

Abstract
During carcinogenesis, tumors induce dysfunctional development of hematopoietic cells. Myeloid lineage cells, in the form of myeloid derived suppressor cells (MDSCs) and alternatively polarized M2 macrophages, influence almost all types of cancers by regulating diverse facets of immunosuppression, angiogenesis, cell proliferation, growth and metastasis. One-third of Americans are obese, and accumulating evidence suggests that obesity is a risk factor for various cancers. However, the relationship between these immune players and obesity are not well-described. In this review, we evaluate potential mechanisms through which different aspects of obesity, namely insulin resistance, increased estrogen, adiposity and low grade chronic inflammation from adipose tissue macrophages, may coalesce to promote MDSC induction and M2 macrophage polarization, thereby facilitating cancer development. Detailed understanding of the interplay between obesity and myeloid mediated immunosuppression may provide novel avenues for therapeutic targeting, with the goal to reduce the challenge obesity presents towards gains made in cancer outcomes.
Keywords: Obesity, inflammation, myeloid derived suppressor cells, alternately activated macrophage, cancer
Introduction
Despite significant advances in screening and therapeutics, cancer remains a major cause of mortality and morbidity worldwide. In the United States alone, 1 in 4 deaths are attributed to cancer, second only to cardiovascular-related deaths [1]. Various indices such as obesity, smoking, metabolic syndrome and genetics are known to not only increase the risk of cancer, but also to complicate response to chemotherapy and radiation [2]. In the past decade, the role of the immunosuppressive and cancer-promoting myelomonocytic compartment within the tumor environment has also received a great deal of attention [3]. By mechanisms that are incompletely understood, cancer promotes the accumulation of a heterogeneous pool of bone marrow-derived immature, poorly differentiated myelomonocytic cells (monocytes, neutrophils, immature macrophages and dendritic cells), called myeloid derived suppressor cells (MDSCs) [3,4]. These cells are marked by CD11b(+)Gr-1(+) expression, but do not express the mature macrophage marker F4/80. In addition, the tumor microenvironment preferentially polarizes monocytes and mature macrophages towards the so-called alternatively activated M2 phenotype [5,6]. Together, MDSCs and M2 macrophages foster an immunosuppressive environment by recruiting other immunosuppressive cells such as regulatory T cells [7], targeting apoptosis of tumor-specific cytotoxic CD8 T cells [8], promoting and facilitating metastasis [9], while producing various chemokines, cytokines and angiogenic factors to support primary tumors [10]. However, the interplay between these complex mechanisms of myeloid immunosuppression and the widely known cancer risk factors are not well delineated. It is imperative to appreciate and address all details of the mechanistic molecular underpinnings of these risk factors, so as to indentify new therapeutic targets and thereby improve survival and outcome.
Recent data indicate that one-third of Americans are obese [11]. A body mass index (BMI) greater than 30 is associated with increased risk for a wide range of cancers including endometrial, esophageal, kidney, pancreatic and other gastrointestinal malignancies [12,13] (Table 1). Obesity also increases cancer-related morbidity and mortality. The risk holds true across race, gender and different geographic groups [12]. Obesity may exert direct, organ-specific effects in certain cancers. For instance, in esophageal cancer, the increased risk may be secondary to increased reflux from obesity leading to chronic local inflammation, Barrett’s esophagus and subsequently adenocarcinoma [14]. In renal cancers, the risk may be secondary to increased risk of hypertension or genetic susceptibility such as VHL in obese individuals [15,16]. The link between obesity and cancer is even broader in colon cancer, because the direct effect of low fiber diet, high caloric intake, energy balance and BMI are intricately connected and difficult to discriminate [17,18]. It has also been proposed that obese patients suffer worse outcome because of delay in cancer detection [2,19], and sub-optimal dosing of chemotherapeutic drugs in order to avoid drug toxicity [20]. The general association between obesity and several cancers suggests plausible underlying biological mechanisms. Different aspects of the pathophysiology of obesity, namely insulin resistance, estrogen, adiposity, and low-grade chronic inflammation, may indeed facilitate a cancer-promoting state. Here, we review the literature to identify myeloid lineage cells as potential conduit by which components of obesity may increase risks for cancers. We place a special emphasis on the myelomonocytic cells, given their increasingly recognized role in several cancers, and show that different aspects of obesity directly and indirectly influence this important regulator of the tumor microenvironment.
Table 1.
Cancers with strong link to obesity
| Men | ||
|
| ||
| RR (95% CI) | p | |
|
| ||
| Esophageal Adenocarcinoma | 1.52 (1.33-1.74) | <0.0001 |
| Thyroid | 1.33 (1.04-1.70) | 0.02 |
| Colon | 1.24 (1.20-1.28) | <0.0001 |
| Renal | 1.24 (1.15-1.34) | <0.0001 |
| Malignant melanoma | 1.17 (1.05-1.30) | 0.004 |
| Multiple myeloma | 1.11 (1.05-1.18) | <0.0001 |
| Rectum | 1.09 (0.99-1.21) | <0.0001 |
|
| ||
| Women | ||
|
| ||
| RR (95% CI) | p value | |
|
| ||
| Endometrial | 1.59 (1.50-1.68) | <0.0001 |
| Gallbladder | 1.59 (1.02-2.47) | 0.04 |
| Esophageal | 1.51 (1.31-1.74) | <0.0001 |
| Renal | 1.34 (1.25-1.43) | <0.0001 |
| Leukemia | 1.17 (1.04-1.32) | 0.01 |
| Thyroid | 1.14(1.06-1.23) | 0.001 |
| Breast | 1.12 (1.08-1.16) | <0.0001 |
Adapted from Renehan et al [12], “Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies” 2008. Lancet 371: 569-578.
