

Background: mRNA decay releases, in addition to the regular nucleotides, 7-methyl GMP derived from the 5′ cap.
Results: We describe new members of the 5′ nucleotidase family degrading 7-methyl GMP to 7-methylguanosine and orthophosphate.
Conclusion: Cells have mechanisms to prevent potential salvage of 7-methyl GMP.
Significance: 7-Methyl GMP degradation may be important to prevent its incorporation into nucleic acids.
Keywords: Enzymes, mRNA Decay, Nucleic Acid, Nucleoside Nucleotide Metabolism, RNA Turnover
Abstract
Turnover of mRNA releases, in addition to the four regular nucleoside monophosphates, the methylated cap nucleotide in the form of 7-methylguanosine monophosphate (m7GMP) or diphosphate (m7GDP). The existence of pathways to eliminate the modified nucleotide seems likely, as its incorporation into nucleic acids is undesirable. Here we describe a novel 5′ nucleotidase from Drosophila that cleaves m7GMP to 7-methylguanosine and inorganic phosphate. The enzyme, encoded by the predicted gene CG3362, also efficiently dephosphorylates CMP, although with lower apparent affinity; UMP and the purine nucleotides are poor substrates. The enzyme is inhibited by elevated concentrations of AMP and also cleaves m7GDP to the nucleoside and two inorganic phosphates, albeit less efficiently. CG3362 has equivalent sequence similarity to two human enzymes, cytosolic nucleotidase III (cNIII) and the previously uncharacterized cytosolic nucleotidase III-like (cNIII-like). We show that cNIII-like also displays 5′ nucleotidase activity with a high affinity for m7GMP. CMP is a slightly better substrate but again with a higher Km. The activity of cNIII-like is stimulated by phosphate. In contrast to cNIII-like, cNIII and human cytosolic nucleotidase II do not accept m7GMP as a substrate. We suggest that the m7G-specific nucleotidases protect cells against undesired salvage of m7GMP and its incorporation into nucleic acids.
Introduction
Turnover of mRNAs in eukaryotes is mainly catalyzed by hydrolytic exonucleases, which release nucleoside 5′-monophosphates (NMPs)4 (1–3). At least in plants and in prokaryotes, phosphorolytic enzymes also participate, producing nucleoside 5′-diphosphates (4, 5). In addition to these “regular” nucleotides, mRNA decay also releases derivatives of the 5′ cap, which consists of a 7-methylguanosine linked to the RNA body by a 5′-5′ triphosphate bridge (6). The cap can be liberated in two different forms by two different types of enzymes; m7GDP is cleaved off long RNA chains by enzymes of the Nudix family like Dcp2 (7–9) or Nudt16 (10, 11). Alternatively, m7GMP can be generated by cleavage of the free cap dinucleotide or short capped oligonucleotides catalyzed by DcpS (12).
NMPs and nucleoside 5′-diphosphates produced by mRNA decay can easily be recycled to NTPs by kinase reactions. In principle, enzymes of the nucleotide salvage or biosynthesis pathways can also act on m7guanine nucleotides; in vitro, m7GDP can be converted to the triphosphate by nucleoside diphosphate kinase (8, 13). If this were happening in the cell, the methylated base would almost certainly be incorporated into RNA (14). As ribonucleotide reductase accepts all four ribonucleoside diphosphates as substrates (15), it seems plausible that this enzyme would also convert m7GDP to the corresponding deoxynucleotide, which after conversion to the triphosphate by nucleoside diphosphate kinase would presumably find its way into DNA (14). Salvage of m7GMP would require conversion to the diphosphate by guanylate kinase; we do not know if this enzyme accepts m7GMP.
7-Methylguanosine in DNA, which can be generated through non-enzymatic methyl transfer from toxins or cellular metabolites, is subject to base excision repair. The methylated base is removed by cleavage of the N-glycosidic bond catalyzed by enzymes like Escherichia coli AlkA (16), Bacillus cereus AlkD (17, 18), or mammalian alkyladenine glycosylase (or methylpurine glycosylase) (19, 20), which act on a large spectrum of modified purine bases. N7-methylation also chemically destabilizes both the N-glycosidic bond and the purine ring itself (21). Whereas spontaneous depurination would circumvent the need for an N-glycosylase to initiate repair, the opened purine ring is removed by a specific enzyme (22). Although repair mechanisms for m7G in RNA have not been described, the modification itself or the chemical instability associated with it would be expected to interfere with RNA function.
Thus, there is probably evolutionary pressure for cells to prevent the incorporation of m7G into nucleic acids by disposing of the cap remnants directly. In fact, m7GDP is converted to m7GMP in extracts of different cells (23). DcpS has been reported to be responsible for this reaction (23, 24), but this has not been confirmed by others (12, 25, 26). Regardless, m7GMP appears to be a universal intermediate of cap degradation but probably not its final product. Its conversion to an unknown compound in yeast extract and release of its phosphate in mammalian extracts have been noted (13, 23), but the enzymes responsible have not been identified.
Conversion of (deoxy)nucleoside monophosphates to nucleoside and orthophosphate is catalyzed by members of the family of 5′-nucleotidases. In addition to a mitochondrial and an extracellular enzyme, six different cytosolic 5′-nucleotidases exist in humans (27): cytosolic 5′(3′)-deoxyribonucleotidase (NT5C gene product) acts on dUMP and dTMP, cytosolic 5′ nucleotidase IA (NT5C1A) prefers AMP and pyrimidine dNMPs, cNIB (NT5C1B) is closely related and may have a similar substrate specificity, cNII (NT5C2) prefers (d)IMP and (d)GMP, and cNIII (NT5C3) dephosphorylates pyrimidine nucleotides. The predicted protein cNIII-like (NT5C3-like) has to our knowledge not been biochemically characterized so far. As members of the haloacid dehalogenase superfamily (28), the cytosolic nucleotidases catalyze the Mg2+-dependent attack of an aspartate side chain on the NMP substrate, resulting in the formation of a phosphoenzyme intermediate and release of the nucleoside. The covalently bound phosphate can then either be liberated by the attack of water or transferred to an acceptor nucleoside (29–32). 5′-Nucleotidases play a role in the regulation of intracellular nucleotide pools. Homozygous mutations in 5′-deoxyribonucleotidase C3 cause hemolytic anemia (33, 34). The 5′ nucleotidases are also of interest because of their role in the metabolism of nucleoside analogs used as antiviral and anti-cancer drugs (27). Here, we report the identification of novel members of the family of 5′-nucleotidases that are specific for the hydrolysis of m7GMP to m7guanosine and inorganic phosphate and may participate in preventing the incorporation of m7-guanosine into cellular nucleic acids.
EXPERIMENTAL PROCEDURES
Extracts, Enzymes, and Other Reagents
Extract was prepared from 0.5–2.5-h-old embryos of wild type Drosophila melanogaster as described (35, 36). Human DcpS fused to glutathione S-transferase was purified from E. coli transformed with an expression plasmid (37), a kind gift of H. Song (National University of Singapore). Purification on glutathione-Sepharose (GE Healthcare) was performed as recommended by the supplier. His-tagged Ulp1 protease was purified by Ni2+-NTA chromatography from E. coli transformed with an expression plasmid (38) obtained from Christopher D. Lima (Memorial Sloan Kettering Cancer Center). Radiolabeled nucleotides were obtained from Hartmann Analytics. Other nucleosides and nucleotides were from Sigma or Jena Bioscience.
