

Background: Mechanisms regulating signaling of mammalian GnRH receptor (GnRHR), which exhibits an atypical structure, are poorly known.
Results: SET interacts with GnRHR, differentially impacts GnRHR coupling to calcium and cAMP signaling, and enhances GnRH stimulation of Gnrhr promoter activity.
Conclusion: This represents the first identification of a GnRHR interacting partner that enhances its coupling to the cAMP pathway.
Significance: SET is a novel regulator of GnRHR signaling.
Keywords: Cyclic AMP (cAMP), G Protein-coupled Receptors (GPCR), Receptor Regulation, Reproduction, Signaling, GPCR-interacting Protein, GnRH Receptor, Gonadotrope, I2PP2A, SET
Abstract
In mammals, the receptor of the neuropeptide gonadotropin-releasing hormone (GnRHR) is unique among the G protein-coupled receptor (GPCR) family because it lacks the carboxyl-terminal tail involved in GPCR desensitization. Therefore, mechanisms involved in the regulation of GnRHR signaling are currently poorly known. Here, using immunoprecipitation and GST pull-down experiments, we demonstrated that SET interacts with GnRHR and targets the first and third intracellular loops. We delineated, by site-directed mutagenesis, SET binding sites to the basic amino acids 66KRKK69 and 246RK247, located next to sequences required for receptor signaling. The impact of SET on GnRHR signaling was assessed by decreasing endogenous expression of SET with siRNA in gonadotrope cells. Using cAMP and calcium biosensors in gonadotrope living cells, we showed that SET knockdown specifically decreases GnRHR-mediated mobilization of intracellular cAMP, whereas it increases its intracellular calcium signaling. This suggests that SET influences signal transfer between GnRHR and G proteins to enhance GnRHR signaling to cAMP. Accordingly, complexing endogenous SET by introduction of the first intracellular loop of GnRHR in αT3-1 cells significantly reduced GnRHR activation of the cAMP pathway. Furthermore, decreasing SET expression prevented cAMP-mediated GnRH stimulation of Gnrhr promoter activity, highlighting a role of SET in gonadotropin-releasing hormone regulation of gene expression. In conclusion, we identified SET as the first direct interacting partner of mammalian GnRHR and showed that SET contributes to a switch of GnRHR signaling toward the cAMP pathway.
Introduction
G protein-coupled receptors (GPCR)3 represent the largest family of membranous receptors with more than 1000 members identified so far (1). GPCR process signals from a great diversity of endogenous and exogenous stimuli, including biogenic amines, peptides, glycoproteins, lipids, nucleotides, ions, proteases, photons, odors, and tastes. Because of their importance in a wide range of physiological and pathophysiological processes and the fact that they represent one of the most important drug targets, understanding the mechanisms regulating the efficacy and specificity of GPCR is a current challenge. GPCR-interacting proteins (GIP) have been shown to influence GPCR function (2). They are cytoplasmic proteins that bind to intracellular domains of GPCR and participate in the assembly of receptors into signal transduction complexes or “receptosomes.” GIP influence signal transfer from the receptor to G proteins, receptor trafficking between plasma membrane and intracellular compartments, and subcellular localization (2–4). The first identified and best characterized GIP, β-arrestin, has been shown to interact with a large number of GPCR following their phosphorylation upon agonist binding. Binding of β-arrestin to GPCR promotes G protein uncoupling from the receptor and receptor internalization, both events contributing to receptor desensitization. In addition, following receptor internalization, β-arrestin anchored to the receptor promotes G protein-independent signaling, such as activation of the MAPK cascade (5). To date, numerous GIP have been identified, and most of them interact with the carboxyl-terminal tail of GPCR (6).
A unique feature of the mammalian type I gonadotropin-releasing hormone receptor (GnRHR) is that, contrasting with other GPCR, it lacks the carboxyl-terminal tail. This atypical receptor is expressed at the surface of pituitary gonadotrope cells, where it interacts with the hypothalamic neuropeptide GnRH to control reproductive function. Activation of GnRHR leads to synthesis and secretion of the two gonadotropin hormones, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which regulate both gametogenesis and steroidogenesis in gonads. GnRH regulates gonadotrope function, notably through its ability to modulate the expression of numerous genes in gonadotrope cells, among them the three gonadotropin subunit genes (Cga, Lhb, and Fshb) as well as genes encoding components of the GnRH signaling pathway, such as the GnRHR itself (7). GnRHR activates a complex signaling network involving notably PKC and PKA pathways. GnRHR is mainly coupled to phospholipase Cβ via Gαq/11, leading to a rapid increase of intracellular calcium concentrations and activation of several isoforms of PKC (8). PKC regulates gene transcription directly or through activation of the MAPK cascade. Recruitment of the cAMP/PKA pathway by GnRH also contributes to the regulation by GnRH of expression of a few genes, including Nos1, Lhb, and Nr4a1 (9–13).
Because of the lack of the carboxyl-terminal tail, the mammalian GnRHR is insensitive to the classical mechanisms involved in GPCR desensitization. Therefore, upon agonist binding, GnRHR is resistant to phosphorylation, does not recruit β-arrestin, and is poorly internalized (14). Consequently, not much is known about the mechanisms controlling the efficacy and specificity of the mammalian GnRHR signaling. Few mechanisms have been identified that take place downstream of the receptor. Among them, a down-regulation of inositol 1,4,5-triphosphate receptors and a decrease of Gq/11 and PKC expression contribute to GnRHR signaling desensitization induced by a long term GnRH treatment (15, 16). Our hypothesis is that the GnRHR itself can be the target of regulatory mechanisms, not yet discovered, notably through interaction of cytoplasmic proteins with specific intracellular domains. Indeed, this is supported by the fact that introduction in gonadotrope cells of synthetic peptides corresponding to intracellular domains of mammalian GnRHR increases GnRHR coupling to the inositol phosphate pathway (17). This suggests that intracellular domains of GnRHR are targeted by cytoplasmic proteins that inhibit its signaling.
To date, no study has reported a direct interaction between proteins and intracellular domains of the mammalian GnRHR. Interestingly, in silico analysis of intracellular domain sequences of this receptor revealed the presence of basic amino acid clusters similar to the one previously identified within the muscarinic receptor. This basic amino acid cluster is located within the third intracellular loop of the M3 muscarinic receptor (M3-MR) and has been shown to interact with the proto-oncogene SET (18). SET was first described as part of the SET-CAN fusion gene, a putative oncogene associated with acute undifferentiated leukemia (19). SET is involved in the control of gene transcription through regulation of chromatin remodeling and as a component of the INHAT (inhibitor of histone acetyltransferase) complex (20, 21). SET, also called I2PP2A (inhibitor 2 of protein phosphatase 2A), is also an inhibitor of protein phosphatase 2A (PP2A) activity (22), a phosphatase involved in various signaling cascades (23). More recently, SET has been found to inhibit M3-MR coupling to the Gq/PLC/calcium pathway (18). Altogether, these data led us to hypothesize that SET may bind to GnRHR intracellular domains and influence its signaling.
Here, we report for the first time a direct interaction between SET and the first and third intracellular loops of the mammalian GnRH receptor. We showed that SET endogenously expressed in gonadotrope cells has a differential impact on GnRHR signaling because it inhibits receptor coupling to calcium signaling, whereas it increases receptor coupling to the cAMP signaling pathway. Moreover, our data demonstrated that SET is critical for cAMP-mediated GnRH regulation of Gnrhr promoter activity. This suggests that SET may impact gonadotrope cell function, notably through changes of the regulation exerted by GnRH on gene expression.
EXPERIMENTAL PROCEDURES
Materials
GnRH agonist ([d-Trp6]GnRH), antide, poly-l-lysine, probenecid, protein G-Sepharose beads, 3-isobutyl-1-methylxanthine (IBMX), gentamicin, l-glutamine, and monoclonal anti-β-actin antibody were from Sigma-Aldrich. The 38-amino acid form of pituitary adenylyl cyclase-activating polypeptide (PACAP38) was from Calbiochem (VWR International, Strasbourg, France). Iodine-125 radionuclide was purchased from PerkinElmer Life Sciences. Glutathione-Sepharose 4B matrix and ECL+ reagent were from GE Healthcare. Ni2+-nitrilotriacetic acid beads were from Qiagen Inc. (Courtaboeuf, France). Protease inhibitors (Complete Mini, EDTA-free) were from Roche Applied Science. DMEM was from Lonza (Verviers, Belgique). Lipofectamine 2000 and Alexa 488- and Alexa 568-conjugated secondary antibodies were from Invitrogen. 96-well clear-bottomed black or white microplates were from Greiner Bio-One (Dutscher, Les Ulis, France), eight-chamber treated glass slides were from BD Falcon (Dutscher, Les Ulis, France), and FBS was from Pan Biotech GmbH (Dutscher, Les Ulis, France). The QuikChange site-directed mutagenesis kit was purchased from Agilent Technologies (Massy, France). The FLIPR calcium 5 assay kit was purchased from Molecular Devices (St. Grégoire, France). The Dual-Luciferase® reporter assay system, pGloSensorTM-22F cAMP plasmid, and GloSensorTM cAMP reagent were purchased from Promega (Charbonnières, France). Anti-hemagglutinin (HA) mouse monoclonal antibody (clone 16B12) was from Covance. Citifluor was from Biovalley (London, UK). Cell-permeable peptides were synthesized by Proteogenix (Oberhausbergen, France).
Recombinant Protein Preparations
The cDNAs encoding the intracellular subdomains of the rat GnRH receptor were subcloned into pGEX-4T-1 vector. To generate GnRHR ICL1 and ICL2 loops, complementary oligonucleotides from these regions (Lys59–Lys77 and Leu137–Gln156, respectively) were synthesized and annealed prior to ligation into the EcoRI and XhoI restriction sites of the PGEX-4T-1 vector. The rat GnRHR gene segment encoding the ICL3 peptide (Lys232–Leu262) was amplified by polymerase chain reaction (forward primer, 5′-CATGAATTCAAAATCATCTTCGCCCTCA-3′; reverse primer, 5′-CATCTCGAGCAGCCGTGCTCTTGGGAT-3′) and subcloned into the EcoRI and XhoI restriction sites of pGEX-4T-1. Mutations of basic amino acids (lysine and arginine) into glutamic acids within the ICL1 and ICL3 were made using the QuikChange site-directed mutagenesis kit according to the manufacturer's instructions.
GST fusion proteins were expressed in BL21 bacteria and purified on a glutathione-Sepharose 4B matrix. Immobilized fusion proteins were stored at 4 °C, and each batch of fusion proteins used for experiments was first analyzed by SDS-PAGE and Coomassie Blue staining.
