cells are shown in Fig. 1Aa. In uninfected cells, ROS is produced at a low rate as a by-product of cellular respiration. However, in KSHV-infected cells, a significantly higher level of ROS was induced, as evidenced by higher fluorescence than that of uninfected cells as early as 30 min p.i. and throughout the 6-h observation period (Fig. 1Aa). We observed a 1.5-fold increase in ROS production at 30 min p.i., as well as maintenance throughout the 6 h p.i. (Fig. 1Ab).
The initial contact of KSHV with target cell heparan sulfate (HS) molecules via its envelope glycoproteins can be blocked with heparin, an analogue of HS (39, 40). We measured ROS production in HMVEC-d cells infected with untreated or heparin-treated KSHV and in control cells mock infected with medium alone or heparin-containing medium, respectively. The fold induction of ROS production in KSHV-infected cells relative to their respective uninfected controls is shown in Fig. 1Ba. ROS production was greatly reduced in heparin-treated KSHV-infected cells during the first 3 h p.i. (Fig. 1Ba), and we observed 75% inhibition in ROS production at 30 min and at 1, 2, and 3 h p.i. (Fig. 1Bb). Together, these results demonstrate that the binding of KSHV to HMVEC-d cells triggered an increase in ROS production during the very early stages of infection.
Increased ROS production is induced during the establishment of KSHV latency after primary infection of HMVEC-d cells.
In contrast to infection with alpha- and betaherpesviruses, in vitro infection with KSHV does not result in a productive lytic cycle. Instead, it is characterized by the expression of latent genes, such as ORF 73, as early as 2 h p.i.; expression of these genes reaches a peak at 24 h p.i. and is steadily maintained thereafter (3). To determine whether ROS production is also induced during the establishment of latency, HMVEC-d cells were infected with KSHV (40 DNA copies/cell) for 2 h; the virus was removed by washing; and the cells were incubated with HMVEC-d medium until 12, 24, or 28 h p.i. The cells were then loaded with the CM-H2DCFDA, and ROS measurement was performed. The virus was removed after 2 h in order to differentiate the ROS induction mediated by the early events of virus binding and immediate signaling from the ROS production at later time points after viral entry and viral gene expression. We observed a significant 2.4-fold increase in ROS levels at 24 h and 48 h p.i. in infected cells (Fig. 1C). These results demonstrated that ROS are not only induced immediately after KSHV binding and entry into HMVEC-d cells (Fig. 1A and B) but are also elevated after 24 h p.i. (Fig. 1C), when latency is established after primary infection; these high ROS levels could be due to viral gene expression. Thus, the increase in intracellular ROS levels could be due to different mechanisms depending on the time of KSHV infection. Since increased production of ROS and its role in latency/the lytic cycle have been studied previously (4–6), we set out here to decipher the role of ROS during the early stages of KSHV infection.
The antioxidant NAC reduced KSHV gene expression during primary infection of HMVEC-d cells.
Early during infection, KSHV induces several preexisting host cell signal molecules, which play roles in the different stages of viral infection (9). We have shown previously that primary infection of HMVEC-d cells is characterized by the sustained expression of ORF 73 and the transient expression of a limited number of lytic genes, including ORF 50 and K8 (3, 19, 20). We first investigated whether blocking the increase in ROS production could have an effect on viral gene expression. To reduce ROS induction, HMVEC-d cells were preincubated with 10 mM N-acetylcysteine (NAC), a ROS scavenger, for 2 h. These cells were then infected with KSHV for 2 h, washed, and maintained in 1 mM NAC for 24 h; then RNA was extracted and analyzed by real-time RT-PCR using ORF 73, ORF 50, and K8 primers (3, 20). Treatment with NAC reduced both latent and lytic gene expression. The expression of the ORF 73 gene was reduced by about 72% in NAC-treated cells (Fig. 2A). We also observed about 45% reductions in ORF 50 and K8 mRNA levels in NAC-treated HMVEC-d cells (Fig. 2B and C).
Fig 2.

ROS participate in primary KSHV infection of HMVEC-d cells. (A, B, and C) ROS induction is necessary for the expression of KSHV genes. Serum-starved (2 h) HMVEC-d cells were either mock treated or pretreated with NAC (10 mM) for 2 h, after which they were infected with KSHV (20 DNA copies/cells) for 2 h. Then the virus was removed, and the cells were mock treated or treated with NAC (1 mM) for 24 h. RNA was extracted with TRIzol reagent (Invitrogen) and was analyzed by real-time RT-PCR using latency-associated ORF 73 (A) or lytic-cycle-associated ORF 50 (B) or K8 (C) gene primers. Each PCR was performed in triplicate, and the data are means for two independent experiments ± standard deviations. (D) ROS induction is necessary for LANA-1 expression. HMVEC-d cells in chamber slides were either mock treated or pretreated with NAC (10 mM) for 2 h and were then infected with KSHV (20 DNA copies/cells) for 2 h. Unbound virus was removed by washing, and cells were either mock treated or treated with NAC (1 mM) for 48 h. Cells were fixed and processed for an IFA using a rabbit anti-LANA-1 antibody. (a) Representative IFA images. (b) LANA-1-positive cells from four different independent fields, each containing at least 15 cells, were counted to calculate the percentage of LANA-1-positive cells. Statistical analysis was carried out using a two-tailed Student test. *, P < 0.05.
To confirm the effect of NAC on KSHV gene expression, NAC-treated and untreated infected cells were reacted with rabbit anti-LANA-1 antibodies after 48 h p.i. and were observed for the characteristic punctate LANA-1 staining by an immunofluorescence assay (IFA). In contrast to untreated samples, in which ∼80% of cells were LANA-1 positive, only 40% of the NAC-treated cells expressed LANA-1 (Fig. 2 Da and Db). These results clearly demonstrate that ROS production early during KSHV infection plays an important role in viral latent and lytic gene expression, thus contributing to KSHV infectivity.
The antioxidant NAC did not block KSHV binding but blocked the entry of KSHV into HMVEC-d cells.
