

Abstract
Noncanonical microRNAs (miRNAs) and endogenous small interfering RNAs (endo-siRNAs) are key gene regulators in eukaryotes. Noncanonical miRNAs, which bypass part of the canonical miRNA biogenesis pathway, can originate from a variety of genomic loci, which include small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs) and introns, whereas endo-siRNAs can arise from repetitive elements, some of which are transposable. The roles of noncanonical miRNAs and endo-siRNAs in complex diseases have yet to be characterized. To investigate their potential expression and function in psoriasis, we carried out a comprehensive, genome-wide search for noncanonical miRNAs and endo-siRNAs in small RNA deep-sequencing data sets from normal and psoriatic human skin. By analyzing more than 670 million qualified reads from 67 small RNA libraries, we identified 21 novel, noncanonical miRNAs (3 snoRNA-derived and 2 tRNA-derived miRNAs and 16 miRtrons) and 39 novel endo-siRNAs that were expressed in skin. The expression of four novel small RNAs was validated by qRT–PCR in human skin, and their Argonaute association was confirmed by co-immunoprecipitation of ectopic small RNAs in HEK293 cells. Fifteen noncanonical miRNAs or endo-siRNAs were significantly differentially expressed in psoriatic-involved versus normal skin, including an Alu-short interspersed element-derived siRNA which was 17-fold up-regulated in psoriatic-involved skin. These and other differentially expressed small noncoding RNAs may function as regulators of gene expression in skin and potentially play a role in psoriasis pathogenesis.
INTRODUCTION
MicroRNAs (miRNAs) and endogenous small interfering RNAs (endo-siRNAs) are classes of small noncoding regulatory RNAs (sncRNAs), which play critical roles in gene regulation for many biological processes in eukaryotic organisms including mRNA cleavage, RNA degradation, translation inhibition and DNA methylation (1). miRNAs in particular have been heavily studied in recent years and shown to be essential gene regulators that broadly contribute to disease pathogenesis.
miRNAs are produced from short-hairpin-forming primary transcripts (pri-miRNAs) which are typically processed through a canonical pathway involving two RNase III enzymes, Drosha and Dicer (double-stranded RNA-specific endoribonuclease), and a double-stranded RNA (dsRNA)-binding protein Dgcr8 (DiGeorge syndrome critical region gene 8) (2). Besides canonical miRNAs, there exist noncanonical miRNAs whose biogenesis requires Dicer but neither Drosha nor Dgcr8 (3). The first example of noncanonical miRNA is the class of miRtrons, which arise from short [60–100 nucleotide (nt)] debranched intron lariats that form stem–loop structures that serve as Dicer substrates (4–7). Noncanonical miRNAs can also arise from local hairpin formation within larger noncoding RNA (ncRNA) species, such as small nucleolar RNAs (snoRNAs) and transfer RNAs (tRNAs) (8,9). Although there have been several snoRNA-derived miRNAs described in eukaryotes, only one example of a tRNA-derived miRNA has been described. In this case, a murine tRNA, IleTAT, folds into an alternative secondary structure consisting of a stem–loop hairpin from which mmu-miR-1983 is derived (10). In contrast to miRNAs, endo-siRNAs arise from long dsRNA transcripts derived from repetitive or transposable elements (11–13), which require Dicer, but not Drosha/Dgcr8 for processing (3,10). Endo-siRNAs have been described in plants (1,14), Caenorhabditis elegans (15), Drosophila melanogaster (16–19) and murine embryonic stem cells (mESCs) and oocytes (10,12,13). There is also evidence suggesting that endo-siRNAs are expressed in murine embryonic skin (20).
Both miRNAs and siRNAs exert their regulatory functions through association with the RNA-induced silencing complex (RISC), which contains an Argonaute (AGO) protein (5,10,21). However, miRNAs and siRNAs have distinct requirements for mRNA target recognition. The base pairing between miRNAs and their targets is degenerate, whereas the pairing between siRNAs and their targets requires high fidelity, typically allowing no more than three mismatches (12). Consequently, most miRNAs lower the expression levels of target genes by mRNA destabilization or translational repression (22), whereas endo-siRNAs elicit direct cleavage of target mRNAs (12).
miRNAs are differentially expressed in myriad human diseases, and in some cases, play a direct role in disease etiologies, but little is known about the expression or function of noncanonical miRNAs and endo-siRNAs in human disease. In the case of the inflammatory skin disorder psoriasis, we have analyzed the expression of the majority of canonical miRNAs with small RNA library construction followed by deep sequencing (23). In the current study, we re-analyzed these deep-sequencing data sets, focusing on the discovery and characterization of noncanonical miRNAs and endo-siRNAs, and identified 60 novel sncRNAs (Table 1). Digital gene expression analyses revealed differential expression of 15 sncRNAs in psoriatic-involved skin versus normal skin. This study broadens our perspective on the diversity of sncRNAs in human skin and reveals a subset of noncanonical miRNAs and endo-siRNAs that are differentially expressed in psoriasis.
Table 1.
Summary of noncanonical miRNAs and endo-siRNAs in the current study
| Total | Conserved | Nonconserved | |
|---|---|---|---|
| Noncanonical miRNA | |||
| snoRNA-derived | 11 | 8 | 3 |
| tRNA-derived | 2 | 2 | – |
| miRtron | |||
| Typical | 19 | 3 | 16 |
| Tailed | 98 | 18 | 80 |
| Endo-siRNA | 39 | – | 39 |
RESULTS
NextGen sequencing of small RNAs
Sequencing and pre-processing of 67 small RNA libraries from normal (NN), psoriatic-uninvolved (PN) and psoriatic-involved (PP) human skin, alignment of reads to the human genome and profiling of canonical miRNAs were described in detail previously (23). Here, we systematically analyzed reads that aligned to introns, various types of ncRNAs, repetitive elements and transposable elements to identify read clusters that were consistent with the expression of Dicer-dependent sncRNAs (Supplementary Material, Fig. S1).