Preparation of [32P]m7GMP
The RNA used as a substrate for the capping reaction was 38 nucleotides long (a tandem repeat of the sequence CCCCACCCUCUUCCCCAAG) and synthesized as described (39). After phenol-chloroform extraction and sodium acetate/ethanol precipitation, 72 ng of the RNA was capped with 20 units of vaccinia virus capping enzyme (ScriptCap m7G capping kit, Epicenter Biotechnology) in a volume of 20 μl as recommended by the supplier, except the concentration of GTP was reduced to 5 μm, and 90 μCi [α-32P]GTP was included. The capped RNA was purified by phenol-chloroform extraction and sodium acetate/ethanol precipitation. 60 ng of the capped RNA was digested to [32P]m7GpppN and 5′-NMPs with one unit of P1 nuclease (Biomol) in 10 mm HEPES-KOH, pH 7.4, for 2 h at 37 °C followed by the addition of 50 ng of DcpS and 2.5 mm magnesium acetate and incubation for 1 h at 37 °C to convert [32P]m7GpppN into [32P]m7GMP. The completeness of digestion was checked by thin-layer chromatography (TLC) as described below. The desired product was not purified so that all preparations of [32P]m7GMP contained contaminating unlabeled 5′-NMPs from the RNA body. Radioactive [32P]m7GMP was diluted with unlabeled m7-GMP (Jena Bioscience) to the desired specific activity.
Nucleotidase Assays
Reactions were carried out in 20 mm HEPES-KOH, pH 7.5, 5 mm MgCl2, 50 mm KCl. Reducing agents were omitted, as they did not increase the activity. Substrate concentrations were as indicated, and reaction temperature was 25 °C for the Drosophila enzyme and 37 °C for human proteins. Reactions were started by enzyme addition. Where indicated, an ATP regenerating system (80 μg/ml creatine kinase (rabbit muscle; Roche Applied Science), 30 mm creatine phosphate) or an ATP depleting system (20 mm glucose, 0.1 units/μl of hexokinase; Sigma) was added. In these cases the extract was preincubated for 10 min under ATP depleting or regenerating conditions, and the ATP status was checked by TLC analysis of a control reaction containing a trace of [α-32P]ATP. When radioactive NMPs were used as substrates, nucleotidase reactions were stopped by the addition of 10 mm EDTA, and 2 μl per time point were analyzed by TLC on polyethyleneimine cellulose plates (Merck). Solvents were 0.5 m LiCl, 1 m HCOOH for separation of adenine nucleotides, 0.3 m LiCl, 1 m HCOOH for separation of m7GMP and related compounds, and 0.8 m NaCl, 1× TBE (90 mm Tris-HCl pH 8.0, 90 mm boric acid, 2 mm EDTA) for GMP and related compounds. Plates were analyzed by phosphorimaging, and results were quantitated with ImageQuantTM (GE Healthcare). When unlabeled nucleotides were used as substrates, a colorimetric assay for orthophosphate was used as described (40) except that Tween 20 was left out. In this case the reaction (20 μl per time point) was stopped by the addition of the malachite green oxalate/ammonium molybdate reagent to the reaction mixture. A phosphate standard curve was used to estimate the phosphate released in the enzymatic reactions. Where indicated, pyrophosphatase (Fermentas) was added at a concentration of 0.1 units/μl.
For the determination of steady-state parameters, initial velocities were determined from progress curves with at least four time points at each substrate concentration. The substrate concentrations were 1 μm–1.5 mm when radioactive substrates were used and 4 μm–3 mm for unlabeled substrates. A hyperbolic fit to the Michaelis-Menten equation was calculated with the help of Sigma Plot 8.0, and kcat was calculated from Vmax. In the case of AMP the V/S curve was fitted to an equation describing a substrate inhibition, v = Vmax/(1+ Km/[S]0 + [S]0/Ki). Each experiment was carried out at least twice with R values ranging from 0.9607 to 0.9940. Kinetic constants derived from individual experiments deviated by up to ± 25%.
HPLC Analysis
Unlabeled GMP or m7GMP were incubated with a partially purified nucleotidase fraction under standard conditions. Reactions were stopped with 10 mm EDTA, and proteins were precipitated by the addition of 5 volumes of ice-cold ethanol. The mixture was centrifuged for 30 min at 20,000 × g, and the supernatant was evaporated in a vacuum centrifuge. The residue was dissolved in 80 μl of water and loaded onto a reversed phase C18 HPLC column (Keystone Scientific Betabasic, 150 × 1 mm, 150 Å, 3 μm). Buffer A was 100 mm potassium phosphate, pH 7.5, and buffer B was 20% (v/v) acetonitrile in buffer A. A linear gradient from buffer A to buffer B was applied (0.03 ml/min, 45 min). The eluate was continuously monitored photometrically either at 258 nm (for reactions containing m7GMP) or 254 nm (for reactions containing GMP). The retention times were compared with standards.
UV Cross-linking
A 15-μl aliquot from the peak fraction from the second hydroxyapatite column in the purification of the Drosophila enzyme (see below) was diluted with 11 μl of 16 mm HEPES-KOH, pH 7.4, 3 mm magnesium acetate, and 4 μl of [32P]GMP (∼15 μCi). Aliquots of 5 μl were irradiated with different intensities of UV light (60–1860 mJ/cm2; UV Stratalinker 1800, Stratagene) and analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography.
Partial Purification of the m7G-specific Nucleotidase from Schneider Cells
Suspension cultures of Schneider 2 cells were grown in Schneider's Drosophila medium (Invitrogen) at 25 °C with 0.05% (v/v) of F-68 Pluronic solution (Invitrogen). 50 g of cells (wet weight) were resuspended in 20 ml of hypotonic buffer (10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT) and, after 15 min, lysed with a tight-fitting Dounce homogenizer (41). The lysate was centrifuged for 1 h at 120,000 × g. The supernatant was collected, adjusted to 50 mm KCl, and loaded onto a 150-ml DEAE-Sepharose column. This column, in contrast to the one shown in Fig. 2A, resulted in two activity peaks, the first of which was used for further purification. Activity in the second peak was not specific for m7GMP, and Western blots later showed that these fractions did not contain the m7G-specific nucleotidase (data not shown). Additional purification steps were chromatography on Mono S, hydroxyapatite with phosphate elution, Sephacryl S200, hydroxyapatite with KCl elution, and Mono Q. All column materials were from GE Healthcare, except hydroxyapatite (Bio-Rad). A final purification factor of 650 with a yield of 7% was achieved. A preparative SDS-polyacrylamide gel was run with the peak fraction of the last Mono Q column, and the protein band of interest was cut out and analyzed by protease digestion and MALDI-TOF mass spectrometry as described (42). Protein concentrations were measured with a Bradford assay (Rotiquant, Roth).
FIGURE 2.