KSHV has been shown to interact with its target cells through several molecules. The first contact involves the binding of viral glycoproteins gB, gpK8.1A, and gH/gL to heparan sulfate on the cell surface, and integrins and xCT have been identified as entry receptors in HMVEC-d cells (9, 24). To determine whether the inhibition of KSHV gene expression by NAC was due to blockage of viral binding to the target cells, we next determined the role of ROS in the binding of KSHV to the target cells. HMVEC-d cells either were infected with KSHV (20 DNA copies/cell) for 1 h at 4°C in the presence of H2O2 (100 μM) (Fig. 3A) or were treated with NAC (10 mM) for 2 h prior to infection (Fig. 3B). After infection, the cells were washed; total DNA was isolated; and KSHV binding was quantitated by real-time DNA PCR using ORF 73 gene primers. Neither the H2O2 nor the NAC treatment affected the binding of KSHV to the cells (Fig. 3A and B).
Fig 3.

ROS induction during primary infection of endothelial cells promotes KSHV entry. (A and B) ROS have no effect on KSHV binding. HMVEC-d cells were infected with KSHV (20 DNA copies/cell) for 1 h at 4°C in the presence of H2O2 (100 μM) (A) or were treated with NAC (10 mM) for 2 h prior to infection (B). Then the cells were washed; total DNA was isolated; and KSHV binding was determined by real-time DNA PCR for the ORF 73 gene. (C and D) ROS are involved in KSHV entry. HMVEC-d cells were infected with KSHV (20 DNA copies/cell) for 1 h at 37°C in the presence of H2O2 (100 μM) (C) or were treated with NAC (1, 5, and 10 mM) for 2 h prior to infection (D). After washing, cells were treated with 0.25% trypsin-EDTA for 5 min at 37°C to remove bound but noninternalized virus and were then washed; total DNA was isolated; and KSHV entry was determined by real-time DNA PCR for the ORF 73 gene. Each PCR was carried out in triplicate, and the data are means for three independent experiments ± standard errors of the means. Statistical analysis was carried out using a two-tailed Student test. *, P < 0.05; **, P < 0.01.
To determine whether the inhibition of KSHV gene expression by NAC was due to blockage of virus internalization, HMVEC-d cells that either were left untreated or were treated with H2O2 (100 μM) or NAC (1, 5, or 10 mM) were infected with KSHV (20 DNA copies/cell) for 1 h at 37°C. Cells were washed with PBS to remove unbound virus and were treated with 0.25% trypsin-EDTA to remove bound but noninternalized virus. The viral DNA in the extracted total DNA was quantitated by real-time DNA PCR using ORF 73 primers (3). As shown in Fig. 3C, treatment with H2O2 significantly increased the quantity of internalized KSHV DNA copy numbers by 60%. In contrast, NAC treatment inhibited KSHV entry in a dose-dependent manner (Fig. 3D), with about 57, 66, and 77% inhibition at 1, 5, and 10 mM NAC, respectively. These results demonstrate that the decreased viral LANA-1 expression observed with NAC was due to a decrease in the entry of KSHV into the target cells.
Treatment with the antioxidant NAC blocked the translocation of integrin αVβ3 into the lipid rafts of KSHV-infected HMVEC-d cells.
One of the first events observed during KSHV infection of HMVEC-d cells is the translocation of integrins αVβ3 and α3β1 and of the xCT molecule into the lipid-raft (LR) domain of the plasma membrane, followed by virus entry via macropinocytosis and viral gene expression (24, 35). In contrast, virus bound to integrin αVβ5 does not translocate into the LRs but enters via the clathrin-mediated pathway and is associated with lysosomes (24, 35). To further determine the role of ROS in KSHV entry, we analyzed the localization of integrins αVβ3 and αVβ5 in the LRs of infected HMVEC-d cells by using specific antibodies against integrins and flotillin-1, a well-established marker of LRs (Fig. 4A and B). As observed previously, flotillin-1 staining was diffuse in uninfected cells, whereas KSHV induced the clustering of LRs early during infection (Fig. 4A and B, left) (35). In addition, we observed the colocalization of integrin αVβ3 with clustered flotillin-1 in KSHV-infected cells (Fig. 4A, center). In contrast, this colocalization was completely absent in cells pretreated with NAC, and the flotillin-1 staining pattern was not affected (Fig. 4A, bottom). As in our earlier studies (35), integrin αVβ5 did not colocalize with LRs in infected cells with or without NAC (Fig. 4B). These results suggest that ROS induced by KSHV early during infection are necessary for the translocation of integrin αVβ3 into the LRs of infected cells.
Fig 4.

ROS induced by primary infection with KSHV participate in the translocation of integrin αVβ3 into lipid rafts. HMVEC-d cells were starved for 2 h, pretreated with NAC (10 mM) for 2 h, infected with KSHV (20 DNA copies/cell) for 5 min at 37°C, and then washed. The cells were then processed for IFA using the following antibodies: anti-flotillin-1 (with an anti-goat secondary antibody conjugated to Alexa Fluor 584 [A] or Alexa Fluor 488 [B]) and anti-integrin αVβ3 (with an Alexa Fluor 488-conjugated anti-mouse secondary antibody) (A) or anti-integrin αVβ5 (with an Alexa Fluor 584-conjugated anti-mouse secondary antibody) (B). Boxed areas are enlarged. Arrowheads indicate the colocalization of integrin αVβ3 with lipid rafts. Magnification, ×40.
ROS are necessary for KSHV-induced EphA2 activation during primary infection of HMVEC-d cells.
Our studies have shown that very early during infection, αVβ3 and α3β1 integrin-bound KSHV translocates into LRs and interacts with the tyrosine kinase ephrin-A2 receptor (EphA2) (14). Studies, including ours, have demonstrated that KSHV infection induces EphA2 phosphorylation (12, 14). Pretreatment of KSHV with soluble EphA2, knockdown with EphA2 short hairpin RNA (shRNA), or pretreatment of the cells with anti-EphA2 monoclonal antibodies inhibited KSHV entry and infection (12, 14). EphA2 association with integrins α3β1 and αVβ3 in LRs early during infection was essential for macropinocytosis-associated signaling events (14). Since EphA2 plays a crucial role in KSHV infection, we next evaluated the effect of ROS on EphA2 activation during primary infection of HMVEC-d cells with KSHV (Fig. 5).
Fig 5.

ROS participate in EphA2 activation induced by KSHV infection. HMVEC-d cells were serum starved for 5 h. Uninfected serum-starved cells were treated either with 5 mM NAC for 2 h or with 100 μM H2O2 for 10 min. For KSHV infection, either serum-starved cells were pretreated with 5 mM NAC for 2 h or 100 μM H2O2 was added simultaneously with KSHV. Cells were infected with KSHV (20 DNA copies/cell) for 10 min at 37°C, washed, and processed for Western blot analysis (A) or IFA (B) using the indicated antibodies. (A) pEphA2 induction was calculated by assigning the value of 1 to levels in uninfected cells. tEphA2, total EphA2. (B) Arrowheads indicate colocalization of KSHV (gpK8.1A) with pEphA2, whereas arrows indicate the viral particle outside the cells, not colocalized with pEphA2. UN, uninfected cells. Magnification, ×40.
