

Abstract
Transforming growth factor-β1 (TGF- β1) and thrombospondin-1 (TSP-1) are hypoxia-responsive mitogens that promote vascular smooth muscle cell (SMC) proliferation, a critical event in the pathogenesis of pulmonary hypertension (PH). We previously demonstrated that hypoxia-induced human pulmonary artery smooth muscle (HPASMC) cell proliferation and expression of the NADPH oxidase subunit, Nox4, were attenuated by the peroxisome proliferator-activated receptor γ (PPARγ) agonist, rosiglitazone. The current study examines the hypothesis that rosiglitazone regulates Nox4 expression and HPASMC proliferation by attenuating TSP-1 signaling. Selected HPASMC were exposed to normoxic or hypoxic (1% O2) environments or TSP-1 (0-1 μg/ ml) for 72 hours ± administration of rosiglitazone (10 μM). Cellular proliferation, Nox4, TSP-1, and TGF-β1 expression and reactive oxygen species generation were measured. Mice exposed to hypoxia (10% O2) for three weeks were treated with rosiglitazone (10 mg/kg/day) for the final 10 days, and lung TSP-1 expression was examined. Hypoxia increased TSP-1 and TGF-β1 expression and HPASMC proliferation, and neutralizing antibodies to TSP-1 or TGF-β1 attenuated proliferation. Rosiglitazone attenuated hypoxia-induced HPASMC proliferation and increases in mouse lung and HPASMC TSP-1 expression, but failed to reduce increases in TGF-β1 expression or Nox4 expression and activity caused by direct TSP-1 stimulation. Transfecting HPASMC with siRNA to Nox4 attenuated hypoxia- or TSP-1-stimulated HPASMC proliferation. These findings provide novel evidence that TSP-1-mediated Nox4 expression plays a critical role in hypoxia-induced HPASMC proliferation. PPARγ activation with exogenous ligands attenuates TSP-1 expression to reduce Nox4 expression. These results clarify mechanisms of hypoxia-induced SMC proliferation and suggest additional pathways by which PPARγ agonists may regulate critical steps in the pathobiology of PH.
Keywords: hypoxia, Nox4, PPARγ, rosiglitazone, thrombospondin-1
Pulmonary hypertension (PH) is a progressive disorder with poor survival. Although, advances in the modern drug treatment era have led to improvements in survival, morbidity is unacceptably high, and the disease prognosis remains poor.[1] The pathogenesis of PH is complex and involves derangements of molecular mediators of vascular tone and patency, as well as enhanced proliferation of cells in the vascular wall. These changes result in pulmonary arterial remodeling, a process that increases pulmonary vascular resistance, right ventricular pressure, and leads to cor pulmonale.[2–4]
Chronic hypoxia is one of the most common causes of PH, and belongs to the Group III category of the revised World Health Organization classification of PH.[5] The mechanisms by which chronic hypoxia exposure mediates PH are complex and multifactorial.[6] Several recent studies have implicated NADPH oxidase-generated reactive oxygen species (ROS) in PH pathogenesis. NAPDH oxidase is a hypoxia-responsive, multisubunit protein complex which produces ROS which alters pulmonary vascular structure and function and may contribute to the pathogenesis of vascular diseases such as atherosclerosis and pulmonary hypertension.[7–10] NADPH oxidase was originally described in phagocytic cells where its activation led to the generation of ROS critically involved in host defense.[11,12] Nox2, also known as gp91Phox, the catalytic moiety of the NADPH oxidase complex, was the first member of the Nox family to be discovered in phagocytic cells.[8]
Nox4 is a constitutively active homologue of Nox2 expressed in a variety of non-phagocytic cells.[13–15] For example, Nox4 is abundantly expressed in pulmonary vascular wall cells,[12,15] and its expression is selectively upregulated in the lungs of hypoxia-exposed mice, as well as in the vascular media of patients with idiopathic pulmonary arterial hypertension (IPAH).[15] Emerging evidence demonstrates that hypoxic increases in Nox4 expression are mediated in part by increased transforming growth factor β (TGF-β)-1,[16] a multifunctional matrix protein that contributes to the pathogenesis of many vascular diseases,[17,18] pulmonary vascular remodeling, and PH.[19,20] Recently, NF-kB,[14] and hypoxia-inducible factor-1α (HIF1-α)[21] were shown to modulate the transcriptional expression of Nox4 by binding to its promoter. Upregulation of Nox4 in the hypoxic pulmonary vasculature is associated with increased ROS generation, and vascular smooth muscle cell (VSMC) and endothelial cell (EC) proliferation, which contribute to the development of vascular remodeling and dysfunction in PH.[15]
The matrix-associated glycoprotein, thrombospondin (TSP)-1, regulates TGF-β1 activation,[22] and smooth muscle proliferation (SMC).[23] Detailed mechanism studies revealed that TSP-1 interacts with the latency associated protein (LAP) of TGF-β1, inducing a conformational change that allows the active site of TGF-β1 to bind to its receptor.[24,25] Although, TSP-1 could plausibly modulate Nox4 expression through its ability to activate latent TGF-β1, a detailed understanding of its role on redox signaling pathways in PH is lacking. TSP-1 also functions as a potent mitogen and chemoattractant for SMC.[26,27] While little is known about the role of TSP-1 in the development of PH, its expression is increased in the lungs of patients with sickle cell disease and idiopathic pulmonary arterial hypertension (IPAH).[28] Hypoxia stimulates TSP-1 expression in the mouse pulmonary artery,[29] and TSP-1 null mice exposed to chronic hypoxia were protected from the development of pulmonary vascular remodeling and PH.[30] These studies suggest that hypoxia-induced TSP-1 expression contributes to the development of PH, whereas its absence is protective.