The full-length encoding sequence of human SET cloned into the pQE30 vector was kindly provided by Dr R. Z. Qi (Hong Kong University of Science and Technology). The SET protein containing a histidine tag at its amino-terminal extremity was expressed in M15 bacteria and purified on Ni2+-nitrilotriacetic acid beads according to the manufacturer's instructions.
Protein Interaction Assays
Human recombinant His-tagged SET protein (30 nm) were gently mixed for 1 h at 4 °C with GST or GST-ICL1, GST-ICL2, or GST-ICL3 fusion protein (∼300 nm) bound to glutathione-Sepharose 4B (15 μl) in 750 μl of buffer A (25 mm Tris-HCl, pH 7.5, 1 mm EDTA, 100 mm NaCl, 1 mm DTT, 0.1% Nonidet P-40). Resins were washed three times with 1 ml of buffer A. The retained proteins were eluted from the resin with 2× loading buffer (120 mm Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 20% β-mercaptoethanol), placed in a boiling water bath for 5 min, and applied to a 10% SDS-polyacrylamide gel. Separated proteins were then transferred to a nitrocellulose membrane and processed for immunoblotting with a polyclonal anti-SET antibody (1:2000) kindly provided by Dr. T. D. Copeland (NCI-Frederick, National Institutes of Health, Frederick, MD). Membranes were systematically stained in Ponceau Red to confirm equal amounts of GST fusion proteins.
For GST pull-down assays with cell lysates, gonadotrope αT3-1 or LβT2 cells were lysed in buffer B (50 mm Tris-HCl, pH 8.8, 5 mm EDTA, 150 mm NaCl, 1% Nonidet P-40), incubated for 1 h on ice, and centrifuged at 10,000 × g for 30 min at 4 °C. The supernatants were collected, and 750 μg of proteins were gently mixed for 1 h at 4 °C with GST or GST-ICL1, GST-ICL2, or GST-ICL3 fusion protein (∼300 nm) bound to glutathione-Sepharose 4B (15 μl) and processed as described above. All buffers contained a protease inhibitor mixture (Complete Mini, EDTA-free).
Cell Culture
Pituitary gonadotrope cell lines αT3-1 and LβT2 generated by Pamela Mellon (University of California San Diego, La Jolla, CA) (24, 25) were grown in DMEM supplemented with glucose (4.5 g/liter) containing 10% FBS, 2% l-glutamine, and 0.1% gentamicin. Cells were passaged weekly and incubated at 37 °C in a humidified atmosphere with 5% CO2.
Immunocytochemistry
Gonadotrope αT3-1 cells were seeded in DMEM containing 10% FBS, 2% l-glutamine, and 0.1% gentamicin into 8-well chamber slides (250,000 cells/chamber) precoated with 250 μg/ml poly-l-lysine. They were transfected at 70–90% confluence with an HA-tagged GnRHR construct kindly provided by Dr. K. A. Eidne (University of Western Australia) using Lipofectamine 2000 according to manufacturer's instructions. 48 h later, cells were rinsed with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were then washed with PBS and permeabilized for 30 min in blocking solution (PBS with 0.1% Triton X-100 and 1% BSA). Subsequently, cells were incubated with anti-HA (dilution 1:200) and anti-SET (dilution 1:150) primary antibodies for 1 h at room temperature. Unbound antibodies were removed by extensive washes in PBS, and cells were incubated with Alexa 488- and Alexa 568-conjugated secondary antibodies (dilution 1:500) for 1 h at room temperature. Cells were washed in PBS, slides were mounted in Citifluor, and cells were observed under an oil immersion objective (×63) using a Zeiss LSM 700 confocal laser microscope.
Immunoprecipitation
Gonadotrope cells transiently transfected with HA-GnRHR as described above were lysed in 750 μl of ice-cold lysis buffer (75 mm Tris, 2 mm EDTA, 5 mm MgCl2, 0.5% digitonin, protease inhibitor mixture, pH 8). Cell lysates were sonicated, incubated overnight at 4 °C, and centrifuged at 12,000 × g (20 min, 4 °C). Supernatants were precleared with protein G-Sepharose beads and then incubated (1.5 mg of protein in 750 μl of lysis buffer) with monoclonal anti-HA antibody (dilution 1:150) for 1 h at 4 °C. Protein G-Sepharose beads (50 μl) were added, and incubation was continued for 2 h. The resin was pelleted and washed three times with lysis buffer. Immunoprecipitated proteins were eluted with 4× protein sample buffer, subjected to SDS-PAGE, and immunoblotted with monoclonal anti-HA antibody (dilution 1:1000) and polyclonal anti-SET antibody (dilution 1:2000).
siRNA-mediated Gene Silencing of SET
Gonadotrope αT3-1 cells were transfected at 70–90% confluence with SET siRNA duplexes (5′-CAGAAGAGGUCAGAAUUGAUCGCCA-3′, bp 268–292; Invitrogen) targeting SET mRNA (accession number 45198) using Lipofectamine 2000 according to the manufacturer's instructions. In control experiments, cells were transfected with the corresponding predicted oligonucleotide control for the siRNA duplex (Invitrogen). The transfection mixture was removed 5 h later and replaced by DMEM containing 2% FBS and 0.4% l-glutamine.
To assess SET knockdown, cells were homogenized 72 h after transfection in lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40) and incubated for 1 h on ice. Cell homogenates were then centrifuged at 10,000 × g at 4 °C, and 10 μg of the supernatant were separated on 10% SDS-PAGE and transferred to a nitrocellulose membrane. Expression of SET was assessed by immunoblotting with the polyclonal anti-SET antibody. Equal protein loading was verified by reprobing blots with an anti-β-actin antibody.
Quantification of Cell Surface GnRH Receptor Number
The [His5-DTyr6]GnRH was kindly provided by Dr. R. P. Millar (Edinburgh, UK). Five micrograms of [His5-d-Tyr6]GnRH were radioiodinated in the presence of 1 mCi of iodine-125 radionuclide using a modified oxidative reaction catalyzed by chloramine-T and purified by chromatography on a Sephadex G25 column (26, 27).
Saturation binding assays were carried out on intact αT3-1 cells as described previously (27). Briefly, cells transfected with siRNA duplexes in 96-well plates were incubated at 25 °C for 75 min in DMEM containing 0.1% BSA, 25 mm HEPES, and 0.075–5 nm 125I-[His5-d-Tyr6]GnRH. Nonspecific binding was determined using a 10−3 m concentration of the GnRH agonist (GnRHa) and was <3% of the total binding. Binding was stopped by placing the plates on ice, followed by two washes with ice-cold PBS containing 0.1% BSA. Cells were then scraped with 0.2 m NaOH, 0.1% SDS solution, and radioactivity was measured using a γ-counter. All assay points were performed in duplicate, and independent experiments were repeated at least four times.
Real-time Measurement of GnRH Receptor Signaling Using Calcium and cAMP Biosensors in Living αT3-1 Cells
To evaluate GnRH receptor coupling to calcium signaling, αT3-1 cells were loaded with a calcium-sensitive dye (FLIPR calcium 5 assay kit). Briefly, 72 h following siRNA transfection, αT3-1 cells were seeded in DMEM containing 2% FBS and 0.4% l-glutamine (200,000 cells/100 μl/well) in 96-well clear-bottomed black microplates precoated with 250 μg/ml poly-l-lysine (Sigma-Aldrich). After 4 h, cells were dye-loaded with 100 μl of the dye-loading buffer containing 2.5 mm probenecid for 1 h at 37 °C in a 5% CO2 incubator. During a data run, cells in individual wells were exposed to various concentrations of drugs, and fluorescent signals were recorded every 1.5 s for 2 min using the FlexStation III microplate reader. Increases in intracellular calcium were observed as sharp peaks above the basal fluorescent levels typically ∼10 s after drug addition. Increases in intracellular calcium levels were determined by subtracting basal values from peak values. Data were plotted, and t½ to peak values were determined using GraphPad Prism (version 4.0). Data are an average from three wells and are representative of three independent experiments.
To monitor GnRH receptor coupling to cAMP signaling, αT3-1 cells were transfected with a plasmid encoding an engineered cAMP-sensitive luciferase (pGloSensorTM-22F cAMP plasmid). Briefly, αT3-1 cells were seeded in DMEM containing 10% FBS (100,000 cells/100 μl/well) in 96-well clear-bottomed white microplates, precoated with 250 μg/ml poly-l-lysine. The next day, cells were co-transfected with pGloSensorTM-22F cAMP plasmid (200 ng) and SET siRNA duplexes using Lipofectamine 2000 according to the manufacturer's instructions. The transfection mixture was then removed 5 h later and replaced by DMEM containing 2% FBS. Twenty-four hours following transfection, medium was removed, and cells were incubated for 2 h at room temperature in 45 μl of the equilibration medium, a substrate-containing medium (GloSensorTM cAMP reagent) diluted at 4% in DMEM red phenol-free containing 15 mm HEPES and 10% FBS. Cells were incubated with 250 μm IBMX for 10 min, and luminescence was recorded every minute for 15 min before and after the addition of increasing concentrations of drugs on a Centro XS3 microplate luminometer LB 960 plate reader (Berthold Technologies, 150-ms integration). Increases in intracellular cAMP levels were determined by subtracting basal from maximal luminescence values obtained after drug addition. Data were plotted, and t½ to maximal values were determined using GraphPad Prism (version 4.0). Data are an average from three wells and are representative of three independent experiments.
Introduction of ICL1 Peptide in αT3-1 Cells
In order to reduce SET interaction with GnRHR, αT3-1 cells were incubated with peptide corresponding to the first intracellular loop of GnRHR (KLQKWTQKRKKGKKLSRMK). A control experiment was performed using a peptide mutated on the SET binding site (KLQKWTQEEEEGKKLSRMK). To allow efficient peptide delivery into αT3-1 cells, each peptide was covalently attached in its amino-terminal part to a 16-amino acid peptide (RQIKIWFQNRRMKWKK) corresponding to the third helix homeodomain of Drosophila Antennapedia protein (28). Synthesis of peptides was performed by Proteogenix. Peptide quality was controlled by mass spectrometry and HPLC. The use of FITC-coupled peptides allowed us to ensure efficient delivery of wild type and mutated peptides in αT3-1 cells. Cells transfected with pGloSensorTM-22F cAMP plasmid were preincubated for 75 min at 37 °C with each peptide (wild type or mutated, 50 μm), rinsed, and processed to measure cAMP production in response to GnRHa and PACAP38 as described above.