The addition of 100 μM H2O2 for 10 min to serum-starved (5 h) uninfected HMVEC-d cells increased the phosphorylation of EphA2 by 2.1-fold (Fig. 5A, lanes 1 and 3). In contrast, treatment of serum-starved uninfected cells for 2 h with 5 mM NAC reduced basal EphA2 phosphorylation by about 40% (Fig. 5A, lanes 1 and 2), indicating a potential regulation of EphA2 at the oxidative level. As reported previously, KSHV infection induced a 2-fold increase in EphA2 phosphorylation (Fig. 5A, compare lanes 4 and 1) (12, 14). However, when serum-starved HMVEC-d cells were pretreated with 5 mM NAC for 2 h and were then infected, KSHV-induced EphA2 phosphorylation was reduced by about 60% (1.2-fold; Fig. 5A, compare lanes 4 and 5).
To confirm the results presented above, we carried out IFA experiments (Fig. 5B). As reported previously (14), at 10 min p.i., phospho-EphA2 (pEphA2) colocalized with KSHV on the cell surfaces, as well as inside infected HMVEC-d cells (Fig. 5B, third panel, arrowheads). In contrast, in cells pretreated with NAC, we observed (i) an almost complete absence of pEphA2 immunostaining (Fig. 5B, bottom), (ii) the absence of KSHV colocalization with pEphA2, and (iii) the failure of cell surface-bound virus to enter the cells (Fig. 5B, bottom, arrows). These results further confirm that ROS are critical for the first event of KSHV/αVβ3 translocation into the lipid rafts, which leads to EphA2 phosphorylation, and clearly demonstrate that induction of ROS is essential for the activation of the EphA2 receptor early during KSHV infection of HMVEC-d cells.
Treatment with the antioxidant NAC blocked the actin-dependent membrane protrusions induced by KSHV in HMVEC-d cells.
Our studies have demonstrated that EphA2 is a KSHV receptor that coordinates the formation of an active signaling complex inducing macropinocytosis, leading to KSHV entry (14, 41). Since the results presented above demonstrate that KSHV-induced ROS are essential for the activation of EphA2 during KSHV entry, we next deciphered whether ROS modulated the formation of membrane blebs, a characteristic of KSHV macropinocytosis (14, 41). We pretreated HMVEC-d cells with NAC (10 mM) for 2 h prior to KSHV infection and costained with phalloidin, which binds to filamentous actin, and with an antibody against KSHV envelope gpK8.1A. In untreated infected cells (Fig. 6A, top), we observed bleb formations (white arrows), the colocalization of virus with blebs, and internalized viral particles (red arrows). In contrast, in NAC-treated KSHV-infected cells, we did not detect bleb formation (Fig. 6A, bottom). In addition, viral particles bound to cell surfaces that typically had few or no surface protrusions were detected, and they did not appear to enter the cells (Fig. 6viii, yellow arrows). When we calculated the percentage of cells showing bleb formation in uninfected and KSHV infected cells in the presence or absence of NAC (Fig. 6B), an 80% increase in bleb formation after KSHV infection was detected, a finding similar to those of our earlier studies (14, 41). In contrast, NAC treatment resulted in an almost complete absence of membrane bleb formation, which was statistically significantly decreased to the basal level (Fig. 6B). Our results are very similar to those of a recent study demonstrating increased levels of superoxide generation during the initial event of integrin-mediated blebbing (42). Taken together, these results demonstrate that ROS are necessary for KSHV infection-induced actin cytoskeleton rearrangement, bleb formation, and subsequent macropinocytic entry of virus into HMVEC-d cells.
Fig 6.

The antioxidant NAC blocks the actin reorganization and membrane bleb formation observed during KSHV entry. (A) HMVEC-d cells were starved for 2 h, pretreated with NAC (10 mM) for 2 h, infected with KSHV (20 DNA copies/cell) for 5 min at 37°C, washed, and processed for IFA using phalloidin (Alexa Fluor 488 conjugate) and an anti-KSHV gpK8.1A antibody (with an Alexa Fluor 594-conjugated anti-mouse secondary antibody). A representative image is shown. Boxed area is enlarged. White arrows indicate bleb formations. Red arrows indicate the colocalization of blebs and internalized viral particles. Yellow arrows indicate viral particles outside the cells. Magnification, ×40. (B) The percentage of cells bearing membrane projections was determined by counting such cells in a field containing at least 10 cells. Three independent fields were chosen for uninfected (UN) cells, KSHV-infected cells, and KSHV-infected cells pretreated with NAC. Statistical analysis was performed using a two-tailed Student test. **, P < 0.01; ***, P < 0.005.
ROS induced by KSHV during primary infection of HMVEC-d cells are involved in FAK activation.
Early during infection of HMVEC-d cells, the binding of KSHV to integrins induces FAK activation, which is one of the initial important integrin-induced signaling events participating in KSHV entry (9, 24). Interestingly, ROS have been shown to activate FAK tyrosine phosphorylation (30, 43–47). To decipher the mechanism by which ROS play a role in KSHV infection, we first evaluated the effects of NAC and H2O2 on KSHV-induced FAK activation (Fig. 7A). In findings similar to our earlier results (15, 16, 21), KSHV induced a 5-fold increase in FAK phosphorylation as early as 10 min p.i. (Fig. 7A, compare lane 4 to lane 1). However, treatment of HMVEC-d cells with NAC prior to and during KSHV infection abolished the phosphorylation of FAK (Fig. 7A, compare lanes 4 and 5). As reported previously (30, 43–47), H2O2 induced phosphorylation of FAK 2.7-fold (Fig. 7A, lane 3). In addition, H2O2 treatment during KSHV infection further increased the phosphorylation of FAK to 6.5-fold (Fig. 7A, compare lanes 4 and 6).
Fig 7.