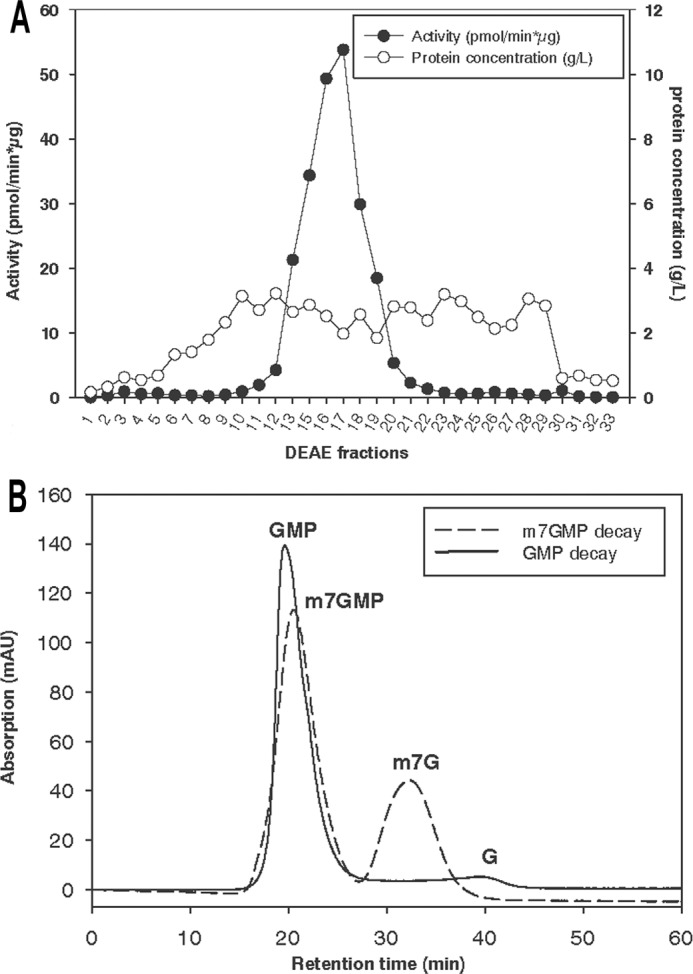
Dephosphorylation of m7GMP is catalyzed by a specific nucleotidase. A, Schneider 2 cells were disrupted as described under “Experimental Procedures,” and cytosolic proteins were fractionated on a DEAE-Sepharose column. Nucleotidase activity of the fractions was determined by an assay with 40 μm m7GMP trace-labeled with [32P]m7GMP. B, an aliquot of fraction 16 of the DEAE column in A was incubated with either GMP or m7GMP (400 μm) for 25 min at 25 °C. The reaction was stopped by the addition of EDTA, and HPLC analysis was carried out as described under “Experimental Procedures.” mAU, milliabsorbance units.
Cloning of Nucleotidase cDNAs
The cDNA of D. melanogaster CG3362 (FlyBase database) was obtained via reverse transcription of RNA extracted from S2 cells with Moloney murine leukemia virus reverse transcriptase (Promega) and a specific oligonucleotide primer followed by PCR amplification. The cDNAs of the human cytosolic 5′ nucleotidases II, III, and III-like were obtained similarly via reverse transcription from RNA of HEK293 cells and PCR with specific oligonucleotide primers. Restriction sites for endonuclease BsaI (5′ end) and XhoI (3′ end) were introduced into the cDNAs by way of the PCR primers. The cDNA of the nucleotidases were cloned into a pET-SUMOadapt vector (43). Primers used and accession numbers are listed in Table 1. All expression clones were checked by sequencing.
TABLE 1.
Accession numbers and primers used for cloning of nucleotidases
RT, primer used for reverse transcription.
| Gene | Accession number | Oligonucleotides | Restriction sites |
|---|---|---|---|
| CG3362 | FBgn0034988 | RT: GAATTCGTGAACTCCCTG | |
| Forward: AAAGGTCTCTATGGGCTTTGACGAGAAG | BsaI | ||
| Reverse: TTTCTCGAGTTATAGGGAGCTCTGCTTG | XhoI | ||
| cN-III-L | NM_052935 | RT: TGGAGCCTGCGCCTTCAG | |
| Forward: AAAGGTCTCTATGGCAGAGGAGGTGAG | BsaI | ||
| Reverse: TTTCTCGAGTCAGGGGCCTTGCATC | XhoI | ||
| cN-III | NM_001166118 | RT: GAGAGGTCTTCTTGGAG | |
| Forward: AAAGGTCTCTTGGTATGAGGGCCCCGTCC | BsaI | ||
| Reverse: TTTCTCGAGTTATAGAATCTTCTGTAAAATAG | XhoI | ||
| cN-II | NM_012229 | RT: CGTTTGTTCCTGTGAGTCCTG | |
| Forward: AAAGGTCTCATGGTTCACCTCCTGGAGTGATCG | BsaI | ||
| Reverse: TTTAAGCTTGGTTTTGGTTTTCCTCCTTATTC | HindIII |
Purification of Overproduced Nucleotidases from E. coli
E. coli Rosetta 2 cells (Novagen) were transformed with the expression plasmids and grown in 1.2 liters of TB medium (44) at 37 °C to an A600 of 1. Expression was induced by the addition of 0.5 mm isopropyl β-d-1-thiogalactopyranoside, and cells were further incubated for 2 h at room temperature. After harvesting, cells were resuspended in 10 ml of lysis buffer (20 mm Tris-HCl, pH 8.0, 400 mm KCl, 10 mm imidazole, 10% (w/v) glycerol). Just before disruption with a French press, 5 mg of lysozyme, 2 mg of DNase I, 3 mm MgCl2, and 20 mg PMSF were added. The lysate was centrifuged at 30,000 × g for 40 min, and the supernatant was loaded onto a Ni2+-NTA-agarose column (1 ml; Qiagen) equilibrated in lysis buffer. Bound protein was eluted with an imidazole gradient (0–500 mm in lysis buffer). Fractions containing the overexpressed protein, identified by SDS-polyacrylamide gel electrophoresis, were pooled and dialyzed again 20 mm Tris-HCl, pH 8.0, 400 mm KCl, 5 mm MgCl2, 20 mm imidazole, 10% (w/v) glycerol. The SUMO tag was cleaved off with Ulp1 protease (substrate:enzyme mass ratio 100:1) for 4 h at 8 °C, and the Ni2+-NTA-agarose column chromatography was repeated. The flow-through fraction was loaded onto a hydroxyapatite column equilibrated in 20 mm imidazole, pH 7.5, 200 mm KCl, 5% (w/v) glycerol (5 mg of protein per ml column volume). Bound protein was eluted with a gradient from equilibration buffer to 0.5 m potassium phosphate pH 7.5 (7 column volumes). Fractions were pooled on the basis of activity assays and dialyzed into 20 mm HEPES-KOH, pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.2 mm EDTA, 5% glycerol. Concentrations were determined from the UV spectrum with extinction coefficients calculated from the amino acid sequence. Human SUMO-cNIII was purified by Ni2+-NTA chromatography as above, dialyzed against 20 mm Tris, pH 8.0, 75 mm KCl, 5 mm MgCl2, 10% (w/v) glycerol, loaded onto a 1-ml Mono Q column and eluted with a gradient up to 1 m KCl in the same buffer. An aliquot from the activity peak fraction was used for enzyme assays. The remaining material was pooled, the SUMO tag was cleaved off, and a second Ni2+-NTA column was run. Concentration of cNIII was determined by densitometry of a Coomassie-stained SDS-polyacrylamide gel and comparison to a BSA standard.
Analytical Ultracentrifugation
Proteins were dialyzed overnight against 20 mm HEPES, pH 7.4 (20 °C),100 mm KCl, 5 mm MgCl2, 0.2 mm EDTA, 5% (w/v) glycerol and centrifuged in a Beckmann X-LA centrifuge at 12,000 rpm, 20 °C, for 3 days. Absorption profiles were fitted to the equation
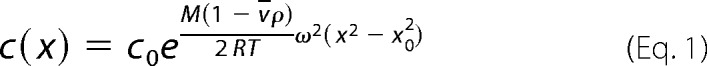 |
where c is the macromolecule concentration, m is the apparent molar mass of the macromolecule, ν is the partial specific volume, ρ is the density, R is the ideal gas constant, T is the temperature, ω is the angular frequency, and x is the distance from the rotation center.