Noncanonical miRNAs derived from snoRNAs and tRNAs
Overall, the small RNA reads derived from ncRNAs account for 26.4, 30.2 and 26.8% of the qualified reads in PP, PN and NN skin, respectively (Supplementary Material, Table S1). Although many of these reads reflect random degradation products from abundant ncRNAs, several read clusters exhibited characteristic features of Dicer-dependent sncRNA expression. In total, 58 million (8.8%) qualified reads aligned to snoRNAs, including 8 out of 8 of the previously described miRNA-producing snoRNAs in humans (9; Supplementary Material, Table S2) and three novel miRNA-producing snoRNAs: U60, ACA25 and ACA44 (Supplementary Material, Table S2, Fig. 1). Folding structure analysis revealed that the snoRNAs could form miRNA-like hairpin structures (Fig. 1, Supplementary Material, Fig. S2A). Alignment of the most abundant small RNA reads to individual arms of the hairpins resulted in 22-nt RNA–RNA duplexes with 3′ overhangs (Fig. 1, Supplementary Material, Fig. S2B). Unexpectedly, ∼79% of the reads aligned to snoRNAs aligned to U60, a C/D box snoRNA that has not been previously implicated as an miRNA locus. Of these reads, 98.9% were 21 or 22 nt long. Remarkably, the 22 nt miRNA-like RNA was ranked as the second most highly expressed miRNA in human skin based on read counts. However, qRT–PCR indicated a more moderate expression level in the skin (see what follows), perhaps indicating that the very high abundance of U60 reads in small RNA libraries was due to a library preparation bias (24,25).
Figure 1.

Example of snoRNA-derived miRNA. (A) The hairpin structures of ACA25, with the miRNA and miRNA* sequences shown in red. (B) Alignment of reads mapped to ACA25 with the number of sequencing reads, length of reads, the number of genomic loci which the read can be mapped to with no mismatches and the percentage of the read counts.
We next examined small RNA reads that aligned to tRNAs. First, we recognized a cluster of 228 sequencing reads (21 or 22 nt) aligning to human tRNA-IIeTAT, which is consistent with the expression of an mmu-miR-1983 homolog in human skin (Supplementary Material, Fig. S3A and B). A second tRNA candidate with short-hairpin-forming potential, PseudoTTA, was associated with a cluster of 14 small RNA reads (Supplementary Material, Fig. S4C and D). We initially categorized this as a putative miRNA candidate since it was associated with so few sequencing reads and we did not detect miRNA* species, but we were later able to confirm its expression by qRT–PCR (see what follows). Detailed alignments of small RNAs derived from snoRNAs and tRNAs are provided as Supplementary Material, File S1.
miRNAs derived from introns: miRtrons
The miRNA subclass of miRtrons includes typical miRtrons which are 60–100 nt introns that form miRNA precursors, and tailed miRtrons, whose precursors are generated from one end of a longer intron (5,26). Fourteen typical miRtrons have been previously described in humans (4) and were also detected in the current study (Supplementary Material, Table S3). Further, we identified an additional 26 known human miRNAs that are likely to be processed by the Drosha/Dgcr8-independent miRtron biogenesis pathway (2 typical miRtrons and 24 tailed miRtrons; Supplementary Material, Table S3). Although the currently annotated precursors for the majority of these miRNAs extend beyond their respective host intron regions, we found that, on average, 80% of the reads aligning to the intron/exon boundary terminated within 2 nt of the AG splice acceptor site, which provides strong evidence that these pre-miRNAs were derived directly from debranched introns (Supplementary Material, Fig. S4).
A search for novel miRtrons led to the discovery of 16 miRtron candidates, which were not reported in our previous study (23) due to their low abundance and/or atypical folding structures (Supplementary Material, Table S4). The lengths of the host introns varied from 70 to several thousand bases, suggesting that long introns can also give rise to miRtrons. In total, we detected 117 miRtron candidates in human skin, including 75 previously described miRtrons (4,23), 26 known miRNAs which our new data suggest are processed by the miRtron biogenesis pathway and 16 novel miRtrons; these can be further classified into 19 typical miRtrons and 98 tailed miRtrons.
In the set of known human miRtrons (4,23), the most highly expressed in skin was miR-877, with 4827 reads (Supplementary Material, Table S3). Four additional miRtrons were expressed at levels comparable with miR-877: miRtrons #80 and #103 (Supplementary Material, Fig. S2C and D), which we previously discovered (23), and miR-1292 and miR-3605, known miRNAs that we re-classified as miRtrons. Detailed read alignments for novel and re-classified miRtrons are provided as Supplementary Material, File S2.
Endo-siRNAs from repetitive and transposable elements
The expression of endo-siRNAs in adult human tissues is not well understood. Our data set of deeply sequenced small RNAs accommodates a genome-wide search for putative endo-siRNAs in human skin. A total of 27 million (4%) qualified small RNA reads from human skin aligned to repetitive and transposable elements, such as short interspersed elements (SINEs), long interspersed elements (LINEs) and long terminal repeat (LTRs) (Supplementary Material, Table S1).
The locus with the most aligned reads was on chromosome 1 within an intron of the gon-4-like (GON4L) gene, where three Alu-SINEs appear in tandem (Fig. 2A). A total of 5534 reads (53.4% of which were 21–23 nt) mapped to this locus; 2800 of these reads (83.7% of which were 21–23 nt) uniquely clustered within the first Alu-SINE element (E1; Fig. 2A), suggesting that this repetitive element may harbor a novel endo-siRNA. Notably, this Alu-SINE/GON4L endo-siRNA was the most strongly up-regulated siRNA in psoriatic-involved skin compared with normal skin (see what follows). In contrast, the expression level of the host gene GON4L showed little variation between psoriatic-involved and normal skin (data not shown), indicating that the endo-siRNA and the host gene may be transcribed or processed independently. To identify the minimal locus encoding the siRNA precursor, we ectopically expressed various combinations of the three Alu-SINE elements in HEK293 cells (E1–3, Fig. 2A). We found that transcription of the E1 segment is sufficient for siRNA production (Fig. 2B), whereas the segment of E1 and E2 together has a slightly enhanced siRNA production (Fig. 2B). Since E1 does not form an intrinsic hairpin structure, the mechanism of siRNA biogenesis from this locus remains unclear. Alternatively, transcription of E1 and E2 together may produce a long-hairpin-structured dsRNA for siRNA production (Supplementary Material, Fig. S5A).
Figure 2.

(A) Endo-siRNA derived from tandem Alu SINE transposable elements in an intron of the gon-4-like (GON4L) gene on chromosome 1. Alignment of reads to the Alu SINEs (hg19), with the red box indicating the reads uniquely mapped to this locus. (B) Abundance of siRNAs in six constructs that include six combinations of the three Alu SINEs. The y-axis indicates the normalized expression relative to the wild-type.