Mechanisms of MDSC induction M2 macrophage polarization
The transcriptional pathways leading to MDSC and M2 macrophage polarization are an area of current intense research due to their implication in cancer [21]. In brief, hematopoietic stem cells give rise to a clonogenic common myeloid progenitor cell, from which arises all myeloid lineage cells, including granulocytes, monocytes and myeloid dendritic cells through their respective precursor cells [22]. During cancer development, dysfunctional myelopoiesis arises, and some of these precursors are directed towards the myeloid derived suppressor cells category. Macrophages are instructed by their environmental cues to two major classes of polarization [23]. M1 macrophages are instructed by bacterial antigens such as lipopolysacharide (LPS) and gamma interferon (IFNγ), and produce pro-inflammatory cytokines such as IL-12, IL-6, iNOS and TNF-α. These cells are required for robust clearance of tumors and bacterial infections. In contrast, M2 macrophages are generated by cell factors such as IL-4 and prostaglandins to mediate tissue repair and dampen inflammatory response through cytokines such as IL-10 and TGF-β [23]. Unfortunately, some of the effects of these cytokines promote tumor growth through angiogenesis, matrix degradation (which enables metastasis) and cell survival [23].
Monocytes and MDSCs may also differentiate preferentially towards the alternatively activated M2 macrophage phenotype [24,25]. Among the transcriptional factors described include C/EBP α, C/EBPβ, PPARγ, NF-kβ, STAT-3, IRF-4-Jmjd3 and COX-2 (See Figure 1). The C/EBP (CCAAT/enhancer-binding protein) family is a group of transcription factors that can bind as a homodimer to promoters and enhancers involved in myelopoiesis, monocyte differentiation, and macrophage polarization as well as adipogenesis [26]. The perixosome proliferator activated receptors (PPARs) are a family of nuclear receptor proteins that play essential role in regulation of genes responsible for metabolism of carbohydrates, lipids and fats, as well as cellular differentiation and development. Recent data suggest a key role of PPARγ in alternative macrophage polarization [27]. These various pathways provide therapeutic opportunities for eliminating immunosuppression during carcinogenesis. In addition, chemokines such as monocyte chemotactic protein 1 (MCP-1, or CCL-2) that recruit MDSCs, monocytes and M2 macrophages from the bone marrow to the tumor microenvironment are important avenues of therapy.
Figure 1.
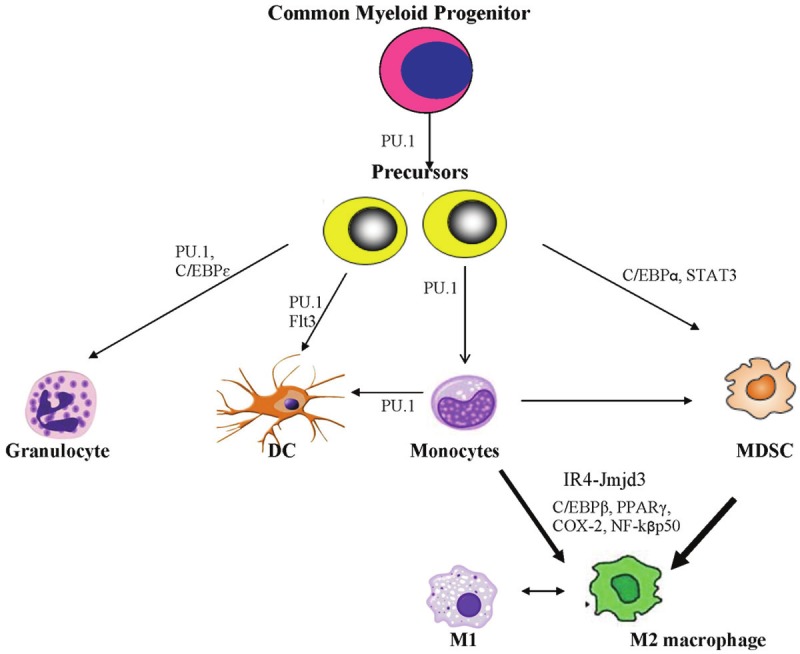
Schematics of development of myeloid lineage cells. Common myeloid progenitor cells give rise to precursors that then differentiate into granulocytes, dendritic cells and monocytes. The major transcription factor involved is PU.1, which collaborate with other lineage specific factors (such as Flt3 and C/EBPε) for commitment to the final myeloid cell types. Depending on cytokines and other cell factors, monocytes may differentiate into spectrum of macrophages (classically activated M1 or alternatively activated M2 macrophages) or dendritic cells. During tumorigenesis, cancer cells usurp aspects of differentiation of these myeloid cells. Precursor cells, under the influence of transcription factors such as C/EBPα, STAT3, are re-directed to form myeloid derived suppressor cells (MDSCs). Monocytes and MDSCs preferentially differentiate into the tumor-promoting M2 macrophages. Transcription factors such as Jmjd3-IR4C/EBPβ, PPARγ, NF-kβp50 have been implicated in the M2 macrophage polarization.