Our lab recently reported that activation of the nuclear hormone receptor peroxisome proliferator-activated receptor gamma (PPARγ) attenuated hypoxia-induced Nox4 expression, PH, and pulmonary vascular remodeling.[31] In a separate report, we confirmed that Nox4 was an essential mediator of human pulmonary artery smooth muscle cell (HPASMC) proliferation caused by hypoxia.[14] PPARγ is expressed in the lung and pulmonary vasculature and participates in vascular homeostasis. Available evidence suggests that plexiform lesions in patients with PH have decreased PPARγ expression, a finding that was confirmed in an in vivo model of severe PH in rats.[32] Conversely, PPARγ ligands attenuated or reversed PH in several models including chronic hypoxia-exposed mice,[31] and Apo E-/- mice fed high fat diets,[33] as well as in hypoxia-[34] and monocrotaline-treated[35] rats. Because PPARγ activation attenuated TGF-β1-induced vascular injury,[36] the current study was performed to further examine the role of TGF-β1 and TSP-1 in the induction of Nox4 by hypoxia in PASMC. We hypothesized that PPARγ activation would attenuate hypoxia-induced increases in TSP-1 and TGF-β1 and thereby attenuate hypoxia-induced increases in HPASMC Nox4 expression.
MATERIALS AND METHODS
Cell culture and in vitro hypoxia studies
Monolayers of HPASMC (Lonza, Walkersville, Md.) were grown and maintained at 37°C in a 5% CO2 atmosphere in smooth muscle growth medium (Lonza) containing 5% fetal bovine serum, 10 ng/ml human epidermal growth factor, 1.0 mg/ml hydrocortisone, 12 mg/ml bovine brain extract, 50 mg/ml gentamicin, and 50 ng/ ml amphotericin. HPASMC were placed in hypoxic (1% O2, 5% CO2) or normoxic conditions (21% O2, 5% CO2) in a cell culture incubator at 37°C for 72 hours unless otherwise specified, as reported.[14] The synthetic PPARγ ligand, rosiglitazone (10 μM in vehicle) or an equal volume of vehicle 1% dimethyl sulfoxide (DMSO, Fisher Scientific, Fair Lawn, N.J.) was added to the HPASMC culture media during the final 24 hours of exposure to normoxic or hypoxic conditions as indicated. We previously reported that this hypoxia regimen stimulated Nox4 expression and activity and proliferation in HPASMC and that treatment with this rosiglitazone regimen attenuated these hypoxia-induced derangements.[14]
Assays of HPASMC proliferation
Proliferation of HPASMC was determined using an 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT assay; ATCC, Manassas, Va.) as we recently reported.[14] In selected studies, neutralizing antibodies to TGF-β1 (1 μg/ml, Santa Cruz Biotechnology, Santa Cruz, Calif.) or TSP-1 (1 μg/ml, Thermo Scientific, Fremont, Calif.) were added to the culture media three hours prior to hypoxia exposure.
TGF-β1 enzyme-linked immunosorbent assay
An enzyme-linked immunosorbent assay (ELISA) kit (Promega, Madison, Wisc.) was used to detect bioactive TGF-β1 protein secreted by HPASMC into the culture media. Cells (5 × 104) were propagated in a 6-well plate to 80% confluence in growth media supplemented with 5% FBS. The cells were then washed with PBS, and placed in fresh media supplemented with 1% FBS. The ELISA was performed according to the manufacturer's protocol. Samples or standards containing known amounts of TGF-β1 were incubated in a 96-well plate precoated with antibodies to TGF-β1. Horseradish peroxidase-conjugated secondary antibodies were then added. The amount of TGF-β1 was determined by measuring the formation of the colored product at 450 nm on a plate reader. In parallel experiments, the media was acid treated prior to performing the ELISA to detect total (bioactive + latent) TGF-β1.
Smad 3 reporter construct, transfection techniques, and luciferase assays
The canonical TGF-β1 signaling pathway involves phosphorylation of the Smad 3 transcription factor which leads to binding and activation of the promoter elements of target genes. Purified plasmid containing the Smad 3 binding element with attached luciferase reporter was provided as a gift from Dr. Alan Ramirez (University of Louisville). The plasmid (0.5 μg) was transfected using the TransIT-2020 Transfection Reagent according to the manufacturer's protocol (Mirus Bio LLC, Madison, Wisc.). After transfection, the cells were allowed to incubate for 24 hours in an incubator at 37°C. To control for transfection efficiency, cells were cotransfected with (0.1 mg/well) Renilla luciferase (Promega, Madison, Wisc.). The cells were then treated with purified recombinant human TGF-β1 (R and D Systems, Minneapolis, Minn.) for 24 hours. To assess the effect of PPARγ activation on early TGF-β1 induced Smad3 DNA binding, rosiglitazone (10 μM) or vehicle DMSO was added to selected wells after six hours of incubation with TGF-β1 (2 ng/ml). Cells were then washed with 1x PBS and placed in passive lysis buffer (300 μl, Promega). Luciferase activities were measured with the Luciferase Assay System (Promega, Madison, Wisc.). Light detection was recorded with a luminometer (PerkinElmer). Relative light units in each sample were normalized to renilla, and all samples were examined in triplicate.
Western blot analysis
After treatment, HPASMC monolayers were washed, collected, and lysed as previously reported.[37] Protein content of the lysate was determined using a bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Rockford, Ill.). Equal amounts of protein were then loaded onto 4-12% gradient gels (Invitrogen, Carlsbad, Calif.) and resolved with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by electroblotting onto polyvinylidene fluoride (PVDF) or nitrocellulose membranes. The membranes were blocked with 5% powdered nonfat dry milk for one hour and incubated overnight with primary antibodies to TSP-1 (Thermo Scientific, Fremont, Calif.; 1:1000) or Nox4 (Dr. David Lambeth, Emory University; 1:1000). After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) raised against the species in which the primary antibodies were developed. Immunodetection was then performed using chemiluminescence (SuperSignal, Pierce Biotechnology, Rockford, Ill.). Relative immunoreactive protein levels were quantified using the Chemidoc XRS imaging system and Quantity One software (Bio-Rad Laboratories). All samples were normalized to their respective content of cyclin-dependent kinase 4 (CDK 4).