RESULTS
Dephosphorylation of m7GMP in Drosophila Embryo Extract
In the course of experiments dealing with the regulation of the Drosophila nanos mRNA (35, 45), the fate of the mRNA cap structure was examined in Drosophila embryo extracts. For this purpose, cap-labeled RNA was generated containing [32P]phosphate in the α position with respect to the methylated guanosine (see “Experimental Procedures”). The fate of the cap structure upon incubation in extract was followed by TLC. When an ATP regenerating system was present during the reaction, m7GMP was produced, but most of the radioactivity released co-migrated with inorganic phosphate (Fig. 1A). When the same reaction was carried out after ATP depletion with hexokinase and glucose, a similar amount of radioactivity was released from the RNA, but m7GMP was the major product, with minor quantities of inorganic phosphate. An additional compound that was present in the substrate RNA and disappeared in the ATP-containing reaction remained unidentified. The expected product of Dcp2 activity, m7GDP, was not detectable independently of the ATP status of the reaction (Fig. 1A). M7GDP might have been generated in the reaction but was rapidly turned over. However, even when excess unlabeled m7GDP or m7GpppG were added to compete with potential turnover of labeled m7GDP, accumulation of 32P-m7GDP was not observed (data not shown). Thus, m7GDP was probably not an intermediate in the production of m7GMP. Together, the results suggest that the cap structure was liberated by exo- and/or endonucleolytic degradation of the RNA body followed by the release of m7GMP due to DcpS activity and further conversion of m7GMP to the nucleoside and orthophosphate. In fact, DcpS activity in the extract was directly demonstrated by its ability to degrade m7GpppG produced by nuclease P1 digestion of cap-labeled RNA (data not shown). The decapping enzyme Dcp1/Dcp2 is active in mRNA decay in Drosophila embryos at 2–4 h of development (46), a time window bordering on the one from which our extracts were derived, but Dcp2 activity was not apparent in the extract.
FIGURE 1.

A, shown is release of phosphate during incubation of 32P-labeled capped RNA with Drosophila embryo extract. The reaction mixtures containing 20% embryo extract were preincubated for 10 min with hexokinase and glucose for ATP depletion or complemented with creatine kinase and creatine for ATP regeneration as indicated. The reactions were started by the addition of the 32P-labeled capped RNA (200 nm). At different time points (0, 5, 10, 20, 40 min) 2-μl aliquots were removed and mixed with EDTA. The reaction products were separated by TLC and detected by autoradiography. For details, see “Experimental Procedures.” The first lane contains the substrate at time point 0. Migration of standards is indicated on the left. Labeled m7GMP and m7GpppG were generated by digestion of 32P-labeled capped RNA with P1 nuclease with or without DcpS, respectively. M7GTP was an unlabeled standard detected by UV absorbance, and 32P-orthosphosphate, shown in the last lane, was obtained commercially. All standards were analyzed after the addition to a reaction mixture lacking labeled RNA. An unidentified compound is labeled X. B, release of phosphate during incubation of 32P-labeled m7GMP with Drosophila embryo extract is shown. The reaction mixtures contained 20% embryo extract. ATP-depleting or -regenerating conditions were as in A. The reactions were started by the addition of 32P-labeled m7GMP (200 nm). After 0, 5, 10, 20, and 40 min, 2 μl were removed, and the reactions were stopped with EDTA. Products were separated by TLC and detected by autoradiography. The first lane represents the substrate m7GMP analyzed directly from water. Its migration is thus different from that of samples taken from the reaction mixture. Standards indicated on the left were analyzed from a reaction mixture.
To confirm the suspected dephosphorylation of m7GMP, we generated [32P]m7GMP by enzymatic degradation of cap-labeled RNA (see “Experimental Procedures”) and incubated this in the embryo extract. Indeed, the extract dephosphorylated m7GMP in a time-dependent manner (Fig. 1B). Interestingly, a small amount of m7GMP was also converted to a product tentatively identified as m7GTP; the labeled product co-migrated with an unlabeled m7GTP standard at two different LiCl concentrations in the TLC solvent, it appeared only when an ATP-regenerating system was included in the reaction, and the product was not obtained when [32P]orthophosphate was used as the source of radioactivity (Fig. 1B and data not shown). The observation supports the hypothesis that m7GMP, if not degraded, can enter the nucleotide salvage pathway.
Dephosphorylation of m7GMP Is Specific for the Methylated Nucleotide
With [32P]m7GMP as a substrate, several other extracts were assayed for their ability to hydrolyze this nucleotide. Phosphate release was observed in extracts of several mammalian cell types (CHO cells, mouse ES cells, K562 cells) and of Drosophila Schneider 2 (S2) cells but was barely detectable in yeast cell extract (data not shown). When S2 cell cytosolic extract was fractionated over a DEAE column, the activity eluted in a single peak (Fig. 2A). The peak fraction was used to investigate the specificity of the nucleotidase reaction; unlabeled GMP or m7GMP was incubated with the fraction, the reactions were stopped by he addition of EDTA, and products were analyzed by reversed-phase HPLC. Under the conditions used, approximately one-third of m7GMP was converted to the nucleoside, whereas GMP was barely degraded (Fig. 2B). The same DEAE column fraction was also tested with different concentrations of [32P]m7GMP or its unmethylated counterpart, and phosphate release was analyzed by TLC. The apparent Km for m7GMP estimated in these experiments was 25 times lower than for GMP (∼ 4 μm versus ∼ 100 μm), and the apparent Vmax for m7GMP was 2-fold higher than for GMP (data not shown). Together, the two assays show that m7GMP is indeed the substrate of a nucleotidase with m7guanosine and orthophosphate as products and that this activity is specific for the methylated variant of GMP. The ATP dependence of the activity that was apparent in the embryo extract was not observed in DEAE column fractions derived either from S2 cells or embryo extract and could not be restored by a combination of column fractions (data not shown).
Identification of the m7GMP Degrading Nucleotidase
The enzyme degrading m7GMP was partially purified from S2 cell extract by conventional column chromatography (see “Experimental Procedures”). The fractions of the final Mono Q column were analyzed by SDS-polyacrylamide gel electrophoresis. Several bands could be discerned by the profiles of that corresponded to the nucleotidase activity profile (Fig. 3, A and B). Among these, a ∼ 40-kDa protein matched the molecular mass of a GMP-binding protein, as determined by UV-cross linking with [32P]GMP (Fig. 3C) and the apparent native molecular mass of the partially purified nucleotidase in gel filtration (data not shown). Analysis of this protein band by trypsin digestion and mass spectrometry revealed peptides covering 43% of the amino acid sequence encoded by the predicted gene CG3362; no other protein was detectable (data not shown). The protein encoded by CG3362 has a predicted molecular mass of 36.3 kDa and a similar extent of sequence identity (∼ 35%) both with the human cytosolic 5′ nucleotidase III (SwissProt Q9H0P0.3) (47) and the human cNIII-like protein (SwissProt Q969T7.3). The catalytic motifs for nucleotidase activity (30, 48) are well conserved in CG3362 (Fig. 4) (see “Discussion”).
FIGURE 3.

Identification of the m7GMP-specific 5′ nucleotidase from S2 cells. A, the enzyme was partially purified as described under “Experimental Procedures.” The figure shows the activity peak of the final Mono Q column, determined with 400 μm trace-labeled m7GMP as a substrate. B, 10 μl of the indicated fractions of the Mono Q column in A were loaded onto a 12% SDS-gel. Proteins were detected by silver staining. M, marker; L, load. Molecular weights of marker proteins (in kDa) are given on the left. C, a fraction from the second hydroxyapatite column was mixed with [32P]GMP, and different UV intensities were used to cross-link the nucleotide with proteins (see “Experimental Procedures”). The samples were analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography. Molecular masses of marker proteins (in kDa) are given on the left.
FIGURE 4.