Insulin resistance, MDSCs and M2 macrophages
Insulin resistance secondary to obesity is a crucial component of mechanisms leading to cancer susceptibility. Insulin resistance leads to increased levels of circulating insulin, which in turn increases the bioactivity insulin growth factor (IGF-1), IGF-1 receptors, and insulin receptors (IR) in normal and cancerous cells. The multiple role of IGF-1 in cancers has received recent thorough review [28,29]. In brief, IGF-1 induces vascular endothelial growth factor (VEGF) expression in tumor cells, thereby inducing angiogenesis [30]. IGF-1 also induces CD147, a transmembrane protein whose expression on tumor cells equally leads to angiogenesis and increased glycolysis (required for energy metabolism) in tumor cells [31,32]. Furthermore IGF-1 signaling activates beta catenin, a key regulator of epithelial mesenchymal transition, and potentiates other cell growth stimulants. Both insulin and IGF-1 signaling through IGF-1R and IR mediate angiogenesis, cell differentiation and cell proliferation, thereby promoting cancer burden [33,34]. Accordingly, individuals with elevated IGF-1 have increased risk of several malignancies, including bladder, colorectal, endometrial, lung and prostate cancer [35-38]. Indeed, elevated IGF-1 has been implicated in the increased susceptibility to cancer in patients with acromegaly [39].
Further investigations by other groups have recently revealed that in addition to the mechanism of direct stimulation of angiogenesis, insulin resistance may inadvertently promote carcinogenesis through MDSCs and M2 macrophages. An exciting observation has been described in the laboratory of Dr Qi et al, whose group showed that MDSC and M2 macrophage induction may be a physiological response to promote insulin sensitivity [40]. The obese (ob/ob) mouse has remained a standard model to understand the interconnection of genetics and diet in obesity [41]. They present with hypercholesterolemia, increased IGF-1 and increased adiposity [42,43]. Xia et al showed that these mice have profound accumulation of MDSC and M2 macrophage as they are fed a high fat diet. It appears that increased accumulation of MDSCs and M2 macrophages was associated with increased response to insulin. In fact, adoptive transfer of MDSCs into mice fed high fat diet improved the response of the recipient mice to insulin [40]. In contrast, depletion of these cells increased their susceptibility to obesity and further worsened their insulin insensitivity. Yin et al have also described the ability of MDSCs to delay onset of diabetes and insulin resistance [44].
Although mechanisms by which MDSCs enhance insulin sensitivity are unknown, there are insights into to how upregulated IGF-1 in the setting of insulin resistance may lead to the accumulation of MDSCs and M2 macrophages. During acute skeletal muscle injury, monocytes are recruited by way of the CC chemokine ligand 2 (CCL2) to facilitate repair of damaged muscle tissue. These recruited monocytes differentiate towards the alternatively activated M2 phenotype, which is required for resolution of inflammation and muscle repair [45]. Interestingly, Lu et al have recently provided evidence that upregulation of local IGF-1 is necessary for the polarization of these recruited myeloid cells towards the M2 phenotype [46,47]. In fact, the potent M2 response essential for limiting overt acute tissue damage during experimental helminth infection is not only dependent on IL-10, but also on IGF-1 [48]. Altogether, these data suggest that not only does insulin resistance induce physiological response for MDSC and M2 macrophage expansion, but that insulin may also modulate direct gene transcriptional control of these cells. Further work is required to determine the mechanisms by which IGF-1 drives monocyte differentiation. These data suggest that pharmacologic enhancement of insulin sensitivity in obese individuals may preemptively hinder the development of MDSCs which inadvertently contribute to immune escape of cancer cells from an effective host response.
One pharmacotherapy that improves insulin sensitivity and has found recent implication in cancer is metformin. It is a guanide based antidiabetic agent that works by activating AMP-activated protein kinase (AMPK), an enzyme which plays an important role in insulin signaling, whole body energy balance, and the metabolism of glucose and fats [49,50]. Metformin suppresses hepatic gluconeogenesis and is the mainstay therapy for type 2 diabetes particularly in obese patients [51]. Interestingly, a series of studies have shown that diabetic patients on metformin have a reduced risk of cancers such as breast, pancreatic and ovarian cancer [52-54]. The effects are probably not due to anti-diabetic mechanisms, because other similar agents do not reduce- and may even increase-risk for pancreatic and other cancers [52,55]. The mechanisms of the anticancer effect of metformin remain largely unknown, but anti-proliferative effects on several cancer lines have been demonstrated in vitro, with AMPK signaling the proposed primary pathway [56-58]. However, the doses required to achieve this in vitro effect were several folds higher than those used in the clinical setting. Furthermore, recent work by Bonani et al revealed equivalent proliferative index between breast cancer surgical tissues of control and metformin-treated patients, suggesting possible alternative tumor cell-independent mechanisms [59]. In addition, several AMPK-independent effects have been described, raising questions as to the true mechanism mediating the effect of metformin [60-62]. Importantly, recent work by different groups revealed reduced macrophage infiltration and cytokine production within tumors after metformin treatment in vivo and in vitro [54,63]. Further work is needed to unravel the likely complex mechanisms by which metformin affects cancer development. In that effort, the potential role of metformin via IGF-1 in modulating the myeloid-cell inflammatory response through MDSC and M2 macrophages should be closely explored.
Estrogen, MDSCs and M2 macrophages
Obesity is associated with increased estrogen production through conversion of androgens in adipocytes by aromatase [64,65]. The role of estradiol, the predominant circulating estrogen, in breast, endometrial and other hormone responsive cancers is well known and thoroughly described elsewhere [66-68]. Increasing new evidence also indicates an interesting association between estradiol and myeloid cell differentiation and polarization. Estradiol may drive differentiation of myelomonocytic cells from the bone marrow towards ineffective antigen presenting cells. This is thought to be mediated by the increased activation of the interferon response factor -4 (IRF4) [69], which collaborates with the jmjd3 axis as a potent pathway in differentiation of myeloid cells towards the M2 phenotype [70]. Estrogen is an important growth factor for bone marrow stimulation of myelomonocytic cell production, such that estrogen excess results in hematopoietic dysfunction and inability to produce mature dendritic cells from the bone marrow [71-73]. Thus, estrogen excess in obese individuals may be another mechanism by which immunosuppression is promoted through MDSCs and M2 macrophages. Estrogen antagonism with agents such as tamoxifen and raloxifen may improve hormone-responsive cancer survival. Recently, the role of these selective estrogen receptor modulators (SERMs) in suppressing the inflammatory response and decreasing the recruitment of monocytes has been described [74,75]. It is noteworthy that IRF-4 pathway involved in macrophage polarization is also implicated in adipocyte handling of lipids [76]. Furthermore, estrogen also increases the function of CD4+CD25+ regulatory T cells, another immunosuppressive cell type associated with immune evasion by cancer cells [77,78].