We also uncovered two piRNA loci for which we observed a bimodal distribution of read sizes at 21–23 and 27–30 nt (Supplementary Material, Fig. S6A). This distribution would be indicative of siRNA and piRNA expression, respectively. Notably, these loci are also annotated as Alu-SINE elements and, like the Alu-SINE/GON4L endo-siRNA locus, no hairpin structures could be detected by folding the corresponding flanking sequences. Interestingly, the siRNAs from these two loci share sequence similarity with miR-1285 and miR-1303 (Supplementary Material, Fig. S6B), which also overlap Alu-SINE elements. A search for paralogs of miR-1285 and miR-1303 mature miRNAs revealed more than 80 loci with exact sequence identity in the human genome, indicating that miR-1285 and miR-1303 may be more accurately classified as repeat-derived siRNAs.
We also identified putative endo-siRNAs derived from repetitive elements with long-hairpin-forming potential. In one case, 12 reads (83.3% were 21–23 nt in length) uniquely aligned to inverted repeats of tandem L1 elements on chromosome 16 (Supplementary Material, Fig. S5B). In another case, a cluster of 72 reads (79.2% were 21–23 nt in length) aligned to an LTR element on chromosome 4 (75% were uniquely mapped) (Supplementary Material, Fig. S5C). Transcripts from these loci were predicted to form long-hairpin structures of 98 and 135 nt, respectively, which could serve as Dicer substrates. Despite the low number of reads within these clusters, the reads exhibited clear 5′ homogeneity, suggesting that the small RNAs were authentic endo-siRNAs rather than random degradation products.
A striking approximately 11 000 reads uniquely aligned to Made1 elements. These are a human-specific subclass of miniature-inverted repeat transposable elements, which encode the large miR-548 family (27). We confirmed that 34 Made1 elements (Supplementary Material, Table S5), which were not previously associated with miRNA production, were predicted to form hairpin structures (Supplementary Material, Fig. S7). Alignment of small RNA reads to these hairpins revealed Dicer-cleavage signatures: 21–22 nt RNA duplexes along the hairpin stem with 3′ overhangs (Supplementary Material, Fig. S7). These small RNA reads shared extensive sequence identity with the 58 annotated miR-548 family members in miRBase (v17). Thus, based on their genomic origin within a common transposable element, we preferred to classify miR-548 family members as siRNAs. Detailed read alignments for all putative endo-siRNAs are provided as Supplementary Material, File S3.
Conservation of noncanonical miRNAs and endo-siRNAs
We performed a comparative genomic analysis of all 130 noncanonical miRNAs (13 derived from tRNAs or snoRNAs and from 117 miRtrons) and 39 endo-siRNAs that were expressed in human skin across 8 vertebrate species (Supplementary Material, File S4). For 48 (37%) noncanonical miRNAs, the mature miRNA sequence was human specific. For 51 noncanonical miRNAs (39%), the mature sequence was present in Macaca mulatta (macaque) but not in Mus musculus (mouse), suggesting that they might be primate specific. The sequences of the remaining 31 (23%) mature noncanonical miRNAs (21 miRtrons, 8 snoRNA- and 2 tRNA-derived miRNAs) were conserved in one or more species beyond M. mulatta. The homologs of two snoRNAs, ACA25 and ACA44, and the PesudoTTA tRNA in the mouse genome are shown in Supplementary Material, Figure S8A. Furthermore, small RNA reads derived from the corresponding murine homologs (Supplementary Material, Fig. S8B) have been found to be Dicer-dependent but Dgcr8-independent (Supplementary Material, Fig. S8C) (10). The presence of snoRNA- and tRNA-derived miRNAs in nonprimate mammals may suggest potential conserved functions for these sncRNAs. The 39 endo-siRNA loci described here were present only in human and M. mulatta, which was largely due to the absence of hosting repetitive elements in nonprimates (28). Overall, the majority of the noncanonical miRNAs and endo-siRNAs we studied were not conserved beyond primates, suggesting that these recently evolved sncRNAs may serve primate-specific functions.
qRT–PCR validation of noncanonical miRNAs and endo-siRNAs
We selected three noncanonical miRNAs and one endo-siRNA for validation with qRT–PCR: the U60 snoRNA-derived miRNA, the pseudoTTA-derived miRNA, miRtron #103 (23) and the Alu-SINE/GON4L-derived siRNA. We first attempted to validate their endogenous expression in skin. All four sncRNAs produced consistent qRT–PCR signals that were both RNA- and reverse transcriptase-dependent (Fig. 3A), and we were able to obtain visible products for the U60- and PseudoTTA-derived miRNAs and the Alu-SINE/GON4L-derived siRNA (Fig. 3B). We next assessed whether the ectopic expression of ∼200 nt genomic DNA segments harboring these sncRNAs was sufficient for the accumulation of mature sncRNAs in HEK293 cells. For each of the four loci tested, there was >5-fold enrichment in mature sncRNA expression when the appropriate genomic segment was ectopically expressed, suggesting that these loci are indeed sufficient to generate sncRNAs (data not shown). Finally, we assessed whether the ectopically expressed sncRNAs co-immunoprecipitated with AGO2 or AGO3 by performing RNA-binding protein immunoprecipitation followed by qRT–PCR (RIP-PCR; Fig. 3C). Indeed, ectopically expressed miRtron #103 was significantly enriched in both AGO2 and AGO3 immunoprecipitates compared with IgG; the pseudoTTA-derived miRNA and the Alu-SINE/GON4L siRNA were significantly enriched in AGO2 immunoprecipitates; and the U60-derived miRNA was modestly enriched in AGO2 and AGO3 immunoprecipitates (not significant). This suggested that these novel sncRNAs may function through association with AGO effectors.
Figure 3.

Endogenous expression of mature novel miRNAs in skin. (A) qRT–PCR amplification traces of sncRNAs from human skin biopsy RNA (black lines) and negative controls (gray lines). An arbitrary threshold used for relative quantitation of expression levels (not shown) is indicated in green. (B) qRT–PCR product bands from human skin biopsy RNA and negative controls. A band size of ∼50 bp corresponds to an ∼21 nt RNA species due to the addition of arbitrary sequence during the reverse transcription step (see Materials and Methods). (C) Abundance of sncRNAs in AGO2 and AGO3 immunoprecipitates relative to IgG. (*P < 0.05, **P < 0.01, ***P < 0.001). RT, reverse transcriptase.
Differential expression of noncanonical miRNAs and endo-siRNAs
In order to investigate the potential dysregulation of noncanonical miRNAs and endo-siRNAs in psoriatic skin, we performed a digital gene expression analysis in PP, PN and NN skin samples. A total of 15 out of 169 (8.9%) noncanonical miRNAs and endo-siRNAs exhibited significant (P < 0.001) differential expression with at least a 1.5-fold change in one or more comparisons (PP versus NN; PP versus PN; or PN versus NN). Ten sncRNAs were up-regulated in PP versus PN and NN skin, whereas five were down-regulated in PP versus PN and NN (Table 2). Four of the 15 differentially expressed sncRNAs were expressed at intermediate levels in PN skin versus PP or NN (Table 2). Notably, the Alu-SINE/GON4L-derived siRNA was ∼17-fold increased in PP versus NN skin, and 2.4-fold increased in PN versus NN skin. The differential expression of this siRNA and miR-937 (re-classified as a typical miRtron) was confirmed by qRT–PCR (Fig. 4A). The clustering of samples based on the expression of the 15 differentially expressed small RNAs distinguished the majority of PP samples from PN and NN samples (Fig. 4B).
