

Background: Aberrant activation of autophagy (proteinopathy) leads to neurodegeneration in various neurological disorders.
Results: Compensatory basal autophagy is activated, and genotoxins deregulate autophagy in the ubiquitin pathway-compromised ataxia telangiectasia (A-T) cells.
Conclusion: Deregulation of autophagy is due to the elevated expression of ISG15 in A-T cells.
Significance: Results highlight a causal contribution of a novel “ISG15 proteinopathy” in A-T neurodegeneration.
Keywords: Ataxia, Autophagy, Interferon, Lysosomes, Neurodegeneration, Proteasome, Protein Degradation, Ataxia Telangiectasia, ISG15
Abstract
Ataxia-telangiectasia (A-T) is a cerebellar neurodegenerative disorder; however, the basis for the neurodegeneration in A-T is not well established. Lesions in the ubiquitin and autophagy pathways are speculated to contribute to the neurodegeneration in other neurological diseases and may have a role in A-T neurodegeneration. Our recent studies revealed that the constitutively elevated ISG15 pathway impairs targeted proteasome-mediated protein degradation in A-T cells. Here, we demonstrate that the basal autophagy pathway is activated in the ubiquitin pathway-compromised A-T cells. We also show that genotoxic stress triggers aberrant degradation of the proteasome and autophagy substrates (autophagic flux) in A-T cells. Inhibition of autophagy at an early stage using 3-methyladenine blocked UV-induced autophagic flux in A-T cells. On the other hand, bafilomycin A1, which inhibits autophagy at a late stage, failed to block UV-induced autophagic flux, suggesting that overinduction of autophagy may underlie aberrant autophagic flux in A-T cells. The ISG15-specific shRNA that restored proteasome function restores autophagic function in A-T cells. These findings suggest that autophagy compensates for the ISG15-dependent ablation of proteasome-mediated protein degradation in A-T cells. Genotoxic stress overactivates this compensatory mechanism, triggering aberrant autophagic flux in A-T cells. Supporting the model, we show that autophagy is activated in the brain tissues of human A-T patients. This highlights a plausible causal contribution of a novel “ISG15 proteinopathy” in A-T neuronal cell death.
Introduction
Ataxia-telangiectasia (A-T)2 is a childhood disease with an incident of 1 in 40,000 children in the U.S. and 1 in 200,000 worldwide each year (1–4). Ataxia refers to uncoordinated movements, such as walking, and telangiectasia is the enlargement of capillaries just below the surface of the skin, a feature characteristically exhibited by A-T patients (3). It is a rare inherited disorder that mainly affects nervous and immune systems (3). A-T patients are also at an increased risk of developing cancer (5). Affected individuals are very sensitive to radiation, including medical x-rays (6, 7). This feature has been attributed to the defective Atm (ataxia telangiectasia mutated) gene in A-T patients (8). ATM is a serine/threonine protein kinase that is activated upon DNA damage (9). Activated ATM kinase phosphorylates several key proteins that initiate activation of the DNA damage checkpoints, cell cycle arrest, and DNA repair to favor cell survival (10). Therefore, a defect in ATM has severe consequences in DNA-damaged cells, especially in terminally differentiated cells, such as neurons (11). Indeed, a defective DNA repair pathway has been linked to the progressive neurodegeneration in A-T patients (11–14). However, whether the defect in DNA repair is solely responsible for neurodegeneration in A-T is still unclear.
Altered expression/mutations in genes involved in protein turnover pathways have been linked to neurodegeneration in other neurological diseases (15, 16). Accumulation of misfolded protein deposits in affected brain regions is reported in neurodegenerative diseases including Alzheimer, Parkinson, Creutzfeldt-Jakob, and Huntington disease (15, 16). In most cases, proteinaceous deposits were composed of ubiquitin conjugates, suggesting a failure in their degradation via the ubiquitin/26S proteasome, the major cellular proteolytic machinery responsible for targeted destruction of short lived and abnormal proteins in mammalian cells (17). Surprisingly, very little is known about the potential accumulation of non-degraded ubiquitylated proteins in neurons of A-T patients despite the fact that progressive neurodegeneration is a hallmark of A-T (12, 13). Although some documented reports in the literature support such a possibility (18, 19), the molecular mechanisms leading to the accumulation of non-degradable ubiquitylated proteins and the potential causal relationship with neuronal degeneration in A-T patients are largely unknown. On the other hand, our recent studies have demonstrated that the targeted proteasome-mediated degradation is impaired in A-T cells (20). The reduced protein turnover in A-T cells is associated with elevated expression of ISG15 (interferon-stimulated gene of 15 kDa), a ubiquitin-like protein demonstrated to antagonize the ubiquitin pathway (20).
The ISG15 protein is a member of the UBL (ubiquitin-like) class of proteins (21, 22). It is induced upon interferon treatment (23). Intracellular ISG15 exists in two forms: (i) free and (ii) conjugated to target proteins. ISG15 is conjugated to its target proteins in an enzymatic cascade involving an E1 (UbE1L), E2 (UbcH8), and E3 (HERC5 and others) (21–23). Free ISG15 has been suggested to have cytokine-like activity (23). Conjugated ISG15 exerts its biological effect by inhibiting polyubiquitylation of cellular proteins (24–26). Several groups have now demonstrated that ISG15 inhibits the ubiquitin pathway by modulating the activities of the ubiquitin E2/E3 ligases (27–30). Concurrently, ISG15 inhibits polyubiquitylation and degradation of cellular proteins in breast cancer and A-T cells (24). In addition, the ISG15/ubiquitin inclusions were found in A-T human post-mortem brain tissues (20). Together, these results point toward the possibility that, as in other neurological diseases, ISG15-mediated defects in targeted protein turnover by the proteasome may contribute to the neuronal atrophy in A-T.
Compelling evidence now demonstrates that under conditions where proteasome function is compromised, the large ubiquitin-containing protein aggregates are cleared by autophagy (31–34), a second major proteolytic pathway that targets destruction of long lived cellular proteins, larger macromolecular complexes, and defective organelles through lysosomes (35, 36). In the current study, we demonstrate that the basal autophagy is activated in A-T cells impaired in the ubiquitin pathway due to the constitutively elevated ISG15 pathway. Additionally, genotoxins (UV and camptothecin) (37, 38) lead to the aberrant autophagic flux (increased degradation of autophagy substrates) in A-T cells. Inhibition of autophagy at an early stage using 3-methyladenine (3-MA) blocked UV-induced autophagic flux. In contrast, bafilomycin A1 (Bafl), which inhibits autophagy at a late stage, failed to block UV-induced autophagic flux in A-T cells. These results revealed that the overinduction of autophagy may underlie aberrant autophagic flux in A-T cells. ISG15 shRNA that restored the proteasome function attenuated both basal and UV-activated autophagy in A-T cells. These findings in A-T fibroblast cells suggest that, in addition to the ATM-mediated defects in the DNA repair pathways, autophagic stress caused due to the “ISG15 proteinopathy” (ISG15-mediated defects in protein turnover pathways) in other lineages of A-T cells, such as neurons, may contribute to the A-T phenotype of neurodegeneration.
EXPERIMENTAL PROCEDURES
Cells
FT169A (A-T) and FT169A (ATM+) fibroblast cells, were obtained from Dr. Y. Shiloh (Tel Aviv University, Ramat Aviv, Israel). FT169A (A-T) cells were derived from FT169A cells (ATM null) by stable transfection with the expression vector alone (39). FT169A (ATM+) cells were derived from FT169A cells by stable transfection with full-length ATM cDNA (39). Both FT169A (A-T) and FT169A (ATM+) fibroblast cells were cultured in complete DMEM supplemented with hygromycin B (100 μg/ml).
Human Tissues and Ethics Statement
Human brain tissues and tissue sections were obtained from the NICHD, National Institutes of Health, Brain and Tissue Bank for Developmental Disorders at the University of Maryland (supported by NICHD Contracts N01-HD-4–3368 and N01-HD-4–3383) under ethics protocols approved by the University of Maryland Institutional Review Board.
Tissues
Frozen human mid-brain tissues containing specifically substantia nigra were obtained post-mortem from patients with confirmed A-T disease and control individuals (without any known disease).
Tissue Sections
Slides with paraffin-embedded sections of the same midbrain tissues described above were used in an immunofluorescence study.