Chronic inflammation, MDSCs and M2 macrophages
Clinically, obese patients have an increased circulating pro-inflammatory cytokines such as TNF-α, IL-6, IL-1β and IL-12 [79]. The levels of these cytokines may correlate with BMI [80]. The importance of these cytokines is underscored in animal models, where their deficiency leads to improved response to insulin and reduced obesity [81,82]. Furthermore, pharmacologic or genetic inhibition of these cytokines also improves severity of some obesity-induced cancers [83,84]. The source of these cytokines may be from the adipocytes themselves or recruited bone-marrow derived monocytes [85], although the relative contribution from these sources are unclear.
The mechanisms by which obesity-associated chronic inflammation induces M2 macrophages and MDSCs are arguably stepwise. In general, the initial pro-inflammatory cytokine mileu induced by obesity may actually bear the trademark of the broadly anti-cancer, classically activated M1 macrophage features [86,87]. However, it is still important to note that some of these cytokines can have dual, complex pro-tumor contributions as well. For example, IL-1β is capable of inducing MDSC function [88], while at the same time, directly stimulating angiogenesis and metastasis [89,90]. In addition, TNFα is capable of supporting tumor growth through NF-kβ while at the same time increasing insulin resistance [115]. However, it is the long-standing chronic inflammation that may precipitate the induction of MDSCs and M2 macrophages by the following mechanism. In several well-described models of inflammation, MDSCs are induced in an attempt to curtail overt responses [3]. For example, chronic inflammation in the setting of viruses, complete freud’s adjuvant and other infections induces accumulation of MDSCs [91,92]. It is therefore conceivable that in a similar paradigm, obesityrelated low-grade chronic inflammation may stimulate the deployment of immature myelomonocytic cells from the bone marrow, which eventually accumulate as MDSCs and M2 macrophages [93]. Fujikawa et al have attributed this subsequent M1 to M2 switch in recruited adipose tissue macrophage to IL-10 [94]. Thus, a possible scenario is set where the initial proinflammatory state created in early obesity induces adipogenesis and insulin resistance. This in turn leads to accumulation of MDSC in an attempt to curtail overt inflammation and improve insulin sensitivity (Figure 2).
Figure 2.
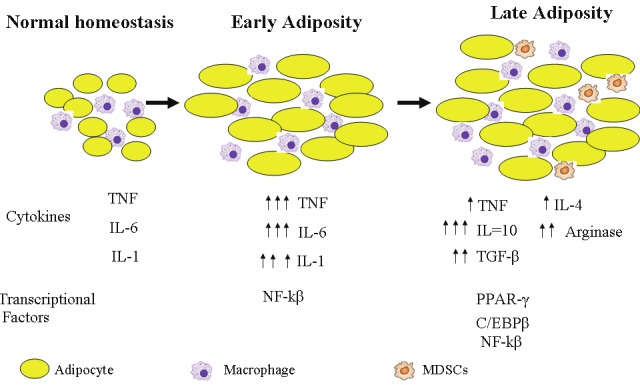
Schematic of stepwise effect of chronic inflammation and MDSC and M2 macrophages in the setting of obesity. In normal (non-obese) state, few macrophages are associated with fat cells in adipose tissues, with minimal inflammation. During early obesity, more macrophages are recruited. These cells produce pro-inflammatory cytokines, in a M1 macrophage inflammatory response pattern. As obesity progresses, insulin resistance develops. A physiologic as well as immune response to this chronic insulin resistance and chronic inflammation is the induction of the M2 macrophage response.
The importance of the myeloid compartment of the chronic grade inflammation has been bolstered by recent work in which regulation of the alternative activation of bone marrow derived monocytes was sufficient to curtail inflammation secondary to high fat diet, and thereby protect mice from diabetes and the complicated sequelae. In brief, Dr Friedman’s group have shown that genetic deletion of C/EBPβ specifically in the myeloid compartment successfully abrogate obesity, inflammation, insulin insensitivity [95]. As one might predict, C/EBPβ is a key regulator of alternative activation of macrophages, myeloid derived suppressor cells, and cancer [26,96,97].
Adiposity, MDSCs and M2 macrophages
Adiposity is associated with recruitment of macrophages that initiate chronic inflammation. In fact, the amount of visceral fat accumulation is a good measure of recruited adipose tissue macrophages [98]. It is important to recognize that increased lipid bioavailability is an excellent resource for cellular proliferation, growth and development. Any of these processes may be usurped by cancer cells to facilitate tumor growth. One such candidate is the fatty acid synthase (FASN). It is key to lipogenesis in obesity [99], and has received great deal of attention recently in carcinogenesis [100-102]. However, FASN via the COX-2 pathway, may induce MDSC accumulation as well as M2 macrophage differentiation [103]. Indeed, targeting FASN has become an attractive anti-cancer target [104].