Table 2.
Top 15 differentially expressed sncRNAs between psoriatic-involved skin (PP), psoriatic-uninvolved skin (PN) and normal skin (NN)
| ID | Normalized read count |
Fold change |
P-value | ||||
|---|---|---|---|---|---|---|---|
| PP | PN | NN | PP/NN | PP/PN | PN/NN | ||
| Alu-SINE-siRNA | 1614.78 | 220.54 | 91.81 | 17.59 | 7.32 | 2.40 | 0.00E + 00 |
| miRtron #14 | 11.58 | 2.05 | 1.00 | 11.58 | 5.65 | 2.05 | 9.41E − 04 |
| miR-937 | 20.40 | 2.05 | 3.19 | 6.40 | 9.95 | −1.56 | 4.25E − 06 |
| tRNAIle/miR-1983 | 117.40 | 25.16 | 23.98 | 4.90 | 4.67 | 1.05 | 3.33E − 23 |
| Made1_#1-siRNA | 1182.67 | 354.99 | 293.12 | 4.03 | 3.33 | 1.21 | 2.72E − 176 |
| miRtron #144 | 79.48 | 25.16 | 22.88 | 3.47 | 3.16 | 1.10 | 3.24E − 11 |
| miRtron #323 | 32.75 | 11.50 | 17.41 | 1.88 | 2.85 | −1.67 | 2.88E − 03 |
| miRtron #375 | 87.42 | 34.61 | 31.63 | 2.76 | 2.53 | 1.09 | 4.46E − 09 |
| U60 miRNA-5p | 1.08E + 7 | 7.64E + 6 | 4.67E + 6 | 2.30 | 1.41 | 1.64 | 0.00E + 00 |
| miRtron #72 | 143.86 | 82.93 | 74.30 | 1.94 | 1.73 | 1.12 | 6.03E − 07 |
| miRtron #145-5p | 10.70 | 64.03 | 42.57 | −4.00 | −5.88 | 1.50 | 1.01E − 08 |
| miRtron #252 | 10.70 | 20.96 | 34.92 | −3.23 | −1.96 | −1.67 | 1.28E − 03 |
| miRtron #80 | 739.11 | 1483.14 | 2179.30 | −2.94 | −2.0 | −1.47 | 2.84E − 154 |
| miRtron #145-3p | 11.58 | 33.56 | 28.35 | −2.43 | −2.86 | 1.18 | 4.59E − 03 |
| Made1_#47-siRNA | 225.87 | 435.87 | 340.16 | −1.52 | −1.92 | 1.28 | 4.22E − 15 |
Figure 4.

Differential expression of sncRNAs in psoriatic-involved skin. (A) qRT–PCR levels of differentially expressed sncRNAs in 10 normal (NN), 10 uninvolved psoriatic (PN) and 10 involved psoriatic (PP) skin samples. Lines indicate matched uninvolved and involved samples from the same patient. Relative expression was calculated with respect to the endogenous snoRNA Z30 (*P < 0.05, **P < 0.01, ***P < 0.001). (B) Heat map of 15 differentially expressed sncRNAs across 67 skin samples.
Putative functions of targets of differentially expressed miRNAs
Our recent study reported 98 differentially expressed canonical miRNAs in PP versus NN skin (23). To further appreciate the roles of these miRNAs as well as the noncanonical miRNAs and endo-siRNAs identified in the current study in regulating the expression of their targets, we searched for putative mRNA targets of differentially expressed miRNAs with TargetScan (29) and the targets of endo-siRNAs with an in-house siRNA target-finding method (see Materials and Methods). We then identified predicted targets that were differentially expressed in psoriatic skin, based on previously published microarray data (27,30). From this list of psoriasis-relevant targets, we focused on those that were inversely correlated with their respective targeting sncRNAs. This analysis revealed 59 mRNAs that were inversely correlated with 9 differentially expressed noncanonical miRNAs (7 up-regulated and 2 down-regulated) identified in the current study (Fig. 5 Supplementary Material, Table S6) and 287 mRNAs that were inversely correlated with 55 differentially expressed canonical miRNAs (43 up-regulated and 12 down-regulated; Supplementary Material, Table S6). As for endo-siRNAs, four targets were inversely correlated with two differentially expressed siRNAs, including the Alu-SINE/GON4L siRNA (Fig. 5; Supplementary Material, Table S7). The low number of predicted siRNA targets may reflect an incomplete understanding of their functions; for example, we only queried known human cDNAs as putative targets, but it is possible that siRNAs target unannotated repetitive transcripts, other ncRNAs or even genomic DNA. Taken together, these data suggest that some differentially expressed miRNAs and endo-siRNAs influence psoriasis pathogenesis by regulating the expression of target genes in the skin.
Figure 5.

Anti-correlation of differentially expressed target mRNAs with miRNAs and siRNAs. The color bar indicates the range of fold changes.
Finally, we performed a gene ontology analysis on the sets of inversely correlated target mRNAs of both noncanonical and canonical miRNAs. Interestingly, the targets of these two types of miRNAs have different enriched biological processes and molecular functions (Supplementary Material, Table S8), indicating that they may have distinct roles in gene regulation. For the targets of noncanonical miRNAs, the most significantly enriched biological processes included ‘growth’, ‘developmental process’ and ‘multicellular organismal development’ (Benjamini-corrected P-values < 0.01). In contrast, the most significantly enriched biological processes for the targets of canonical miRNAs included ‘response to cytokine stimulus’, ‘organic substance’ and ‘estrogen stimulus’ (Benjamini-corrected P-value < 0.02). These results suggest that noncanonical and canonical miRNAs have distinguishable biological functions, some of which are related to the pathogenesis of psoriasis.
DISCUSSION
We have systematically identified novel sncRNAs from diverse origins in the human genome, characterized their expression in normal and psoriatic skin and interrogated their potential psoriasis-relevant targets. Newly identified sncRNA species include an snoRNA-derived miRNA that was moderately abundant in human skin, the first example of a tRNA-derived miRNA in an adult human tissue, and an siRNA derived from an intronic Alu-SINE that was highly up-regulated in psoriatic skin.