GnRHR Gene Promoter and Luciferase Assay
Transcriptional activity of the Gnrhr was measured using a pLuc0.44Gnrhr promoter fusion construct as described previously (11). Briefly, this construct was obtained by inserting the −475/−32 fragment of the 5′-upstream sequence of the rat Gnrhr promoter into the pGL3-Basic vector containing the firefly luciferase reporter gene. Luciferase constructs containing promoters mutated on cAMP-response element-binding protein (CREm) and/or AP1 (AP1m) elements have been described previously (29). The day before transfection, αT3-1 cells were plated at 100,000 cells/well in 96-well plates in DMEM containing 10% FBS. Cells were transfected with pLuc0.44Gnrhr plasmid (100 ng/well) and co-transfected either with empty or pCMV-PKI vector (10 ng/well) (30) or with control or SET siRNA duplexes using Lipofectamine 2000 according to the manufacturer's instructions. The pRL-SV plasmid expressing Renilla luciferase reporter gene was systematically used (10 ng/well) as an internal control to normalize for transfection efficiency. The next day, cells were incubated overnight with IBMX (250 μm) in the presence or absence of GnRHa (10 nm) or PACAP38 (20 nm). Following stimulation, firefly and Renilla luciferase activities were measured in cell lysates using the Dual-Luciferase reporter assay system according to the manufacturer's instructions.
Statistical Analysis
Results are expressed as mean ± S.E. Statistical analyses were performed using a one- or two-way analysis of variance test. If the F test was significant, the means were compared using least significant difference test. Values were considered statistically different when p was <0.05.
RESULTS
SET Protein Interacts Directly with the First and Third Intracellular Loops of the GnRH Receptor
We first asked if SET interacts with intracellular domains of mammalian GnRHR. For this attempt, we generated GST fusion proteins encompassing each intracellular loop (ICL) of the rat GnRHR (GST-ICL) and used them as affinity matrices in GST pull-down assays. We generated and purified a His-SET recombinant protein (rSET) that was incubated with each GST fusion protein (Fig. 1). Results showed that SET interacts directly with the first (ICL1) and third intracellular (ICL3) loops of GnRHR, whereas no interaction was apparent with the second intracellular loop (ICL2) or GST protein alone used as control (Fig. 1A). Quantification of the amount of bound SET to each ICL revealed that whereas all of the rSET introduced in the pull-down assay bound to ICL1, only ∼20% bound to ICL3 (Fig. 1B), suggesting a better affinity of rSET for ICL1. Altogether, our results demonstrate for the first time a direct interaction between SET protein and intracellular domains of GnRHR. Because GnRHR is expressed in pituitary gonadotrope cells, we then asked whether SET was coexpressed in these cells. We thus screened by immunoblotting the expression of SET in the two gonadotrope cell lines, LβT2 and αT3-1, and showed that SET was endogenously expressed in both cell lines (Fig. 2A, top). We also verified by co-detection of SET and LHβ subunit in rat anterior pituitary gland using immunohistochemistry that SET was expressed in native gonadotrope cells as well (data not shown). We then analyzed the subcellular distribution of GnRHR and SET in αT3-1 cells by immunocytochemistry (Fig. 2A, bottom). As described for other cell types, SET mainly localized into the nucleus of αT3-1 cells (green signal). SET antibody also recognized a fair amount of endogenous SET in the cytoplasm. Interestingly, a fraction of cytoplasmic SET localized in close vicinity to plasma membrane, where the GnRHR is expressed (red signal). Furthermore, we could also detect some overlapping signals within plasma membrane. The close proximity of the two proteins suggests that SET interacts with cell surface GnRHR in αT3-1 cells (Fig. 2A, bottom; see merge and magnification panels). We then performed a co-immunoprecipitation experiment and showed that SET was co-immunoprecipitated with GnRHR expressed in gonadotrope cells (Fig. 2B), indicating that the two proteins in their native conformations interact with each other.
FIGURE 1.

Recombinant SET interacts with the first and third intracellular loops of the GnRHR. A, purified GST fusion proteins encompassing the first, second, or third intracellular loop of rat GnRHR (GST-ICL1, -2, and -3) were incubated with rSET. The bound proteins were then eluted, separated on SDS-PAGE (10%), transferred to nitrocellulose membrane, and immunoblotted with a polyclonal anti-SET antibody. Fusion proteins were detected after membrane staining with Ponceau Red. B, quantification of rSET binding to intracellular domains of GnRHR. SET immunoreactivity and Ponceau Red-stained protein signals were quantified using MultiGauge software. SET signals were normalized to the amount of GST proteins used for each GST pull-down assay, and the results are expressed as the percentage of total rSET used in the pull-down assay. The data are presented as the mean ± S.E. of three independent experiments. ***, p < 0.001 compared with GST alone.
FIGURE 2.

Interaction between SET, endogenously expressed in gonadotrope cells, and GnRHR. A, top, 10 μg of protein homogenates from LβT2 and αT3-1 gonadotrope cells were separated onto 10% SDS-PAGE, transferred onto nitrocellulose membrane, and probed with a polyclonal anti-SET antibody. Bottom, αT3-1 cells were transfected with the HA-GnRHR construct, and subcellular distribution of HA-GnRHR (red) and endogenous SET (green) was determined by immunocytochemistry as described under “Experimental Procedures.” Images were acquired using the ×63 objective of a Zeiss LSM 700 confocal laser microscope. The magnification panel is a magnification of the white box in the merge panel. Scale bar, 2 μm. B, co-immunoprecipitation of SET and HA-GnRHR. Gonadotrope cells transiently transfected (+) or not (−) with HA-GnRHR were processed for immunoprecipitation with anti-HA antibody (IP: anti-HA) as described under “Experimental Procedures.” Immunoprecipitates were subjected to SDS-PAGE followed by immunoblotting (IB) with anti-HA and anti-SET antibodies. 1/20 of the lysate volume prior to immunoprecipitation were also processed for Western blotting with anti-SET antibody. Data are representative of two independent experiments. C, purified GST fusion protein encompassing the first, second, or third intracellular loop of rat GnRHR was incubated with αT3-1 cell lysates, and the bound proteins were probed with anti-SET antibody as described in the legend to Fig. 1 and under “Experimental Procedures.” The same result was obtained with LβT2 cell lysates (data not shown). D, purified GST-ICL3 fusion proteins were preincubated (+) or not (−) with αT3-1 lysates prior to performing a GST pull-down assay in the presence of rSET as described in the legend to Fig. 1. The immunoblot shown is representative of three independent experiments.
We then asked which intracellular domains of GnRHR were involved in GnRHR interaction with endogenous SET. Using GST pull-down assays with gonadotrope lysates, we showed that SET endogenously expressed in gonadotrope αT3-1 cells interacts with the first intracellular loop of GnRHR (Fig. 2C). Surprisingly, no interaction of gonadotrope SET could be detected with ICL3, contrasting with the results obtained with rSET (Fig. 2C). This suggests that a factor present in gonadotrope cells prevents SET binding to ICL3 either by a direct competition with SET or by complexing SET and selectively precluding its interaction with ICL3 but not ICL1. To better understand SET-ICL3 interaction, we evaluated if GST-ICL3 fusion protein, preincubated with gonadotrope cell lysates, still retains its capacity to bind rSET (Fig. 2D). Our results show that whereas GST-ICL3 is able to interact with rSET, preincubation of GST-ICL3 with gonadotrope lysate abrogates rSET binding (Fig. 2D). This result strongly suggests that interaction of SET with ICL3 might be regulated in gonadotrope cells by a protein competing with SET for binding.
To delineate SET binding sites within ICL1 and ICL3 of GnRHR, we generated mutants in which basic amino acids (arginine and lysine) within ICL1 and ICL3 were replaced by glutamic acid residues (Fig. 3). We targeted basic amino acids based on our recent characterization of SET binding sites on M3-MR ICL3 (31). This strategy was strengthened by the fact that whereas ICL1 and ICL3 contain such basic amino acid clusters, they are absent in ICL2, which does not bind SET (Fig. 1). Mutation of the basic cluster “KRKK” into “EEEE” completely abolished SET binding to ICL1 (mutant A; Fig. 3A). Mutation of only one amino acid did not significantly change the amount of bound SET (mutants C and D). However, mutation of two amino acids severely disrupted SET interaction (mutants E–G). In this case, only ∼20% of SET interaction with ICL1 was retained at the most. These results indicate that within the KRKK cluster, at least three basic amino acids are needed for a full interaction of SET with ICL1. This may explain the weaker interaction observed between SET and ICL3, which contains only the two basic residues RK (Figs. 1 and 3B). Additionally, we demonstrated that the two basic amino acids KK following the KRKK sequence in ICL1 have a role in stabilizing SET interaction because their mutation into glutamic acids decreased by ∼50% the interaction of SET with ICL1 (mutant B; Fig. 3A). As expected from ICL1 data, mutation into glutamic acid of the RK amino acids abolished SET binding to ICL3 (Fig. 3B).
FIGURE 3.

Localization of SET binding sites within GnRHR intracellular loops. Basic amino acids lysine and arginine located in the first (A) and third (B) intracellular loops of GnRHR (boxed sequence) were mutated into glutamic acid by site-directed mutagenesis. Each mutant ICL1 (A–G) and ICL3 (EE) construct was generated and tested in GST pull-down assays with rSET. The amount of rSET associated with each construct was quantified as described in the legend to Fig. 1. The immunoblot shown is representative of three independent experiments. **, p < 0.01; ***, p < 0.001 compared with wild type ICL (WT). The data are presented as the mean ± S.E. of 3 to 5 independent experiments performed in triplicate.
Modulation of SET Expression in αT3-1 Gonadotrope Cells
To address the role of SET in GnRHR signaling, we evaluated the effect of a decreased expression of SET in αT3-1 gonadotrope cells. To do this, we used siRNA targeting a specific sequence of SET mRNA (bp 268–292) as described previously (31) to transfect αT3-1 cells. Endogenous SET expression was consistently decreased by 51 ± 7% in cells transfected with SET siRNA as compared with those transfected with control siRNA (Fig. 4A). Prior to addressing the role of SET in GnRHR signaling, we first evaluated whether SET knockdown may impact cell surface expression of GnRHR. Saturation binding studies using a radioiodinated GnRH ligand (125I-[His5-d-Tyr6]GnRH) on αT3-1 cells transfected either with control or SET siRNA indicated that there was no apparent effect of SET knockdown on cell surface GnRHR number or ligand affinity (Fig. 4B, top). Scatchard analyses indeed revealed similar Bmax and Kd values in both control and SET knock-down cells (control siRNA and SET siRNA: 6.2 ± 1.4 and 5.5 ± 1.4 fmol/well for Bmax and 1.1 ± 0.2 and 1.0 ± 0.1 nm for Kd, respectively) (Fig. 4B, bottom).
FIGURE 4.