Construction of Lentiviral ISG15 shRNA Stable Transfectants of FT169A (A-T) Cells
Preparation of lentiviral particles has been described (40). Briefly, five shRNA constructs (TRCN00000042 (0-4)) for the ISG15 in a pLKO1 vector and one control non-targeting shRNA lentiviral vector (SHC002V) were purchased from Sigma-Aldrich. Among the five shRNA constructs tested, TRCN0000007422 NM_005101.1-295S1C1 shRNA that showed efficient ISG15 knocked down (>75%) in FT169A cells was used for the production of lentiviral particles. Lentiviral particles were generated by transfecting HEK293T cells with the lentiviral shRNA vector (pLKO.1-Puro harboring ISG15 or SHC002V vector harboring control shRNA), together with the packaging (psPax2) and an envelope (pMD2.G) vector (Addgene) using standard calcium phosphate precipitation as described (41). 6–8 h post-transfections, cells were washed once and replenished with the fresh DMEM and allowed to grow for an additional 48 h. The viral supernatants were then harvested and filtered through a 0.45-μm pore size filter. For transduction, FT169A (A-T) cells (65,000 cells/ml) were plated in a 6-well tissue culture plate 24 h prior to the lentiviral infection. The next day, culture medium was replaced with 1 ml of fresh medium containing 6.5 μg/ml of Polybrene (Chemicon International). Cells were infected with lentiviral particles containing ISG15 or control shRNA and incubated in a tissue culture incubator overnight. After 12 h of incubation, all transduced cells were replenished with the fresh culture medium without Polybrene. Two days post-transduction, cells were split (1:5) and allowed to grow under normal conditions (37 °C and 5% CO2). Selection medium that contained 6.5 μg/ml puromycin dihydrochloride (Sigma) was then added to the cells 48 h after replating. Individual colonies were picked following 5 weeks of puromycin selection and screened for ISG15 expression by Western blotting analysis using anti-ISG15 antiserum.
Immunoblotting and Immunofluorescence Analysis and Immunoblotting Analysis of Proteins in Cultured Cells
Cells (5 × 105) were cultured in 35-mm tissue culture plates. After various experimental treatments, cells were lysed using an SDS-PAGE sample buffer. Cell lysates were then analyzed by SDS-PAGE (10% for p62 or 15% for LC3 (microtubule-associated protein 1A/1B-light chain 3) and polyubiquitin conjugates) and immunoblotting analysis using either anti-ISG15 (raised against human ISG15 (23)), anti-ubiquitin (Sigma), anti-HA (gift from Dr. Walworth, UMDNJ-Robert Wood Johnson Medical School), anti-LC3 (MBL International Corp.), or p62 (Sigma) antibodies, as indicated, using the ECL Western procedure (Pierce) and the Bio-Rad VersaDoc imaging system.
Immunoblotting Analysis of HA-Ubiquitin-conjugated Proteins in Cells Exposed to UV Radiation
Cells (5 × 105) were transfected with a hemagglutinin (HA)-ubiquitin plasmid using the PolyFect transfection reagent (Qiagen) as described (24). 24 h after transfection, cells were treated with either proteasome inhibitor MG132 (1 μm) (Boston Biochemicals), autophagy inhibitor Bafl (1 nm) (Sigma), or 3-MA (100 nm) for 18 h as indicated. Cells were exposed to UV radiation (25 mJ, using a Bio-Rad GS Gene LinkerTM UV chamber) and/or left untreated. Cells were then allowed to recover in the presence of inhibitors for 3 h. Cell lysis, SDS-PAGE, and immunoblotting analysis to detect HA-ubiquitin-conjugated proteins using anti-HA antibodies were carried out as described above.
Immunofluorescence Analysis in Cells and LC3 Expression
Cells (100,000/point) were cultured on fibronectin-coated glass coverslips. The next day, cells were fixed in 4% paraformaldehyde. After washing with phosphate-buffered saline (PBS) (2 × 5 min), cells were incubated with 100 μg/ml digitonin for 15 min at room temperature. Cells were then washed with PBS (2 × 5 min) and immunostained for LC3 (MBL International Corp.) for 1 h at room temperature. After washing with PBS (2 × 5 min), cells were incubated with Alexa Fluor 488 goat anti-mouse IgG secondary antibody (1:100) (Invitrogen) for 1 h. Cells were then washed with PBS and mounted on slides in anti-fade mounting medium with DAPI (Invitrogen). Images were taken using a ×63 oil immersion objective with a Leica DMRA2 upright microscope run through SlideBook software (Intelligent Imaging Innovations).
Autophagasome, Lysosome, and Autophagolysosomes Staining
Cells (100,000/point) were cultured on fibronectin-coated glass coverslips. Cells were treated with autophagy inhibitor Bafl (1 nm) (Sigma) for 18 h. Cells were then exposed to UV radiation and allowed to recover in the presence of inhibitors for 3 h as indicated. Cells were then washed (twice for 1 min each) with PBS and co-stained with the Cyt-ID® (Cyt-ID® autophagy detection kit from Enzo Lifesciences) and Lyso Tracker® Red DND-99 (Invitrogen) for 30 min at 37 °C in a CO2 incubator following the manufacturer's protocol. Stained cells were then washed (twice for 1 min each) with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. After washing with PBS (3 × 10 min), cells were mounted on slides in anti-fade mounting medium with DAPI (Invitrogen). Images were taken using a ×63 oil immersion objective with a Leica DMRA2 upright microscope run through SlideBook software (Intelligent Imaging Innovations).
Immunoblotting Analysis of LC3 Expression in Brain Tissues of A-T Patients by Western Blotting
Frozen tissues were stored at −80 °C until use. For detecting LC3, frozen tissues were weighed, cut into small pieces, and placed in test tubes containing SDS sample buffer. Tissue samples were then sonicated with a tissue homogenizer (Biospec Products, Inc.). Sonicated samples were immediately boiled for 10 min at 100 °C and then centrifuged at 13,000 × g for 10 min. Cleared supernatants containing SDS-solubilized protein extracts were analyzed by 15% SDS-PAGE and immunoblotted using anti-LC3 as described above.
Immunofluorescence Analysis in A-T Brain Tissue Sections
For double immunofluorescence, tissue sections were deparaffinized in xylene and incubated with the GFAP (Abcam) and LC3 (MBL International Corp.) primary antibodies (1:100) for 1 h. After washing in PBS, sections were stained with Alexa Fluor 488 goat anti-rabbit IgG secondary antibody (Invitrogen) and goat polyclonal secondary antibody to mouse IgG (Cy5®) (Abcam). Sections were mounted in gold anti-fade mounting medium (Invitrogen) and examined using a Nikon E600 epifluorescence microscope. All of the operations were performed at room temperature.
RESULTS
UV Light Induces Degradation of Polyubiquitylated Proteins in A-T but Not in ATM+ Cells
Previous studies using the FT169A (A-T) (ATM null; henceforth referred to as A-T) and FT169A (ATM+) (ATM-reconstituted FT169A; henceforth referred to as ATM+) isogenic pair of fibroblast cells have demonstrated that ISG15, a ubiquitin-like protein known to antagonize the ubiquitin pathway, is elevated and inhibits the ubiquitin pathway in A-T cells (24).
The ubiquitin pathway plays a key role in ATM-dependent DNA repair (42). Because A-T cells are defective in both the DNA repair (due to the defective ATM kinase) (43) and ubiquitin (due to the constitutively elevated ISG15 pathway) pathways (20), we opted to examine the effect of UV, a genotoxic stressor known to induce DNA damage, on global protein polyubiquitylation and its subsequent degradation in A-T cells. As shown in Fig. 1A, the steady state levels of the endogenous polyubiquitylated proteins and free ubiquitin rapidly decreased in A-T cells exposed to different doses of UV radiation and detected 3 h after radiation treatment (Fig. 1A, left, compare lane 1 with lanes 2 and 3). By contrast, very little decrease in the steady state levels of polyubiquitylated proteins was seen in ATM+ cells under the same conditions (Fig. 1A, right, compare lane 1 with lanes 2 and 3). These results revealed that the steady state levels of polyubiquitylated proteins are decreased in UV-treated A-T but not in ATM+ cells.
FIGURE 1.

Genotoxic stress induces aberrant degradation of the polyubiquitylated proteins in A-T cells. A, A-T and ATM+ cells were exposed to different doses of UV and allowed to recover for 3 h. Cells were lysed, and lysates were analyzed by Western blotting for polyubiquitylated proteins and tubulin using anti-ubiquitin and anti-tubulin antibodies, respectively. B, A-T and ATM+ cells were transfected with a HA-ubiquitin construct. Cells were then treated with MG132 (1 μm) or Bafl (1 nm) for 18 h and exposed to UV radiation (25 mJ/m2). After recovery in the presence of inhibitors for an additional 3 h, cells were lysed. Cell extracts were analyzed by Western blotting for HA-polyubiquitylated proteins and actin using anti-HA and anti-actin antibodies, respectively. Intensity of the total HA-polyubiquitylated proteins was quantitated using Bio-Rad Quantity One software. The bar graph shows average values ± S.E. (error bars) of percentage degradation of HA-polyubiquitylated proteins from three independent experiments. C, A-T and ATM+ cells were transfected with a HA-Lys48-only ubiquitin construct. The inhibitor and UV treatments, cell lysis, SDS-PAGE, and immunoblotting analysis to detect HA-ubiquitin-conjugated proteins using anti-HA antibodies was carried out as described in B. The experiment was repeated two times with reproducible results. D, A-T cells were treated with camptothecin (10 μm) in the presence or absence of Bafl (1 nm) for 24 h. Ubiquitin-conjugated proteins using anti-ubiquitin antibodies were detected as described in A. The experiment was repeated three times, and the two-tailed p values are presented under “Results.”