Common features of noncanonical miRNAs and endo-siRNAs from diverse genomic origins
The noncanonical miRNAs and endo-siRNAs that we identified exhibit several distinctive characteristics that set them apart from RNA degradation products and other transcription products. First, these sncRNAs exhibited a size distribution centered around 21–23 nt, which is distinct from other classes of small RNAs and random degradation products. Second, noncanonical miRNAs and siRNAs both require Dicer but not Drosha/Dgcr8 for their biogenesis. In agreement with this, the murine homologs of the ACA44 and ACA25 snoRNA-derived miRNAs, the pseudoTTA-derived miRNA and the re-classified tailed miRtron, miR-3064, were present in deep-sequencing data sets from Dgcr8-knockout mice but absent from Dicer-knockout mice (10,20,21). Other sncRNAs were absent from these data sets, due to shallow sequencing depth or to their lack of conservation in mouse. Third, the 5′ nucleotide identity of noncanonical miRNAs and endo-siRNAs was nonrandom, with an overrepresentation of A as the 5′ terminal nucleotide (47.1 and 72.8%, respectively). Although canonical miRNAs have a stronger bias toward a 5′ terminal U (78.8%), the presence of either a U or A nucleotide has been shown to facilitate interactions within the middle domain of the human AGO2 protein (31). Another interesting characteristic that distinguished noncanonical miRNAs from most canonical miRNAs was the prevalence of 5′ heterogeneity, which is consistent with previous studies of noncanonical miRNAs (30,32–35). In particular, the majority of snoRNA-derived miRNAs, including those that were previously described, exhibited 5′ heterogeneity, suggesting that the biogenesis of snoRNA-derived miRNAs is not fully understood.
Association with protein components of the small RNA silencing machinery
miRNAs and siRNAs exert their functions through association with RISC complexes, which include an AGO protein. In humans, there are four AGO family members, but little direct evidence has been collected for the association of specific sncRNA classes with particular AGO family members (36). Here, we show that three noncanonical miRNAs and one endo-siRNA associate with AGO2 and AGO3 proteins, indicating that they may function through AGO effector complexes. Our findings are largely consistent with recently published small RNA deep sequencing of AGO1/2/3/4 immunoprecipitates from HEK293 cells (37; GSE21918). In that data set, the pseudoTTA-derived miRNA, 28/56 miRtrons, 6/34 MADE1-derived siRNAs, the Alu-SINE/GON4L siRNA and the piRNA-locus-derived siRNAs, all associated with one or more AGO proteins. Interestingly, sequences derived from the U60 snoRNA were present in the AGO immunoprecipitates, but there were no exact matches to the abundant 22 nt species we detected; instead, reads were consistent with the 5′ terminus of the 22 nt species but were typically 28 nt in length. Also, the abundant miRtron, #103, was not present in AGO1/2/3/4 immunoprecipitates, but we later found that miRtron #103 is not endogenously expressed in HEK293 cells (data not shown). Thus, it is likely that cell-type-specific expression underlies the absence of miRtron #103 and perhaps other sncRNA species from the AGO immunoprecipitation data sets as well.
Genomic origin of siRNAs and miRNAs
Although miRNAs and siRNAs associate with many of the same cellular proteins for their biogenesis and function, they are thought to differ in their genomic origins and their mechanism of target regulation. miRNAs tend to arise from nonrepetitive regions of the genome encoding short, intramolecular hairpin sequences, whereas siRNAs tend to originate from longer dsRNAs derived from repetitive or transposable elements. This may be one reason that the number of annotated miRNAs greatly exceeds siRNAs; if, by definition, siRNAs originate from repetitive regions, many legitimate siRNA reads may be discarded during analysis due to promiscuous alignment. Although endo-siRNA loci typically exhibit potential to form long RNA hairpins, we identified three putative endo-siRNA loci from which no RNA hairpin is likely. We speculate that precursor transcripts from these repetitive loci may hybridize in trans with highly similar repeat-derived transcripts, forming a long dsRNA that could be recognized by Dicer.
Based on the distinct genomic origins of miRNAs and siRNAs, our findings suggested that a small subset of annotated miRNAs should be annotated as siRNAs. One example is the miR-548 family; many of its members are derived from Made1 transposable elements. The characterization of the miR-548 family members as siRNAs would also be consistent with the finding that Tcl1 elements, of which Made1 elements are a human-specific subclass, are known to give rise to siRNAs in C. elegans (11). We also acknowledge that distinction of miRNAs and siRNAs based solely on genomic origin is challenging. In mammals, it has been proposed that the insertion of transposable elements into new genomic sites could be one of the driving forces that create new miRNAs during mammalian evolution (38). Thus, we may have captured evolutionarily transient sncRNA species that blur boundaries between the two sncRNA species. Future studies regarding the mechanism of target regulation by individual sncRNAs will likely improve classification.
Low expression of miRtrons and tRNA-derived miRNAs in skin
Given that we analyzed 670 million qualified reads, all of the novel noncanonical miRNAs and endo-siRNAs discovered in this study were expressed at low levels, with the exception of the U60-derived siRNA (which was shown to be over-represented in deep sequencing data compared with the result from qRT–PCR). In the case of miRtrons, their low abundance may result from multiple factors, including the presence of bulges and unpaired bases in their precursors that lower Dicer-processing efficiency, susceptibility to endogenous lariat degradation pathways and, in many cases, their recent evolution. For snoRNA and tRNA-derived miRNAs, the larger ncRNAs that harbor them are typically abundant, but it appears that only a small fraction of those ncRNAs are processed by Dicer. For example, the cluster of reads associated with the novel hsa-miR-1983 derived from tRNA-IleTAT encompassed only 2% of the total number of reads aligning to the tRNA. In contrast, 85% of small RNA reads aligning to murine tRNA-IIeTAT in mESCs clustered within mature mmu-miR-1983 (10). Overall, low expression of hsa-miR-1983 and the pseudoTTA-derived miRNA in human skin might indicate that the alternative hairpin structure is less favorable than the tRNA structure in human tissues, or in nonembryonic tissues in general. Differences in sample preparation between the two studies may also account for some of the relative differences in abundance.
Implications for psoriasis and other human diseases
For 11 out of the 15 differentially expressed noncanonical miRNAs and endo-siRNAs, we observed inversely correlated expression of a subset of their predicted psoriasis-relevant targets. In several cases, these predicted targets were also predicted to be regulated by canonical miRNAs that are differentially expressed in psoriasis, suggesting that multiple sncRNAs may contribute to psoriasis pathogenesis through their interactions with a single target.