Amplex red H2O2 assay
TSP-1 (0.1-1 μg/ml; Athens Research and Technology, Athens, Ga.) was added to the culture media of HPASMC for 24-72 hours, and H2O2 production was measured with Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen, Molecular Probes, Eugene, Oreg.) as previously described.[14] The assay is based on the detection of H2O2 which reacts with 1:1 stoichiometry with Amplex® Red reagent in combination with horseradish peroxidase to produce highly fluorescent resorufin red. H 2O2 released from HPASMC was then quantified by fluorometric detection on a plate reader (λex = 590 nm and λem = 560 nm), followed by plotting sample values against a standard curve containing serial dilutions of known concentrations of H2O2.
RNA isolation, reverse transcription, and quantitative PCR
Total RNA was isolated from HPASMC using the RNeasy Mini Kit (Quiagen, Valencia, Calif.) and quantitated using NanoDrop spectrophotometry (Thermo Scientific, Wilmington, Del.). cDNA was prepared using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, Calif.) and reverse transcribed from 1 μg of total RNA. Real-time PCR was then performed to characterize the expression of target genes with primers based on human RNA sequences using iQ SYBR Green Supermix and the iCycler Real-Time PCR Detection System (Bio-Rad). Primer sequences were as follows: TGF-β1, forward primer (CAGAAATACAGCAACAATTCCTGG) and reverse primer (TTGCAGTGTGTTATCCCTGCTGTC); No×4, forward primer (GCTGACGTTGCATGTTTCAG) and reverse primer (CGGGAGGGTGGGTATCTAA); glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward primer (AGCCACATCGCTCAGACAC) and reverse primer (GCCCAATACGACCAAATCC); 9S, forward primer (CTGACGCTTGATGAGAAGGAC) and reverse primer (CAGCTTCATCTTGCCCTCAT; Eurofins MWG Operon, Huntsville, Ala.). Expression of target mRNA in each sample was normalized to its GAPDH or 9S content. The relative abundance of target mRNA in each sample was calculated using the ΔΔCt method.[38]
RNA interference
Nox4 gene expression was reduced using Nox4 small interfering RNA (siRNA, Qiagen) as described previously.[14] HPASMC monolayers were transfected with 35 nM siNox4 or si control for 24 hours before treatment with TSP-1 (1 μg/ml) or hypoxia (1% O2, 5% CO2). Real-time PCR was employed to confirm Nox4 knock down by at least 50% in all studies. HPASMC proliferation was measured with MTT assays at the conclusion of each study.
Mouse model of chronic hypoxia exposure
Eight-week-old male C57Bl/6 mice were exposed to normoxic (21% O2) or hypoxic (10% O2) conditions for three weeks as we reported.[31] During the final 10 days of exposure to hypoxic or normoxic conditions, each animal was given rosiglitazone (10 mg/kg/day) or an equal volume of vehicle (methylcellulose) daily by oral gavage. We previously reported that this hypoxia regimen stimulated increased right ventricular systolic pressures, right ventricular hypertrophy, and pulmonary vascular remodeling and that these hypoxic derangements were attenuated by this rosiglitazone regimen.[31] All animals had access to standard mouse chow and water ad libitum, and all procedures were reviewed and approved by the Atlanta VA Medical Center Institutional Animal Care and Use Committee.
Statistical analysis
For all experiments, statistical analysis was performed by one-way ANOVA followed by a Tukey's post-hoc analysis to detect differences among experimental groups. The level of statistical significance was set at an alpha value of P ≤ 0.05.
RESULTS
Rosiglitazone or neutralizing antibodies to either TSP-1 or TGF-β1 attenuated hypoxia-induced HPASMC proliferation
As illustrated in Figure 1, hypoxia increased HPASMC proliferation, and treatment with rosiglitazone or with neutralizing antibodies to either TSP-1 or TGF-β1 attenuated proliferation in both control and hypoxia-exposed cells. These results suggest that the constitutive expression of TSP-1 and TGF-β1 contribute to autocrine stimulation of HPASMC proliferation under basal conditions, and that enhanced production of TGF-β1 and TSP-1 contribute to hypoxic increases in HPASMC proliferation.
Figure 1.
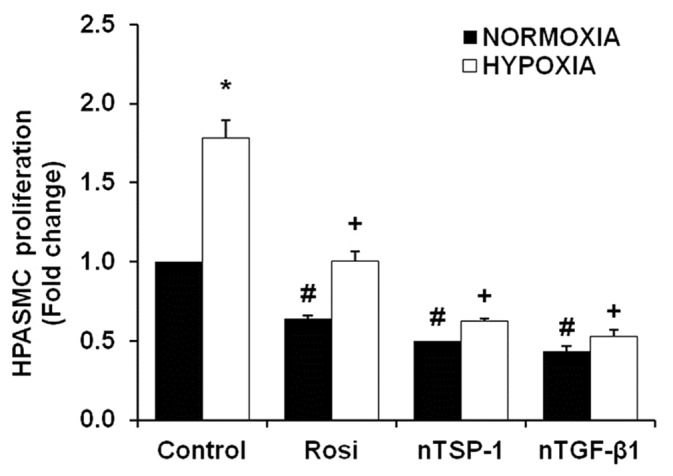
Rosiglitazone or neutralizing antibodies to TSP-1 or TGF-β1 attenuated hypoxia-induced HPASMC proliferation. HPASMC exposed to normoxic (21% O2) or hypoxic (1% O2) conditions for 72 hours were treated with neutralizing antibodies to either TSP-1 (nTSP-1, 1 μg/ml) or TGF-β1 (nTGF-b1, 1 μg/ml) 3 hours prior to exposure, or rosiglitazone (Rosi, 10 μM) was administered during the last 24 hours of exposure. After treatment, an MTT assay was performed to measure cellular proliferation (n = 6). *P< 0.05 vs. Control-Normoxia, +P< 0.05 vs. Control-Hypoxia, #P< 0.05 vs. Control-Normoxia.