The decrease in the steady state levels of polyubiquitylated proteins could either be due to their increased deubiquitylation or increased degradation via the 26S proteasome. Also, the ubiquitin antibody used in the above experiment is known to cross-react with free ISG15/UCRP (44), and ISG15 protein is elevated in A-T cells (20). We therefore transfected an HA-ubiquitin construct and then assessed the steady state levels of the HA-polyubiquitylated proteins (to rule out the possibility of protein polyubiquitylation versus protein polyISGlyation), in the absence or presence of the proteasome inhibitor MG132 (to rule out the possibility of protein deubiquitylation versus protein degradation), in UV-treated A-T and ATM+ cells. Consistent with the results shown in Fig. 1A, UV also induced degradation of HA-polyubiquitylated proteins in A-T cells (Fig. 1B, left, compare lanes 1 and 2). Intriguingly, MG132 failed to protect the UV-induced decrease of HA-polyubiquitylated proteins in A-T cells (Fig. 1B, left, lanes 3 and 4). UV also induced moderate degradation of HA-polyubiquitylated proteins in ATM+ cells (Fig. 1B, right, compare lanes 1 and 2). However, unlike in A-T cells, MG132 completely blocked the decrease of HA-polyubiquitylated proteins in ATM+ cells exposed to UV (Fig. 1B, right, lanes 3 and 4). Inhibition of protein disappearance in MG132-treated ATM+ cells suggested that the UV-induced disappearance of HA-polyubiquitylated proteins in A-T cells is not due to their deubiquitylation but due to their degradation via the 26S proteasome. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay for cell survival revealed that the degradation of polyubiquitylated proteins was not due to the decreased viability of UV-treated A-T cells under our experimental conditions (data not shown). These results were intriguing because we previously demonstrated that degradation of polyubiquitylated proteins via the 26S proteasome is impaired in A-T but not in ATM+ cells (20).
The autophagy pathway is induced as a compensatory mechanism to degrade cellular proteins in cells defective in the ubiquitin pathway (31–34). In addition, the MG132 proteasome inhibitor induces autophagy (45, 46). We therefore tested whether the UV-induced degradation of polyubiquitylated proteins is via autophagy in the ubiquitin pathway-ablated A-T cells. To test the involvement of autophagy, we employed the autophagy inhibitor Bafl (47) in this study. Similar to MG132, Bafl also failed to block UV-mediated degradation of polyubiquitylated proteins in A-T cells (Fig. 1B, left, lanes 5 and 6). In contrast, Bafl significantly blocked the decrease of HA-polyubiquitylated proteins in ATM+ cells exposed to UV (Fig. 1B, right, lanes 5 and 6). We also assessed protein degradation in the presence of 50 nm Bafl and observed that even this high concentration of Bafl failed to protect UV-induced degradation of cellular proteins in A-T cells (data not shown). The results using ATM+ cells and inhibitors imply that the failure of Bafl and MG132 to block UV-induced degradation of the polyubiquitylated proteins in A-T cells is not due to the limiting concentration of MG132 and Bafl used in our experiment because these inhibitors efficiently blocked the degradation of polyubiquitylated proteins in ATM+ cells. The bar graph shows average values ± S.E. of percentage degradation of polyubiquitylated proteins measured from three independent experiments, confirming reproducibility of the qualitative results shown in Fig. 1B. Together, these results revealed that UV induces MG132- and bafilomycin-resistant degradation of polyubiquitylated proteins in A-T but not in ATM+ cells.
To complement the results shown in Fig. 1B, we used another construct that expresses HA-ubiquitin that can preferentially make polyubiquitin chains linked through Lys48 on the substrates (20). Similar results were obtained using this distinct HA-ubiquitin construct (Fig. 1C); we found that UV induces MG132- and bafilomycin-resistant degradation of HA-Lys48-linked polyubiquitylated proteins in A-T but not in ATM+ cells.
To test the generality of this observation, we examined if the anticancer drug camptothecin (CPT), a genotoxic agent (37, 48–50), known to sensitize A-T cells (37), like UV, can induce degradation of polyubiquitylated proteins in A-T cells. Similar to UV, CPT also induced degradation of endogenous polyubiquitylated proteins (Fig. 1D, lanes 1 and 2) (p < 0.0001), and Bafl failed to protect CPT-mediated degradation of polyubiquitylated proteins (Fig. 1D, lanes 3 and 4) in A-T cells (p < 0.0001). These results imply that genotoxins, such as UV and CPT, induce aberrant degradation of polyubiquitylated cellular proteins in the proteasome function-ablated A-T cells.
Basal Autophagy Is Activated in A-T Cells Impaired in the Ubiquitin Pathway
Basal autophagy is activated in Atm knock-out mouse brains (51). It is possible that basal autophagy is also activated and genotoxins deregulate activated autophagy, leading to aberrant degradation of polyubiquitylated proteins in human A-T cells. To test this hypothesis, we first examined the status of endogenous LC3 puncta, a biological marker commonly used to trace induction of autophagy in mammalian cells (52–54). As shown in Fig. 2A, A-T cells showed a significant increase in LC3 puncta as compared with ATM+ cells (left panels). The bar graph in the right panel shows the mean number ± S.E. of LC3-puncta counted per cell using ImageJ software. These results revealed that, like in Atm knock-out mice, basal autophagy is activated in human A-T cells that are impaired in the ubiquitin pathway.
FIGURE 2.

Basal autophagy is activated in A-T cells. A, representative immunofluorescence images of LC3 puncta in A-T and ATM+ cells are shown (left) (scale bar, 10 μm); the average number ± S.E. (error bars) of puncta counted in 50 cells in different fields is shown in the bar graph (right). B, representative images of A-T (panels 1–3) and ATM+ (panels 4–6) cells co-stained with Cyt-ID® and LysoTracker Red® dyes are shown (scale bar, 10 μm). C, green (autophagosomes; from Cyt-ID®-stained panels) and yellow (autophagolysosomes; from merged panels) dots in cells were counted manually using the ImageJ plug-in cell counter. The average number of dots/cell is shown in the bar graph. Experiments were repeated two times with similar results.
We also examined autophagy using Cyto-ID® and LysoTracker Red stains. Cyto-ID® selectively labels autophagic vacuoles (preautophagosomes, autophagosomes, and autophagolysosomes), and a fluorescent acidotropic probe, LysoTracker Red, labels acidic organelles, such as lysosomes and autophagolysosomes (54). The appearance of green dots indicated the formation of autophagosomes; red dots indicated lysosomes; and yellow dots in merged images (green dots that overlay red dots in merged images) indicated autophagolysosomes (autophagosomes fused with lysosomes). As shown in Fig. 2B, increased numbers of bright green (compare panels 1 and 4) and red (compare panels 2 and 5) dots were seen in A-T compared with ATM+ cells, suggesting increased autophagic activity in A-T cells. In addition, there was a significant increase in yellow dots, indicating that there was a marked constitutive increase in autophagolysosomes in A-T compared with ATM+ cells (compare panels 3 and 6 and the quantitative bar graph in Fig. 2C). Together, immunofluorescence data using anti-LC3 antibodies and Cyto-ID/LysoTrack Red dyes revealed that basal autophagy is activated in A-T cells. To the best of our knowledge, this is the first report demonstrating the detection of autophagolysosomes using a combination of the Cyto-ID® and LysoTrack Red dyes in live cells.
Degradation of Autophagy Substrates Is Deregulated in UV Light-exposed A-T Cells
Because degradation of proteasome substrates is deregulated in A-T cells, we assessed the degradation of the autophagy substrates (autophagic flux) LC3 and p62 in UV-exposed A-T and ATM+ cells (55). As shown in Fig. 3, UV-induced degradation of LC3 and p62 (Fig. 3, A and B, left panels, lanes 1 and 2) in A-T cells. The autophagy inhibitor Bafl failed to protect UV-mediated degradation of LC3 and p62 in A-T cells (Fig. 3, A and B, left panels, lanes 3 and 4). On the other hand, no apparent changes in LC3 and p62 levels were detected in ATM+ cells treated with UV in the absence or presence of Bafl (Fig. 3, A and B, right panels). The bar graphs (far right panels) show average ± S.E. degradation of LC3 (LC3-I and -II) and p62 proteins in UV-exposed A-T and ATM+ cells treated with Bafl from three independent experiments. These results revealed that, like the proteasome substrates (Fig. 1), UV also induces aberrant degradation of autophagy substrates in A-T cells.
FIGURE 3.