Although all of the predicted targets have been implicated in psoriasis pathogenesis by their differential expression in psoriatic-involved skin, many are expressed at low levels in human skin, and their fold changes in psoriatic versus normal skin are relatively low. Moreover, these targets have not yet been strongly implicated in psoriasis pathogenesis. Despite this, a few have particular functional relevance. First, the single predicted target for the highly up-regulated Alu-SINE-derived siRNA was MDM4 (mouse double minute 4 homolog). This gene is an inhibitor of p53 that is frequently up-regulated in cancers (39,40), but modestly down-regulated in psoriasis (Supplementary Material, Table S7) (41). Thus, the down-regulation of MDM4 in psoriasis, which may be mediated by the novel Alu-SINE-derived siRNA we identified, might be one factor that distinguishes the benign hyperplasia in psoriatic-involved skin from cancerous tumors. A second target with particular relevance to psoriasis pathogenesis was SDC4 (syndecan 4), which was predicted to be targeted by the tailed miRtron #72. SDC4 knockout mice display delayed wound healing and impaired angiogenesis (42). Thus, down-regulation of SDC4, which may be mediated by miRtron #72, might contribute to the chronic regenerative phenotype observed in psoriatic-involved skin. Importantly, both MDM4 and SDC4 are also predicted targets of canonical miRNAs that are up-regulated in psoriasis (Supplementary Material, Table S6) (23).
We have shown that the sncRNAs described here may regulate genes that are differentially expressed in psoriatic lesions. However, many previously described sncRNAs exhibit developmentally restricted expression patterns (23). Novel sncRNAs described here may also exhibit this property, which would be consistent with their low expression in adult skin. Thus, it is important to consider the possibility that aberrant expression of certain sncRNAs during development may underlie innate skin barrier or immune defects that contribute to disease susceptibility.
CONCLUSIONS
In summary, our discovery and analysis of noncanonical miRNAs and endo-siRNAs from a variety of genomic loci have substantially increased our appreciation for the diversity and prevalence of sncRNA expression in adult human tissues, provided insights into their biogenesis and identified additional small RNAs that are differentially expressed in psoriatic versus normal skin.
MATERIALS AND METHODS
Skin samples and small RNA library preparation and sequencing
Four to 6 mm punch skin biopsies were collected from healthy controls and from the uninvolved and involved skin of psoriasis patients. Psoriasis patients enrolled in this study received no systemic, photo or topical therapy in the 4 weeks prior to sample collection. Biopsies were stored in RNAlater (Qiagen) at −80°C prior to RNA extraction. RNA was extracted with the miRNeasy Mini Kit (Qiagen), with on-column DNase I digestion. RNA was prepared for sequencing on the Illumina GAIIx platform with the Small RNA Sample Prep Kit (Illumina) according to manufacturer's instructions (protocol v1.5). This protocol required the use of a proprietary 3′ adapter that has a high affinity for Dicer cleavage products. Briefly, 3′ and 5′ adapters were ligated to 1 μg of total RNA. cDNA was synthesized with SuperScript II Reverse Transcriptase (Invitrogen) and subjected to 12 cycles of PCR amplification with high-fidelity Phusion Polymerase (Finnzymes Oy). Each library was loaded on a single Illumina lane at 20 pM and subjected to 36 cycles of sequencing.
Read processing and mapping
Each deep sequencing library was processed independently. Reads with a 3′ adapter substring <6 nt or trimmed sequence length <17 nt were removed from the analysis. Trimmed reads were mapped to the reference human genome sequences (UCSC human genome, hg19 build) and sequences in many noncoding and small RNA databases, which include UCSC genome browser tables (tRNA and snoRNA/miRNA), miRNA (miRBase v.17, http://www.mirbase.org/ftp.shtml), piwi-RNA (fRNAdb database, http://www.ncrna.org/frnadb/) and repeats regions (UCSC RepeatMasker Table, hg19 build).
Identification of noncanonical miRNA
Qualified reads that aligned to every locus of human snoRNAs, tRNAs and intro-exon boundaries were subjected to our novel miRNA prediction method, which was tailored for the identification of noncanonical miRNAs. Any reads that mapped to previously specified miRNA loci were removed. For each snoRNA and tRNA, 50 nt extensions on both sides were added for secondary structure analysis with RNAfold and mFold (43,44). Since the two programs can predict multiple secondary structures, many of which are different, we merged all the predictions from both programs for downstream analysis. Structures lacking stems of at least 18 nt and lacking reads that mapped to any of their stems were excluded. To accommodate atypical stem structures that often appear in noncanonical miRNAs, more unpaired bases, in some cases up to 9 bp, were allowed. Candidate miRNAs were then prioritized based on (i) the occurrence of sequencing reads on the stem of a predicted hairpin structure with minimum free energy less than −18 kcal/mol; (ii) possible presence of miRNA* reads on the opposite stem of the hairpin; (iii) the presence of ∼2 nt 3′ overhangs on the highest likelihood miRNA/miRNA* duplex; and (iv) read length distribution peaks around ∼22 nt. To identify miRtrons, 160 nt intronic sequence segments adjacent to exon/intron boundaries were extracted for introns analyzed. The same procedure as described earlier, without a sliding window, was then followed to identify miRtrons. In addition, we examined the presence of sequencing reads ending with AG dinucleotides as a characteristic feature of miRtron. We also applied RNALfold (44) to find local secondary structures or manually examined the structures based on the sequencing reads.
Identification of endogenous siRNAs from multiple genomic origins
We applied EINVERTED (45) on each 10 kb segment of DNA sequences of whole human genome (hg 19 build) to search for inverted repeats and long-hairpin structures they can fold into. We chose inverted repeats by setting a minimum score threshold of 70. Next, we filtered out those predicted inverted repeats that have no more than 10 mappable reads. We also analyzed the genomic annotation of the predicted inverted repeats, i.e. checking them if they overlap with SINEs, LINEs or pseudogenes. We next blasted these mappable reads by allowing mismatches to the annotated human ncRNAs to rule out the possibility that such sequencing reads were byproducts of other types of ncRNAs. Finally, we examined genomic regions in which the mappable reads have lengths of dominantly 21–23 nt, expected for the length of siRNAs. The amount of the normalized reads having unique genomic loci represented the expression levels of such inverted repeats in each skin category.