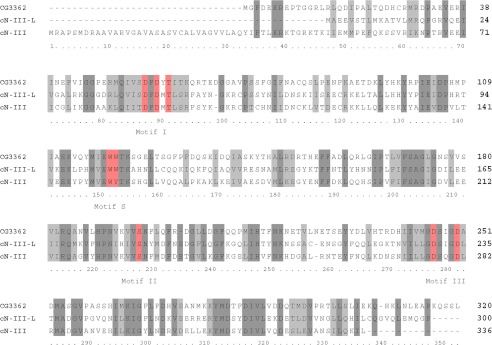
Alignment of the amino acid sequences of CG3362, human cNIII, and cNIII-like proteins. Amino acids constituting the catalytic motifs I, II, and III (24, 27) are shown in red and marked below the sequence. The lysine at position 268 of cytosolic nucleotidase III is also considered part of motif III (24), but the corresponding amino acid in other enzymes is not immediately obvious from the sequence. The WW or WY motif at position 152/153 of cytosolic nucleotidase III, also shown in red, is involved in binding the base of the substrate. Sequence identities are highlighted by dark gray shading, and amino acid similarities are marked by light gray shading. Protein sequences were retrieved from the PubMed server (blast.ncbi.nlm.nih.gov) (cNIII, CAG38549; cNIII-like, Q969T7; CG3362, AAF47180), and the alignment was done with MUSCLE (53) with preset restraints.
The Drosophila CG3362 Gene Product Is an m7G-specific Nucleotidase
The CG3362 protein was overproduced in E. coli and purified to homogeneity (Fig. 5A). Analytical ultracentrifugation showed the enzyme to be a monomer (native molecular weight 36 kDa) at a concentration of 27.5 μm (Fig. 5B). Nucleotidase activity of the recombinant protein was confirmed by a colorimetric assay in which the release of orthophosphate from various NMPs was determined. Phosphate release was linear with time and enzyme concentration (Fig. 6A and data not shown). Enzyme activity was reduced to non-detectable levels (less than 7%) by the addition of EDTA to the reaction buffer (data not shown). Tests with the partially purified enzyme preparation from S2 cells showed a pH optimum near 7.5 (range tested, pH 5.0–8.0). Compared with standard reaction conditions (50 mm KCl), the activity was reduced to 50% by an increase in KCl concentration to 520 mm or by the addition of 650 mm potassium acetate or 60 mm potassium phosphate (data not shown).
FIGURE 5.

A, Purified proteins used in this study. Lanes 1 and 5, molecular weight standards; lane 2, purified recombinant human cN II; lane 3, purified recombinant human cNIII-L; lane 4, purified recombinant Drosophila CG3362 protein; lane 6, partially purified recombinant human cNIII after SUMO cleavage. The band representing cNIII is marked with an asterisk. Molecular weight standards (M; kDa) are given in the margin. B, molecular weight determination of CG3362 by equilibrium sedimentation is shown. The experiment was carried out as described under “Experimental Procedures” with an initial protein concentration of 27.5 μm. The calculated and the experimentally determined molecular weights are given as an inset in the figure. r, radius. C, molecular weight determination of human cNIII-like by equilibrium sedimentation is shown. The experiment was carried out as in B, with an initial protein concentration of 29 μm.
FIGURE 6.

Nucleotidase activity of Drosophila CG3362 measured by a colorimetric assay for inorganic phosphate. A, time course of phosphate release with m7GMP and GMP is shown. Enzyme was used at a concentration of 13.8 nm and substrates at 200 μm. The standard deviation, based on three independent experiments, is shown. B, titration of m7GMP and GMP is shown. Enzyme concentrations of 13.8–55 nm were used to determine initial rates. Curves were fitted to the Michaelis-Menten equation. C, titration of AMP is shown. Enzyme was used at a concentration of 1.4 μm. The data were fitted as described under “Experimental Procedures.” D, time courses of phosphate release from m7GDP are shown. Enzyme was used at 2.8 μm and substrate at 200 μm. The reactions were carried out with or without the addition of 1 unit of pyrophosphatase per 100 μl reaction, as indicated.
Initial reaction velocities at different concentrations of m7GMP, GMP, CMP, and UMP could be fitted to the Michaelis-Menten equation (Fig. 6B and data not shown). As expected, the enzyme preferred m7GMP over GMP by about 50-fold, as measured by kcat/Km (Fig. 6B and Table 2). The kinetic constants were in reasonable agreement with the preliminary data obtained for the partially purified enzyme from S2 cells (see above). CMP was also a good substrate, but the Km for m7GMP was the lowest. AMP and UMP were nearly as poor substrates as GMP (Table 2). Titration of AMP revealed an inhibition at higher substrate concentrations, which could be modeled with a Km of 32 μm and a Ki of 2 mm (Fig. 6C and Table 2). The addition of ATP (up to 4 mm) had no effect on the rate of m7GMP dephosphorylation (data not shown).
TABLE 2.
Steady-state kinetic parameters for CG3362
| Substrate | Km | Vmax | kcat | Ki | kcat/Km |
|---|---|---|---|---|---|
| μm | μmol/min/mg | 1/s | mm | ||
| m7GMP | 13 | 11 | 6.3 | 0.46 | |
| CMP | 48 | 21 | 12.0 | 0.25 | |
| GMP | 102 | 1.5 | 0.88 | 0.01 | |
| AMP | 32 | 0.49 | 0.28 | 2.1 | 0.01 |
| UMP | 91 | 4.0 | 2.3 | 0.03 |
Surprisingly, CG3362 also released orthophosphate from m7GDP, albeit at a rate 32-fold lower than with m7-GMP (Fig. 6D). Appropriate controls showed that pyrophosphate did not react in the colorimetric assay for orthophosphate, the addition of pyrophosphatase did not accelerate the production of orthophosphate by CG3362, and the enzyme preparation was not contaminated with pyrophosphatase. Upon nearly complete degradation of m7GDP, an ∼2-fold molar excess of phosphate was released (Fig. 6D and data not shown). Thus, the enzyme first releases the β-phosphate from m7GDP and then dephosphorylates the remaining m7GMP. Nucleoside 5′-diphosphate dephosphorylation was specific for m7GDP; GDP did not serve as a substrate (40-fold lower rate; data not shown). The specificity for the methylated nucleotide suggests that CG3362 rather than a contaminating protein was responsible for the activity. Because the enzyme can accommodate nucleoside diphosphates in its active site, we considered the possibility that the AMP inhibition (Fig. 6C) might be due to the nucleotide acting as a phosphate acceptor and being converted to the diphosphate. However, the addition of 1 units/μl of apyrase, which would have digested the hypothetical product ADP, did not relieve the inhibition at higher AMP concentrations (data not shown); thus the inhibition was not due to ADP production.
The Human cNIII-like Protein Is Also an m7G-specific Nucleotidase
One of two potential human orthologues of CG3362, the cNIII-like protein, was also purified from E. coli (Fig. 5A). At a concentration of 29 μm, the enzyme behaved like a monomer in analytical ultracentrifugation (native molecular mass, 34 kDa; predicted monomeric molecular mass, 34.4 kDa) (Fig. 5C). As predicted from the amino acid sequence, cNIII-like also had nucleotidase activity. Under normal reaction conditions, the time course of phosphate production by cNIII-like was not linear, but linearity could be restored when 50 μm phosphate was added to the reaction buffer (Fig. 7A). The substrate preference of cNIII-like was similar to that of Drosophila CG3362 inasmuch as it had the lowest Km for m7GMP, 10-fold lower than for CMP, the second best. However, in this case Vmax was higher for CMP, so that this substrate was overall slightly better than m7GMP. Likewise, UMP was characterized by a poor Km but high kcat so that it was a reasonable substrate. Unmethylated GMP and AMP were about equally poor (Fig. 7 and Table 3). In summary, human cNIII-like is a pyrimidine- and m7guanosine-specific nucleotidase and, therefore, the orthologue of CG3362. As cNIII-like and Drosophila CG3362 have similar substrate specificities and orthologues can be identified in many organisms (see “Discussion”), we suggest that CG3362 be classified as Drosophila cNIII-like.