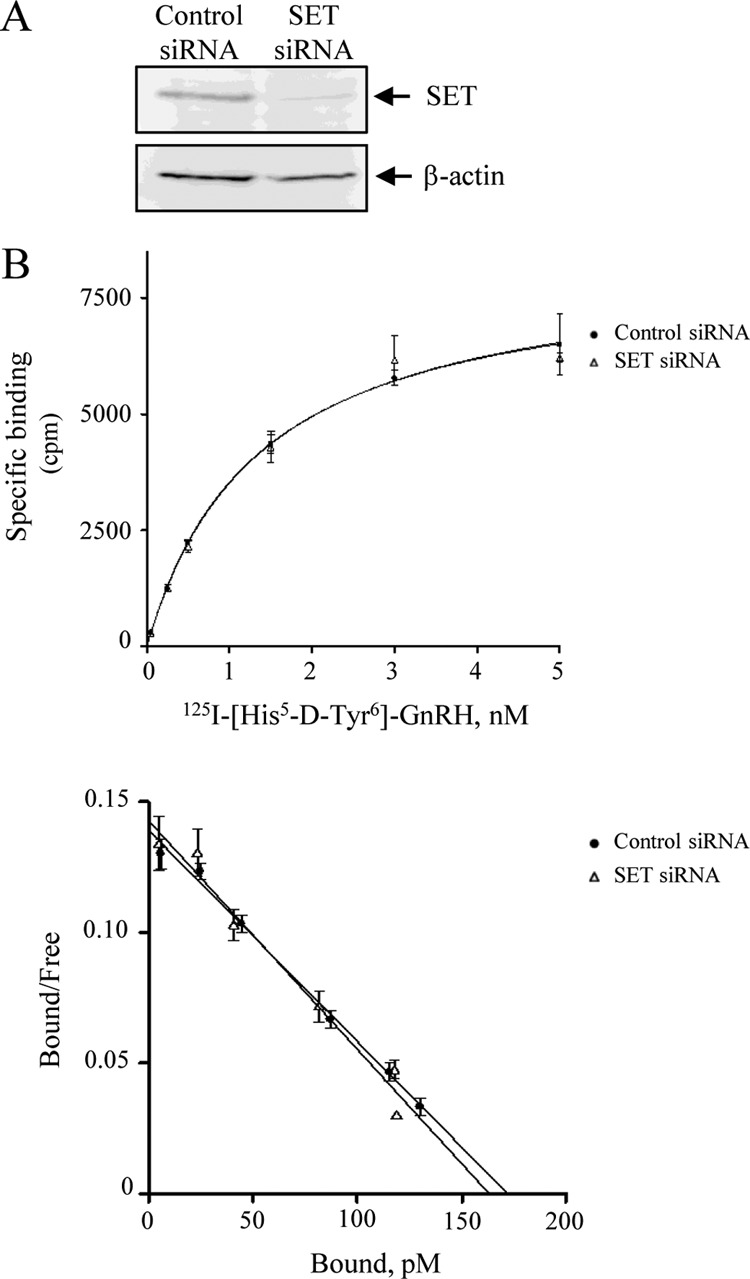
SET knockdown in αT3-1 cells. A, 10 μg of protein homogenates from αT3-1 cells transfected with control or SET siRNA were electrophoresed on 10% SDS-PAGE, membrane-transferred, and probed with anti-SET or anti-β-actin antibodies. B, effect of SET knockdown on the expression of GnRHR at the cell surface. Cell surface GnRHR was measured in αT3-1 cells transfected with control or SET siRNA by radioligand binding as described under “Experimental Procedures.” Top, saturation binding curve. Bottom, Scatchard analysis. The data are presented as the mean ± S.E. of one experiment representative of 4–5 independent experiments performed in duplicate.
Impact of SET on GnRHR Coupling to Calcium Pathway in Gonadotrope Cells
To measure calcium variations, gonadotrope αT3-1 cells were loaded with a calcium-sensitive fluorescent dye, and changes in intracellular calcium concentrations were recorded in living cells after stimulation with GnRHa or with oxytocin, a hypophysiotropic hormone known to signal through the calcium pathway. No significant effect of SET knockdown was observed on basal intracellular calcium concentrations (Fig. 5A; 249 ± 22 and 259 ± 38 arbitrary units for control siRNA and SET siRNA, respectively). In control siRNA cells, the addition of GnRHa induced a rapid and significant increase of intracellular calcium concentration. Calcium peak was reached ∼5 s after the GnRHa addition and corresponded to a 1.7 ± 0.1-fold increase over basal level. Following the peak, intracellular calcium concentration decreased progressively (∼70% decrease) and reached a plateau, which remained stable until at least 5 min of stimulation (Fig. 5A) (data not shown). The same calcium profiles were observed in untransfected cells (data not shown) and were similar to profiles described previously in αT3-1 cells (32). Interestingly, decreasing SET expression significantly increased the calcium peak value by 44 ± 14% in response to GnRHa. All other parameters of the calcium response (kinetics, decay profiles, and plateau values) were unaffected by SET siRNA (see the legend to Fig. 5). As shown in Fig. 5B, the amplitude of the calcium peak was dose-dependently increased by GnRHa in both control and SET siRNA-transfected cells. However, decreasing SET expression significantly potentiated GnRHa-induced calcium mobilization for each concentration of GnRHa (Fig. 5B) without affecting significantly the EC50 value (38 ± 17 versus 17 ± 5 nm in control and SET siRNA transfected cells, respectively). In contrast, SET knockdown did not alter calcium mobilization elicited by the oxytocin receptor, an endogenously expressed Gq-coupled receptor in αT3-1 cells (Fig. 5C). Altogether, our results demonstrate that SET specifically inhibits GnRHR coupling to the Gq/calcium signaling pathway.
FIGURE 5.

Effect of SET knockdown on GnRH-induced calcium mobilization in gonadotrope cells. αT3-1 cells transfected with control or SET siRNA were plated in 96-well black plates precoated with poly-l-lysine and loaded with calcium fluorescent dye. Excitation fluorescence was set at 485 nm, and emission was detected at 525 nm (515-nm emission cut-off filter) using the FlexStation III microplate reader (Molecular Devices). A, cells were stimulated with 1 μm GnRHa, and intracellular calcium mobilization was measured every 1.5 s during the following 2 min. Results are expressed as relative fluorescence units (RFU). Data are representative of three independent experiments performed in triplicate. Kinetics of calcium mobilization (t½ = 2.5 ± 0.5 and 3 ± 0.9 s for control siRNA and SET siRNA, respectively), decay profiles (64 ± 1.3 and 70 ± 15% decrease for control siRNA and SET siRNA, respectively), and plateau values (318 ± 8 and 329 ± 8 arbitrary units for control siRNA and SET siRNA, respectively) were unaffected by SET siRNA. B, cells were treated with increasing concentrations of GnRHa (10−10 to 10−5 m), and increases in intracellular calcium were determined by subtracting the base-line values from peak values. Results were expressed as the percentage of the maximal response in control siRNA-transfected cells. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate. **, p < 0.01; ***, p < 0.001 compared with control at respective GnRHa concentrations. C, intracellular calcium mobilization in αT3-1 cells transfected with control siRNA or SET siRNA was measured in response to increasing concentrations of oxytocin (10−10 to 10−5 m) as described in B. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate.
Impact of SET on GnRHR Coupling to cAMP Pathway in Gonadotrope Cells
Coupling of GnRHR to the cAMP signaling pathway was addressed by introducing in αT3-1 cells a plasmid encoding an engineered cAMP-sensitive luciferase, which becomes active upon cAMP binding (see “Experimental Procedures”). Transfected cells were challenged with GnRHa, and the luminescence intensity emitted in real time by luciferase was measured in living cells (Fig. 6A). Our results showed that GnRHa induces a significant increase in luciferase activity, reflecting an increase in intracellular cAMP concentration. Intracellular cAMP concentration increased progressively upon GnRHa stimulation and reached a maximum at 10 min. Mobilization of cAMP increased dose-dependently upon GnRHa stimulation (Fig. 6B) and was completely blocked by preincubation with antide, a GnRH antagonist (data not shown), thus demonstrating that GnRHR couples to the cAMP signaling pathway in the gonadotrope αT3-1 cell line. Knockdown of SET in αT3-1 cells did not change basal cAMP levels but dramatically reduced by 47 ± 9% intracellular cAMP mobilization induced by GnRHa (Fig. 6A). Decreasing SET expression did not significantly affect kinetics (Fig. 6A, t½ = 4.3 ± 0.9 and 3.2 ± 0.9 min for control siRNA and SET siRNA, respectively) or the EC50 values (Fig. 6B, EC50 = 1.4 ± 0.5 and 1.2 ± 0.5 nm for control siRNA and SET siRNA, respectively). It is noteworthy that cAMP mobilization by an endogenously expressed Gs-coupled receptor, such as the PACAP type I receptor, was not altered by decreased SET expression (Fig. 6C). Altogether, our results demonstrate that SET specifically enhances GnRHR coupling to the Gs/cAMP signaling pathway.
FIGURE 6.