Genotoxic stress induces aberrant autophagic flux in A-T cells; Western blot analysis. A and B, A-T and ATM+ cells were treated with Bafl (1 nm for 18 h) and then exposed to UV as indicated (25 mJ/m2). 3 h after recovery in the presence of inhibitors, cells were lysed. Cell lysates were analyzed by Western blotting for LC3 (A), p62 (B), and actin (bottom panels in A and B) using their specific antibodies. Intensity of the total LC3 (LC3-I + II) and p62 proteins was quantitated using Bio-Rad Quantity One software. The bar graphs in A and B show average values ± S.E. (error bars) of percentage degradation of LC3 and p62 from three independent experiments. All control values (−UV and +Bafl) are normalized to 100%, and values for experimental treatments are expressed as percentage variations over control.
We also monitored autophagic flux using Cyto-ID® and LysoTrack Red dyes. Immunofluorescence results are shown in Fig. 4A, and the quantitation of the immunofluorescence data is shown in Fig. 4B. The appearance of yellow dots in merged images (green dots (autophagosomes) that overlay red dots (lysosomes) in merged images) indicated the formation of autophagolysosomes in UV/Bafl-treated/untreated cells. The green dots that do not overlay red dots and appear as green in merged images indicated a failure of fusion between autophagosomes and autolysosomes in UV/Bafl-treated/untreated cells. A decrease in green, red, and yellow dots was taken as an indication of increased autophagic flux in UV/Bafl-treated/untreated cells as the autophagolysosomes break down and disappear at the end of autophagy. As shown in Fig. 4, A-T cells displayed an increased number of green (compare panels 1 and 13), bright red (compare panels 3 and 15), and yellow (compare panels 5 and 17) dots as compared with ATM+ cells. These results revealed that autophagic activity is increased in A-T cells. However, upon UV treatment, all green (panel 2), red (panel 4), and yellow (panel 6) dots disappeared in A-T cells. In contrast, the number of green (panel 14), red (panel 16), and yellow (panel 18) dots was markedly increased in UV-treated ATM+ cells (compare quantitative bar graphs in B (i and iv)). The disappearance of the autophagic organelles in A-T and appearance of the autophagic organelles in ATM+ cells revealed that UV induces autophagic flux in A-T but not in ATM+ cells.
FIGURE 4.
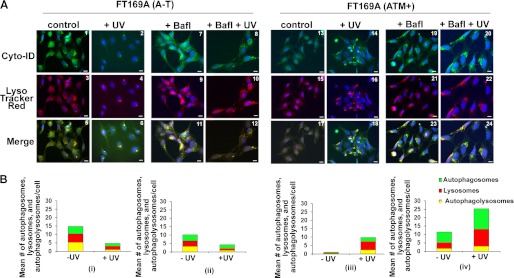
Genotoxic stress induces aberrant autophagic flux in A-T cells; immunofluorescence analysis. A, A-T (panels 1–12) and ATM+ (panels 13–24) cells were treated with Bafl (1 nm for 18 h) and then exposed to UV (25 mJ/m2) as indicated. 3 h after recovery in the presence of inhibitors, cells were co-stained with Cyt-ID® and LysoTracker Red® dyes. Representative fluorescence images of Cyt-ID®- and LysoTracker Red®-stained cells are shown (scale bar, 10 μm). B, green (autophagosomes; from Cyt-ID®-stained panels), red (lysosomes; from LysoTracker Red-stained panels), and yellow (autophagolysosomes; from merged panels) dots in A-T (left panel) and ATM+ (right panel) cells were counted manually using the ImageJ plug-in Cell Counter. The mean number of dots/cell is shown in the bar graphs. Experiments were repeated two times with similar results.
Bafilomycin inhibits autophagic flux by blocking fusion between autophagosomes and autolysosomes. We therefore expected a decreased appearance of yellow dots in cells treated with Bafl. Surprisingly, we consistently found increased yellow dots in Bafl-treated A-T cells as compared with the Bafl-treated ATM+ cells (compare panels 11 and 23). In contrast, more green dots were seen in bafilomycin-treated ATM+ cells as compared with A-T cells (compare panels 7 and 19). These results suggested that Bafl blocked fusion between autophagosomes and lysosomes in ATM+ cells but failed to do so in A-T cells.
Additionally, as shown in Fig. 4A, UV/Bafl co-treatment decreased both green (compare panels 7 and 8) and yellow (compare panels 11 and 12) dots in A-T cells as compared with A-T cells treated with Bafl alone. The disappearance of yellow dots representing autophagolysosomes in panel 12 suggested that UV induced autophagic flux in A-T cells, and Bafl failed to protect autophagic flux in UV/Bafl-treated A-T cells. Because lysosomal number and size decreases upon autophagy maturation, the decrease in lysosomal red dots in UV-treated A-T cells (compare panels 3 and 4) further supports our conclusion that UV-mediated induction of autophagy leads to increased autophagic flux in A-T cells. Although UV/Bafl co-treatment increased green dots (compare panels 19 and 20), the number of yellow dots (compare panels 23 and 24) remained unaltered in UV/Bafl-treated ATM+ cells as compared with ATM+ cells treated with Bafl alone. This result suggested that UV induced formation of autophagosomes, but Bafl blocked their fusion with lysosomes (i.e. formation of autophagolysosomes) in ATM+ cells. In addition, the unaltered number of yellow dots in Bafl-treated versus UV/Bafl-treated ATM+ cells (panels 23 and 24) provided evidence that UV did not induce autophagic flux in Bafl-treated ATM+ cells.
Together, results using A-T and ATM+ cells revealed that (a) UV induces aberrant degradation of the proteasome substrates in A-T cells; (b) basal autophagy is activated in A-T cells; (c) UV induces aberrant autophagic flux in A-T cells; (d) bafilomycin blocked formation of autophagolysosomes and, consequently, autophagic flux in UV-treated ATM+ cells; and (e) bafilomycin failed to block fusion between autophagosomes and lysosomes, resulting in sustained formation of autophagolysosomes and, thus, increased autophagic flux in UV-treated A-T cells. Similar findings were observed in cells treated with another autophagy inhibitor, NH4Cl, in A-T cells (data not shown).
Induction of Basal Autophagy Is a Consequence of Constitutively Elevated ISG15 in A-T Cells
Previously, we have shown that ISG15 siRNA restores impaired proteasome function, suggesting the involvement of the constitutively elevated ISG15 pathway in inhibiting the ubiquitin pathway in A-T cells (20). We speculated that if induction of basal autophagy compensates ISG15-impaired proteasome function, ISG15 siRNA should restore the proteasome function and suppress activated autophagy in A-T cells. To test whether this is indeed the case, we generated stable clones of FT169A (A-T) cells expressing lentiviral ISG15 shRNA (A-T/LV-ISG15 shRNA) or control shRNA (A-T/LV-control shRNA). The Western blot in Fig. 5A shows efficient knockdown of ISG15 expression in A-T/LV-ISG15 shRNA cells.
FIGURE 5.

Autophagy activation is a consequence of the elevated expression of the ISG15 pathway in A-T cells. A, extracts of A-T/LV-control and ISG15 shRNA cells were analyzed by Western blotting for ISG15 and actin. B, representative immunofluorescence images of LC3 puncta in A-T/control (left) and ISG15 (right) shRNA cells are shown (scale bar, 10 μm). C, representative images of A-T/control (panels 1–3) and ISG15 (panels 4–6) shRNA cells co-stained with Cyt-ID® and LysoTracker Red® (red; for lysosomes) dyes are shown. Yellow color in the merged images indicates autophagolysosomes. Scale bar, 10 μm. The mean number of green (autophagosomes) and yellow (autophagolysosomes) dots/cell counted manually using the ImageJ plug-in Cell Counter is shown in the bar graphs. Experiments were repeated two times with similar results.
To test if the autophagy pathway is restored, we measured LC3 puncta in ISG15-silenced A-T cells. As shown in Fig. 5B, A-T/LV-control shRNA cells exhibited an increased number of LC3-positive puncta (average number of 67 puncta/cell) as compared with A-T/LV-ISG15 shRNA cells (average number of 5 puncta/cell). These results revealed that basal autophagy is activated and that activated autophagy is due to the elevated expression of ISG15 in A-T cells.
To further test whether silencing of ISG15 expression attenuated autophagy, we stained these cells with Cyto-ID® and LysoTracker Red dye as described in Fig. 2. The same criteria were used to judge autophagic activity in immunofluorescence analysis as described in Fig. 2. As shown in Fig. 5C, a decreased number of green and yellow dots was seen in A-T/LV-ISG15 shRNA (panels 4 and 6 and quantitative bar graph in Fig. 5C) as compared with A-T/LV-control shRNA (panels 1 and 3 and quantitative bar graph in Fig. 5C) cells, suggesting attenuation of autophagic activity in A-T/ISG15-shRNA cells. Together, immunofluorescence data using anti-LC3, Cyto-ID, and LysoTrack Red dyes revealed that, as in FT169A (A-T) cells (Fig. 3), basal autophagic activity is increased in A-T/LV-control shRNA cells, and activated autophagy is due to the elevated expression of ISG15 in A-T/LV-control cells.