FIGURE 7.
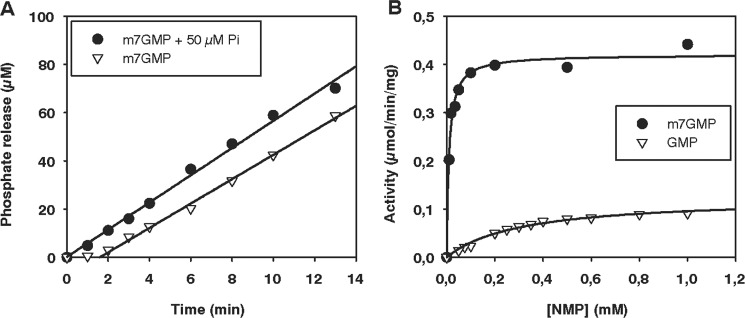
Nucleotidase activity of human cytosolic nucleotidase III-like. The colorimetric assay for phosphate release was used. A, shown is the time course of m7GMP dephosphorylation with and without initial addition of 50 μm potassium phosphate. Enzyme was used at a concentration of 49.4 nm and substrate at 250 μm. B, titration of GMP and m7GMP is shown. Enzyme was used at concentrations of 29–251 nm to determine initial rates of phosphate release. The reaction mixtures contained an initial potassium phosphate concentration of 50 μm.
TABLE 3.
Steady-state kinetic parameters for human cNIII-like
| Substrate | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| μm | μmol/min/mg | 1/s | ||
| m7GMP | 7.8 | 0.41 | 0.24 | 0.03 |
| CMP | 79 | 12.0 | 7 | 0.09 |
| GMP | 355 | 0.13 | 0.07 | 0.0002 |
| AMP | 456 | 0.07 | 0.04 | 0.0001 |
| UMP | 439 | 10.7 | 6.2 | 0.01 |
Human cNII and cNIII Do Not Prefer m7GMP
Human cNII has been characterized as an IMP/GMP-specific enzyme (see the Introduction). The enzyme was purified from E. coli (Fig. 5A), and dephosphorylation time courses measured at fixed substrate concentrations showed that it dephosphorylated GMP, as expected, but did not catalyze any detectable reaction with m7GMP (kobs at least 350-fold lower than with GMP) (Fig. 8A).
FIGURE 8.
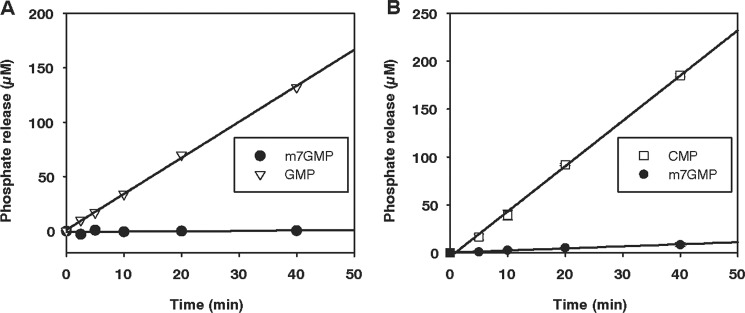
Human cytosolic nucleotidases II and III do not act on m7GMP. Phosphate release was measured with a colorimetric assay. A, purified human cNII protein (154 nm) was incubated with 250 μm m7GMP or GMP. B, the human cNIII protein was incubated with CMP or m7GMP as substrates (500 μm each). Protein with an N-terminal SUMO tag was used because of solubility problems during the purification procedure. Protein concentration was 73 nm. Faster dephosphorylation of CMP was also seen at lower substrate concentrations and after cleavage of the SUMO tag.
Human cNIII prefers pyrimidine nucleotides as substrates (see the Introduction). Because of solubility problems, the enzyme was only available in a partially purified state (Fig. 5A and “Experimental Procedures”). Time courses at several substrate concentrations revealed the expected dephosphorylation of CMP. The turnover rate of m7GMP was much lower than that of CMP at any substrate concentration tested (10–500 μm; Fig. 8B and data not shown). From a limited number of experiments, the Km value for CMP was estimated as ∼30 μm, in approximate agreement with a previous report (47). The Km value for m7GMP was estimated as >100 μm. Thus, cNIII strongly prefers CMP; dephosphorylation of m7GMP is a specific property of the cNIII-like enzyme.
DISCUSSION
Each mRNA cap is synthesized de novo by a co-transcriptional reaction using GTP and S-adenosylmethionine as substrates (49). As there is no known reaction by which m7G nucleotides released during mRNA decay could be recycled into new caps, these nucleotides are likely to be disposed of. Here, a Drosophila 5′ nucleotidase, encoded by the gene CG3362, was shown specifically to hydrolyze m7-GMP to orthophosphate and the corresponding nucleoside. Of the two human nucleotidases related to CG3362, cytosolic nucleotidase III-like, a previously uncharacterized enzyme, proved to be the true orthologue, having a substrate specificity similar to CG3362, whereas cytosolic nucleotidase III did not accept m7GMP as a substrate. We suggest that the m7GMP-specific enzymes act as sanitizing agents that participate in the removal of N7-methylated guanine nucleotides to prevent their entry into the nucleotide salvage pathway and incorporation into nucleic acids. To examine the biological role of these enzymes in detail, we have generated a null allele of Drosophila CG3362; preliminary analysis indicates a semi-lethal phenotype.5 Among the many problems to be addressed in the analysis of this mutant will be the question of whether failure to degrade m7GMP or the other preferred substrate, CMP, is responsible for the phenotype.
The conserved active site of the nucleotidase family, located in the globular core domain, is easily recognized in the primary structures of CG3362 and human cNIII-like (Fig. 4). By sequence alignment, Asp-55 (CG3362 numbering) at the beginning of motif I is predicted to form the covalent phosphoenzyme intermediate and Asp-57 to act as a general acid/base catalyst, protonating the 5′-OH leaving group of the first step and activating the attacking water in the second step. Conserved residues in motifs II and III have mostly roles in coordinating the catalytic Mg2+ ion and phosphate (27, 30, 32).
Recognition of the base moiety of the substrate is mediated by the α-helical lid domain covering the active site. In the crystal structure of murine cNIII complexed with UMP, the base is coordinated between 3 π electron-containing amino acids (His-68, Trp-113, Tyr-114) and is also approached by Asn-69, which is thought to sterically interfere with binding of the larger purine nucleotides (50). In both CG3362 and cNIII-like, the Trp-113, Tyr-114 motif is replaced by two tryptophans. Database searches revealed that the WW motif is common to almost all sequences classified as cytosolic nucleotidase III-like, whereas those classified as cytosolic nucleotidase III have a WY motif. In contrast, changes in the positions corresponding to His-68 and Asn-69 of murine cNIII are not specific to either of the two classes (supplemental Fig. 1). Thus, the WW motif may be important for the recognition of m7GMP. The two types of enzymes, cNIII and cNIII-like, are closely related by primary structure and by the overlap in substrate specificity, both acting on CMP. Genes encoding the two enzymes are simultaneously present in most vertebrates except fish, which only seem to have cNIII (supplemental Fig. 1). In contrast, as mentioned above, Drosophila has only one gene, CG3362, with comparable sequence similarity to both cNIII and cNIII-like, which based on its substrate specificity and possession of the WW motif, is classified as cNIII-like in this paper. Drosophila also has a second 5′ nucleotidase family member related to cNII, CG32549. Neither Drosophila nor human cNIII-like is strictly specific for m7GMP; both degrade CMP approximately as efficiently as m7GMP and, with lower efficiency, also UMP. Dephosphorylation of some fraction of the CMP and UMP pools may be a price organisms pay for the ability to eliminate m7GMP. Saccharomyces cerevisiae does not appear to contain an orthologue of cNIII-like, in agreement with very weak dephosphorylation of m7GMP in extract (see above). As yeast extract converts m7GMP into an unknown compound (23), these cells may have an alternative pathway for eliminating the modified nucleotide.