ROS participate in KSHV-induced FAK activation. HMVEC-d cells were starved for 5 h. Either cells were treated with 5 mM NAC for 2 h prior to KSHV infection or 100 μM H2O2 was added simultaneously with KSHV. Cells were infected with KSHV (20 DNA copies/cell) for 10 min at 37°C, washed, and processed for Western blot analysis (A) or IFA (B) using the antibodies indicated. (A) pFAK induction was calculated by assigning the value of 1 to levels in uninfected cells. Tubulin was used as a loading control. (B) Boxed areas are enlarged. Arrowheads indicate colocalization of KSHV (gB) with pFAK. Arrows indicate viral particles outside the cells, not colocalized with pFAK. Magnification, ×40.
The effect of NAC on KSHV-induced FAK phosphorylation was confirmed by IFA (Fig. 7B). As observed before (9, 24), compared to the basal level of phospho-FAK (pFAK) staining observed in untreated uninfected cells and NAC-treated uninfected cells (Fig. 7B, top two panels), increased levels of pFAK staining were observed in KSHV-infected HMVEC-d cells at 10 min p.i. (Fig. 7B, third panel). Viral particles colocalized with pFAK were also observed inside the cells (Fig. 7B, third panel, arrowheads). However, when the cells were treated with NAC, the pFAK staining was reduced to basal levels (Fig. 7B, bottom panel). In addition, viral particles were detected only on the cell surfaces and did not colocalize with pFAK (Fig. 7B, bottom panel, arrows). Taken together, these results demonstrate that ROS induced by KSHV early during infection are necessary for the amplification of the KSHV-induced FAK signal pathway.
ROS induced by KSHV during primary infection of HMVEC-d cells are involved in Src activation.
Another important early integrin-induced signaling event participating in KSHV entry is Src activation (9, 24). We have also demonstrated recently that EphA2 participates in an amplification loop leading to the activation of Src (14). Our studies have shown that EphA2 interacts with Src upon KSHV infection and that knockdown of EphA2 inhibited KSHV-induced Src activation (14). As shown in Fig. 5, EphA2 activation induced by KSHV infection is reduced by NAC treatment. In addition, Src kinases could also be directly regulated by oxidative stress (48). Therefore, we set out to determine the effects of NAC and H2O2 on KSHV-induced Src activation. KSHV induced a small but consistent 1.2-fold increase in the phosphorylation of Src as early as 10 min p.i. (Fig. 8A, compare lane 4 to lane 1). However, treatment of HMVEC-d cells with NAC prior to and during KSHV infection reduced the phosphorylation of Src to basal levels (Fig. 8A, compare lanes 4 and 5). In contrast, H2O2 treatment during KSHV infection increased Src phosphorylation by 1.4-fold over basal levels (Fig. 8A, compare lanes 4 and 6).
Fig 8.

ROS participate in KSHV-induced Src activation. HMVEC-d cells were starved for 5 h. Either cells were treated with 5 mM NAC for 2 h prior to KSHV infection or 100 μM H2O2 was added simultaneously with KSHV. Cells were infected with KSHV (20 DNA copies/cell) for 10 min at 37°C, washed, and processed for Western blot analysis (A) or IFA (B) using the antibodies indicated. (A) pSrc induction was calculated by assigning the value of 1 to levels in uninfected cells. Tubulin was used as a loading control. (B) Boxed areas are enlarged. Arrowheads indicate colocalization of KSHV (gpK8.1A) with pSrc. Arrows indicate viral particles outside the cells, not colocalized with pSrc. Magnification, ×40.
The results presented above, demonstrating the effect of NAC on KSHV-induced Src activation, were further confirmed by IFA (Fig. 8B). As reported before (14), compared to the basal level of phospho-Src (pSrc) staining observed in untreated and NAC-treated uninfected cells (Fig. 8B, top two panels), we observed a significant increase in pSrc staining in KSHV-infected cells at 10 min p.i. (Fig. 8B, third panel). KSHV particles colocalized with pSrc staining inside the cells (Fig. 8B, third panel, arrowheads). However, with NAC treatment, the level of pSrc staining was greatly reduced (Fig. 8B, bottom panel), and viral particles were detected only on the cell surfaces and did not colocalize with pSrc (Fig. 8B, bottom panel, arrows). Overall, pSrc activity was more readily seen by IFA than by Western blotting, a difference that could be due to the sensitivity of the assay and the ability of antibodies to recognize pSrc in a Western blot. These results demonstrate that KSHV-induced ROS are necessary for the amplification of the Src signal pathway early during KSHV infection of HMVEC-d cells.
ROS induced by KSHV during primary infection of HMVEC-d cells are involved in Rac1 activation.
Numerous studies have linked the small GTPase protein Rac1 to ROS, integrin, or EphA2 (25, 28–31, 33, 49–51). During the engagement of integrin with the extracellular matrix, ROS production is increased in a Rac1-dependent manner (25, 28–31, 33). In addition, Rac1 activation is facilitated by ROS, creating an amplification loop (52). Interestingly, Rac1 has been found to be activated in latently infected endothelial cells and in KS tumors (7, 53). Several KSHV genes, as well as prostaglandin E2 (PGE2) secretion, are involved in Rac1 activation (7, 54, 55). Our previous study indicated that RhoA and Rac1-GTPases were activated during primary infection of fibroblasts through the engagement of integrin α3β1 (17). In addition, the general small GTPase inhibitor Clostridium difficile toxin B (CdTxB) inhibited KSHV entry (22). Finally, several studies have also shown that Rac1 is also activated downstream of EphA2 activation (49–51). Hence, we next evaluated the effect of NAC on Rac1 activation during primary infection of endothelial cells with KSHV.
Rac1 activation was first measured by a pulldown assay as described previously (17) (Fig. 9A). Cell lysates were incubated with beads containing the GST-Rac1-binding domain of PAK1 (PBD), and the resulting bound Rac1-GTP complexes were analyzed by Western blotting for Rac1. Total Rac1 and actin were used as loading controls. We observed that oxidative stress slightly increased Rac1 activation (1.2-fold) (Fig. 9A, compare lane 3 to lane 1). In addition, KSHV also activated Rac1 (1.4-fold) (Fig. 9A, compare lane 4 to lane 1). Interestingly, NAC treatment reduced both basal and KSHV-induced Rac1 activation (Fig. 9A, compare lanes 1 and 4 with lanes 2 and 5, respectively).
Fig 9.