Degradation of Autophagy Substrates Is Restored in the ISG15-silenced A-T Cells
We next assessed whether ISG15 gene knockdown restores autophagy and rescues UV-induced autophagic flux in A-T/LV-control/ISG15-shRNA stable clones. We found that UV also induced MG132- and Bafl-resistant degradation of LC3 and p62 in A-T/control-shRNA cells but not in A-T/ISG15-shRNA cells (Fig. 6, A and B). The bar graphs in Fig. 6, A and B (far right panels), show average ± S.E. degradation of LC3 (LC3-I and -II) and p62 proteins in UV-exposed A-T/LV-control/ISG15 shRNA cells treated with Bafl from three independent experiments. These results further revealed that the constitutively elevated ISG15 pathway contributes to the UV-induced aberrant autophagic flux in A-T cells.
FIGURE 6.

UV-induced autophagic flux is attenuated in ISG15-silenced A-T cells; Western blot analysis. A and B, A-T/LV-control and ISG15 shRNA cells were treated with Bafl (1 nm for 18 h) or left untreated. Cells were then exposed to UV (25 mJ/m2). 3 h after recovery in the presence of inhibitors, cells were lysed, and lysates were analyzed by Western blotting for LC3 (A), p62 (B), and actin (bottom panels of A and B) using their specific antibodies. Intensity of the total LC3 (LC3-I + II) and p62 proteins was quantitated using Bio-Rad Quantity One software. The bar graphs in A and B show average values ± S.E. (error bars) of percentage degradation of LC3 and p62 from three independent experiments. All control values (−UV and +Bafl) are normalized to 100%, and values for experimental treatments were expressed as percentage variations over control.
We also assessed autophagic flux using Cyto-ID® and LysoTrack Red dyes as described in the legend to Fig. 4. The same criteria were used to judge autophagic activity in immunofluorescence analysis as described for Fig. 4. As seen in Fig. 7A, UV induced disappearance of autophagosomes (green dots) and autophagolysosomes (yellow dots) in bafilomycin-untreated (compare panels 1 and 2 (green dots), panels 5 and 6 (yellow dots), and quantitative bar graph) and bafilomycin-treated (compare panels 7 and 8 (green dots), panels 11 and 12 (yellow dots), and quantitative bar graph) A-T/control shRNA cells. Because the disappearance of autophagolysosomes indicates increased autophagic flux, these results suggested that bafilomycin failed to protect UV-mediated autophagic flux in A-T/LV-control shRNA cells. In agreement, in Western analysis, UV treatment induced degradation of HA-polyubiquitylated proteins in bafilomycin-untreated (Fig. 7B) and -treated (Fig. 7C) A-T/LV-control cells. This degradation was not due to the proteasome because MG132, a proteasome inhibitor, failed to block UV-mediated degradation of proteins in A-T/LV-control shRNA cells (Fig. 7D).
FIGURE 7.

Inhibition of autophagy at an early stage using 3-MA blocks UV-induced aberrant autophagic flux in A-T cells. A, A-T/LV-control shRNA cells were either left untreated (panels 1, 3, and 5) or treated with Bafl (1 nm for 18 h) (panels 7, 9, and 11) or treated with 3-MA (10 nm for 18 h) (panels 13, 15, and 17). Cells were then exposed to UV (25 mJ/m2) (panels 2, 4, 6, 8, 10, 12, 14, 16, and 18). 3 h after recovery in the presence of the respective inhibitors, cells were co-stained with Cyt-ID® and LysoTracker Red® dyes. Representative fluorescence images of Cyt-ID®- and LysoTracker Red®-stained cells are shown. The mean number of green (autophagosomes) and yellow (autophagolysosomes) dots/cell counted manually using the ImageJ plug-in Cell Counter is shown in the bar graphs. Experiments were performed twice and yielded similar results (scale bar, 10 μm). B, HA-ubiquitin-transfected A-T/LV-control shRNA cells were exposed to UV (25 mJ/m2). After 3 h of recovery, assessment of HA-polyubiquitylated proteins was carried out as described in the legend to Fig. 1B. C, HA-ubiquitin-transfected A-T/LV-control shRNA cells were treated with Bafl (1 nm for 18 h). Cells were then exposed to UV (25 mJ/m2). After 3 h of recovery in the presence of the inhibitor, assessment of HA-polyubiquitylated proteins was carried out as described in B. D, HA-ubiquitin-transfected A-T/LV-control shRNA cells were treated with MG132 (1 nm for 18 h). Cells were then exposed to UV (25 mJ/m2). After 3 h of recovery in the presence of the inhibitor, assessment of HA-polyubiquitylated proteins was carried out as described in B. E, HA-ubiquitin-transfected A-T/LV-control shRNA cells were treated with 3-MA (10 nm for 18 h). Cells were then exposed to UV (25 mJ/m2). After 3 h of recovery in the presence of the inhibitor, assessment of HA-polyubiquitylated proteins was carried out as described in B. All experiments shown in B–E were performed at least three times and yielded similar results.
Bafilomycin inhibits autophagy at a late stage (47). We therefore tested whether 3-MA, an autophagy inhibitor known to inhibit autophagy at an early stage by inhibiting formation of autophagosomes (56), could block UV-induced autophagic flux in A-T/LV-control shRNA cells. As shown in Fig. 7A, there was a marked decrease in both green (compare panels 1 and 13 and quantitative bar graph) and yellow (compare panels 5 and 17 and quantitative bar graph) dots, suggesting decreased formation of autophagosomes and autophagolysosomes in 3-MA-treated A-T/LV-control shRNA cells. Moreover, both green dots and yellow dots remained unaltered in A-T/LV-control shRNA cells co-treated with 3-MA and UV (compare panels 13 and 14, panels 17 and 18, and quantitative bar graphs). These results suggested that 3-MA blocked autophagic activity and UV-mediated autophagic flux in A-T/LV-control shRNA cells. Consistent with these results, UV-induced degradation of HA-polyubiquitylated proteins was markedly blocked in 3-MA/UV-treated A-T/LV/control shRNA cells (Fig. 7E). Our results using 3-MA and Bafilomycin thus revealed that UV overactivates autophagy in A-T cells. Bafilomycin is unable to block the overeactivated autophagy, leading to aberrant autophagic flux in A-T cells. In contrast, 3-MA, which inhibits autophagosome formation, markedly blocked autophagic flux in A-T cells.
We next tested the effect of 3-MA on UV-induced autophagic flux in A-T/LV-ISG15 shRNA cells. Results are shown in Fig. 8. No apparent change was noted in autophagic activity in ISG15-silenced A-T and 3-MA-treated ISG15-silenced A-T cells exposed to UV (compare panels 1 and 2, 5 and 6, 7 and 8, and 11 and 12 and quantitative bar graphs). However, although autophagy was attenuated, UV induced degradation of HA-polyubiquitylated proteins in both 3-MA-untreated and -treated ISG15-silenced cells (Fig. 8, B and C). We presumed that the degradation of HA-polyubiquitylated proteins could be due to the restoration of proteasome function in ISG15-silenced A-T cells. Indeed, we found that the MG132 proteasome inhibitor completely blocked degradation of HA-polyubiquitylated proteins, suggesting that UV-mediated degradation of HA-polyubiquitylated proteins in 3-MA-treated A-T cells was due to their degradation via the proteasome in ISG15-silenced A-T cells.
FIGURE 8.

In the absence of the functional autophagy pathway, ubiquitylated proteins are degraded via proteasome in UV-treated ISG15-silenced A-T cells. A, A-T/LV-ISG15 shRNA cells were either left untreated (panels 1, 3, and 5) or treated with 3-MA (10 nm for 18 h) (panels 7, 9, and 11). Cells were then exposed to UV (25 mJ/m2) (panels 2, 4, 6, 8, 10, and 12). 3 h after recovery in the presence of the inhibitor, cells were co-stained with Cyt-ID® and LysoTracker Red® dyes. Representative fluorescence images of Cyt-ID®- and LysoTracker Red®-stained cells are shown. The mean number of green (autophagosomes) and yellow (autophagolysosomes) dots/cell counted manually using the ImageJ plug-in Cell Counter is shown in the bar graphs. Experiments was performed twice and yielded similar results (scale bar, 10 μm). B, HA-ubiquitin-transfected A-T/LV-ISG15 shRNA cells were exposed to UV (25 mJ/m2). After 3 h of recovery, assessment of HA-polyubiquitylated proteins was carried out as described in the legend to Fig. 1B. C, HA-ubiquitin-transfected A-T/LV-ISG15 shRNA cells were treated with 3-MA (10 nm for 18 h). Cells were then exposed to UV (25 mJ/m2). After 3 h of recovery in the presence of the inhibitor, assessment of HA-polyubiquitylated proteins was carried out as described in B. D, HA-ubiquitin-transfected A-T/LV-control shRNA cells were treated with MG132 (1 nm for 18 h). Cells were then exposed to UV (25 mJ/m2). After 3 h of recovery in the presence of inhibitor, assessment of HA-polyubiquitylated proteins was carried out as described in B. All experiments shown in B–D were performed at least three times and yielded similar results.