Activation of other concomitant transcriptional programs in adipocytes and monocytes may also result in M2 polarization. As mentioned earlier, PPARs are a family of nuclear receptor proteins that regulate metabolism of carbohydrates and lipids. Overall, PPARγ activation may confer anti-cancer effect via apoptosis, growth arrest, and reciprocal downregulation of tumor cell IGF signaling. Accordingly, PPARγ agonists have emerged as a potential therapeutic target in a number of malignancies, while remaining mainstay therapy for diabetes and insulin resistance. However, activation of both adipocyte PPARδ and PPARγ can induce differentiation of macrophages towards the alternately activated M2 phenotype [27,105]. Thus, adipogenesis requires upregulation of PPAR, which is an important transcription factor for alternate activation of macrophages and cancer development [106]. Interestingly, PPARγ agonist also may have cancer risks, particularly bladder cancer [107]. It is perhaps the untoward polarization of macrophages to the M2 phenotype that raises the risk of these drugs to cancer, as well as the disappointing results of these agents in clinical trials [108]. Thus, the details of these complex mechanisms must be investigated in order to harness the potential benefit of PPAR agonists. The PPAR example illustrates that the mere presence of excess adiposity may result in associated macrophages acting as bystanders that differentiate into M2 macrophages, while the excess availability of lipids themselves can drive cellular differentiation and growth. Whether adiposity alone is sufficient to drive carcinogenesis remains to be proven.
Conclusion
The obesity epidemic imposes an increasing burden on global health [109]. Accumulating evidence indicates that obesity represents a risk factor for various cancers and threatens to undercut the gains in the therapeutic advances made towards improving prognosis and survival in cancer-related morbidity and mortality. Obesity increases circulating estrogen, insulin, IGF, and cause chronic low-grade inflammation. These diverse pathways directly or indirectly converge to induce accumulation of myeloid derived suppressor cells, while programming macrophages to the alternatively activated M2 phenotype (Figure 3). MDSCs and M2 macrophages are a major source of immunosuppression that allows for tumor-escape from effective anti-cancer responses. The induction and preferential tilting of macrophages towards the M2 phenotype may be a primary physiologic and metabolic response to insulin insensitivity, as well a secondary consequence of an immune process to control overt, chronic, low grade inflammation. In any event, these processes may be inadvertently modulated by tumor cells to promote angiogenesis, metastasis and overall poor outcome.
Figure 3.
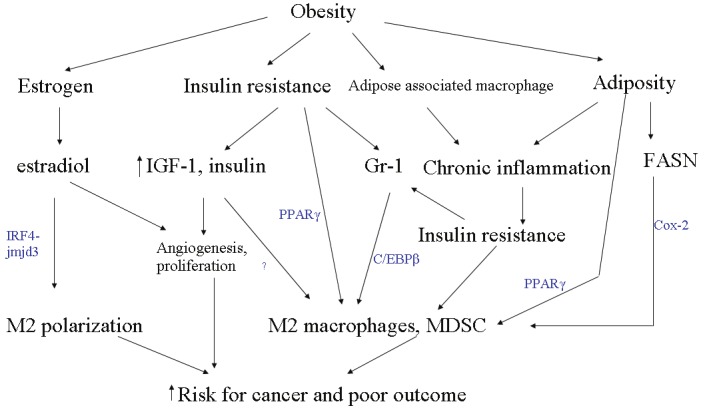
Crosstalk between components of obesity and MDSC and M2 induction. Different aspects of obesity, namely elevated estrogen, increased insulin resistance, increased inflammation secondary to recruited adipose tissue macrophages and adiposity employ a variety of mechanisms, all of which coalesce on M2 macrophage and MDSC induction to facilitate cancer development.
The link between myeloid cells and obesity in cancer provides several avenues for therapeutic targeting. One approach is to reduce the recruitment of myeloid cells within the tumor microenvironment, so as to starve tumor cells from MDSCs and M2 macrophages which provide pro-tumor milieu. CCL2 is a chemokine responsible for recruitment of monocytes and macrophages from the bone marrow into the tumor microenvironment. Targeting CCL2 and CCL2 receptors is currently a promising mechanism in breast cancer therapy, and will likely be found to be critical in several other cancers as well [110-112]. Theoretically, CCL2 inhibitors could be used as combination therapy with other chemotherapeutic regimens that influence different aspects of carcinogenesis.
Another area of potential therapeutic targeting is the use of anti-inflammatory agents to reduce the low grade chronic inflammation that leads to the insulin insensitivity and subsequent physiologic response of MDSC and M2 macrophage induction. Some of these anti-inflammatory therapies are already in use for cardiovascular disease prevention in obese individuals. The use of aspirin is currently under trial for use against certain cancers, and is showing some promise [110,113].