ROS participate in Rac1 activation induced by KSHV. HMVEC-d cells were starved for 5 h. Either cells were treated with 5 mM NAC for 2 h prior to KSHV infection or 100 μM H2O2 was added simultaneously with KSHV. Cells were infected with KSHV (20 DNA copies/cell) for 10 min at 37°C, washed, and processed for a pulldown assay (A) or IFA (B) using the antibodies indicated. (A) Rac1 induction was calculated by assigning the value of 1 to levels in uninfected cells. (B) Boxed areas are enlarged. Arrowheads indicate colocalization of KSHV (gpK8.1A) with Rac1-GTP. Arrows indicate viral particles outside the cells, not colocalized with Rac1-GTP. Magnification, ×40.
To confirm the results presented above, we performed IFA using a Rac1-GTP-specific antibody (Fig. 9B). Whereas Rac1-GTP staining is low and diffuse in uninfected cells, we observed an increase in Rac1-GTP staining in infected cells (Fig. 9B, top three left panels). This staining was clustered at the peripheries of infected cells and colocalized with KSHV particles (Fig. 9B, third panel, arrowheads). In contrast, in NAC-treated infected cells, Rac1-GTP staining was substantially reduced and appeared diffuse, like that in uninfected cells (Fig. 9B, bottom panel). In addition, the viral particles in NAC-treated cells were localized only at the cell surfaces, indicating an impairment of entry into the cells (Fig. 9B, bottom panel, arrows). The effect of KSHV on Rac1 activation was more dramatic in the IFA experiment than in the Western blot analysis. This could be due to differences in the techniques and tools used. For the Western blot analysis, we performed a pulldown assay followed by Western blotting using an antibody against total Rac1. In contrast, for IFA, we stained the cells with an anti-Rac1-GTP specific antibody. Hence, the difference in the results observed could be due to a low efficiency of the pulldown assay/Western blot versus a high affinity/sensitivity of the antibody in IFA. Taken together, these results demonstrate that KSHV-induced ROS participate in virus infection by inducing the signaling necessary for entry via macropinocytosis.
DISCUSSION
The studies presented here demonstrate for the first time that during primary infection of endothelial cells, KSHV-induced ROS promote KSHV entry and the amplification of the initial host signal cascade, including EphA2, FAK, and Src. This amplification loop is probably essential for sustained EphA2, FAK, and Src phosphorylation, which in turn contributes to the downstream signal cascades that are essential for the translocation of the virus to LRs, actin cytoskeleton rearrangement, bleb formation, macropinocytic entry of KSHV, and the establishment of infection (Fig. 10). EphA2 has been shown to be associated with integrin-associated signaling during KSHV infection (14), and the involvement of ROS in modulating integrin translocation into LRs, EphA2 activation, FAK and Src signaling, and KSHV entry reveals new insights into the amplification loop necessary for virus entry.
Fig 10.
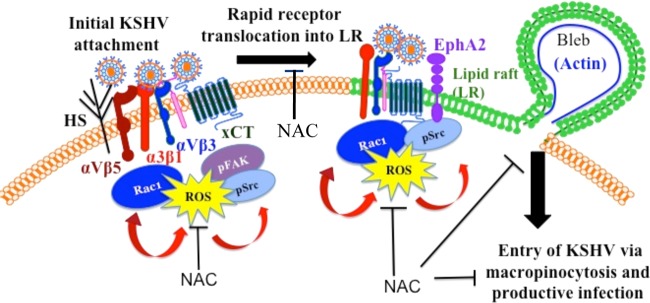
Schematic depicting the induction of ROS by KSHV early during primary infection of endothelial cells to promote virus entry. KSHV initially binds and interacts with heparan sulfate, integrins (α3β1, αVβ3, αVβ5), and x-CT in non-LR regions of HMVEC-d cells and is then rapidly translocated, along with selective integrins (α3β1, αVβ3) and x-CT receptors, into lipid rafts. In LRs, the receptor EphA2 interacts with KSHV and integrins and is phosphorylated. KSHV entry is initiated by the induction of signaling pathways such as FAK, Src, and Rac1 and the concurrent formation of macropinocytic blebs. The studies presented here demonstrate increased ROS production very early during KSHV primary infection. Pretreatment with the antioxidant NAC significantly inhibited KSHV entry, and consequently gene expression, without affecting virus binding. The translocation of KSHV-integrins into LRs, actin-dependent membrane bleb formation, and the activation of the signal molecules ephrin-A2, FAK, Src, and Rac1 were inhibited by NAC. In contrast, H2O2 treatment increased KSHV entry and the phosphorylation of ephrin-A2, FAK, and Src. These studies demonstrate that KSHV infection induces ROS very early during infection to amplify the signaling pathways that are necessary for the efficient entry of viral particles into HMVEC-d cells via macropinocytosis.
Accumulating evidence has suggested an important role for ROS and oxidative stress during viral infection (56). It is known that virus binding and entry activate several secondary messengers and signaling pathways. We observed a 1.5-fold increase in ROS production at early time points following KSHV infection and a 2.5-fold increase at 24 h p.i. The kinetics and fold increases were comparable to those observed in other viral infections. It has been shown that during adenovirus 5 (Ad5) infection, ROS production increases rapidly following lysosomal rupture (57). Within 15 min of infection, Ad5 induced ROS production that was maintained at a ca. 2-fold increase for more than 2 h p.i (58). In addition, a recent study demonstrated that enterovirus 71 (EV71) increased ROS production as early as 30 min p.i. (59). This study also indicated that EV71 increased ROS production following its binding to integrin β1, Rac1, and NADPH oxidase (59). In results similar to ours, ROS were also found to be induced by several herpesviruses. Herpes simplex virus 1 (HSV-1) entry and replication induce ROS production, lasting for a prolonged period, as early as 1 h p.i. (60), and glycoprotein gJ has been shown to induce ROS production (61). HSV-1 infection of microglia induced a rapid 1.5-fold increase in ROS production as early as 3 h p.i., and ROS production was more robust at 24 h p.i., with a 2-fold increase (62, 63). ROS production after HSV-1 infection was dependent on NADPH oxidase activity. Herpes simplex virus 2 (HSV-2) induced a 1.5- to 2-fold increase in ROS production 1 h after the infection of RAW 264.7 cells (64). Similarly, the gamma-1 herpesvirus Epstein-Barr virus (EBV) induced oxidative stress during the early stages of primary infections in B lymphocytes and epithelial and lymphoblastic cell lines (65). In this study, a 2-fold increase in malondialdehyde activity, indicative of lipid peroxidation, was measured.