Rosiglitazone failed to regulate either TGF-β1 expression or mediators of the TGF-β1 canonical signaling pathway
As shown in Figure 2, hypoxia caused small but significant increases in TGF-β1 mRNA levels and secretion of bioactive TGF-β1 from HPASMC. Treatment with rosiglitazone caused small but statistically insignificant increases in TGF-β1 mRNA (Fig. 2A) and protein (Fig. 2B) in control HPASMC and failed to attenuate the hypoxic-induction of TGF-β1 mRNA and protein. Exposure to hypoxic conditions for 72 hours also increased bioactive TGF-β1 secretion that was not reduced by rosiglitazone treatment (data not shown). TGF-β1 binds to its receptor to stimulate Smad 2/3 recruitment, phosphorylation, and colocalization with Smad 4. This complex translocates into the nucleus where it binds to and activates target DNA sequences.[39] To evaluate the effect of PPARγ activation on this canonical TGF-β1 pathway, Smad 3 DNA binding was assessed following stimulation with TGF-β1 and treatment with rosiglitazone. As illustrated in Figure 2C, rosiglitazone (10 μM) did not attenuate TGF-β1-induced increases in Smad 3 DNA binding as measured by a luciferase reporter assay.
Figure 2.
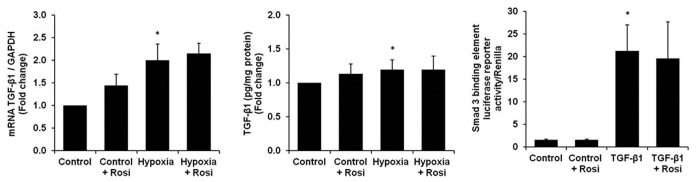
Rosiglitazone treatment did not modulate hypoxia-induced increases in HPASMC TGF-β1 mRNA, protein or TGF-β1-induced activation of the canonical TGF-β1 signaling pathway. HPASMC were exposed to normoxic or hypoxic conditions for 72 hours ± rosiglitazone (Rosi, 10 μM). TGF-β1 expression was measured in cell lysates by (A) qRT-PCR (n = 12), and in the cell media by (B) ELISA (n = 3). (C) Smad 3 binding element luciferase reporter activity was measured in HPASMC treated with TGF-β1 (2 ng/ml) for 24 hours ± rosiglitazone (10 μM) (n = 3). *P< 0.05 vs. Control; **P< 0.01 vs. Control.
Hypoxia increased TSP-1 expression in HPASMC
Because rosiglitazone failed to regulate TGF-β1 expression and signaling, we examined TSP-1 levels in hypoxia-exposed HPASMC. As shown in Figure 3, compared with control conditions, exposure to hypoxia for 72 hours increased HPASMC TSP-1 protein levels as detected by Western blotting. The hypoxic induction of TSP-1 was attenuated by treatment with rosiglitazone during the last 24 hours of hypoxia exposure.
Figure 3.
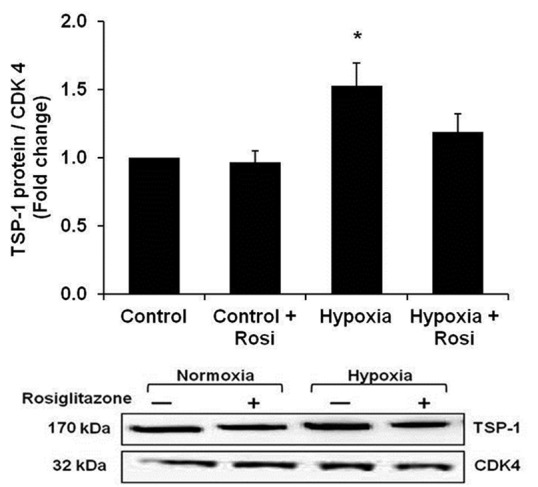
Rosiglitazone attenuated hypoxia-induced increases in TSP-1 protein expression. HPASMC were exposed to normoxic (control) or hypoxic conditions for 72 hours. During the final 24 hours of exposure, HPASMC were treated with rosiglitazone (Rosi 10 μM). HPASMC were collected, and proteins were isolated for Western blot analysis. Each bar represents the mean ± SEM TSP-1 band density normalized to CDK 4 and expressed relative to control samples (n=9). *P< 0.05 vs. Control. A representative TSP-1 immunoblot is presented below the bar graph.
Exogenous administration of TSP-1 increased Nox4 expression and ROS generation
We have previously reported that hypoxia stimulates Nox4 expression and ROS generation in the lung in vivo,[31] and in HPASMC in vitro.[14] Since hypoxia increased TSP-1 expression in HPASMC, the ability of TSP-1 to directly stimulate Nox4 expression and augment the generation of ROS was examined. As shown in Figure 4A, TSP-1 (1 μg/ ml) increased HPASMC Nox4 protein expression. Figure 4B demonstrates that TSP-1 increases Nox4 expression after 48 or 72 hours, but not after 24 hours. Amplex Red assays demonstrate that TSP-1 also stimulates corresponding increases in HPASMC H2O2 production (Fig. 4C). Together, these results suggest that TSP-1 is sufficient to stimulate Nox4 expression and activity in HPASMC.
Figure 4.