Finally, mechanisms that lead to signaling in adipose tissue that influence macrophage polarization are another attractive area of anticancer therapeutics. PPAR agonists have already been described above, with the caveat of potential pro-cancer risk because of their capacity to promote M2 macrophage polarization. COX-2 signaling promotes M2 macrophage differentiation, and is currently a target for chemoprevention in colorectal, pancreatic and other malignancies. This effect of COX-2 partly explains the rationale for the potential therapeutic use of aspirin in cancer prevention [114]. Upregulation of fatty acid synthase (FASN) in adipogenesis also links COX-2 pathway to MDSCs induction. FASN is also a new therapeutic target for colon cancers. Indeed, all the factors involved in macrophage polarization (outlined in Figure 3) are worthy of active research, with the hope to identify effective therapeutic advances against cancer. To this end, detailed mechanistic understanding of the role of obesity in MDSC and M2 biology could translate into novel, potentially convergent targets that may be exploited. Beyond pharmacotherapy, the effect of weight loss on macrophages and MDSCs may also merit investigation. Findings from these studies could help encourage weight loss in the general population, and inexpensively improve health outcomes in our society where cancer and cardiovascular disease account for the majority of deaths.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U. S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 3.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 4.Brandau S, Trellakis S, Bruderek K, Schmaltz D, Steller G, Elian M, Suttmann H, Schenck M, Welling J, Zabel P, Lang S. Myeloid-derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol. 2011;89:311–317. doi: 10.1189/jlb.0310162. [DOI] [PubMed] [Google Scholar]
- 5.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121:4015–4029. doi: 10.1172/JCI45862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 10.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 11.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 12.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 13.Larsson SC, Wolk A. Excess body fatness: an important cause of most cancers. Lancet. 2008;371:536–537. doi: 10.1016/S0140-6736(08)60247-0. [DOI] [PubMed] [Google Scholar]
- 14.Akiyama T, Yoneda M, Maeda S, Nakajima A, Koyama S, Inamori M. Visceral obesity and the risk of Barrett’s esophagus. Digestion. 2011;83:142–145. doi: 10.1159/000321810. [DOI] [PubMed] [Google Scholar]
- 15.McGuire BB, Fitzpatrick JM. BMI and the risk of renal cell carcinoma. Curr Opin Urol. 2011;21:356–361. doi: 10.1097/MOU.0b013e32834962d5. [DOI] [PubMed] [Google Scholar]
- 16.Smits KM, Schouten LJ, Hudak E, Verhage B, van Dijk BA, Hulsbergen-van de Kaa CA, Goldbohm RA, Oosterwijk E, van den Brandt PA. Body mass index and von Hippel-Lindau gene mutations in clear-cell renal cancer: Results of the Netherlands Cohort Study on diet and cancer. Ann Epidemiol. 2010;20:401–404. doi: 10.1016/j.annepidem.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Adams KF, Leitzmann MF, Albanes D, Kipnis V, Mouw T, Hollenbeck A, Schatzkin A. Body mass and colorectal cancer risk in the NIH-AARP cohort. Am J Epidemiol. 2007;166:36–45. doi: 10.1093/aje/kwm049. [DOI] [PubMed] [Google Scholar]
- 18.Slattery ML, Potter J, Caan B, Edwards S, Coates A, Ma KN, Berry TD. Energy balance and colon cancer--beyond physical activity. Cancer Res. 1997;57:75–80. [PubMed] [Google Scholar]
- 19.Cui Y, Whiteman MK, Flaws JA, Langenberg P, Tkaczuk KH, Bush TL. Body mass and stage of breast cancer at diagnosis. Int J Cancer. 2002;98:279–283. doi: 10.1002/ijc.10209. [DOI] [PubMed] [Google Scholar]
- 20.Griggs JJ, Sorbero ME, Lyman GH. Undertreatment of obese women receiving breast cancer chemotherapy. Arch Intern Med. 2005;165:1267–1273. doi: 10.1001/archinte.165.11.1267. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 22.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 23.Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–330. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonda N, Chioda M, Zilio S, Simonato F, Bronte V. Transcription factors in myeloid-derived suppressor cell recruitment and function. Curr Opin Immunol. 2011;23:279–285. doi: 10.1016/j.coi.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 27.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPAR-gamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 29.Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18:40–50. doi: 10.1158/1078-0432.CCR-11-0998. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Gou X, Ke X, Cui H, Chen Z. Human Tumor Cells Induce Angiogenesis through Positive Feedback between CD147 and Insulin-Like Growth Factor-I. PLoS One. 2012;7:e40965. doi: 10.1371/journal.pone.0040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weidle UH, Scheuer W, Eggle D, Klostermann S, Stockinger H. Cancer-related issues of CD147. Cancer Genomics Proteomics. 2010;7:157–169. [PubMed] [Google Scholar]
- 32.Baba M, Inoue M, Itoh K, Nishizawa Y. Blocking CD147 induces cell death in cancer cells through impairment of glycolytic energy metabolism. Biochem Biophys Res Commun. 2008;374:111–116. doi: 10.1016/j.bbrc.2008.06.122. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Yee D. Tyrosine kinase signalling in breast cancer: insulin-like growth factors and their receptors in breast cancer. Breast Cancer Res. 2000;2:170–175. doi: 10.1186/bcr50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 35.Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, Stampfer MJ. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst. 1999;91:620–625. doi: 10.1093/jnci/91.7.620. [DOI] [PubMed] [Google Scholar]
- 36.Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 37.Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91:151–156. doi: 10.1093/jnci/91.2.151. [DOI] [PubMed] [Google Scholar]
- 38.Zhao H, Grossman HB, Spitz MR, Lerner SP, Zhang K, Wu X. Plasma levels of insulin-like growth factor-1 and binding protein-3, and their association with bladder cancer risk. J Urol. 2003;169:714–717. doi: 10.1097/01.ju.0000036380.10325.2a. [DOI] [PubMed] [Google Scholar]