We observed that ROS production was sustained after the establishment of latent KSHV infection in HMVEC-d cells. This result is in accordance with studies showing the production of high levels of ROS in latent EBV-positive Burkitt's lymphomas (66). Several mechanisms are proposed to be involved in ROS production in EBV-infected cells, depending on the type of latency. ROS production is dependent on paracrine secretion of interleukin 10 (IL-10) in latency I, whereas expression of EBV nuclear antigen 2 (EBNA-2) and latent membrane protein 1 (LMP1) is proposed in latency III (66). EBNA-1 could also be involved in ROS production (67, 68). Very recently, Cao et al. have shown that long-term expression of EBNA-1 in nasopharyngeal carcinoma resulted in an increase in ROS production (69). Taken together, these studies indicated that ROS production induced by EBV could play a role in genome instability as well as in metastasis. ROS can also function in KSHV-induced oncogenesis, a notion supported by several studies. Guilluy et al. have shown that ROS production leads to endothelial junction dysregulation and increased vascular permeability (7). Moreover, the antioxidant NAC has been shown to efficiently reduce KSHV-induced oncogenesis in animal models (5, 6, 8).
In addition to a role in tumorigenesis, ROS can play an important role in facilitating viral infection. Studies suggest that RNA and DNA viruses use oxidative stress to regulate their life cycles. Antioxidants have been shown to block the replication of RNA viruses, including influenza virus, EV71, and HIV-1 (59, 70–73). In the herpesvirus family, the oxidative stress induced by HSV-1 is required for efficient viral replication (60). Indeed, the antioxidant compound Ebselen inhibited the replication of HSV-1. In contrast, H2O2 is instrumental in the maintenance of EBV latency, since it inhibits the expression of EBV immediate-early lytic genes (74). Interestingly, it has been shown previously that oxidative stress can lead to KSHV reactivation in PEL cells and endothelial cells (4–6).
In contrast to the earlier studies demonstrating the role of KSHV latency and lytic reactivation, our studies demonstrate for the first time that ROS induction promotes the early stage of KSHV infection and entry into HMVEC-d cells. KSHV has probably evolved to bind to integrin molecules to modulate several downstream signaling events that facilitate entry and infection (10, 13, 24, 35). Interestingly, several reports indicate an increase in ROS production following integrin engagement (33, 75). KSHV gB also interacts with integrins α3β1, αVβ3, and αVβ5 (10, 13), and this interaction is followed by several downstream signaling events, such as the phosphorylation of FAK, Src, PI-3K, and Rho GTPases (RhoA, Cdc42, and Rac1) (21). Since these signal pathways play vital roles in host cell endocytosis and the movement of particulate materials in the cytoplasm, the early stages of KSHV interaction with host cells may provide an environment very conducive to the successful infection of target cells. Interestingly, we observed that the ROS scavenger NAC efficiently reduced FAK activation. Further investigations are needed to determine the identity of the KSHV envelope glycoprotein(s) involved in the ROS induction observed during the early time points of primary infection.
Previous studies have demonstrated an important role for integrin molecules α3β1, αVβ3, and αVβ5 in KSHV entry (10, 13, 35). However, our attempts to block viral infection by preincubating the cells with anti-integrin antibodies before viral infection did not block ROS production (data not shown). This could be due to cross-linking of integrins by these antibodies and, consequently, induction of downstream signaling, including the production of ROS, which has been reported previously (29). In addition, since KSHV interacts with a multitude of integrin subunits, simultaneous knockdown of all the integrin subunits (α3, αV, β1, and β3) by RNA interference is challenging.
An intriguing observation from our studies is that KSHV-induced ROS activate the FAK, Src, and EphA2 signal pathways. However, further detailed studies are essential to decipher the mechanism by which ROS activate these signal pathways. From the available literature, we speculate the following. ROS are essential cellular messengers, since they modulate several kinase and phosphatase activities through their transient and reversible oxidation. Especially, protein tyrosine phosphatases (PTP) contain cysteine residues in their active sites that could be targeted by oxidative stress, leading to their inactivation. One of these phosphatases is the low-molecular-weight protein tyrosine phosphatase (LMW-PTP). Indeed, H2O2 has been shown to inactivate LMW-PTP through the oxidation of cysteine residues (76). Interestingly, LMW-PTP has been shown to dephosphorylate FAK (30, 77). Another substrate of LMW-PTP is EphA2 (78, 79). Therefore, LMW-PTP could be an attractive link between integrin, ROS, FAK, and EphA2 signaling. We speculate that the increased ROS production after integrin engagement in KSHV-infected cells could shift LMW-PTP to an oxidized/inactive status and consequently allow the amplification of FAK and EphA2 phosphorylation. On the other hand, the inhibition of ROS production during NAC treatment could increase the quantity of reduced/active LMW-PTP and consequently decrease FAK and EphA2 phosphorylation early during KSHV infection. Src kinase has also been shown to be directly and indirectly regulated by ROS (80). Directly, oxidation of cysteine residues in the Src homology 2 (SH2) domain and/or kinase domain causes hyperphosphorylation and activation of Src. Indirectly, it is proposed that oxidation of the Csk kinase keeps Src active longer (by blocking the phosphorylation of Src Tyr527). In addition, Src is activated through the inhibition of phosphatases that dephosphorylate Src Try418 (80). Additional studies examining the possibilities discussed above could shed light on the mechanism by which KSHV-induced ROS activate signal pathways.
In summary, our studies show that induction of ROS very early during infection by KSHV is essential for viral entry into HMVEC-d cells. Since the antioxidant NAC blocked viral entry by blocking the recruitment of integrin to the LRs, the phosphorylation of EphA2, the actin remodeling observed during macropinocytosis, and the activation of integrin-associated signaling molecules such as FAK, Src, and Rac1, antioxidants that have already been shown to be attractive drugs against KSHV oncogenesis are also attractive therapeutic drugs for controlling primary infection of endothelial cells with KSHV.
ACKNOWLEDGMENTS
This study was supported in part by Public Health Service grants AI097540 (to V.B.) and CA075911 and CA168472 (to B.C.) and by the RFUMS–H.M. Bligh Cancer Research Fund (to B.C.).
We thank Keith Philibert for critical reading of the manuscript.