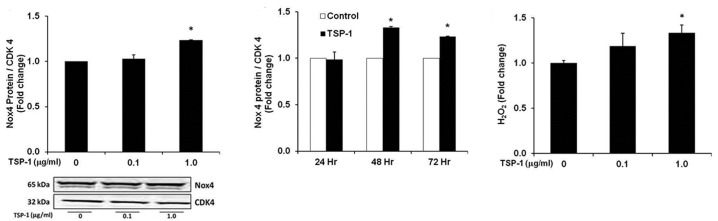
TSP-1 caused dose and time-dependent increases in HPASMC Nox4 expression and ROS generation. Western blot to detect Nox4 protein was performed on protein isolated from HPASMC monolayers treated with (A) graded concentrations of TSP-1 for 72 hours (n = 3), or (B) 1 μg/ml of TSP-1 for 24-72 hours (n = 3). Nox4 immunoblots are presented below the bar graph. (C) HPASMC monolayers were treated with graded concentrations of TSP-1 for 72 hours and H2O2 production was measured by Amplex Red assay (n = 3). (A) *P< 0.01 vs. Control, (B) *P< 0.001 vs. Control, (C) *P< 0.01 vs. Control.
Nox4 siRNA attenuated TSP-1 or hypoxia-induced proliferation of HPASMC
Previous evidence indicated that Nox4 induction was important for hypoxia-mediated increases in HPASMC proliferation.[14,40] We therefore, assessed the effect of Nox4 inhibition on TSP-1-mediated HPASMC proliferation. We employed our recently published Nox4 siRNA protocol to reduce basal Nox4 mRNA and protein levels and hypoxia-induced H2O2 production in HPASMC.[14] As illustrated in Figure 5, both hypoxia and TSP-1 caused comparable increases in HPASMC proliferation over control conditions, and Nox4 siRNA significantly reduced cellular proliferation in response to both stimuli. These results provide novel evidence that both TSP-1 and hypoxia-induced HPASMC proliferation are Nox4-dependent processes.
Figure 5.
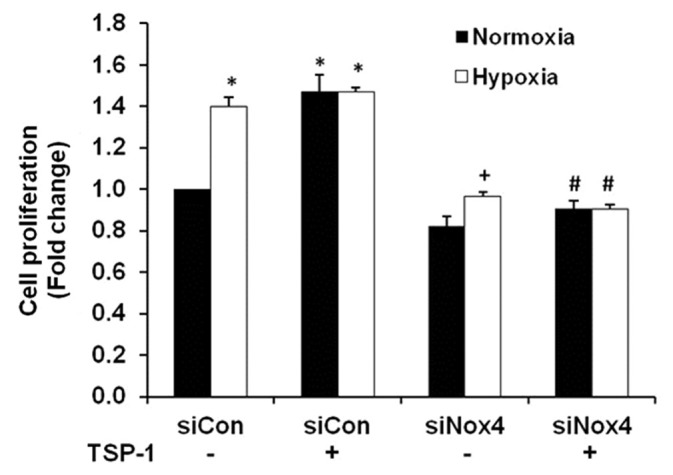
Nox4 siRNA decreased hypoxia or TSP-1-induced HPASMC proliferation. HPASMC were transfected with 35 nM siNox4 or a scrambled control (siCon). Twenty-four hours later, HPASMC were exposed to normoxic or hypoxic conditions for 72 hours ± TSP-1 (1 μg/ml). An MTT assay was then performed. Each bar represents mean ± SEM HPASMC proliferation expressed relative to control samples (n = 3). *P< 0.05 vs. Normoxia-siCon, +P< 0.05 vs. Hypoxia-siCon, #P< 0.05 vs. TSP-1.
Rosiglitazone does not attenuate TSP-1-stimulated Nox4 expression
To further assess the regulation of TSP-1-induced Nox4 expression by PPARγ Nox4 expression was measured in HPASMC following direct stimulation with TSP-1. As shown in Figure 6, TSP-1 (1 μg/ml) increased Nox4 mRNA after 72 hours of treatment, and rosiglitazone (10 μM), administered during the last 24 hours of exposure failed to attenuate TSP-1 stimulated Nox4 expression. Taken along with our previous report,[14] these findings indicate that hypoxic stimulation of TSP-1 contributes to Nox4 induction and that rosiglitazone attenuates hypoxia-induced increases in TSP-1 expression rather than signaling pathways downstream of TSP-1.
Figure 6.
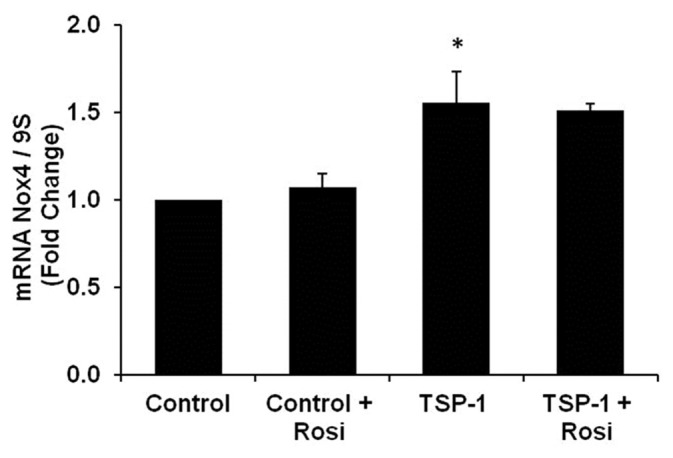
TSP-1 induces Nox4 expression and rosiglitazone fails to attenuate this increase. HPASMC were treated with TSP-1 (1 μg/ml) for 72 hours and rosiglitazone (10 μM) was added to the cell culture media during the final 24 hours of treatment. Total RNA was collected and quantitative real-time PCR of Nox4 transcripts was performed. Each bar represents the mean ± SEM Nox4 transcripts normalized to 9S and expressed as fold change relative to control (n = 3-6). *P < 0.05 vs. Control.