In embryo extract, dephosphorylation of m7GMP was stimulated by ATP. Cytosolic nucleotidase II is a precedent for a nucleotidase that is stimulated by ATP and several other phosphorylated compounds (27, 32). However, in the case of CG3362, the apparent ATP dependence was lost after the first chromatography column, and the recombinant enzyme was not stimulated by ATP either. One plausible explanation for the ATP effect in crude extract may be that ATP depletion led to the accumulation of AMP concentrations sufficient to inhibit the enzyme reaction. We can only speculate whether AMP inhibition of CG3362 is of biological relevance; the Ki is fairly high, probably beyond the physiological concentration range.
Unexpectedly, the active site of CG3362 is flexible enough to accommodate m7GDP as a substrate and able to catalyze cleavage of the anhydride bond to release orthophosphate. Thus, in principle, CG3362 could contribute to the degradation of the Dcp2 product, m7GDP. Given the controversy regarding the role of DcpS in the degradation of m7GDP, the enzyme(s) responsible for this reaction remains to be identified.
Expression of CG3362 mRNA is moderate to moderately high at all developmental stages and detected in most tissues (FlyBase). Likewise, expression of human cNIII-like is found in many different cell types at the RNA (UCSC Genome Bioinformatics) and at the protein level (The Human Protein Atlas), although the specificity of the antibody used for the latter type of data was not entirely clear. Widespread expression is consistent with the anticipated need for cap elimination in all cell types. Relatively high levels in the fly gut and in human intestine might reflect a role of the enzyme in disposing of mRNA caps taken up with food. High expression of CG3362 in early embryonic development, peaking at 2–4 h, may be related to the maternal-to-zygotic transition, which is associated with massive turnover of maternal mRNA (51). The need to dispose of m7GMP may be particularly acute for non-dividing, terminally differentiated cells, as they can persist for a long time, turning over their mRNA without being able to dilute out the methylated nucleotide by growth. In the context of such cells, it is also interesting to consider that tRNA and rRNA also contain large amounts and a wide variety of modified nucleotides. Even these stable RNAs can be degraded (52–55). Enzymes may exist to remove the many different modified nucleotides liberated upon the turnover of stable RNAs.
Acknowledgments
We are grateful to Ralph Golbik for helpful discussions, Haiwei Song and Christopher Lima for gifts of reagents, and Michael Götze for help throughout this work.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (to E. W.).

This article contains supplemental Fig. 1.
J. Buschmann, T. Rudolph, B. Moritz, G. Reuter, and E. Wahle, unpublished data.
- NMP
- nucleoside 5′-monophosphate
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- SUMO
- small ubiquitin-like modifier
- cN
- cytosolic nucleotidase
- m7GMP
- 7-methylguanosine monophosphate
- m7GDP
- 7-methylguanosine diphosphate.
REFERENCES
- 1. Parker R., Song H. (2004) The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11, 121–127 [DOI] [PubMed] [Google Scholar]
- 2. Meyer S., Temme C., Wahle E. (2004) Messenger RNA turnover in eukaryotes. Pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 197–216 [DOI] [PubMed] [Google Scholar]
- 3. Dziembowski A., Lorentzen E., Conti E., Séraphin B. (2007) A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 14, 15–22 [DOI] [PubMed] [Google Scholar]
- 4. Chekanova J. A., Shaw R. J., Wills M. A., Belostotsky D. A. (2000) Poly(A) tail-dependent exonuclease AtRrp41p from Arabidopsis thaliana rescues 5.8 S rRNA processing and mRNA decay defects of the yeast ski6 mutant and is found in an exosome-sized complex in plant and yeast cells. J. Biol. Chem. 275, 33158–33166 [DOI] [PubMed] [Google Scholar]
- 5. Symmons M. F., Williams M. G., Luisi B. F., Jones G. H., Carpousis A. J. (2002) Running rings around RNA. A superfamily of phosphate-dependent RNases. Trends Biochem. Sci. 27, 11–18 [DOI] [PubMed] [Google Scholar]
- 6. Furuichi Y., Shatkin A. J. (2000) Viral and cellular mRNA capping. Past and prospects. Adv. Virus Res. 55, 135–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steiger M., Carr-Schmid A., Schwartz D. C., Kiledjian M., Parker R. (2003) Analysis of recombinant yeast decapping enzyme. RNA 9, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Dijk E., Cougot N., Meyer S., Babajko S., Wahle E., Séraphin B. (2002) Human Dcp2. A catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21, 6915–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z., Jiao X., Carr-Schmid A., Kiledjian M. (2002) The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc. Natl. Acad. Sci. U.S.A. 99, 12663–12668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song M.-G., Li Y., Kiledjian M. (2010) Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell 40, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghosh T., Peterson B., Tomasevic N., Peculis B. A. (2004) Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Mol. Cell 13, 817–828 [DOI] [PubMed] [Google Scholar]
- 12. Liu H., Rodgers N. D., Jiao X., Kiledjian M. (2002) The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 21, 4699–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z., Kiledjian M. (2001) Functional link between the mammalian exosome and mRNA decapping. Cell 107, 751–762 [DOI] [PubMed] [Google Scholar]
- 14. Hendler S., Fürer E., Srinivasan P. R. (1970) Synthesis and chemical properties of monomers and polymers containing 7-methylguanine and an investigation of their substrate or template properties for bacterial deoxyribonucleic acid or ribonucleic acid polymerases. Biochemistry 9, 4141–4153 [DOI] [PubMed] [Google Scholar]
- 15. Reichard P. (2010) Ribonucleotide reductases. Substrate specificity by allostery. Biochem. Biophys. Res. Commun. 396, 19–23 [DOI] [PubMed] [Google Scholar]
- 16. O'Brien P. J., Ellenberger T. (2004) The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J. Biol. Chem. 279, 26876–26884 [DOI] [PubMed] [Google Scholar]
- 17. Alseth I., Rognes T., Lindbäck T., Solberg I., Robertsen K., Kristiansen K. I., Mainieri D., Lillehagen L., Kolstø A.-B., Bjørås M. (2006) A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC, and AlkD. Mol. Microbiol. 59, 1602–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rubinson E. H., Gowda A. S., Spratt T. E., Gold B., Eichman B. F. (2010) An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature 468, 406–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Asaeda A., Ide H., Asagoshi K., Matsuyama S., Tano K., Murakami A., Takamori Y., Kubo K. (2000) Substrate specificity of human methylpurine DNA N-glycosylase. Biochemistry 39, 1959–1965 [DOI] [PubMed] [Google Scholar]
- 20. Lau A. Y., Wyatt M. D., Glassner B. J., Samson L. D., Ellenberger T. (2000) Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl. Acad. Sci. U.S.A. 97, 13573–13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gates K. S., Nooner T., Dutta S. (2004) Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol. 17, 839–856 [DOI] [PubMed] [Google Scholar]