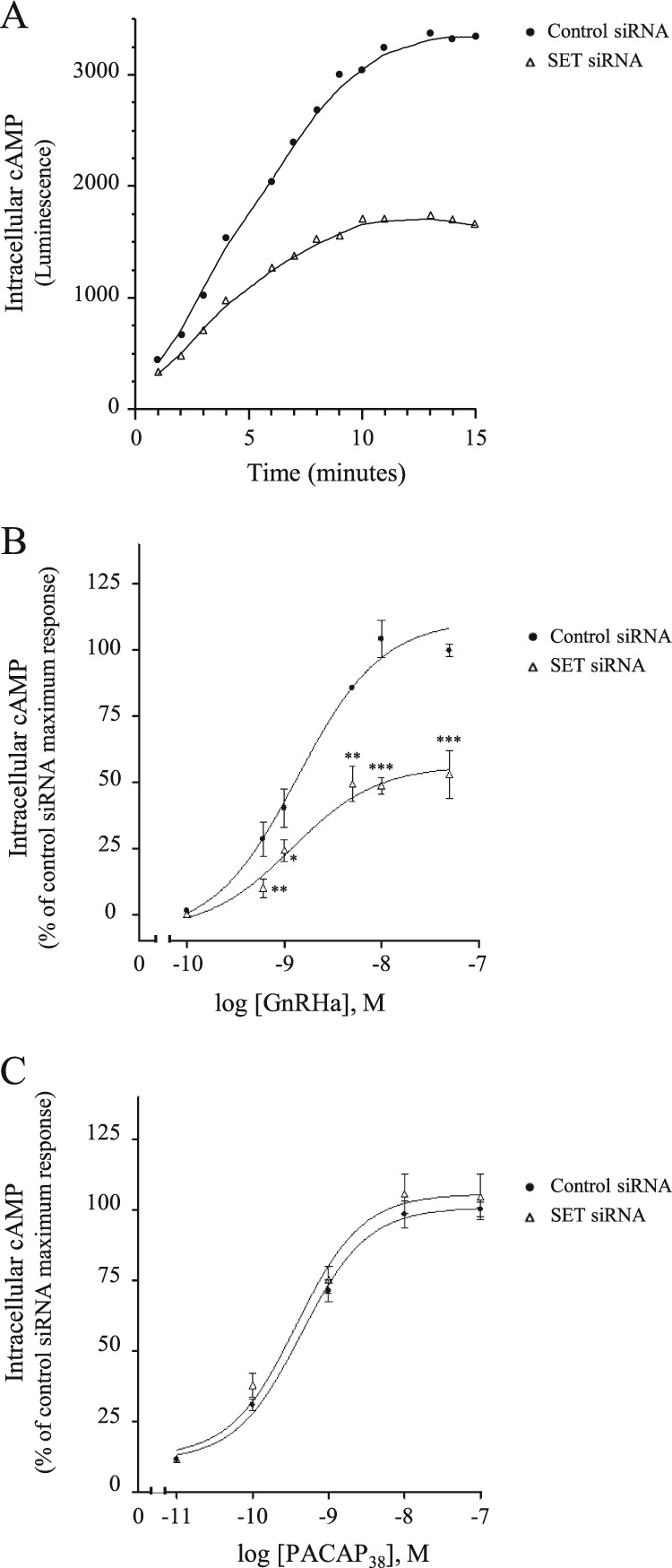
GnRHR coupling to the cAMP pathway in gonadotrope cells. The impact of SET knockdown is shown. αT3-1 cells plated in 96-well white plates precoated with poly-l-lysine were transfected with a plasmid encoding an engineered cAMP-sensitive luciferase (pGloSensorTM-22F cAMP plasmid) together with control or siRNA SET. Twenty-four hours after transfection, cells were incubated in equilibration medium as described under “Experimental Procedures.” Luminescence intensities were measured using a Centro XS3 LB 960 plate reader (150-ms integration). A, cells were stimulated with 10 nm GnRHa, and luminescence intensities were recorded every minute for 15 min. Results are expressed as luminescence arbitrary units and are representative of three independent experiments performed in triplicate. B, cells were stimulated with increasing concentrations of GnRHa (10−10 to 5 × 10−8 m), and increases in intracellular cAMP levels were determined by subtracting basal from maximal luminescence values obtained after GnRHa addition. Results are expressed as the percentage of maximal response in control cells. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with control at respective GnRHa concentrations. C, cells were stimulated with increasing concentrations of PACAP38 (10−11 to 10−7 m), and increases in intracellular cAMP levels were determined. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate.
To confirm these results, we asked if delivery of a peptide corresponding to ICL1 in αT3-1 cells will decrease GnRHR signaling by complexing endogenous SET protein and preventing its action on GnRHR. Cells were preincubated with a cell-permeable ICL1 peptide and challenged either with GnRHa or PACAP38, and intracellular cAMP production was measured in real time in αT3-1 living cells (Fig. 7). Introduction of ICL1 peptide into αT3-1 cells decreased by 33 ± 4% cAMP production in response to GnRHa but did not have any impact on cAMP production by PACAP38 (Fig. 7, ICL1 WT). These results suggest that ICL1 peptide does not disturb cAMP signaling events downstream of GPCR but rather decreases coupling of GnRHR to the cAMP pathway. Importantly, introduction of a peptide corresponding to mutant A of ICL1 (Fig. 3A), which does not interact with SET, did not have any significant effect on GnRHR coupling to cAMP (Fig. 7A, ICL1 Mutant). This indicates that the inhibitory effect of ICL1 on GnRHR signaling is mediated through its interaction with endogenous SET. Altogether, these results, which are in agreement with the siRNA experiments, support a role of SET in GnRHR coupling to the cAMP pathway in αT3-1 cells.
FIGURE 7.
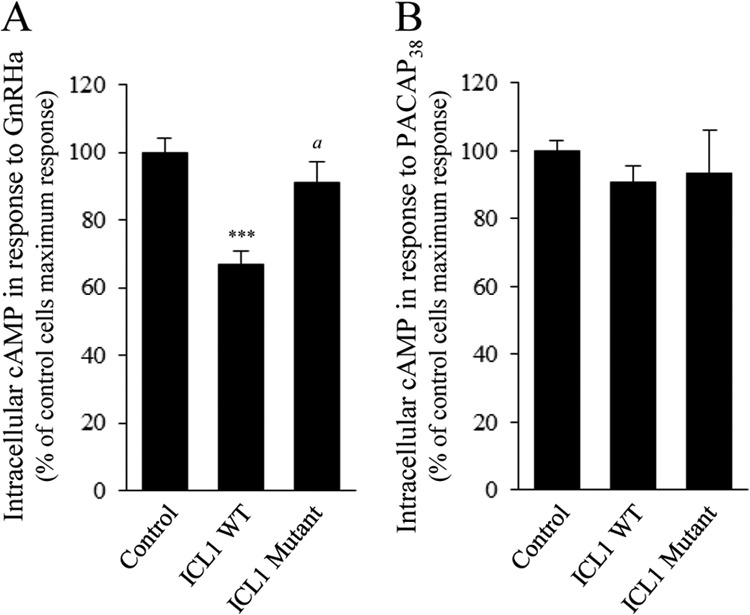
Effect of SET neutralization by ICL1 peptide on GnRHR coupling to cAMP pathway in gonadotrope cells. αT3-1 cells plated in 96-well white plates precoated with poly-l-lysine were transfected with a plasmid encoding an engineered cAMP-sensitive luciferase (pGloSensorTM-22F cAMP plasmid). Twenty-four hours later, cells were preincubated or not (control) at 37 °C with 50 μm peptide corresponding to the first intracellular loop of GnRHR (ICL1 WT). A control experiment was performed using ICL1 peptide mutated on the SET binding site (ICL1 mutant). After 75 min, cells were washed and processed to measure cAMP production in response to GnRHa (100 nm) or PACAP38 (100 nm) as described under “Experimental Procedures.” Increases in intracellular cAMP levels were determined. Results are expressed as the percentage of maximal response in control cells. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate. ***, p < 0.001 compared with control cells. a, p < 0.01 compared with ICL1 WT.
Impact of SET on GnRH-induced Gnrhr Promoter Activity
In gonadotrope cells, the GnRHR gene is known to be regulated by GnRH and by the cAMP pathway (33–35). However, involvement of the cAMP pathway in GnRH regulation of GnRHR gene expression in gonadotrope cells remains unknown. To monitor Gnrhr promoter activity in αT3-1 cells, cells were transfected with a construct encoding the firefly luciferase under the control of the Gnrhr proximal promoter, as described previously (11). Stimulation of αT3-1 cells with GnRHa increases significantly Gnrhr promoter activity (1.4 ± 0.1-fold over basal level; Fig. 8A). Transfection of cells with a vector encoding the PKA inhibitor PKI significantly decreased basal activity of the Gnrhr promoter, confirming the involvement of the PKA pathway in Gnrhr expression. It is noteworthy that the GnRH-mediated Gnrhr promoter activity increase was abolished by overexpression of PKI (Fig. 8A), demonstrating that GnRH regulates Gnrhr promoter activity through the PKA pathway. Further evidence for the mediation by the cAMP/PKA pathway was provided by the fact that mutations of the cAMP-response element and/or AP1 elements within the Gnrhr promoter significantly inhibited the IBMX-induced (Fig. 8B, left) and GnRH-induced increase of Gnrhr promoter activity (Fig. 8B, right). Because we demonstrated that SET increases GnRHR coupling to the cAMP pathway, we evaluated whether SET could potentiate GnRH induction of its own receptor gene. To this end, αT3-1 cells were co-transfected with either control or SET siRNA and then treated with GnRHa (Fig. 8C). In control cells, GnRHa induced a 1.6 ± 0.2-fold increase of Gnrhr promoter activity. Decreasing expression of SET markedly inhibited by 78.5 ± 9.6% GnRH stimulation of promoter activity as compared with control siRNA-transfected cells (Fig. 8C; 1.6 ± 0.2- versus 1.1 ± 0.1-fold increase in control siRNA and SET siRNA transfected cells, respectively), without any significant change of basal Gnrhr promoter activity (Fig. 8C). In αT3-1 cells, PACAP38 also significantly increased Gnrhr promoter activity (Fig. 8D), as reported previously (11). However, contrasting with the drastic effect of SET on the regulation exerted by GnRH, SET siRNA did not interfere with PACAP38 regulation of Gnrhr promoter activity (Fig. 8D). Altogether, this demonstrates that SET is critical for GnRH stimulation of the Gnrhr promoter, probably by enhancing GnRHR coupling to the cAMP signaling pathway.
FIGURE 8.

Involvement of the PKA pathway and SET in basal or GnRH-induced increase of Gnrhr promoter activity in gonadotrope cells. A, αT3-1 cells plated in 96-well plates were co-transfected with the Gnrhr promoter luciferase fusion construct (pLuc0.44Gnrhr) and either the empty vector or the expression vector encoding PKI (pCMV-PKI). Twenty-four hours after transfection, cells were stimulated overnight with 10 nm GnRHa, and firefly and Renilla luciferase activities were measured in cell lysates as described under “Experimental Procedures.” Data are the mean ± S.E. of six independent experiments performed in triplicate. ***, p < 0.001 compared with basal level in cells transfected with empty vector. B, αT3-1 cells were plated in 96-well plates and transiently transfected with the pLuc0.44Gnrhr construct encoding the wild type promoter (WT) or the mutated promoters (AP1m, CREm, or CREm/AP1m). Twenty-four hours after transfection, cells were stimulated overnight with 250 μm IBMX (left) or with 10 nm GnRHa (right), and firefly and Renilla luciferase activities were measured in cell lysates as described under “Experimental Procedures.” Results are expressed as ratios of Firefly to Renilla activities (left) or in -fold over basal level in cells transfected with pLuc0.44Gnrhr WT (right). IBMX and GnRHa increased wild type Gnrhr promoter activity by 5.87 ± 0.47 and 1.32 ± 0.07, respectively. Data are the mean ± S.E. of 5–8 independent experiments performed in triplicate. ***, p < 0.001 compared with WT (left) or with basal level in cells transfected with pLuc0.44Gnrhr WT (right). a, p < 0.001 compared with cells transfected with pLuc0.44Gnrhr CREm or AP1m (left). C and D, αT3-1 cells were plated in 96-well plates and transiently transfected with the wild type pLuc0.44Gnrhr construct together with control or SET siRNA. Twenty-four hours after transfection, cells were stimulated overnight with either 10 nm GnRHa (C) or 20 nm PACAP38 (D). Following stimulation, firefly and Renilla luciferase activities were measured in cell lysates, as described under “Experimental Procedures.” Results are expressed as -fold over basal level in cells transfected with control siRNA. Data are the mean ± S.E. of 4–8 independent experiments performed in triplicate. ***, p < 0.001 compared with respective basal level.