Rosiglitazone attenuated hypoxia-induced TSP-1 expression in mouse lung in vivo
To confirm that rosiglitazone regulates TSP-1 in vivo, C57Bl/6 mice were exposed to control or hypoxic conditions for three weeks and selected mice were treated with rosiglitazone daily by oral gavage during the final 10 days of exposure as reported.[31] As shown in Figure 7, hypoxia increased TSP-1 expression in mouse lung, and treatment with rosiglitazone attenuated hypoxic TSP-1 induction.
Figure 7.
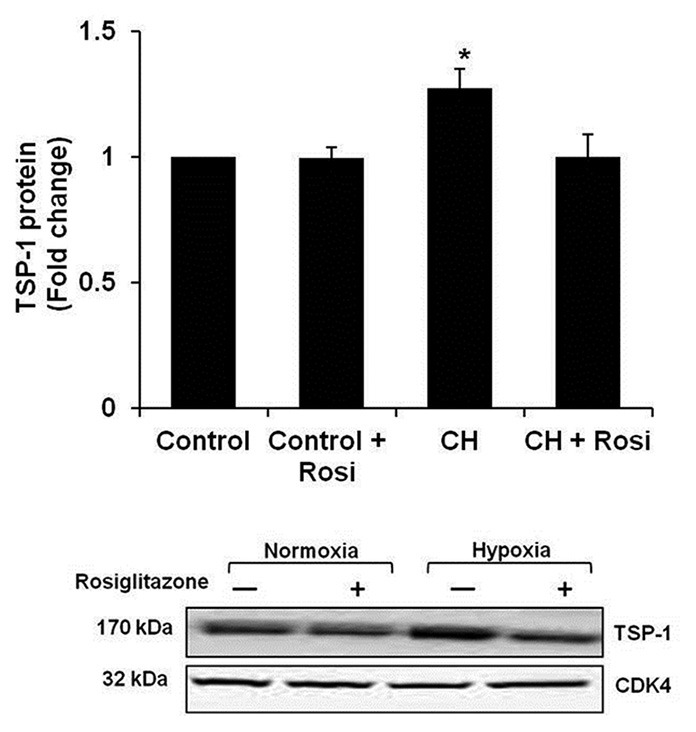
Rosiglitazone attenuates chronic-hypoxia-induced TSP-1 protein expression. C57Bl/6 mice were exposed to normoxic (Control) or chronic hypoxic (CH) (10% O2) conditions for three weeks. During the last 10 days of treatment, selected animals were gavaged with either rosiglitazone (Rosi, 10 mg/kg/day) or vehicle. Protein was prepared from whole lung homogenate for Western blot analysis of TSP-1 expression (n = 8). *P < 0.05 vs. Control. A representative TSP-1 immunoblot is presented below the bar graph.
DISCUSSION
Hypoxia is a clinically important cause of PH that contributes to alterations in pulmonary vascular function. Current evidence demonstrates that Nox4 is an important mediator of pulmonary vascular dysfunction in hypoxic environments.[15] We previously reported that rosiglitazone attenuated hypoxia-induced increases in Nox4 expression and activity, pulmonary vascular remodeling, and PH in the mouse lung.[31] Subsequent reports confirmed that Nox4 was a critical mediator of HPASMC proliferation caused by hypoxia.[14,40] Because hypoxia stimulates TGF-β1 signaling[41] which drives Nox4-mediated ROS generation and HPASMC proliferation,[40] we first examined the ability of rosiglitazone to regulate hypoxia-induced increases in TGF-β1 signaling. However, our findings indicate that rosiglitazone treatment conditions that successfully inhibited hypoxia-induced HPASMC proliferation failed to attenuate hypoxia-stimulated increases in TGF-β1 mRNA, protein, or SMAD 3 nuclear binding in HPASMC. However, TGF-β1 neutralizing antibodies attenuated hypoxia-induced HPASMC proliferation. Taken together, these findings confirm an important role for TGF-β1 in HPASMC responses to hypoxia, but suggest that PPARγ regulates hypoxia-induced Nox4 expression in HPASMC through pathways independent of TGF-β1.
The matricellular protein, TSP-1, is an SMC mitogen[42] whose expression is also upregulated in the hypoxic pulmonary vasculature.[29] Therefore, we hypothesized that rosiglitazone-mediated attenuation of hypoxia-induced Nox4 expression and HPASMC proliferation involved TSP-1. The present study provides novel evidence for the role of TSP-1 in mediating hypoxia-induced Nox4 expression and H2O2 generation, and further defines the impact of rosiglitazone on these signaling pathways. To our knowledge, this is the first to report to establish that TSP-1 is a proximal mediator of Nox4 expression. This adds to our understanding of mechanisms by which Nox4 is upregulated in hypoxic vasculature.
Our studies focused on Nox4, because it is selectively upregulated by hypoxia.[14,15,31] Compared to other NADPH oxidase isoforms, Nox4 also displays several unique characteristics including production of H2O2 rather than superoxide.[43,44] Nox4 is potently stimulated by hypoxia,[21] and unlike other NADPH oxidase isforms, it remains constitutively active and modulates downstream events through effects of H2O2 on redox sensitive targets.[45] Our lab demonstrated that Nox4 expression and activity were essential for hypoxia-induced HPASMC proliferation and that rosiglitazone attenuated increased Nox4 expression, in part by preventing NF-kB-mediated activation of the Nox4 promoter.[14] Although the precise mechanisms by which Nox4-derived H2O2 stimulates HPASMC proliferation remain to be defined, we postulate that activation of kinases and transcription factors participate in the altered expression of growth promoting genes.[8,46,47]
The current findings provide novel evidence for the involvement of hypoxic increases in TSP-1 mediating Nox4 expression and pulmonary vascular SMC proliferation. Previous reports have established the importance of TSP-1 in vascular cell mitogenic responses.[48–50] Our findings extend these reports and suggest that TSP-1-mediated regulation of Nox4 plays a critical role in the proliferative responses of pulmonary vascular wall cells to hypoxia.