- 22. Boiteux S., O'Connor T. R., Lederer F., Gouyette A., Laval J. (1990) Homogeneous Escherichia coli FPG protein. A DNA glycosylase which excises imidazole ring-opened purines and nicks DNA at apurinic/apyrimidinic sites. J. Biol. Chem. 265, 3916–3922 [PubMed] [Google Scholar]
- 23. van Dijk E., Le Hir H., Séraphin B. (2003) DcpS can act in the 5′-3′ mRNA decay pathway in addition to the 3′-5′ pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 12081–12086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malys N., McCarthy J. E. (2006) Dcs2, a novel stress-induced modulator of m7GpppX pyrophosphatase activity that locates to P bodies. J. Mol. Biol. 363, 370–382 [DOI] [PubMed] [Google Scholar]
- 25. Cohen L. S., Mikhli C., Friedman C., Jankowska-Anyszka M., Stepinski J., Darzynkiewicz E., Davis R. E. (2004) Nematode m7GpppG and m3(2,2,7)GpppG decapping. Activities in Ascaris embryos and characterization of C. elegans scavenger DcpS. RNA 10, 1609–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wypijewska A., Bojarska E., Lukaszewicz M., Stepinski J., Jemielity J., Davis R. E., Darzynkiewicz E. (2012) 7-Methylguanosine diphosphate (m7GDP) is not hydrolyzed but strongly bound by decapping scavenger (DcpS) enzymes and potently inhibits their activity. Biochemistry 51, 8003–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hunsucker S. A., Mitchell B. S., Spychala J. (2005) The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther. 107, 1–30 [DOI] [PubMed] [Google Scholar]
- 28. Burroughs A. M., Allen K. N., Dunaway-Mariano D., Aravind L. (2006) Evolutionary genomics of the HAD superfamily. Understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 [DOI] [PubMed] [Google Scholar]
- 29. Allegrini S., Scaloni A., Ferrara L., Pesi R., Pinna P., Sgarrella F., Camici M., Eriksson S., Tozzi M. G. (2001) Bovine cytosolic 5′-nucleotidase acts through the formation of an aspartate 52-phosphoenzyme intermediate. J. Biol. Chem. 276, 33526–33532 [DOI] [PubMed] [Google Scholar]
- 30. Rinaldo-Matthis A., Rampazzo C., Reichard P., Bianchi V., Nordlund P. (2002) Nat. Struct. Mol. Biol. 9, 779–787 [DOI] [PubMed] [Google Scholar]
- 31. Bitto E., Bingman C. A., Wesenberg G. E., McCoy J. G., Phillips G. N., Jr. (2006) Structure of pyrimidine 5′-nucleotidase type 1. Insight into mechanism of action and inhibition during lead poisoning. J. Biol. Chem. 281, 20521–20529 [DOI] [PubMed] [Google Scholar]
- 32. Walldén K., Stenmark P., Nyman T., Flodin S., Gräslund S., Loppnau P., Bianchi V., Nordlund P. (2007) Crystal structure of human cytosolic 5′-nucleotidase II. Insights into allosteric regulation and substrate recognition. J. Biol. Chem. 282, 17828–17836 [DOI] [PubMed] [Google Scholar]
- 33. Valentine W. N., Fink K., Paglia D. E., Harris S. R., Adams W. S. (1974) Hereditary hemolytic anemia with human erythrocyte pyrimidine 5′-nucleotidase deficiency. J. Clin. Invest. 54, 866–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zanella A., Bianchi P., Fermo E., Valentini G. (2006) Hereditary pyrimidine 5′-nucleotidase deficiency. From genetics to clinical manifestations. Br. J. Haematol. 133, 113–123 [DOI] [PubMed] [Google Scholar]
- 35. Jeske M., Meyer S., Temme C., Freudenreich D., Wahle E. (2006) Rapid ATP-dependent deadenylation of nanos mRNA in a cell-free system from Drosophila embryos. J. Biol. Chem. 281, 25124–25133 [DOI] [PubMed] [Google Scholar]
- 36. Jeske M., Wahle E. (2008) Cell-free deadenylation assays with Drosophila embryo extracts. Methods Enzymol. 448, 107–118 [DOI] [PubMed] [Google Scholar]
- 37. Chen N., Walsh M. A., Liu Y., Parker R., Song H. (2005) Crystal structures of human DcpS in ligand-free and m7GDP-bound forms suggest a dynamic mechanism for scavenger mRNA decapping. J. Mol. Biol. 347, 707–718 [DOI] [PubMed] [Google Scholar]
- 38. Mossessova E., Lima C. D. (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 39. Uhlenbeck O. C. (1987) A small catalytic oligoribonucleotide. Nature 328, 596–600 [DOI] [PubMed] [Google Scholar]
- 40. Fisher D. K., Higgins T. J. (1994) A sensitive, high-volume, colorimetric assay for protein phosphatases. Pharm. Res. 11, 759–763 [DOI] [PubMed] [Google Scholar]
- 41. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Temme C., Weissbach R., Lilie H., Wilson C., Meinhart A., Meyer S., Golbik R., Schierhorn A., Wahle E. (2009) The Drosophila melanogaster gene cg4930 encodes a high affinity inhibitor for endonuclease G. J. Biol. Chem. 284, 8337–8348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bosse-Doenecke E., Weininger U., Gopalswamy M., Balbach J., Knudsen S. M., Rudolph R. (2008) High yield production of recombinant native and modified peptides exemplified by ligands for G-protein coupled receptors. Protein Expr. Purif. 58, 114–121 [DOI] [PubMed] [Google Scholar]
- 44. Sambrook J., Russell D. W. (2001) Molecular Cloning. A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 45. Jeske M., Moritz B., Anders A., Wahle E. (2011) Smaug assembles an ATP-dependent stable complex repressing nanos mRNA translation at multiple levels. EMBO J. 30, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin M.-D., Fan S.-J., Hsu W.-S., Chou T.-B. (2006) Drosophila decapping protein 1, dDcp1, is a component of the oskar mRNP complex and directs its posterior localization in the oocyte. Dev. Cell 10, 601–613 [DOI] [PubMed] [Google Scholar]
- 47. Amici A., Magni G. (2002) Human erythrocyte pyrimidine 5′-nucleotidase, PN-I. Arch. Biochem. Biophys. 397, 184–190 [DOI] [PubMed] [Google Scholar]
- 48. Allegrini S., Scaloni A., Careddu M. G., Cuccu G., D'Ambrosio C., Pesi R., Camici M., Ferrara L., Tozzi M. G. (2004) Mechanistic studies on bovine cytosolic 5′-nucleotidase II, an enzyme belonging to the HAD superfamily. Eur. J. Biochem. 271, 4881–4891 [DOI] [PubMed] [Google Scholar]
- 49. Shuman S. (2001) Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acid Res. Mol. Biol. 66, 1–40 [DOI] [PubMed] [Google Scholar]
- 50. Grobosky C. L., Lopez J. B., Rennie S., Skopelitis D. J., Wiest A. T., Bingman C. A., Bitto E. (2012) Structural basis of substrate specificity and selectivity of murine cytosolic 5′-nucleotidase III. J. Mol. Biol. 423, 540–554 [DOI] [PubMed] [Google Scholar]
- 51. Walser C. B., Lipshitz H. D. (2011) Transcript clearance during the maternal-to-zygotic transition. Curr. Opin. Genet. Dev. 21, 431–443 [DOI] [PubMed] [Google Scholar]
- 52. Cole S. E., LaRiviere F. J., Merrikh C. N., Moore M. J. (2009) A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol. Cell 34, 440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fujii K., Kitabatake M., Sakata T., Miyata A., Ohno M. (2009) A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev. 23, 963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chernyakov I., Whipple J. M., Kotelawala L., Grayhack E. J., Phizicky E. M. (2008) Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met-22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev. 22, 1369–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kadaba S., Krueger A., Trice T., Krecic A. M., Hinnebusch A. G., Anderson J. (2004) Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 18, 1227–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]