Together, our results revealed that autophagy is activated in A-T cells, presumably to compensate for the impaired proteasome function in A-T cells. Genotoxic stress overactivates this compensatory mechanism, triggering aberrant autophagic flux in A-T cells. 3-MA attenuated overeactivated autophagy and therefore resulted in attenuation of autophagic flux in genotoxin-treated autophagy.
The Autophagy Pathway Is Activated in Brains of Human A-T Patients
Astroglial cell dysfunction has been implicated in the pathogenesis of various neurological disorders (57), and ISG15 is elevated in A-T astrocytes (20). We therefore looked for evidence of autophagy induction in the A-T human brains. As shown in Fig. 9A, a dramatic increase in LC3 (autophagy marker)/GFAP (astrocyte marker) double-positive stained inclusions was seen in the mid-brain sections obtained from the A-T patient (Fig. 9A, right) (see green (LC3) and red (GFAP) color turning to yellow (GFAP and LC3 double inclusions) in merged images (see arrows)). Although LC3/GFAP inclusions were also present in brain sections of the normal individual (left), the intensity of the LC3/GFAP double-positive staining was much higher in the brain section of the A-T patient as compared with the normal individual. Similar increases in the LC3/GFAP double-positive staining were noted in the brain sections of the two other A-T patients (data not shown).
FIGURE 9.

Evidence for the activation of autophagy in human brains of A-T patients. A, the deparaffinized human brain tissue sections from the normal subject and A-T patient were double-stained with anti-LC3- and anti-GFAP-specific antibodies (scale bar, 100 μm). B, frozen mid-brain post-mortem tissue lysates were analyzed by Western blotting using anti-LC3 antibodies. The positive control for anti-LC3 protein (HA-tagged) (MBL International) was loaded in lane 1.
We also examined tissue lysates of mid-brain regions (specifically containing substantia nigra) obtained post-mortem from A-T human patients with confirmed A-T disease for autophagy induction by Western blotting using anti-LC3 antibodies (Fig. 9B). The presence of the LC3-II form in brain tissue lysates is indicative of a strong induction of autophagy in these patients, as LC3-II form is an indicator of an active autophagy. Together, these results suggested that autophagy is activated/impaired in A-T patients.
DISCUSSION
Proteinopathies caused by molecular lesions in the ubiquitin pathway leading to the neuronal cell death are common in many neurological disorders (15, 16) and recently were implicated in A-T neurodegeneration (18–20). It has been demonstrated that the elevated expression of ISG15, an antagonist of the ubiquitin/26S proteasome pathway (20, 24), impairs the ubiquitin-mediated turnover of cellular proteins in A-T fibroblast cells (20). Hence, it was suggested that, as in other neurological disorders, ISG15-mediated proteinopathy in A-T neurons could lead to their death in A-T patients. The presence of ubiquitin-ISG15 inclusions in brain sections obtained post-mortem from A-T human patients point toward such a possibility (20). In the current study, we show that autophagy is activated in the cells and brains of A-T patients. Interestingly, although a marked increase in LC3-positive inclusions was noted in the brain section of the A-T patient 1459 in immunofluorescence analysis (Fig. 9A), a moderate increase in the LC3-IIp was detected in tissue lysates of the same patient in Western analysis (Fig. 9B, lane 3). Two possibilities could explain these conflicting results: (a) LC3 proteins present in the inclusions are insoluble in the SDS sample buffer and, consequently, are not detected on the Western blot, and/or (b) LC3 protein is degraded during processing of the brain tissue. Nonetheless, the expression of LC3 protein in both immunofluorescence and immunoblotting analysis in this A-T patient reveals that the autophagy pathway is activated in A-T patient 1459 and others. We have previously demonstrated that ISG15 and its conjugates are highly elevated in brain tissue lysates of the A-T patients 1459, 1722, and 4663 shown in Fig. 9B (20). Our current finding that autophagy is activated in the same patients suggest that the induction of autophagy could be the consequence of the ISG15-mediated impairment of the proteasome pathway in the brain of these patients. Similar to A-T, ISG15 is also elevated in the amyotrophic lateral sclerosis neurological disorder (58). Hence, it appears that “ISG15 proteinopathy” may be a common underlying cause of neurological disorders, thus making our current study more significant.
The accumulation of non-degraded proteins has been shown to limit the viability of cells growing in culture in other studies (59). Cellular substrates of the proteasome are also stabilized in A-T cells for which the ubiquitin-proteasome pathway has been compromised by ISG15 overexpression (20). However, A-T cells generally grow normally in culture, suggesting that the non-degraded proteins are eventually removed by an alternate mechanism. Our current findings suggest that the basally activated autophagy may limit the accumulation of non-degraded ubiquitin targets, thus preserving the viability of the A-T cells. This result is consistent with the finding by Barlow et al. (60) that the number of lysosomes is increased in the Atm−/− mouse brains and the finding by Valentin-Vega et al. (51) that basal autophagy is activated in A-T mice brains. Our other findings, of the presence of the ISG15/ubiquitin inclusions (20) and activated autophagy in A-T patient brains, in part have now extrapolated these results in the human model.
Bafilomycin treatment normally accumulates LC3II and p62 proteins in mammalian cells. However, we did not see accumulation of these autophagy substrates in A-T and ATM+ cells treated with bafilomycin alone. However, the same concentration of bafilomycin used in this experiment led to the accumulation of LC3 II in the serum-starved A-T and ATM+ cells.3 These results have revealed the effectiveness of bafilomycin to inhibit induced autophagy A-T cells. Note that there is a difference between induced autophagy and basal autophagy. The induced autophagy is used to produce amino acids following starvation, whereas the basal autophagy is important for constitutive turnover of cytosolic components. It has been demonstrated that induced autophagic activity or total proteolysis is not sustained and decreases during prolonged starvation (35). It is possible that due to the decreased rate of autophagic activity, LC3-II accumulates in serum-starved (starved for 24 h) A-T cells treated with bafilomycin. On the other hand, under normal serum conditions (conditions used in the current work), basal autophagy is sustained, consequently leading to continuous degradation of LC3 in bafilomycin-treated A-T cells. The reason for the failure to accumulate these proteins in bafilomycin-treated ATM+ cells is still not clear, but it could be due to the residual autophagic activity remaining in ATM+ cells; ATM+ cells are derived from A-T cells. Nevertheless, we consistently found a significant amount of LC3II and p62 degradation in UV-treated A-T as compared with ATM+ cells, suggesting increased autophagic flux in UV-treated A-T cells.
Although autophagy is activated, the steady state levels of polyubiquitylated proteins remained unaltered in metabolically stressed (nutrition-deprived) A-T cells.3 In contrast, genotoxins induced aberrant degradation of polyubiquitylated proteins in A-T cells. These results suggest that A-T cells that are impaired in the ubiquitin pathway are able to handle metabolic stress but are unable to cope with the genotoxic stress. It is possible that genotoxic stress (e.g. UV) places a greater burden on autophagy through the accumulation of the non-degraded ubiquitylated proteins in the ubiquitin pathway-compromised A-T cells. Several cellular proteins are ubiquitylated in response to genotoxin treatment. We have shown that the ubiquitin-mediated degradation of topoisomerase I is inhibited in camptothecin (anticancer genotoxic agent)-treated A-T cells (37). Several other proteins (e.g. RNA polymerase II0 and Mcm10) are ubiquitylated and degraded in response to UV treatment (61, 62). Whether the targeted degradation of these proteins is impaired in UV-treated A-T cells is not studied; however, the published results in different cell lineages described above point toward such a possibility. The inability of the dysfunctional ubiquitin pathway to degrade these UV-damaged proteins may increase autophagic demand to degrade these proteins, resulting in overinduction of autophagy in UV-exposed A-T cells. Increased autophagic flux may reduce cellular reserves and hence induce autophagic stress and A-T cell death. Concurrently, autophagy is activated (15, 63, 64), and autophagic stress in neurons leads to neuronal cell death in various proteinopathy-induced neuronal disorders (65, 66).