Few corollary studies supporting the role of TSP-1 in the pathogenesis of hypoxia-induced pulmonary vascular wall proliferation, remodeling, and PH in vivo are available. Existing studies have employed knockout models of TSP-1[28,30] or its cognate receptor, CD47[28] to define the role of TSP-1 in hypoxia-induced PH. TSP-1 null mice were protected from the development of chronic hypoxia-induced PH, RVH, and pulmonary vascular remodeling,[28,30] and displayed decreased responses to the administration of acute pulmonary vasoconstrictors.[30] These studies demonstrate a clear role for TSP-1 in the pathogenesis of PH. We recently demonstrated that GKT137831, a pharmacological Nox4 inhibitor also attenuates chronic hypoxia-induced RVH and vascular remodeling (manuscript in press). In the present study, we show that TSP-1 regulates Nox4 expression, H2O2 production, and hypoxic HPASMC proliferation. Collectively, these lines of evidence suggest the possibility that the TSP-1 knockout mice are protected from chronic hypoxia-induced PH, RVH, and vascular remodeling because of the absence of TSP-1-mediated Nox4 expression and ROS generation. Recent studies confirm the association between TSP-1 and ROS production. In hypoxic environments and in PH, TSP-1- induced CD47 activation led to decreased caveolin-1 (eNOS binding protein) expression, subsequent uncoupling of endothelial nitric oxide synthase (eNOS) and ROS production. These findings mirror other authors conclusions that hypoxia enhances TSP-1 expression in the mouse lung in vivo and in pulmonary vascular wall cells,[29] and suggest that hypoxia-induced PH may develop in response to TSP-1-driven ROS generation from sources that include NADPH oxidases and eNOS.
A major implication of our findings relates to the potential ability to activate the PPARγ receptor with ligands such as rosiglitazone in order to modulate the enhanced production of a variety of mediators involved in pulmonary vascular cell proliferation and PH. For example, we have shown that PPARγ ligands not only attenuate the enhanced expression and activity of Nox4,[31] but also favorably and therapeutically attenuate increases in endothelin signaling in the hypoxic lung.[51] The molecular mechanisms for these effects remain to be completely defined. While we demonstrated that rosiglitazone attenuated hypoxia-induced increases in Nox4 expression by reducing NF-kB binding to the Nox4 promoter,[14] PPARγ regulation of the TSP-1 promoter remains to be established and constitutes an area of active investigation in our laboratory. The presence of NF-kB binding elements in the TSP-1 promoter[25] suggests that rosiglitazone-mediated reductions in TSP-1 expression may also occur through a similar mechanism. Alternatively, rosiglitazone decreased hypoxia-induced PDGFRβ activation,[31] and because PDGF stimulates TSP-1 expression, these findings suggest several candidate pathways for PPARγ-mediated regulation of TSP-1 expression. The presence of a PPAR response element in the TSP-1 promoter further suggests PPARγ could regulate TSP-1 expression through direct interactions with the TSP-1 promoter.
The current study has several important limitations. The examination of hypoxia-induced mechanisms in cultured cells may inadequately model hypoxia-induced PH in vivo. Furthermore, although the hypoxia regimen (1% O2) employed in the current study represents a severe degree of hypoxemia rarely encountered by pulmonary vascular wall cells in common clinical scenarios, recent reports suggest that these levels of hypoxemia may indeed occur in the cells characterizing the pathological lesions in the pulmonary circulation of IPAH patients. Lastly, recent reports have drawn attention to the increased risk of cardiovascular disease in diabetics taking rosiglitazone.[52] Importantly, studies have not demonstrated adverse cardiovascular effects in diabetics taking another synthetic PPARγ agonist, pioglitazone. These observations suggest that the observed cardiovascular complications may be attributable to the specific drug rather than to the TZD class and that only through more detailed mechanistic studies can the true therapeutic potential of targeting this receptor be realized.
In conclusion, our results provide novel evidence that TSP-1 is a proximal mediator of hypoxia-induced Nox4 expression and HPASMC proliferation. In addition, the synthetic thiazolidinedione PPARγ ligand, rosiglitazone, attenuated hypoxic induction of TSP-1 expression and HPASMC proliferation. These findings further clarify mechanisms by which hypoxia stimulates proliferation of pulmonary vascular wall cells contributing to vascular remodeling in PH. Evidence that PPARγ ligands attenuated these hypoxia-induced proliferative signals adds to the literature supporting PPARγ as a therapeutic target in PH and provides additional insights into mechanisms by which PPARγ mediates therapeutic effects (Fig. 8).
Figure 8.
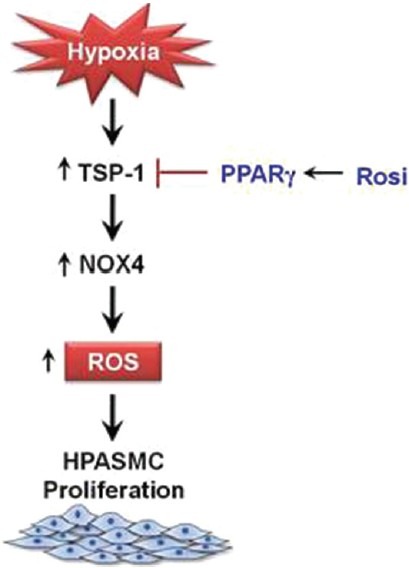
Putative mechanisms by which PPARγ ligands attenuate hypoxia-induced HPASMC proliferation. The current schema depicts relationships that exist between TSP-1, Nox4, and ROS generation in hypoxia-induced HPASMC proliferation. Hypoxia enhances Nox4 expression and ROS generation in a TSP-1-mediated manner. Activation of PPARγ with rosiglitazone attenuates hypoxia-induced HPASMC proliferation through inhibitory effects on TSP-1 expression and signaling.