- 39.Ribeiro-Oliveira A Jr, Barkan A. The changing face of acromegaly-advances in diagnosis and treatment. Nat Rev Endocrinol. 2012;8:605–11. doi: 10.1038/nrendo.2012.101. [DOI] [PubMed] [Google Scholar]
- 40.Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. 2011;286:23591–23599. doi: 10.1074/jbc.M111.237123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trayhurn P. The development of obesity in animals: the role of genetic susceptibility. Clin Endocrinol Metab. 1984;13:451–474. doi: 10.1016/s0300-595x(84)80033-x. [DOI] [PubMed] [Google Scholar]
- 42.Tschop M, Heiman ML. Overview of rodent models for obesity research. Curr Protoc Neurosci. 2002;Chapter 9:Unit 9.10. doi: 10.1002/0471142301.ns0910s17. [DOI] [PubMed] [Google Scholar]
- 43.Tschop M, Heiman ML. Rodent obesity models: an overview. Exp Clin Endocrinol Diabetes. 2001;109:307–319. doi: 10.1055/s-2001-17297. [DOI] [PubMed] [Google Scholar]
- 44.Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, Casares S, Chen SH, Yang WC, Pan PY. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol. 2010;185:5828–5834. doi: 10.4049/jimmunol.0903636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu H, Huang D, Ransohoff RM, Zhou L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 2011;25:3344–3355. doi: 10.1096/fj.10-178939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. 2011;25:358–369. doi: 10.1096/fj.10-171579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, Van Rooijen N, Urban JF Jr, Wynn TA, Gause WC. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;18:260–266. doi: 10.1038/nm.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 50.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137:25–33. doi: 10.7326/0003-4819-137-1-200207020-00009. [DOI] [PubMed] [Google Scholar]
- 52.Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–488. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu B, Li S, Sheng L, Zhu J, Gu L, Shen H, La D, Hambly BD, Bao S, Di W. Metformin inhibits the development and metastasis of ovarian cancer. Oncol Rep. 2012;28:903–908. doi: 10.3892/or.2012.1890. [DOI] [PubMed] [Google Scholar]
- 55.Chong CR, Chabner BA. Mysterious metformin. Oncologist. 2009;14:1178–1181. doi: 10.1634/theoncologist.2009-0286. [DOI] [PubMed] [Google Scholar]
- 56.Gotlieb WH, Saumet J, Beauchamp MC, Gu J, Lau S, Pollak MN, Bruchim I. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol Oncol. 2008;110:246–250. doi: 10.1016/j.ygyno.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 57.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 59.Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS, Galimberti V, Veronesi P, Johansson H, Aristarco V, Bassi F, Luini A, Lazzeroni M, Varricchio C, Viale G, Bruzzi P, Decensi A. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012;30:2593–2600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
- 60.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 61.Janjetovic K, Vucicevic L, Misirkic M, Vilimanovich U, Tovilovic G, Zogovic N, Nikolic Z, Jovanovic S, Bumbasirevic V, Trajkovic V, Harhaji-Trajkovic L. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651:41–50. doi: 10.1016/j.ejphar.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Miller RA, Birnbaum MJ. An energetic tale of AMPK-independent effects of metformin. J Clin Invest. 2010;120:2267–2270. doi: 10.1172/JCI43661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arai M, Uchiba M, Komura H, Mizuochi Y, Harada N, Okajima K. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J Pharmacol Exp Ther. 2010;334:206–213. doi: 10.1124/jpet.109.164970. [DOI] [PubMed] [Google Scholar]
- 64.Bulun SE, Simpson ER. Regulation of aromatase expression in human tissues. Breast Cancer Res Treat. 1994;30:19–29. doi: 10.1007/BF00682738. [DOI] [PubMed] [Google Scholar]
- 65.Sasano H, Miki Y, Nagasaki S, Suzuki T. In situ estrogen production and its regulation in human breast carcinoma: from endocrinology to intracrinology. Pathol Int. 2009;59:777–789. doi: 10.1111/j.1440-1827.2009.02444.x. [DOI] [PubMed] [Google Scholar]
- 66.Nagasaki S, Miki Y, Akahira J, Suzuki T, Sasano H. 17beta-hydroxysteroid dehydrogenases in human breast cancer. Ann N Y Acad Sci. 2009;1155:25–32. doi: 10.1111/j.1749-6632.2008.03682.x. [DOI] [PubMed] [Google Scholar]
- 67.Bulun SE, Mahendroo MS, Simpson ER. Aromatase gene expression in adipose tissue: relationship to breast cancer. J Steroid Biochem Mol Biol. 1994;49:319–326. doi: 10.1016/0960-0760(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 68.Brown KA, Simpson ER. Obesity and breast cancer: progress to understanding the relationship. Cancer Res. 2010;70:4–7. doi: 10.1158/0008-5472.CAN-09-2257. [DOI] [PubMed] [Google Scholar]
- 69.Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, Park CG, Simmons A, Alberola-Ila J, Kovats S. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood. 2010;115:238–246. doi: 10.1182/blood-2009-08-236935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 71.Treves AJ, Fibach E, Kaiser N, Weinstein D, Maoz H, Halimi M, Simon A, Rachmilewitz D. Development of macrophage and granulocyte colonies from human peripheral blood. Int J Radiat Oncol Biol Phys. 1985;11:271–275. doi: 10.1016/0360-3016(85)90149-x. [DOI] [PubMed] [Google Scholar]
- 72.Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligandmediated dendritic cell differentiation. J Immunol. 2008;180:727–738. doi: 10.4049/jimmunol.180.2.727. [DOI] [PubMed] [Google Scholar]
- 73.Kovats S, Carreras E. Regulation of dendritic cell differentiation and function by estrogen receptor ligands. Cell Immunol. 2008;252:81–90. doi: 10.1016/j.cellimm.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harkonen PL, Vaananen HK. Monocyte-macrophage system as a target for estrogen and selective estrogen receptor modulators. Ann N Y Acad Sci. 2006;1089:218–227. doi: 10.1196/annals.1386.045. [DOI] [PubMed] [Google Scholar]