Our results are highly relevant for improving the health status of A-T patients who are constantly exposed to environmental genotoxic agents, such as sunlight, viral infections, high temperature, and human-made mutagenic chemicals, during their lifetime. In addition, A-T patients are also vulnerable to oxidative stress (67), which can also lead to protein damage. Hence, it is reasonable that genotoxic agents and oxidative stress can induce autophagic stress in A-T neurons, which, in turn, lead to their autophagic death. Thus far, the hypersensitivity to the genotoxic stress has been principally linked to defective DNA repair in A-T. Our current results suggest that in addition to the deregulated DNA repair, deregulation of the protein turnover may in part also contribute to the genotoxic stress-mediated hypersensitivity in A-T patients. Knowing that constitutively elevated ISG15 is causally related to the deregulation of both of the major protein turnover pathways in A-T fibroblast cells provides an opportunity to target the ISG15 pathway to reduce neurodegeneration and ataxia associated with it in A-T patients. Also, attenuating autophagy with the pharmacological inhibitors (e.g. 3-MA) could prevent neurodegeneration in A-T. These studies thus have potential clinical applications for treating A-T patients. This is particularly important because there is no cure or preventive medicine available for treating A-T.
Acknowledgment
We thank Dr. Arthur Haas for carefully going through the manuscript and providing insightful comments.
This work was supported, in whole or in part, by National Institutes of Health, NINDS, Grant R21NS060960 (to S. D.). This work was also supported by an A-T Children's Project Foundation grant and Louisiana State University Health Sciences Center start-up funds (to S. D.).
S. D. Desai, unpublished data.
- A-T
- ataxia telangiectasia
- ATM
- ataxia telangiectasia mutated
- 3-MA
- 3-methyladenine
- Bafl
- bafilomycin A1
- CPT
- camptothecin
- GFAP
- glial fibrillary acidic protein.
REFERENCES
- 1. Frappart P. O., McKinnon P. J. (2006) Ataxia-telangiectasia and related diseases. Neuromolecular Med. 8, 495–511 [DOI] [PubMed] [Google Scholar]
- 2. Lavin M. F., Khanna K. K. (1999) ATM. The protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int. J. Radiat. Biol. 75, 1201–1214 [DOI] [PubMed] [Google Scholar]
- 3. Boder E. (1985) Ataxia-telangiectasia. An overview. Kroc Found. Ser. 19, 1–63 [PubMed] [Google Scholar]
- 4. Chun H. H., Gatti R. A. (2004) Ataxia-telangiectasia, an evolving phenotype. DNA Repair 3, 1187–1196 [DOI] [PubMed] [Google Scholar]
- 5. Easton D. F. (1994) Cancer risks in A-T heterozygotes. Int. J. Radiat. Biol. 66, S177–S182 [DOI] [PubMed] [Google Scholar]
- 6. Sun X., Becker-Catania S. G., Chun H. H., Hwang M. J., Huo Y., Wang Z., Mitui M., Sanal O., Chessa L., Crandall B., Gatti R. A. (2002) Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J. Pediatr. 140, 724–731 [DOI] [PubMed] [Google Scholar]
- 7. Taylor A. M., Harnden D. G., Arlett C. F., Harcourt S. A., Lehmann A. R., Stevens S., Bridges B. A. (1975) Ataxia telangiectasia. A human mutation with abnormal radiation sensitivity. Nature 258, 427–429 [DOI] [PubMed] [Google Scholar]
- 8. Lavin M. F., Scott S., Gueven N., Kozlov S., Peng C., Chen P. (2004) Functional consequences of sequence alterations in the ATM gene. DNA Repair 3, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 9. Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D. A., Smith S., Uziel T., Sfez S., Ashkenazi M., Pecker I., Frydman M., Harnik R., Patanjali S. R., Simmons A., Clines G. A., Sartiel A., Gatti R. A., Chessa L., Sanal O., Lavin M. F., Jaspers N. G., Taylor A. M., Arlett C. F., Miki T., Weissman S. M., Lovett M., Collins F. S., Shiloh Y. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753 [DOI] [PubMed] [Google Scholar]
- 10. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 11. Shiloh Y., Rotman G. (1996) Ataxia-telangiectasia and the ATM gene. Linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints. J. Clin. Immunol. 16, 254–260 [DOI] [PubMed] [Google Scholar]
- 12. Katyal S., McKinnon P. J. (2008) DNA strand breaks, neurodegeneration and aging in the brain. Mech. Ageing Dev. 129, 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biton S., Barzilai A., Shiloh Y. (2008) The neurological phenotype of ataxia-telangiectasia. Solving a persistent puzzle. DNA Repair 7, 1028–1038 [DOI] [PubMed] [Google Scholar]
- 14. Rolig R. L., McKinnon P. J. (2000) Linking DNA damage and neurodegeneration. Trends Neurosci. 23, 417–424 [DOI] [PubMed] [Google Scholar]
- 15. Ross C. A., Pickart C. M. (2004) The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 14, 703–711 [DOI] [PubMed] [Google Scholar]
- 16. Schmitt H. P. (2006) Protein ubiquitination, degradation and the proteasome in neuro-degenerative disorders. No clear evidence for a significant pathogenetic role of proteasome failure in Alzheimer disease and related disorders. Med. Hypotheses 67, 311–317 [DOI] [PubMed] [Google Scholar]
- 17. Ciechanover A. (2005) Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover. Interview by CDD. Cell Death Differ. 12, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 18. Eilam R., Peter Y., Groner Y., Segal M. Late degeneration of nigro-striatal neurons in ATM−/− mice. Neuroscience 121, 83–98 [DOI] [PubMed] [Google Scholar]
- 19. Agamanolis D. P., Greenstein J. I. (1979) Ataxia-telangiectasia. Report of a case with Lewy bodies and vascular abnormalities within cerebral tissue. J. Neuropathol. Exp. Neurol. 38, 475–489 [DOI] [PubMed] [Google Scholar]
- 20. Wood L. M., Sankar S., Reed R. E., Haas A. L., Liu L. F., McKinnon P., Desai S. D. (2011) A novel role for ATM in regulating proteasome-mediated protein degradation through suppression of the ISG15 conjugation pathway. PLoS One 6, e16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Narasimhan J., Potter J. L., Haas A. L. (1996) Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J. Biol. Chem. 271, 324–330 [DOI] [PubMed] [Google Scholar]
- 22. Zhang D., Zhang D. E. (2011) Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 31, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haas A. L., Ahrens P., Bright P. M., Ankel H. (1987) Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem. 262, 11315–11323 [PubMed] [Google Scholar]
- 24. Desai S. D., Haas A. L., Wood L. M., Tsai Y. C., Pestka S., Rubin E. H., Saleem A., Nur-E-Kamal A., Liu L. F. (2006) Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 66, 921–928 [DOI] [PubMed] [Google Scholar]
- 25. Lu G., Reinert J. T., Pitha-Rowe I., Okumura A., Kellum M., Knobeloch K. P., Hassel B., Pitha P. M. (2006) ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol. Biol. 52, 29–41 [PubMed] [Google Scholar]
- 26. Okumura A., Pitha P. M., Harty R. N. (2008) ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. U.S.A. 105, 3974–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malakhova O. A., Zhang D. E. (2008) ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 283, 8783–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takeuchi T., Yokosawa H. (2005) ISG15 modification of Ubc13 suppresses its ubiquitin-conjugating activity. Biochem. Biophys. Res. Commun. 336, 9–13 [DOI] [PubMed] [Google Scholar]
- 29. Zou W., Papov V., Malakhova O., Kim K. I., Dao C., Li J., Zhang D. E. (2005) ISG15 modification of ubiquitin E2 Ubc13 disrupts its ability to form thioester bond with ubiquitin. Biochem. Biophys. Res. Commun. 336, 61–68 [DOI] [PubMed] [Google Scholar]
- 30. Zou W., Wang J., Zhang D. E. (2007) Negative regulation of ISG15 E3 ligase EFP through its autoISGylation. Biochem. Biophys. Res. Commun. 354, 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pandey U. B., Batlevi Y., Baehrecke E. H., Taylor J. P. (2007) HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 3, 643–645 [DOI] [PubMed] [Google Scholar]