Conservation analysis of sncRNAs
To analyze conservation of sncRNAs, we retrieved from the UCSC genome browser (46) the multiple alignments of nine species—Homo sapiens (human), M. mulatta (rhesus), M. musculus (mouse), Canis lupus familiaris (dog), Loxodonta Africana (elephant), Monodelphis domestica (opossum), Gallus gallus (chicken), Xenopus tropicalis (frog) and Danio rerio (zebrafish)—for each novel miRNA and siRNA. We calculated the number of insertions, deletions and mismatches when each nonhuman sequence was compared with a human sncRNA (Supplementary Material, File S4) based on the multiple alignments of the nine species. An sncRNA was considered conserved in one species if the sequence in that species had no variation (insertions, deletions and mismatches) in the seed region for an miRNA or less than two variations in the whole region for an siRNA.
qRT–PCR
qRT–PCR of novel sncRNAs was performed with custom TaqMan small RNA assays (Life Technologies), as described previously (23). qRT–PCR of the known miRNA miR-937 and the endogenous control Z30 was performed with TaqMan miRNA assays (Life Technologies). Exact primer and probe sequences are proprietary. Relative expression levels were calculated according to the 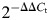 method as follows:
method as follows: 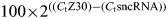 . Significance was determined with one-way ANOVA and post hoc two-tailed t-tests.
. Significance was determined with one-way ANOVA and post hoc two-tailed t-tests.
Cell culture
The cloning of sncRNAs into pEP-miR cloning and expression vectors (Cell Bio Labs) and transfections of HEK293 cells were performed as described previously (23). Primer sequences were as follows: pEP-miR-E1-F/R: 5′-GAGAAGGAGTTGTGGAATCCAAT/5′-CTGTTGCCCAGCTATTCCTC; pEP-miR-E2-F/R: 5′-GAGGAATAGCTGGGCAACAG/5′-AGACAGAGACAGGGTCTCAGATG; pEP-miR-E3-F/R: 5′-CTGTGTCATCTGAGACCCTGTC/5′-TGCATGTCTGCTCCAACAAT; pEP-miR-E1/2-F/R: 5′-CGTGAAAATAGTCTCTACAAGTAATCC/5′-GAGCCTAAGGGTTCAAGATCA; pEP-miR-E2/3-F/R: 5′-CAAATCCTTGGAGGAATAGCTG/5′-ATGCATGTCTGCTCCAACAA; pEP-miR-E1-3-F/R: 5′-TGGTGAGAGAAGGAGTTGTGG/5′-TGCATGTCTGCTCCAACAAT; pEP-miR-U60-F/R: 5′-GCACCACTTTGTCCCTTAGC/5′-GCGACGAAACCTGAAAGTC; pEP-miR-pseudoTTA-F/R: 5′-CTGCCTCACCACCAGCTAC/5′-AGGCCCACACCATCCTTAC; pEP-miRtron #103-F/R: 5′-TTTTCTGTCAGTGCTTTTGGA/5′-GATCTCCAGTGGGAGAGCAG.
RIP-PCR
RIP-PCR was performed as previously described for AGO2 (23). The same protocol was applied with a rabbit polyclonal antibody for AGO3 (Sigma-Aldrich). Significance was determined by one-way ANOVA and post hoc two-tailed t-tests.
Differential expression of miRNAs and siRNAs
Reads that aligned perfectly to the miRNAs and siRNAs of interest were subjected to gene expression analysis. Reads that mapped to multiple genomic loci were attributed to all potential derivative small RNAs. Read counts in each skin category (NN, PN and NN) were normalized to adjust for slight variation in total read count between categories. Let Ncategory be the number of qualified reads that aligned to the human genome (hg19 build) in each category, C the average of Ncategory and Mcategory the number of qualified reads aligned to each miRNA and siRNA in each category. Thus, the normalized number of reads for each miRNA and siRNA in a given category is (Mcategory × C)/(Ncategory). Fold changes were calculated from normalized read counts, and χ2 test was applied to determine significance.
Putative targets of miRNAs and siRNAs
The target mRNA genes of miRNAs of interest were predicted by TargetScan (29). For predicting targets of siRNAs, human cDNA sequences were downloaded from UCSC genome browser (hg19 build), and the reverse complementary sequences of siRNAs were mapped to the cDNA sequences by BLAST (47). Those mapping sites with no more than three mismatches were considered as putative targets of siRNAs.
mRNA gene expression analysis
Microarray data of mRNA genes of psoriatic and normal skin of human were downloaded from Gene Expression Omnibus (GEO) accession number GSE13355, which has a total of 122 samples containing 58 psoriatic patients and 64 normal healthy controls. Differentially expressed genes were identified using rank product (48). The rationale behind rank product is that it is unlikely for a gene to be ranked in the top position in all replicates if the gene was not differentially expressed. It has been shown that RP is less sensitive to noise and has a better performance than other methods when sample size is small (RP_comp). We selected differentially expressed genes with the percentage of false-positive predictions, equivalent to false discovery rate, no greater than 0.05 for 1000 permutations. Differentially expressed probe sets were then mapped to corresponding genes according to the annotation of Affymetrix Human Genome U133 Plus 2.0 Array. We also used the set of differentially expressed genes in PP versus NN skin previously reported in Gudjonsson et al. (49).
GO term and functional enrichment of target genes
The GO term analysis was performed using the online tool DAVID (50). DAVID provides P-values, before and after multiple-test corrections, based on a modified Fisher's exact test.