Footnotes
Published ahead of print 21 November 2012
REFERENCES
- 1. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 2. Ganem D. 2007. Kaposi‘s sarcoma-associated herpesvirus, p 2847–2888 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li X, Feng J, Sun R. 2011. Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 85:715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye F, Gao SJ. 2011. A novel role of hydrogen peroxide in Kaposi sarcoma-associated herpesvirus reactivation. Cell Cycle 10:3237–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, Gao SJ. 2011. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 7:e1002054 doi:10.1371/journal.ppat.1002054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guilluy C, Zhang Z, Bhende PM, Sharek L, Wang L, Burridge K, Damania B. 2011. Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood 118:5344–5354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma Q, Cavallin LE, Leung HJ, Chiozzini C, Goldschmidt-Clermont PJ, Mesri EA. 20 August 2012. A role for virally induced reactive oxygen species in Kaposi's sarcoma herpesvirus tumorigenesis. Antioxid. Redox Signal. [Epub ahead of print.] doi:10.1089/ars.2012.4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J. Virol. 84:2188–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419 [DOI] [PubMed] [Google Scholar]
- 11. Kaleeba JA, Berger EA. 2006. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311:1921–1924 [DOI] [PubMed] [Google Scholar]
- 12. Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F. 2012. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat. Med. 18:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Veettil MV, Sadagopan S, Sharma-Walia N, Wang FZ, Raghu H, Varga L, Chandran B. 2008. Kaposi's sarcoma-associated herpesvirus forms a multimolecular complex of integrins (αVβ5, αVβ3, and α3β1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J. Virol. 82:12126–12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chakraborty S, Veettil MV, Bottero V, Chandran B. 2012. Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. U. S. A. 109:E1163–E1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krishnan HH, Sharma-Walia N, Streblow DN, Naranatt PP, Chandran B. 2006. Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J. Virol. 80:1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naranatt PP, Krishnan HH, Smith MS, Chandran B. 2005. Kaposi's sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 79:1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Caballero A, Sivakumar R, Varga L, Bottero V, Chandran B. 2007. Lipid rafts of primary endothelial cells are essential for Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 81:7941–7959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veettil MV, Sharma-Walia N, Sadagopan S, Raghu H, Sivakumar R, Naranatt PP, Chandran B. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 80:11432–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 79:15027–15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakraborty S, Veettil MV, Chandran B. 20 January 2012. Kaposi's sarcoma associated herpesvirus entry into target cells. Front. Microbiol. 3:6 doi:10.3389/fmicb.2012.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gregg D, de Carvalho DD, Kovacic H. 2004. Integrins and coagulation: a role for ROS/redox signaling? Antioxid. Redox Signal. 6:757–764 [DOI] [PubMed] [Google Scholar]
- 26. Hu CT, Wu JR, Cheng CC, Wang S, Wang HT, Lee MC, Wang LJ, Pan SM, Chang TY, Wu WS. 2011. Reactive oxygen species-mediated PKC and integrin signaling promotes tumor progression of human hepatoma HepG2. Clin. Exp. Metastasis 28:851–863 [DOI] [PubMed] [Google Scholar]
- 27. Svineng G, Ravuri C, Rikardsen O, Huseby NE, Winberg JO. 2008. The role of reactive oxygen species in integrin and matrix metalloproteinase expression and function. Connect. Tissue Res. 49:197–202 [DOI] [PubMed] [Google Scholar]
- 28. Taddei ML, Parri M, Mello T, Catalano A, Levine AD, Raugei G, Ramponi G, Chiarugi P. 2007. Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid. Redox Signal. 9:469–481 [DOI] [PubMed] [Google Scholar]
- 29. Werner E, Werb Z. 2002. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J. Cell Biol. 158:357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G. 2003. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 161:933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. 1998. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science 280:898–902 [DOI] [PubMed] [Google Scholar]
- 32. Finkel T. 2001. Reactive oxygen species and signal transduction. IUBMB Life 52:3–6 [DOI] [PubMed] [Google Scholar]
- 33. Goitre L, Pergolizzi B, Ferro E, Trabalzini L, Retta SF. 2012. Molecular crosstalk between integrins and cadherins: do reactive oxygen species set the talk? J. Signal Transduct. 2012:807682 doi:10.1155/2012/807682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 77:7978–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chakraborty S, Valiyaveettil M, Sadagopan S, Paudel N, Chandran B. 2011. c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi's sarcoma-associated herpesvirus infection in endothelial cells. J. Virol. 85:12410–12430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang FZ, Akula SM, Sharma-Walia N, Zeng L, Chandran B. 2003. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 77:3131–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu L, Puri V, Chandran B. 1999. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 262:237–249 [DOI] [PubMed] [Google Scholar]
- 38. Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84 [DOI] [PubMed] [Google Scholar]
- 39. Akula SM, Pramod NP, Wang FZ, Chandran B. 2001. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284:235–249 [DOI] [PubMed] [Google Scholar]
- 40. Akula SM, Wang FZ, Vieira J, Chandran B. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282:245–255 [DOI] [PubMed] [Google Scholar]
- 41. Valiya Veettil M, Sadagopan S, Kerur N, Chakraborty S, Chandran B. 2010. Interaction of c-Cbl with myosin IIA regulates bleb associated macropinocytosis of Kaposi's sarcoma-associated herpesvirus. PLoS Pathog. 6:e1001238 doi:10.1371/journal.ppat.1001238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yi YH, Chang YS, Lin CH, Lew TS, Tang CY, Tseng WL, Tseng CP, Lo SJ. 2012. Integrin-mediated membrane blebbing is dependent on sodium-proton exchanger 1 and sodium-calcium exchanger 1 activity. J. Biol. Chem. 287:10316–10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ben Mahdi MH, Andrieu V, Pasquier C. 2000. Focal adhesion kinase regulation by oxidative stress in different cell types. IUBMB Life 50:291–299 [DOI] [PubMed] [Google Scholar]
- 44. Gozin A, Franzini E, Andrieu V, Da Costa L, Rollet-Labelle E, Pasquier C. 1998. Reactive oxygen species activate focal adhesion kinase, paxillin and p130cas tyrosine phosphorylation in endothelial cells. Free Radic. Biol. Med. 25:1021–1032 [DOI] [PubMed] [Google Scholar]
- 45. Sonoda Y, Kasahara T, Yokota-Aizu E, Ueno M, Watanabe S. 1997. A suppressive role of p125FAK protein tyrosine kinase in hydrogen peroxide-induced apoptosis of T98G cells. Biochem. Biophys. Res. Commun. 241:769–774 [DOI] [PubMed] [Google Scholar]