- 32. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., Yao T. P., Baehrecke E. H., Taylor J. P. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 33. Nedelsky N. B., Todd P. K., Taylor J. P. (2008) Autophagy and the ubiquitin-proteasome system. Collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rubinsztein D. C. (2007) Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron 54, 854–856 [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N. (2007) Autophagy. Process and function. Genes Dev. 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 36. Klionsky D. J., Emr S. D. (2000) Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Desai S. D., Wood L. M., Tsai Y. C., Hsieh T. S., Marks J. R., Scott G. L., Giovanella B. C., Liu L. F. (2008) ISG15 as a novel tumor biomarker for drug sensitivity. Mol. Cancer Ther. 7, 1430–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu L. F. (1989) DNA topoisomerase poisons as antitumor drugs. Annu. Rev. Biochem. 58, 351–375 [DOI] [PubMed] [Google Scholar]
- 39. Wu X., Rathbun G., Lane W. S., Weaver D. T., Livingston D. M. (2000) Interactions of the Nijmegen breakage syndrome protein with ATM and BRCA1. Cold Spring Harb. Symp. Quant. Biol. 65, 535–545 [DOI] [PubMed] [Google Scholar]
- 40. Desai S. D., Reed R. E., Burks J., Wood L. M., Pullikuth A. K., Haas A. L., Liu L. F., Breslin J. W., Meiners S., Sankar S. (2012) ISG15 disrupts cytoskeletal architecture and promotes motility in human breast cancer cells. Exp. Biol. Med. 237, 38–49 [DOI] [PubMed] [Google Scholar]
- 41. Pear W. S., Nolan G. P., Scott M. L., Baltimore D. (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 90, 8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomson T. M., Guerra-Rebollo M. (2010) Ubiquitin and SUMO signaling in DNA repair. Biochem. Soc. Trans. 38, 116–131 [DOI] [PubMed] [Google Scholar]
- 43. Lavin M. F., Birrell G., Chen P., Kozlov S., Scott S., Gueven N. (2005) ATM signaling and genomic stability in response to DNA damage. Mutat. Res. 569, 123–132 [DOI] [PubMed] [Google Scholar]
- 44. Loeb K. R., Haas A. L. (1992) The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J. Biol. Chem. 267, 7806–7813 [PubMed] [Google Scholar]
- 45. Wu W. K., Wu Y. C., Yu L., Li Z. J., Sung J. J., Cho C. H. (2008) Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 374, 258–263 [DOI] [PubMed] [Google Scholar]
- 46. Ge P. F., Zhang J. Z., Wang X. F., Meng F. K., Li W. C., Luan Y. X., Ling F., Luo Y. N. (2009) Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG-44 glioma cells. Acta Pharmacol. Sin. 30, 1046–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R., Tashiro Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42 [DOI] [PubMed] [Google Scholar]
- 48. Desai S. D., Li T. K., Rodriguez-Bauman A., Rubin E. H., Liu L. F. (2001) Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 61, 5926–5932 [PubMed] [Google Scholar]
- 49. Desai S. D., Liu L. F., Vazquez-Abad D., D'Arpa P. (1997) Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem. 272, 24159–24164 [DOI] [PubMed] [Google Scholar]
- 50. Desai S. D., Zhang H., Rodriguez-Bauman A., Yang J. M., Wu X., Gounder M. K., Rubin E. H., Liu L. F. (2003) Transcription-dependent degradation of topoisomerase I-DNA covalent complexes. Mol. Cell Biol. 23, 2341–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Valentin-Vega Y. A., Maclean K. H., Tait-Mulder J., Milasta S., Steeves M., Dorsey F. C., Cleveland J. L., Green D. R., Kastan M. B. (2012) Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., Bamber B. A., Bassham D. C., Bergamini E., Bi X., Biard-Piechaczyk M., Blum J. S., Bredesen D. E., Brodsky J. L., Brumell J. H., Brunk U. T., Bursch W., Camougrand N., Cebollero E., Cecconi F., Chen Y., Chin L. S., Choi A., Chu C. T., Chung J., Clarke P. G., Clark R. S., Clarke S. G., Clavé C., Cleveland J. L., Codogno P., Colombo M. I., Coto-Montes A., Cregg J. M., Cuervo A. M., Debnath J., Demarchi F., Dennis P. B., Dennis P. A., Deretic V., Devenish R. J., Di Sano F., Dice J. F., Difiglia M., Dinesh-Kumar S., Distelhorst C. W., Djavaheri-Mergny M., Dorsey F. C., Droge W., Dron M., Dunn W. A., Jr., Duszenko M., Eissa N. T., Elazar Z., Esclatine A., Eskelinen E. L., Fesus L., Finley K. D., Fuentes J. M., Fueyo J., Fujisaki K., Galliot B., Gao F. B., Gewirtz D. A., Gibson S. B., Gohla A., Goldberg A. L., Gonzalez R., Gonzalez-Estevez C., Gorski S., Gottlieb R. A., Haussinger D., He Y. W., Heidenreich K., Hill J. A., Hoyer-Hansen M., Hu X., Huang W. P., Iwasaki A., Jaattela M., Jackson W. T., Jiang X., Jin S., Johansen T., Jung J. U., Kadowaki M., Kang C., Kelekar A., Kessel D. H., Kiel J. A., Kim H. P., Kimchi A., Kinsella T. J., Kiselyov K., Kitamoto K., Knecht E., Komatsu M., Kominami E., Kondo S., Kovacs A. L., Kroemer G., Kuan C. Y., Kumar R., Kundu M., Landry J., Laporte M., Le W., Lei H. Y., Lenardo M. J., Levine B., Lieberman A., Lim K. L., Lin F. C., Liou W., Liu L. F., Lopez-Berestein G., Lopez-Otin C., Lu B., Macleod K. F., Malorni W., Martinet W., Matsuoka K., Mautner J., Meijer A. J., Melendez A., Michels P., Miotto G., Mistiaen W. P., Mizushima N., Mograbi B., Monastyrska I., Moore M. N., Moreira P. I., Moriyasu Y., Motyl T., Munz C., Murphy L. O., Naqvi N. I., Neufeld T. P., Nishino I., Nixon R. A., Noda T., Nurnberg B., Ogawa M., Oleinick N. L., Olsen L. J., Ozpolat B., Paglin S., Palmer G. E., Papassideri I., Parkes M., Perlmutter D. H., Perry G., Piacentini M., Pinkas-Kramarski R., Prescott M., Proikas-Cezanne T., Raben N., Rami A., Reggiori F., Rohrer B., Rubinsztein D. C., Ryan K. M., Sadoshima J., Sakagami H., Sakai Y., Sandri M., Sasakawa C., Sass M., Schneider C., Seglen P. O., Seleverstov O., Settleman J., Shacka J. J., Shapiro I. M., Sibirny A., Silva-Zacarin E. C., Simon H. U., Simone C., Simonsen A., Smith M. A., Spanel-Borowski K., Srinivas V., Steeves M., Stenmark H., Stromhaug P. E., Subauste C. S., Sugimoto S., Sulzer D., Suzuki T., Swanson M. S., Tabas I., Takeshita F., Talbot N. J., Talloczy Z., Tanaka K., Tanida I., Taylor G. S., Taylor J. P., Terman A., Tettamanti G., Thompson C. B., Thumm M., Tolkovsky A. M., Tooze S. A., Truant R., Tumanovska L. V., Uchiyama Y., Ueno T., Uzcategui N. L., van der Klei I., Vaquero E. C., Vellai T., Vogel M. W., Wang H. G., Webster P., Wiley J. W., Xi Z., Xiao G., Yahalom J., Yang J. M., Yap G., Yin X. M., Yoshimori T., Yu L., Yue Z., Yuzaki M., Zabirnyk O., Zheng X., Zhu X., Deter R. L. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Komatsu M., Ichimura Y. (2010) Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 584, 1374–1378 [DOI] [PubMed] [Google Scholar]
- 56. Seglen P. O., Gordon P. B. (1982) 3-Methyladenine. Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maragakis N. J., Rothstein J. D. (2006) Mechanisms of disease. Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689 [DOI] [PubMed] [Google Scholar]
- 58. Wang R., Yang B., Zhang D. (2011) Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59, 946–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Figueiredo-Pereira M. E., Cohen G. (1999) The ubiquitin/proteasome pathway. Friend or foe in zinc-, cadmium-, and H2O2-induced neuronal oxidative stress. Mol. Biol. Rep. 26, 65–69 [DOI] [PubMed] [Google Scholar]
- 60. Barlow C., Ribaut-Barassin C., Zwingman T. A., Pope A. J., Brown K. D., Owens J. W., Larson D., Harrington E. A., Haeberle A. M., Mariani J., Eckhaus M., Herrup K., Bailly Y., Wynshaw-Boris A. (2000) ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc. Natl. Acad. Sci. U.S.A. 97, 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bregman D. B., Halaban R., van Gool A. J., Henning K. A., Friedberg E. C., Warren S. L. (1996) UV-induced ubiquitination of RNA polymerase II. A novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11586–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sharma A., Kaur M., Kar A., Ranade S. M., Saxena S. (2010) Ultraviolet radiation stress triggers the down-regulation of essential replication factor Mcm10. J. Biol. Chem. 285, 8352–8362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Metcalf D. J., García-Arencibia M., Hochfeld W. E., Rubinsztein D. C. (2012) Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 238, 22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lehman N. L. (2009) The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 118, 329–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cherra S. J., 3rd, Chu C. T. (2008) Autophagy in neuroprotection and neurodegeneration. A question of balance. Future Neurol. 3, 309–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chu C. T. (2006) Autophagic stress in neuronal injury and disease. J. Neuropathol. Exp. Neurol. 65, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guo Z., Kozlov S., Lavin M. F., Person M. D., Paull T. T. (2010) ATM activation by oxidative stress. Science 330, 517–521 [DOI] [PubMed] [Google Scholar]