Data deposition
All 76 small RNA sequencing libraries from psoriatic lesion and normal human skin biopsy samples have been deposited into NCBI/GEO databases under GEO31037.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (5RC1AR058681 to A.M.B. and W.Z. and R01GM100364 to W.Z.); the National Science Foundation (DBI-0743797 to W.Z.); and the NHGRI (T32HG000045 to C.E.J.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Genome Technology Access Center at Washington University for GAIIx data generation, and Xiang Zhou for preprocessing of sequencing data.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Carthew R.W., Sontheimer E.J. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim V.N., Han J., Siomi M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 3.Babiarz J.E., Blelloch R., Eli T., Broad E. Small RNAs – their biogenesis, regulation and function in embryonic stem cells. StemBook. 2009:1–16. 10.3824/stembook.1.47.1. [PubMed] [Google Scholar]
- 4.Berezikov E., Chung W.-J., Willis J., Cuppen E., Lai E.C. Mammalian mirtron genes. Mol. Cell. 2007;28:328–336. doi: 10.1016/j.molcel.2007.09.028. 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010:992–1009. doi: 10.1101/gad.1884710. 10.1101/gad.1884710.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruby J.G., Jan C.H., Bartel D.P. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okamura K., Hagen J.W., Duan H., Tyler D.M., Lai E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brameier M., Herwig A., Reinhardt R., Walter L., Gruber J. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 2011;39:675–686. doi: 10.1093/nar/gkq776. 10.1093/nar/gkq776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ender C., Krek A., Friedländer M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G. A human snoRNA with microRNA-like functions. Mol. Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 10.Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamura K., Lai E.C. Endogenous small interfering RNAs in animals. Nat. Rev. Mol. Cell Biol. 2008;9:673–678. doi: 10.1038/nrm2479. 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tam O.H., Aravin A.A, Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M., et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe T., Totoki Y., Toyoda A., Kaneda M., Kuramochi-Miyagawa S., Obata Y., Chiba H., Kohara Y., Kono T., Nakano T., et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 14.Chapman E.J., Carrington J.C. Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 2007;8:884–896. doi: 10.1038/nrg2179. 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- 15.Ruby J.G., Jan C., Player C., Axtell M.J., Lee W., Nusbaum C., Ge H., Bartel D.P. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans . Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 16.Czech B., Malone C.D., Zhou R., Stark A., Schlingeheyde C., Dus M., Perrimon N., Kellis M., Wohlschlegel J.A., Sachidanandam R., et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghildiyal M., Seitz H., Horwich M.D., Li C., Du T., Lee S., Xu J., Kittler E.L.W., Zapp M.L., Weng Z., et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamura K., Balla S., Martin R., Liu N., Lai E.C. Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nat. Struct. Mol. Biol. 2008;15:581–590. doi: 10.1038/nsmb.1438. 10.1038/nsmb.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamura K., Chung W.-J., Ruby J.G., Guo H., Bartel D.P., Lai E.C. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008;453:803–806. doi: 10.1038/nature07015. 10.1038/nature07015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi R., Pasolli H.A., Landthaler M., Hafner M., Ojo T., Sheridan R., Sander C., O'Carroll D., Stoffel M., Tuschl T., et al. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc. Natl Acad. Sci. USA. 2009;106:498–502. doi: 10.1073/pnas.0810766105. 10.1073/pnas.0810766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babiarz J.E., Hsu R., Melton C., Thomas M., Ullian E.M., Blelloch R. A role for noncanonical microRNAs in the mammalian brain revealed by phenotypic differences in Dgcr8 versus Dicer1 knockouts and small RNA sequencing. RNA. 2011;17:1489–1501. doi: 10.1261/rna.2442211. 10.1261/rna.2442211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce C.E., Zhou X., Xia J., Ryan C., Thrash B., Menter A., Zhang W., Bowcock A.M. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum. Mol. Genet. 2011;20:4025–4040. doi: 10.1093/hmg/ddr331. 10.1093/hmg/ddr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker M. MicroRNA profiling: separating signal from noise. Nat. Methods. 2010;7:687–692. doi: 10.1038/nmeth0910-687. 10.1038/nmeth0910-687. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W., Gao S., Zhou X., Chellappan P., Chen Z., Zhou X., Zhang X., Fromuth N., Coutino G., Coffey M., et al. Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol. Biol. 2011;75:93–105. doi: 10.1007/s11103-010-9710-8. 10.1007/s11103–010–9710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynt A.S., Greimann J.C., Chung W.-J., Lima C.D., Lai E.C. MicroRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol. Cell. 2010;38:900–907. doi: 10.1016/j.molcel.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piriyapongsa J., Jordan I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PloS One. 2007;2:e203. doi: 10.1371/journal.pone.0000203. 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurka J., Smith T. A fundamental division in the Alu family of repeated sequences. Proc. Natl Acad. Sci. USA. 1988;85:4775–4778. doi: 10.1073/pnas.85.13.4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. 1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martí E., Pantano L., Bañez-Coronel M., Llorens F., Miñones-Moyano E., Porta S., Sumoy L., Ferrer I., Estivill X. A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010;38:7219–7235. doi: 10.1093/nar/gkq575. 10.1093/nar/gkq575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frank F., Sonenberg N., Nagar B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010;465:818–822. doi: 10.1038/nature09039. 10.1038/nature09039. [DOI] [PubMed] [Google Scholar]
- 32.Lee L.W., Zhang S., Etheridge A., Ma L., Martin D., Galas D., Wang K. Complexity of the microRNA repertoire revealed by next-generation sequencing. RNA. 2010;16:2170–2180. doi: 10.1261/rna.2225110. 10.1261/rna.2225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berezikov E., Robine N., Samsonova A., Westholm J.O., Naqvi A., Hung J., Okamura K., Dai Q., Bortolamiol-Becet D., Martin R., et al. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 2011;21:203–215. doi: 10.1101/gr.116657.110. 10.1101/gr.116657.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M., et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morin R.D., Connor M.D.O., Griffith M., Kuchenbauer F., Delaney A., Prabhu A., Zhao Y., McDonald H., Zeng T., Hirst M., et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. 10.1101/gr.7179508.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Czech B., Hannon G.J. Small RNA sorting: matchmaking for Argonautes. Nat. Rev. Genet. 2011;12:19–31. doi: 10.1038/nrg2916. 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A.-C., Munschauer M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smalheiser N.R., Torvik V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–326. doi: 10.1016/j.tig.2005.04.008. 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Terzian T., Torchia E.C., Dai D., Robinson S.E., Murao K., Stiegmann R.A., Gonzalez V., Boyle G.M., Powell M.B., Pamela M., et al. p53 prevents progression of nevi to melanoma predominantly through cell cycle regulation. Pigment Cell Melanoma Res. 2010;23:781–794. doi: 10.1111/j.1755-148X.2010.00773.x. 10.1111/j.1755-148X.2010.00773.x.p53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danovi D., Meulmeester E., Pasini D., Capra M., Frenk R., Graaf P.D., Gasparini P., Gobbi A., Helin K., Pelicci P.G., et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Society. 2004 24 10.1128/MCB.24.13.5835. [Google Scholar]
- 41.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.-J., et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Echtermeyer F., Streit M., Wilcox-Adelman S., Saoncella S., Denhez F., Detmar M., Goetinck P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Invest. 2001;107:R9–R14. doi: 10.1172/JCI10559. 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rice P. EMBOSS: The European Molecular Biology Open Software Suite. Science. 2000;16:2–3. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 46.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Breitling R., Armengaud P., Amtmann A., Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. doi: 10.1016/j.febslet.2004.07.055. 10.1016/j.febslet.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 49.Gudjonsson J.E., Ding J., Johnston A., Tejasvi T., Guzman A.M., Nair R.P., Voorhees J.J., Abecasis G.R., Elder J.T. Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J. Invest. Dermatol. 2010;130:1829–1840. doi: 10.1038/jid.2010.36. 10.1038/jid.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dennis G. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.