DISCUSSION
A unique feature of the mammalian GnRHR is that, unlike other receptors belonging to the GPCR family, it lacks the carboxyl-terminal tail classically involved in GPCR regulation. Therefore, mechanisms regulating the efficacy of its signaling are poorly known. In this study, we report for the first time an interaction between the intracellular domains of GnRHR and a protein distinct from heterotrimeric G proteins, the proto-oncogene SET. We show here that SET co-localizes with cell surface GnRHR in gonadotrope cells and interacts with GnRHR through two distinct regions of the receptor, the ICL1 and to a lesser extent the ICL3. There is now growing evidence that SET is an accessory protein for GPCR signaling. Its interaction was first reported with the ICL3 of the M3-MR (18). Since then, other receptors have been found to interact with SET (i.e. the M1, M2, and M5 subtypes of muscarinic receptor (36), the type 1A angiotensin receptor (37), and the β1-adrenergic receptor (38)). Using site-directed mutagenesis, we delineated SET binding sites to basic amino acid clusters contained within the ICL1 and ICL3 of GnRHR. Importantly, these sites were absent from the ICL2 of GnRHR, which does not bind SET, and their mutation in the ICL3 of M3-MR precluded SET binding (31). Interestingly, in silico analyses revealed the presence of similar basic amino acid clusters within intracellular domains of receptors known to interact with SET, such as the muscarinic (ICL3 and carboxyl-terminal domain) and the β1-adrenergic receptors (ICL3), but also within other GPCR, such as rhodopsin and melatonin receptors. This suggests that SET could interact with other GPCR through these particular sites and influence their signaling.
To assess the potential role of SET in GnRHR signaling in gonadotrope cells, we took advantage of calcium and cAMP biosensors that allowed us to monitor with high sensitivity and in real time calcium and cAMP mobilization in gonadotrope αT3-1 living cells. To reduce interaction between GnRHR and SET in αT3-1 cells, we developed several experimental strategies. In a first series of experiments, we introduced mutations in the first and third intracellular domains of the full-length GnRHR to abolish SET binding to the receptor. However, whereas wild type GnRHR was efficiently targeted at the plasma membrane of αT3-1 and CHO cells, the mutant receptor did not reach the plasma membrane but instead remained in the cytoplasm of both cell types (Fig. 9A). Consequently, GnRHa did not induce any calcium signaling in CHO cells expressing the mutant GnRHR (Fig. 9B). These results are in accordance with other studies showing that targeting of mammalian GnRHR at the plasma membrane is very often impaired by mutations, leading in some cases to the development of disease, such as hypogonadotropic hypogonadism in humans (for a review, see Ref. 39). Nevertheless, this precluded us from addressing the role of SET in GnRHR signaling with this strategy. We then used the RNAi antisense strategy to decrease efficiently SET endogenous expression in αT3-1 cells. With this strategy, we demonstrated for the first time that GnRHR mobilization of calcium signaling is markedly inhibited by SET in gonadotrope αT3-1 cells. Indeed, decreasing expression of SET by siRNA significantly increased calcium signaling in response to GnRHa. However, SET did not have any significant impact on intracellular calcium mobilization by the Gq-coupled oxytocin receptor, endogenously expressed in αT3-1 cells. This demonstrates that SET does not regulate signaling events downstream of the receptor but rather acts at the level of the receptor itself to specifically inhibit its coupling to Gq/PLC/calcium signaling pathway. A similar inhibitory action of SET on M3-MR coupling to calcium signaling has been reported (18). Interestingly, a recent study addressing some of the mechanisms involved in SET action on M3-MR signaling showed that SET decreased Gq protein engagement by the M3-MR, possibly through a competition for binding to the receptor, given the close proximity of the two protein binding sites within ICL3 (31). Similarly, in ICL3 of GnRHR, SET binding sites (246RK247) are next to amino acids involved in coupling to the Gq/PLC/calcium pathway (259RAR261) (40, 41), suggesting that SET may also inhibit Gq protein binding to GnRHR in gonadotrope αT3-1 cells. SET has been described as an inhibitor of PP2A activity (22). There is very recent evidence showing that SET may regulate GPCR signaling by influencing the phosphorylation of proteins involved in GPCR signaling cascades through the inhibition of PP2A. For example, SET inhibits dephosphorylation of the M3-MR as well as of the β1- and β2-adrenergic receptors (31, 38). Inhibition of β1-adrenergic receptor dephosphorylation contributes to the attenuation of its signaling, probably by preventing receptor resensitization (38). SET can also influence the phosphorylation of proteins recruited by GPCR. This is the case for Akt kinase, which is recruited by the type 1A angiotensin receptor (37). SET association with this receptor leads to sustained Akt phosphorylation through PP2A inhibition, resulting in the attenuation of glycogen synthase 3β signaling (37). A role of PP2A inhibition in the attenuation of GnRHR coupling to calcium signaling is an attractive hypothesis, especially because we identified by affinity chromatography and mass spectrometry analysis both the catalytic subunit of PP2A and the protein SET associated with the ICL3 of GnRHR.4 Although it is unlikely that SET will have an impact on GnRHR because of its resistance to phosphorylation (42), we could hypothesize that docking of SET onto intracellular domains of GnRHR may control the phosphorylation of proteins associated directly with the GnRHR and/or present in the signaling complex activated by the receptor. Phosphorylation of Gαq/11 protein has been shown to decrease coupling to GPCR and induce receptor desensitization, as demonstrated for the 5-HT2A receptor (43). Therefore, the inhibition of GnRHR coupling with the calcium pathway mediated by SET might result from a competition between SET and Gq protein and/or could be the consequence of increased phosphorylation of Gq or other signaling entities.
FIGURE 9.

Impact of SET binding site mutations on GnRHR subcellular localization and signaling. αT3-1 and CHO cells were transfected with HA-tagged GnRHR construct carrying (mutant) or not (WT) mutations precluding SET binding onto intracellular domains of GnRHR (mutations corresponding to mutant A of Fig. 3A were generated on full-length GnRHR by site-directed mutagenesis). A, expression and subcellular localization of GnRHR were monitored by immunocytochemistry by staining either permeabilized or non-permeabilized cells with monoclonal anti-HA antibody (red). Nuclei were stained with DAPI (blue). Images were acquired using a Zeiss LSM 700 confocal laser microscope. B, CHO cells transfected either with WT or mutant GnRHR were incubated with increasing concentrations of GnRHa, and intracellular calcium was determined as described under “Experimental Procedures.” Results were expressed as the percentage of the maximal response in cells transfected with WT GnRHR. The data are presented as the mean ± S.E. of three independent experiments performed in triplicate.
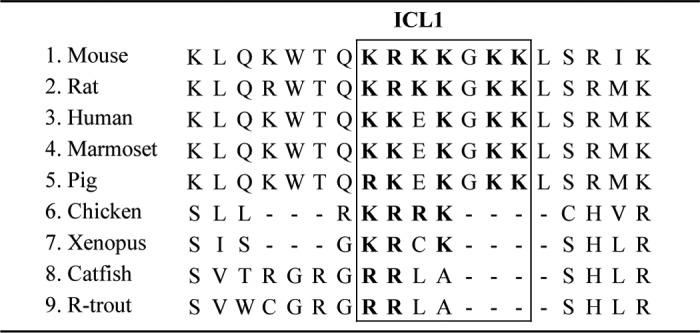
Interestingly, we found that in addition to ICL3, SET interacts also with another intracellular domain of GnRHR (i.e. the ICL1), demonstrating for the first time that SET binding to GPCR is not restricted to ICL3. Our results suggest that SET binding to ICL1 is stronger than with ICL3. This might be due to the presence of more basic amino acids within ICL1. SET binds to the sequence 66KRKKGKK72 within ICL1, whereas it binds to the 246RK247 sequence within ICL3. It is noteworthy that in mammals, the 66KRKKGKK72 sequence is part of a putative binding site for heterotrimeric G protein 71KKLSR75, and mutations of the amino acids 73LSR75 markedly reduced cAMP production in response to GnRH in COS-7 cells (44). In gonadotrope cells, GnRHR coupling to cAMP has remained controversial for a long time, and it is only recently that this coupling has been demonstrated in the LβT2 gonadotrope cells (16, 45) as well as in rat pituitary cell culture and in vivo (12). However, coupling of GnRHR to the cAMP pathway could not be demonstrated in the αT3-1 gonadotrope cell line (46). To address this issue, we initially attempted to measure intracellular cAMP concentrations by using classical biochemical and immunological assays. Despite a clear increase of cAMP production in response to PACAP38 or forskolin, no significant increase could be detected in response to GnRHa,5 consistent with data previously reported by others using these cells (46). We further re-explored GnRHR signaling to cAMP pathway in αT3-1 cells by taking advantage of a recent highly sensitive technology based on the use of a cAMP biosensor to monitor cAMP mobilization in living cells (47). This strategy allowed us to demonstrate for the first time that GnRHa increases cAMP production in αT3-1 cells. This increase was dose-dependent with an EC50 in the range of 1 nm. Furthermore, GnRHa-induced cAMP increase was completely blocked by preincubation of αT3-1 cells with a GnRH antagonist. Altogether, this is the first study demonstrating that, in addition to the calcium pathway, GnRHR couples to the cAMP pathway in the αT3-1 gonadotrope cell line. We then looked for a role of SET in the ability of GnRHR to couple to the cAMP pathway. For this attempt, we developed two complementary experimental strategies to decrease endogenous interaction between GnRHR and SET (i.e. decreasing endogenous SET expression with RNAi antisense and introducing in αT3-1 cells a peptide corresponding to the ICL1 sequence). The ICL1 peptide was used to block SET action on GnRHR through its ability to complex endogenous SET. Using both strategies, we demonstrated that, contrasting with its inhibitory action on GnRHR coupling to the calcium pathway, SET significantly increased GnRHR coupling to the cAMP pathway. Importantly, the effect of SET on GnRHR signaling was specific because it did not have any significant impact on signaling of another GPCR, the PACAP type I receptor, known to activate efficiently the cAMP pathway in gonadotrope cells. This demonstrates that SET does not have any effect on the cAMP-signaling components but rather acts at the level of GnRHR to increase its coupling to the cAMP pathway. The unexpected finding that SET increases Gs/cAMP signaling in response to GnRHa indicates that rather than disrupting receptor interaction with Gs, SET may stabilize or favor its interaction with GnRHR. Alternatively, SET could interact with other components of the cAMP signaling cascade to tether downstream effectors in close proximity to GnRHR, thus creating an efficient signaling complex that increases the amplitude of response. Such a role has been described for INAD (inactivation-no-after-potential D) protein that associates with rhodopsin. Interaction of INAD with rhodopsin increases the amplitude of response to light by interacting with PLCβ and PKC (48). In the case of the parathyroid hormone receptor, an interaction between the NHERF2 (Na/H exchanger regulatory factor 2) protein and the Gq protein has been proposed to explain enhanced receptor coupling to the calcium signaling pathway (49). SET has been shown to interact with a myriad of proteins (50), and among them, some are involved in the GPCR signaling cascade. For example, SET interacts with PI3K (38), Rac1 (51), casein kinase II (50), and β-arrestin (37). Our hypothesis is that SET could act as a scaffold protein and interact with some components of the cAMP signaling, allowing an efficient and robust coupling of GnRHR to the cAMP pathway. Alignment of the ICL1 sequence of a number of GnRHR (52) reveals that, whereas mammalian GnRHR contain the SET binding sequence, such sequence is only partial or even absent in non-mammalian GnRHR, such as Xenopus or catfish (Table 1). This strongly suggests that SET may not bind to ICL1 of non-mammalian GnRHR. It is thus tempting to speculate that, in addition to the unique absence of the carboxyl-terminal tail, the mammalian GnRHR has acquired another structural peculiarity through evolution, giving the capacity to couple efficiently to the cAMP pathway.