- 46. Sonoda Y, Watanabe S, Matsumoto Y, Aizu-Yokota E, Kasahara T. 1999. FAK is the upstream signal protein of the phosphatidylinositol 3-kinase-Akt survival pathway in hydrogen peroxide-induced apoptosis of a human glioblastoma cell line. J. Biol. Chem. 274:10566–10570 [DOI] [PubMed] [Google Scholar]
- 47. Vepa S, Scribner WM, Parinandi NL, English D, Garcia JG, Natarajan V. 1999. Hydrogen peroxide stimulates tyrosine phosphorylation of focal adhesion kinase in vascular endothelial cells. Am. J. Physiol. 277:L150–L158 [DOI] [PubMed] [Google Scholar]
- 48. Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. 1997. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J. Clin. Invest. 100:1813–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. 2004. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J. Cell Sci. 117:2037–2049 [DOI] [PubMed] [Google Scholar]
- 50. Lin KT, Gong J, Li CF, Jang TH, Chen WL, Chen HJ, Wang LH. 2012. Vav3-rac1 signaling regulates prostate cancer metastasis with elevated Vav3 expression correlating with prostate cancer progression and posttreatment recurrence. Cancer Res. 72:3000–3009 [DOI] [PubMed] [Google Scholar]
- 51. Tanaka M, Ohashi R, Nakamura R, Shinmura K, Kamo T, Sakai R, Sugimura H. 2004. Tiam1 mediates neurite outgrowth induced by ephrin-B1 and EphA2. EMBO J. 23:1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heo J. 2011. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid. Redox Signal. 14:689–724 [DOI] [PubMed] [Google Scholar]
- 53. Ma Q, Cavallin LE, Yan B, Zhu S, Duran EM, Wang H, Hale LP, Dong C, Cesarman E, Mesri EA, Goldschmidt-Clermont PJ. 2009. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi's sarcoma. Proc. Natl. Acad. Sci. U. S. A. 106:8683–8688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Montaner S, Sodhi A, Servitja JM, Ramsdell AK, Barac A, Sawai ET, Gutkind JS. 2004. The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood 104:2903–2911 [DOI] [PubMed] [Google Scholar]
- 55. Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 6:e1000777 doi:10.1371/journal.ppat.1000777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwarz KB. 1996. Oxidative stress during viral infection: a review. Free Radic. Biol. Med. 21:641–649 [DOI] [PubMed] [Google Scholar]
- 57. McGuire KA, Barlan AU, Griffin TM, Wiethoff CM. 2011. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J. Virol. 85:10806–10813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barlan AU, Griffin TM, McGuire KA, Wiethoff CM. 2011. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 85:146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tung WH, Hsieh HL, Lee IT, Yang CM. 2011. Enterovirus 71 induces integrin β1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J. Cell. Physiol. 226:3316–3329 [DOI] [PubMed] [Google Scholar]
- 60. Kavouras JH, Prandovszky E, Valyi-Nagy K, Kovacs SK, Tiwari V, Kovacs M, Shukla D, Valyi-Nagy T. 2007. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 13:416–425 [DOI] [PubMed] [Google Scholar]
- 61. Aubert M, Chen Z, Lang R, Dang CH, Fowler C, Sloan DD, Jerome KR. 2008. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J. Virol. 82:617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hu S, Sheng WS, Schachtele SJ, Lokensgard JR. 2011. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflammation 8:123 doi:10.1186/1742-2094-8-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schachtele SJ, Hu S, Little MR, Lokensgard JR. 2010. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 7:35 doi:10.1186/1742-2094-7-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, Hartmann R, Paludan SR. 2011. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 7:e1002250 doi:10.1371/journal.ppat.1002250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lassoued S, Ben Ameur R, Ayadi W, Gargouri B, Ben Mansour R, Attia H. 2008. Epstein-Barr virus induces an oxidative stress during the early stages of infection in B lymphocytes, epithelial, and lymphoblastoid cell lines. Mol. Cell. Biochem. 313:179–186 [DOI] [PubMed] [Google Scholar]
- 66. Cerimele F, Battle T, Lynch R, Frank DA, Murad E, Cohen C, Macaron N, Sixbey J, Smith K, Watnick RS, Eliopoulos A, Shehata B, Arbiser JL. 2005. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr virus (EBV)-positive versus EBV-negative Burkitt's lymphoma. Proc. Natl. Acad. Sci. U. S. A. 102:175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. 2009. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 106:2313–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gruhne B, Sompallae R, Masucci MG. 2009. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 28:3997–4008 [DOI] [PubMed] [Google Scholar]
- 69. Cao JY, Mansouri S, Frappier L. 2012. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein-Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses. J. Virol. 86:382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cai J, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP. 2003. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 34:928–936 [DOI] [PubMed] [Google Scholar]
- 71. Mihm S, Ennen J, Pessara U, Kurth R, Droge W. 1991. Inhibition of HIV-1 replication and NF-κB activity by cysteine and cysteine derivatives. AIDS 5:497–503 [DOI] [PubMed] [Google Scholar]
- 72. Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H. 1989. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 244:974–976 [DOI] [PubMed] [Google Scholar]
- 73. Staal FJ, Roederer M, Herzenberg LA. 1990. Intracellular thiols regulate activation of nuclear factor κB and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 87:9943–9947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Osipova-Goldberg HI, Turchanowa LV, Adler B, Pfeilschifter JM. 2009. H2O2 inhibits BCR-dependent immediate early induction of EBV genes in Burkitt's lymphoma cells. Free Radic. Biol. Med. 47:1120–1129 [DOI] [PubMed] [Google Scholar]
- 75. Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, Raugei G, Ramponi G, Chiarugi P. 2008. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 15:867–878 [DOI] [PubMed] [Google Scholar]
- 76. Chiarugi P. 2001. The redox regulation of LMW-PTP during cell proliferation or growth inhibition. IUBMB Life 52:55–59 [DOI] [PubMed] [Google Scholar]
- 77. Rigacci S, Rovida E, Dello Sbarba P, Berti A. 2002. Low Mr phosphotyrosine protein phosphatase associates and dephosphorylates p125 focal adhesion kinase, interfering with cell motility and spreading. J. Biol. Chem. 277:41631–41636 [DOI] [PubMed] [Google Scholar]
- 78. Kikawa KD, Vidale DR, Van Etten RL, Kinch MS. 2002. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J. Biol. Chem. 277:39274–39279 [DOI] [PubMed] [Google Scholar]
- 79. Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, Chiarugi P. 2005. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J. Biol. Chem. 280:34008–34018 [DOI] [PubMed] [Google Scholar]
- 80. Knock GA, Ward JP. 2011. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal. 15:1531–1547 [DOI] [PubMed] [Google Scholar]