- 75.Yasui T, Uemura H, Hyodo S, Yamada M, Yamamoto S, Maegawa M, Tsuchiya N, Noguchi M, Yuzurihara M, Kase Y, Irahara M. Raloxifene reduces circulating levels of interleukin-7 and monocyte chemoattractant protein-1 in postmenopausal women. Atherosclerosis. 2009;204:471–475. doi: 10.1016/j.atherosclerosis.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 76.Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo CY, Wang L, Sun C, Li DJ. Estrogen enhances the functions of CD4(+)CD25(+) Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. 2011;8:50–58. doi: 10.1038/cmi.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Valor L, Teijeiro R, Aristimuno C, Faure F, Alonso B, de Andres C, Tejera M, Lopez-Lazareno N, Fernandez-Cruz E, Sanchez-Ramon S. Estradiol-dependent perforin expression by human regulatory T-cells. Eur J Clin Invest. 2011;41:357–364. doi: 10.1111/j.1365-2362.2010.02414.x. [DOI] [PubMed] [Google Scholar]
- 79.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 80.Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. 2000;85:3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- 81.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 82.Yamakawa T, Tanaka S, Yamakawa Y, Kiuchi Y, Isoda F, Kawamoto S, Okuda K, Sekihara H. Augmented production of tumor necrosis factor-alpha in obese mice. Clin Immunol Immunopathol. 1995;75:51–56. doi: 10.1006/clin.1995.1052. [DOI] [PubMed] [Google Scholar]
- 83.Flores MB, Rocha GZ, Damas-Souza DM, Osorio-Costa F, Dias MM, Ropelle ER, Camargo JA, de Carvalho RB, Carvalho HF, Saad MJ, Carvalheira JB. Obesity-Induced Increase in Tumor Necrosis Factor-alpha Leads to Development of Colon Cancer in Mice. Gastroenterology. 2012;143:741–53. e1–4. doi: 10.1053/j.gastro.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 84.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity (Silver Spring) 2010;18:1918–1925. doi: 10.1038/oby.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, Vosshenrich CA. IL-1beta regulates a novel myeloidderived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. 2010;40:3347–3357. doi: 10.1002/eji.201041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumorhost interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 90.Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med. 2006;4:48. doi: 10.1186/1479-5876-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vollbrecht T, Stirner R, Tufman A, Roider J, Huber RM, Bogner JR, Lechner A, Bourquin C, Draenert R. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. AIDS. 2012;26:F31–37. doi: 10.1097/QAD.0b013e328354b43f. [DOI] [PubMed] [Google Scholar]
- 93.Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet--induced obesity in mice. Diabetes. 2010;59:1171–1181. doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, Tsuneyama K, Nagai Y, Takatsu K, Urakaze M, Kobayashi M, Tobe K. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rahman SM, Janssen RC, Choudhury M, Baquero KC, Aikens RM, de la Houssaye BA, Friedman JE. CCAAT/enhancer binding protein beta (C/EBPbeta) expression regulates dietary-induced inflammation in macrophages and adipose tissue in mice. J Biol Chem. 2012;287:34349–60. doi: 10.1074/jbc.M112.410613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 97.Huber R, Pietsch D, Panterodt T, Brand K. Regulation of C/EBPbeta and resulting functions in cells of the monocytic lineage. Cell Signal. 2012;24:1287–1296. doi: 10.1016/j.cellsig.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 98.Michaud A, Drolet R, Noel S, Paris G, Tchernof A. Visceral fat accumulation is an indicator of adipose tissue macrophage infiltration in women. Metabolism. 2012;61:689–698. doi: 10.1016/j.metabol.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 99.Berndt J, Kovacs P, Ruschke K, Kloting N, Fasshauer M, Schon MR, Korner A, Stumvoll M, Bluher M. Fatty acid synthase gene expression in human adipose tissue: association with obesity and type 2 diabetes. Diabetologia. 2007;50:1472–1480. doi: 10.1007/s00125-007-0689-x. [DOI] [PubMed] [Google Scholar]
- 100.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 101.Wang D, Dubois RN. Associations between obesity and cancer: the role of fatty acid synthase. J Natl Cancer Inst. 2012;104:343–345. doi: 10.1093/jnci/djs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nguyen PL, Ma J, Chavarro JE, Freedman ML, Lis R, Fedele G, Fiore C, Qiu W, Fiorentino M, Finn S, Penney KL, Eisenstein A, Schumacher FR, Mucci LA, Stampfer MJ, Giovannucci E, Loda M. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J. Clin. Oncol. 2010;28:3958–3964. doi: 10.1200/JCO.2009.27.0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010;6:551–562. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, Lee CH. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li H, Sorenson AL, Poczobutt J, Amin J, Joyal T, Sullivan T, Crossno JT Jr, Weiser-Evans MC, Nemenoff RA. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS One. 2011;6:e28133. doi: 10.1371/journal.pone.0028133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mamtani R, Haynes K, Bilker WB, Vaughn DJ, Strom BL, Glanz K, Lewis JD. Association Between Longer Therapy With Thiazolidinediones and Risk of Bladder Cancer: A Cohort Study. J Natl Cancer Inst. 2012;104:1411–21. doi: 10.1093/jnci/djs328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, Slovin S, Spiegelman B, Small E, Kantoff PW. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer. 2004;101:1569–1574. doi: 10.1002/cncr.20493. [DOI] [PubMed] [Google Scholar]
- 109.Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, Farzadfar F, Riley LM, Ezzati M. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–1613. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 111.Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009;284:29087–29096. doi: 10.1074/jbc.M109.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 114.Mione M, Zon LI. Cancer and inflammation: an aspirin a day keeps the cancer at bay. Curr Biol. 2012;22:R522–525. doi: 10.1016/j.cub.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. 1994;91:4854–4858. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]