Footnotes
Source of Support: Research Service of the Atlanta Veterans Affairs Medical Center and the National Institutes of Health (R01 DK 074518). NHLBI T32 training grant (HL076118-06).
Conflict of Interest: None declared.
REFERENCES
- 1.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 2.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation. 2010;121:2045–66. doi: 10.1161/CIRCULATIONAHA.108.847707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–65. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 4.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 6.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 8.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–97. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 9.Lassegue B, Griendling KK. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–61. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 11.Lambeth JD, Cheng G, Arnold RS, Edens WA. Novel homologs of gp91phox. Trends Biochem Sci. 2000;25:459–61. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- 12.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 13.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–83. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 14.Lu X, Murphy TC, Nanes MS, Hart CM. PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–66. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–67. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 16.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L73. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 17.Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–27. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 18.Upton PD, Morrell NW. TGF-beta and BMPR-II pharmacology-implications for pulmonary vascular diseases. Curr Opin Pharmacol. 2009;9:274–80. doi: 10.1016/j.coph.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, et al. Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol. 2008;295:L86–95. doi: 10.1152/ajplung.00534.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen YF, Feng JA, Li P, Xing D, Zhang Y, Serra R, et al. Dominant negative mutation of the TGF-beta receptor blocks hypoxia-induced pulmonary vascular remodeling. J Appl Physiol. 2006;100:564–71. doi: 10.1152/japplphysiol.00595.2005. [DOI] [PubMed] [Google Scholar]
- 21.Diebold I, Petry A, Hess J, Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010;21:2087–96. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy-Ullrich JE, Schultz-Cherry S, Hook M. Transforming growth factor-beta complexes with thrombospondin. Mol Biol Cell. 1992;3:181–8. doi: 10.1091/mbc.3.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawler J. The functions of thrombospondin-1 and-2. Curr Opin Cell Biol. 2000;12:634–40. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 24.Ribeiro SM, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE, et al. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J Biol Chem. 1999;274:13586–93. doi: 10.1074/jbc.274.19.13586. [DOI] [PubMed] [Google Scholar]
- 25.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 26.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, et al. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–92. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel MK, Lymn JS, Clunn GF, Hughes AD. Thrombospondin-1 is a potent mitogen and chemoattractant for human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:2107–14. doi: 10.1161/01.atv.17.10.2107. [DOI] [PubMed] [Google Scholar]
- 28.Bauer PM, Bauer EM, Rogers NM, Yao M, Feijoo-Cuaresma M, Pilewski JM, et al. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc Res. 2012;93:682–93. doi: 10.1093/cvr/cvr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwapiszewska G, Wilhelm J, Wolff S, Laumanns I, Koenig IR, Ziegler A, et al. Expression profiling of laser-microdissected intrapulmonary arteries in hypoxia-induced pulmonary hypertension. Respir Res. 2005;6:109. doi: 10.1186/1465-9921-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ochoa CD, Yu L, Al-Ansari E, Hales CA, Quinn DA. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J Cardiothorac Surg. 2010;5:32. doi: 10.1186/1749-8090-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, et al. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–90. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–9. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- 33.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–84. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 34.Kim EK, Lee JH, Oh YM, Lee YS, Lee SD. Rosiglitazone attenuates hypoxia-induced pulmonary arterial hypertension in rats. Respirology. 2010;15:659–68. doi: 10.1111/j.1440-1843.2010.01756.x. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, et al. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi. 2005;43:283–8. [PubMed] [Google Scholar]
- 36.Fu M, Zhang J, Zhu X, Myles DE, Willson TM, Liu X, et al. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J Biol Chem. 2001;276:45888–94. doi: 10.1074/jbc.M105490200. [DOI] [PubMed] [Google Scholar]
- 37.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic Biol Med. 2010;48:1618–25. doi: 10.1016/j.freeradbiomed.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 39.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 40.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, et al. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–99. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Dai A, Li Q, Hu R. Hypoxia induces transforming growth factor-beta1 gene expression in the pulmonary artery of rats via hypoxia-inducible factor-1alpha. Acta Biochim Biophys Sin (Shanghai) 2007;39:73–80. doi: 10.1111/j.1745-7270.2007.00249.x. [DOI] [PubMed] [Google Scholar]
- 42.Stouffer GA, Hu Z, Sajid M, Li H, Jin G, Nakada MT, et al. Beta3 integrins are upregulated after vascular injury and modulate thrombospondin- and thrombin-induced proliferation of cultured smooth muscle cells. Circulation. 1998;97:907–15. doi: 10.1161/01.cir.97.9.907. [DOI] [PubMed] [Google Scholar]
- 43.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 44.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK, et al. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–51. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–83. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 47.Griendling KK, Ushio-Fukai M. Redox control of vascular smooth muscle proliferation. J Lab Clin Med. 1998;132:9–15. doi: 10.1016/s0022-2143(98)90019-1. [DOI] [PubMed] [Google Scholar]
- 48.Majack RA, Majesky MW, Goodman LV. Role of PDGF-A expression in the control of vascular smooth muscle cell growth by transforming growth factor-beta. J Cell Biol. 1990;111:239–47. doi: 10.1083/jcb.111.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majack RA, Goodman LV, Dixit VM. Cell surface thrombospondin is functionally essential for vascular smooth muscle cell proliferation. J Cell Biol. 1988;106:415–22. doi: 10.1083/jcb.106.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sajid M, Lele M, Stouffer GA. Autocrine thrombospondin partially mediates TGF-beta1- induced proliferation of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2000;279:H2159–65. doi: 10.1152/ajpheart.2000.279.5.H2159. [DOI] [PubMed] [Google Scholar]
- 51.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARgamma ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2011;301:L881–91. doi: 10.1152/ajplung.00195.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]