TABLE 1.
Amino acid sequence alignment of the first intracellular loop from various vertebrate GnRH receptors
The amino acid sequence involved in SET binding is boxed, and basic residues are shown in boldface type. This table was adapted from Millar et al. (52).
Altogether, our results strongly suggest that SET binding generates a signaling switch of GnRHR from calcium to cAMP signaling, thus highlighting a new role for the accessory protein SET in the regulation of GPCR signaling. This constitutes an original finding regarding GPCR regulation because apart from NHERF2, which reduces parathyroid hormone receptor coupling to Gs but increases its coupling to Gq (49), only very few GIP have been shown to operate a switch of GPCR coupling to G protein.
We have demonstrated here that GnRH regulation of rat Gnrhr promoter activity is mediated through the cAMP pathway. Our study thus identifies Gnrhr as an additional gene among the few genes known to be regulated by GnRH through this pathway. In addition, our results indicate that SET is critical to increase GnRH stimulation of Gnrhr expression. In native gonadotrope cells, recruitment of the cAMP pathway by GnRHR occurs preferentially at the proestrus stage of the reproductive cycle in the female rat, a few h prior to the LH surge and ovulation (12, 53, 54). The mechanisms underlying such activation are not yet known. Our previous studies in LβT2 cells and in cultured rat pituitary cells demonstrated that mobilization of the cAMP pathway by GnRHR is dependent on the mode of GnRH stimulation because it only occurs under sustained and not pulsatile stimulation (12, 45). This may explain why the cAMP pathway is massively recruited by GnRHR, in vivo at proestrus, when pituitary gonadotrope cells are challenged with GnRH at a very high pulse frequency. This suggests that GnRH input contributes to the specificity of GnRHR coupling. This is in line with a previous study showing that increasing GnRH concentrations from nanomolar to micromolar induces a switch in GnRHR coupling from Gs to Gi proteins in hypothalamic cells and GT1-7 neurons (55). Because we demonstrated in the present study that SET enhances GnRHR coupling to the cAMP pathway in gonadotrope cells, it is tempting to speculate that SET could play a role in vivo by favoring cAMP pathway recruitment by GnRH, notably at proestrus. In particular, SET may contribute to the increased GnRHR expression at proestrus (56, 57) and hence to the increased sensitivity of gonadotrope cells leading to LH surge. Thus, GnRHR signaling switch induced by SET could be of physiological importance at this specific period of the estrus cycle.
In conclusion, we identified SET as a novel interacting partner of the mammalian GnRHR. We demonstrated that SET binds directly to ICL1 and ICL3 of GnRHR and regulates the efficacy and specificity of GnRHR signaling. Our study highlights a new role for the accessory protein SET, which behaves as a switch molecule to change GnRHR coupling specificity from calcium to the cAMP pathway. Furthermore, our data suggest that SET may impact gonadotrope cell function, notably through changes of the regulation exerted by GnRH on its receptor gene expression.
Acknowledgments
We thank Dr. Terry D. Copeland (NCI-Frederick, National Institutes of Health, Frederick, MD), Dr. Robert Z. Qi (Hong Kong University of Science and Technology), Dr. R. P. Millar (University of Edinburgh), Dr. K. A. Eidne (University of Western Australia), and Dr. P. Mellon (University of California San Diego, La Jolla, CA) for kind gifts of materials. We are grateful to Dr. C. Bleux (Université Paris 7) for a contribution to GnRH radiodination and to Dr. S. M. Lanier (Medical University of South Carolina) for helpful discussions. Measurements of calcium mobilization were performed at the Flexstation III facility of the Biologie Fonctionnelle et Adaptative laboratory.
This work was supported by grants from Paris 7 University and CNRS.
V. Simon and J. Cohen-Tannoudji, unpublished data.
C. Avet and V. Simon, unpublished observations.
- GPCR
- G protein-coupled receptor(s)
- GIP
- GPCR-interacting protein(s)
- AP1
- activator protein 1
- GnRH
- gonadotropin-releasing hormone
- GnRHa
- gonadotropin-releasing hormone agonist
- GnRHR
- gonadotropin-releasing hormone receptor(s)
- IBMX
- 3-isobutyl-1-methylxanthine
- ICL
- intracellular loop
- LH
- luteinizing hormone
- PACAP
- pituitary adenylyl cyclase-activating polypeptide
- PKI
- PKA inhibitor
- PP2A
- protein phosphatase 2A
- rSET
- His-tagged recombinant SET
- PLC
- phospholipase C.
REFERENCES
- 1. Lefkowitz R. J. (2007) Seven transmembrane receptors. Something old, something new. Acta Physiol. (Oxf.) 190, 9–19 [DOI] [PubMed] [Google Scholar]
- 2. Bockaert J., Fagni L., Dumuis A., Marin P. (2004) GPCR interacting proteins (GIP). Pharmacol. Ther. 103, 203–221 [DOI] [PubMed] [Google Scholar]
- 3. Sato M., Blumer J. B., Simon V., Lanier S. M. (2006) Accessory proteins for G proteins. Partners in signaling. Annu. Rev. Pharmacol. Toxicol. 46, 151–187 [DOI] [PubMed] [Google Scholar]
- 4. Maurice P., Guillaume J. L., Benleulmi-Chaachoua A., Daulat A. M., Kamal M., Jockers R. (2011) GPCR-interacting proteins, major players of GPCR function. Adv. Pharmacol. 62, 349–380 [DOI] [PubMed] [Google Scholar]
- 5. Luttrell L. M., Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 6. Ritter S. L., Hall R. A. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Counis R., Laverrière J. N., Garrel G., Bleux C., Cohen-Tannoudji J., Lerrant Y., Kottler M. L., Magre S. (2005) Gonadotropin-releasing hormone and the control of gonadotrope function. Reprod. Nutr. Dev. 45, 243–254 [DOI] [PubMed] [Google Scholar]
- 8. Naor Z. (2009) Signaling by G-protein-coupled receptor (GPCR). Studies on the GnRH receptor. Front. Neuroendocrinol. 30, 10–29 [DOI] [PubMed] [Google Scholar]
- 9. Starzec A., Jutisz M., Counis R. (1989) Cyclic adenosine monophosphate and phorbol ester, like gonadotropin-releasing hormone, stimulate the biosynthesis of luteinizing hormone polypeptide chains in a nonadditive manner. Mol. Endocrinol. 3, 618–624 [DOI] [PubMed] [Google Scholar]
- 10. Horton C. D., Halvorson L. M. (2004) The cAMP signaling system regulates LHbeta gene expression. Roles of early growth response protein-1, SP1 and steroidogenic factor-1. J. Mol. Endocrinol. 32, 291–306 [DOI] [PubMed] [Google Scholar]
- 11. Pincas H., Laverrière J. N., Counis R. (2001) Pituitary adenylate cyclase-activating polypeptide and cyclic adenosine 3′,5′-monophosphate stimulate the promoter activity of the rat gonadotropin-releasing hormone receptor gene via a bipartite response element in gonadotrope-derived cells. J. Biol. Chem. 276, 23562–23571 [DOI] [PubMed] [Google Scholar]
- 12. Garrel G., Simon V., Thieulant M. L., Cayla X., Garcia A., Counis R., Cohen-Tannoudji J. (2010) Sustained gonadotropin-releasing hormone stimulation mobilizes the cAMP/PKA pathway to induce nitric oxide synthase type 1 expression in rat pituitary cells in vitro and in vivo at proestrus. Biol. Reprod. 82, 1170–1179 [DOI] [PubMed] [Google Scholar]
- 13. Hamid T., Malik M. T., Millar R. P., Kakar S. S. (2008) Protein kinase A serves as a primary pathway in activation of Nur77 expression by gonadotropin-releasing hormone in the LβT2 mouse pituitary gonadotroph tumor cell line. Int. J. Oncol. 33, 1055–1064 [PubMed] [Google Scholar]
- 14. McArdle C. A., Franklin J., Green L., Hislop J. N. (2002) Signalling, cycling, and desensitisation of gonadotrophin-releasing hormone receptors. J. Endocrinol. 173, 1–11 [DOI] [PubMed] [Google Scholar]
- 15. Willars G. B., Royall J. E., Nahorski S. R., El-Gehani F., Everest H., McArdle C. A. (2001) Rapid down-regulation of the type I inositol 1,4,5-trisphosphate receptor and desensitization of gonadotropin-releasing hormone-mediated Ca2+ responses in αT3-1 gonadotropes. J. Biol. Chem. 276, 3123–3129 [DOI] [PubMed] [Google Scholar]
- 16. Liu F., Austin D. A., Webster N. J. (2003) Gonadotropin-releasing hormone-desensitized LβT2 gonadotrope cells are refractory to acute protein kinase C, cyclic AMP, and calcium-dependent signaling. Endocrinology 144, 4354–4365 [DOI] [PubMed] [Google Scholar]
- 17. Shacham S., Cheifetz M. N., Fridkin M., Pawson A. J., Millar R. P., Naor Z. (2005) Identification of Ser153 in ICL2 of the gonadotropin-releasing hormone (GnRH) receptor as a phosphorylation-independent site for inhibition of Gq coupling. J. Biol. Chem. 280, 28981–28988 [DOI] [PubMed] [Google Scholar]
- 18. Simon V., Guidry J., Gettys T. W., Tobin A. B., Lanier S. M. (2006) The proto-oncogene SET interacts with muscarinic receptors and attenuates receptor signaling. J. Biol. Chem. 281, 40310–40320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. (1992) Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes. Characterization of the set gene. Mol. Cell. Biol. 12, 3346–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsumoto K., Nagata K., Miyaji-Yamaguchi M., Kikuchi A., Tsujimoto M. (1999) Sperm chromatin decondensation by template activating factor I through direct interaction with basic proteins. Mol. Cell. Biol. 19, 6940–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo S. B., McNamara P., Heo S., Turner A., Lane W. S., Chakravarti D. (2001) Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the SET oncoprotein. Cell 104, 119–130 [DOI] [PubMed] [Google Scholar]
- 22. Li M., Makkinje A., Damuni Z. (1996) The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 271, 11059–11062 [DOI] [PubMed] [Google Scholar]
- 23. Lechward K., Awotunde O. S., Swiatek W., Muszyńska G. (2001) Protein phosphatase 2A. Variety of forms and diversity of functions. Acta Biochim. Pol. 48, 921–933 [PubMed] [Google Scholar]
- 24. Thomas P., Mellon P. L., Turgeon J., Waring D. W. (1996) The LβT2 clonal gonadotrope. A model for single cell studies of endocrine cell secretion. Endocrinology 137, 2979–2989 [DOI] [PubMed] [Google Scholar]
- 25. Turgeon J. L., Kimura Y., Waring D. W., Mellon P. L. (1996) Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol. Endocrinol. 10, 439–450 [DOI] [PubMed] [Google Scholar]
- 26. Ma Q. P., Woolf C. J. (1995) Involvement of neurokinin receptors in the induction but not the maintenance of mechanical allodynia in rat flexor motoneurones. J. Physiol. 486, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chauvin S, Bérault A, Lerrant Y, Hibert M, Counis R. (2000) Functional importance of transmembrane helix 6 Trp279 and exoloop 3 Val299 of rat gonadotropin-releasing hormone receptor. Mol. Pharmacol. 57, 625–633 [DOI] [PubMed] [Google Scholar]
- 28. Derossi D., Joliot A. H., Chassaing G., Prochiantz A. (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 269, 10444–10450 [PubMed] [Google Scholar]
- 29. Granger A., Bleux C., Kottler M. L., Rhodes S. J., Counis R., Laverrière J. N. (2006) The LIM-homeodomain proteins Isl-1 and Lhx3 act with steroidogenic factor 1 to enhance gonadotrope-specific activity of the gonadotropin-releasing hormone receptor gene promoter. Mol. Endocrinol. 20, 2093–2108 [DOI] [PubMed] [Google Scholar]
- 30. Olsen S. R., Uhler M. D. (1991) Inhibition of protein kinase A by overexpression of the cloned human protein kinase inhibitor. Mol. Endocrinol. 5, 1246–1256 [DOI] [PubMed] [Google Scholar]
- 31. Simon V., Oner S. S., Cohen-Tannoudji J., Tobin A. B., Lanier S. M. (2012) Influence of the accessory protein set on M3 muscarinic receptor phosphorylation and G protein coupling. Mol. Pharmacol. 82, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McArdle C. A., Willars G. B., Fowkes R. C., Nahorski S. R., Davidson J. S., Forrest-Owen W. (1996) Desensitization of gonadotropin-releasing hormone action in αT3-1 cells due to uncoupling of inositol 1,4,5-trisphosphate generation and Ca2+ mobilization. J. Biol. Chem. 271, 23711–23717 [DOI] [PubMed] [Google Scholar]
- 33. Kaiser U. B., Jakubowiak A., Steinberger A., Chin W. W. (1993) Regulation of rat pituitary gonadotropin-releasing hormone receptor mRNA levels in vivo and in vitro. Endocrinology 133, 931–934 [DOI] [PubMed] [Google Scholar]
- 34. Sadie H., Styger G., Hapgood J. (2003) Expression of the mouse gonadotropin-releasing hormone receptor gene in αT3-1 gonadotrope cells is stimulated by cyclic 3′,5′-adenosine monophosphate and protein kinase A, and is modulated by steroidogenic factor-1 and Nur77. Endocrinology 144, 1958–1971 [DOI] [PubMed] [Google Scholar]
- 35. Lerrant Y., Kottler M. L., Bergametti F., Moumni M., Blumberg-Tick J., Counis R. (1995) Expression of gonadotropin-releasing hormone (GnRH) receptor gene is altered by GnRH agonist desensitization in a manner similar to that of gonadotropin β-subunit genes in normal and castrated rat pituitary. Endocrinology 136, 2803–2808 [DOI] [PubMed] [Google Scholar]
- 36. Borroto-Escuela D. O., Correia P. A., Romero-Fernandez W., Narvaez M., Fuxe K., Ciruela F., Garriga P. (2011) Muscarinic receptor family interacting proteins. Role in receptor function. J. Neurosci. Methods 195, 161–169 [DOI] [PubMed] [Google Scholar]
- 37. Kendall R. T., Strungs E. G., Rachidi S. M., Lee M. H., El-Shewy H. M., Luttrell D. K., Janech M. G., Luttrell L. M. (2011) The β-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J. Biol. Chem. 286, 19880–19891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vasudevan N. T., Mohan M. L., Gupta M. K., Hussain A. K., Naga Prasad S. V. (2011) Inhibition of protein phosphatase 2A activity by PI3Kγ regulates β-adrenergic receptor function. Mol. Cell 41, 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conn P. M., Ulloa-Aguirre A., Ito J., Janovick J. A. (2007) G protein-coupled receptor trafficking in health and disease. Lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol. Rev. 59, 225–250 [DOI] [PubMed] [Google Scholar]
- 40. Myburgh D. B., Millar R. P., Hapgood J. P. (1998) Alanine-261 in intracellular loop III of the human gonadotropin-releasing hormone receptor is crucial for G-protein coupling and receptor internalization. Biochem. J. 331, 893–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Roux N., Young J., Misrahi M., Genet R., Chanson P., Schaison G., Milgrom E. (1997) A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 337, 1597–1602 [DOI] [PubMed] [Google Scholar]
- 42. Willars G. B., Heding A., Vrecl M., Sellar R., Blomenröhr M., Nahorski S. R., Eidne K. A. (1999) Lack of a C-terminal tail in the mammalian gonadotropin-releasing hormone receptor confers resistance to agonist-dependent phosphorylation and rapid desensitization. J. Biol. Chem. 274, 30146–30153 [DOI] [PubMed] [Google Scholar]
- 43. Shi J., Damjanoska K. J., Singh R. K., Carrasco G. A., Garcia F., Grippo A. J., Landry M., Sullivan N. R., Battaglia G., Muma N. A. (2007) Agonist-induced phosphorylation of Galpha11 protein reduces coupling to 5-HT2A receptors. J. Pharmacol. Exp. Ther. 323, 248–256 [DOI] [PubMed] [Google Scholar]
- 44. Arora K. K., Krsmanovic L. Z., Mores N., O'Farrell H., Catt K. J. (1998) Mediation of cyclic AMP signaling by the first intracellular loop of the gonadotropin-releasing hormone receptor. J. Biol. Chem. 273, 25581–25586 [DOI] [PubMed] [Google Scholar]
- 45. Larivière S., Garrel G., Simon V., Soh J. W., Laverrière J. N., Counis R., Cohen-Tannoudji J. (2007) Gonadotropin-releasing hormone couples to 3′,5′-cyclic adenosine-5′-monophosphate pathway through novel protein kinase Cδ and -ϵ in LβT2 gonadotrope cells. Endocrinology 148, 1099–1107 [DOI] [PubMed] [Google Scholar]
- 46. Horn F., Bilezikjian L. M., Perrin M. H., Bosma M. M., Windle J. J., Huber K. S., Blount A. L., Hille B., Vale W., Mellon P. L. (1991) Intracellular responses to gonadotropin-releasing hormone in a clonal cell line of the gonadotrope lineage. Mol. Endocrinol. 5, 347–355 [DOI] [PubMed] [Google Scholar]
- 47. Pantel J., Williams S. Y., Mi D., Sebag J., Corbin J. D., Weaver C. D., Cone R. D. (2011) Development of a high throughput screen for allosteric modulators of melanocortin-4 receptor signaling using a real time cAMP assay. Eur. J. Pharmacol. 660, 139–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scott K., Zuker C. S. (1998) Assembly of the Drosophila phototransduction cascade into a signalling complex shapes elementary responses. Nature 395, 805–808 [DOI] [PubMed] [Google Scholar]
- 49. Wang B., Ardura J. A., Romero G., Yang Y., Hall R. A., Friedman P. A. (2010) Na/H exchanger regulatory factors control parathyroid hormone receptor signaling by facilitating differential activation of Gα protein subunits. J. Biol. Chem. 285, 26976–26986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vera J., Estanyol J. M., Canela N., Llorens F., Agell N., Itarte E., Bachs O., Jaumot M. (2007) Proteomic analysis of SET-binding proteins. Proteomics 7, 578–587 [DOI] [PubMed] [Google Scholar]
- 51. ten Klooster J. P., Leeuwen I., Scheres N., Anthony E. C., Hordijk P. L. (2007) Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 26, 336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Millar R. P., Lu Z. L., Pawson A. J., Flanagan C. A., Morgan K., Maudsley S. R. (2004) Gonadotropin-releasing hormone receptors. Endocr. Rev. 25, 235–275 [DOI] [PubMed] [Google Scholar]
- 53. Kimura F, Kawakami M, Nakano H, McCann SM. (1980) Changes in adenosine 3′,5′-monophosphate and guanosine 3′,5′-monophosphate concentrations in the anterior pituitary and hypothalamus during the rat estrous cycle and effects of administration of sodium pentobarbital in proestrus. Endocrinology 106, 631–635 [DOI] [PubMed] [Google Scholar]
- 54. Cohen-Tannoudji J., Avet C., Garrel G., Counis R., Simon V. (2012) Decoding high gonadotropin-releasing hormone pulsatility. A role for GnRH receptor coupling to the cAMP pathway? Front. Endocrinol. (Lausanne) 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krsmanovic L. Z., Mores N., Navarro C. E., Arora K. K., Catt K. J. (2003) An agonist-induced switch in G protein coupling of the gonadotropin-releasing hormone receptor regulates pulsatile neuropeptide secretion. Proc. Natl. Acad. Sci. U.S.A. 100, 2969–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Savoy-Moore R. T., Schwartz N. B., Duncan J. A., Marshall J. C. (1980) Pituitary gonadotropin-releasing hormone receptors during the rat estrous cycle. Science 209, 942–944 [DOI] [PubMed] [Google Scholar]
- 57. Bauer-Dantoin A. C., Hollenberg A. N., Jameson J. L. (1993) Dynamic regulation of gonadotropin-releasing hormone receptor mRNA levels in the anterior pituitary gland during the rat estrous cycle. Endocrinology 133, 1911–1914 [DOI] [PubMed] [Google Scholar]