

Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease share similar clinical features and mechanisms; very rarely, the two diseases coexist in the same patient. This case report presents such a patient, who was first diagnosed with Hand-Schüller-Christian disease (HSC), a type of LCH.
Keywords: Langerhans cell histiocytosis, Erdheim-Chester disease, Hand-Schüller-Christian disease, Central diabetes insipidus
CME Learning Objectives
Distinguish Erdheim-Chester disease from Langerhans cell histiocytosis.
Cite the keys to diagnosis of Hand-Schüller-Christian disease in a patient with only central diabetes insipidus.
List the signs linking a Hand-Schüller-Christian disease patient to coexisting ECD.
Abstract
Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) share similar clinical features and mechanisms. In very rare circumstances, the two diseases coexist in the same patient. Here we report such a patient, who was first diagnosed with Hand-Schüller-Christian disease (HSC), a type of LCH. Several years later, the patient presented with severe exophthalmos and osteosclerosis on radiograph. New biopsy revealed ECD. We also analyze 54 cases of LCH and 6 cases of ECD diagnosed in our hospital, as well as their progression during a follow-up period of 8 years. In five cases of HSC (9.3% of LCH), a triad of central diabetes insipidus, hyperprolactinemia, and pituitary stalk thickening on magnetic resonance imaging (MRI) preceded the typical bone lesions by 4–9 years. In addition, LCH was featured as elevated plasma alkaline phosphatase (ALP), which was normal in ECD. Combined with a literature review, several features are summarized to differentiate ECD from HSC. In patients with diabetes insipidus, concomitant hyperprolactinemia and pituitary stalk thickening on MRI indicate a possible HSC. Additionally, if osteosclerosis is observed in a patient with LCH, the coexistence of ECD should be considered.
Implications for Practice:
Central diabetes insipitus (CDI) is usually the first or one of the first symptoms of Hand-Schüller-Christian disease (HSC). It is difficult to determine whether CDI is part of HSC at its onset. We propose a new triad of symptoms including central diabetes insipitus, hyperprolactinemia, and pituitary stalk thickening on MRI. If a patient is present with the triad, HSC should be considered. Bone scans are very useful to reveal HSC in the absence of bone pain. Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) are featured with osteolytic lesions and osteosclerosis, respectively. If osteosclerosis is observed in a patient with LCH, coexistence of ECD should be considered. A new biopsy is helpful for the diagnosis.
Introduction
Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD), a non-Langerhans form of histiocytosis, have similar clinical presentation. Both of them are very rare and the incidence of ECD is even lower. LCH typically affects children [1]. In contrast, the onset of ECD usually is seen in middle-aged adults [2].
LCH originates from clonal proliferation of Langerhans cells, which are specialized dendritic cells [3]. Langerhans cells are usually located in skin and mucosa [4]. In LCH, the cells can be found in other tissues along with eosinophils and lymphocytes [5]. Thus, the disease should be considered hematological. However, the main form of LCH, also called eosinophilic granuloma, only affects bones [6]. Patients often are admitted to the orthopedic department because of bone pain and enucleation of bone tumor is performed. LCH is often diagnosed based on postoperative histopathological examination [7]. Unlike eosinophilic granuloma, another form of LCH called Hand-Schüller-Christian disease (HSC) includes diabetes insipidus, exophthalmos, and lytic bone lesions—known as the HSC triad [8].
ECD is characterized by an infiltration of lipid-laden macrophages and multinucleated giant cells [9]. It can be difficult to distinguish ECD from LCH because they share the same symptoms. However, radiography may demonstrate the osteolytic lesions and osteosclerosis that are present with LCH and ECD, respectively. Postoperative histopathological examination can establish the final diagnosis.
In very rare circumstances, LCH and ECD occur in the same patient. In this report, one such case is described in detail. In addition, all cases of LCH and ECD diagnosed in the past 8 years in our hospital are reviewed. A triad of diabetes insipidus, hyperprolactinemia and pituitary stalk thickening shown on magnetic resonance imaging (MRI) are highlighted in the discussion.
Case Report
A previously healthy 27-year-old woman presented to the endocrinology service with severe polyuria and extreme thirst. Additionally, she had suffered from menopause and mild galactorrhea for several months. A water deprivation and desmopressin test revealed central diabetes insipidus (CDI). She was also diagnosed with hyperprolactinemia and MRI showed pituitary stalk thickening (Fig. 1A). Vasopressin tannate and bromocriptine were prescribed. However, bromocriptine was abandoned 1 year later because of severe gastrointestinal side effects, such as nausea and vomiting. Four years later, the patient was admitted with a new-onset scalp mass. Neuroimaging revealed that the lesion invaded the occipital bone. The scalp mass and involved skull were excised. Microscopic examination indicated LCH. Because the patient also suffered from CDI, diagnosis of HSC was established.
Figure 1.

Exophthalmos and skull images. (A): Pituitary stalk thickening on magnetic resonance imaging (arrow). (B): Orbit computed tomography (CT) showed a granuloma (arrow) in the left retrobulbar space, and bilateral thickening of rectus muscles. (C): Diffuse bone destruction and hyperosteogeny in the skull CT. (D, E): The patient suffered from severe exophthalmos (left, 29 mm; right, 24 mm). Bilateral periorbital xanthomas accompanied the exophthalmos.
Five years after diagnosis of HSC, the patient was admitted again with upper left thigh pain. Plain radiography revealed a lesion located in the upper fourth of the left femur. A bone tumor was enucleated and a steel plate was implanted. One year later, the implanted steel plate was removed. However, a spontaneous fracture of the left femur occurred 1 month after removal. A technetium-99-m bone scan demonstrated abnormal enhanced radionuclide uptake in both femurs. Abnormal uptake was also seen in the other regions of skeleton. Debridement and bone graft substitute was performed. LCH was confirmed in the postoperative histopathological examination. The patient then received 6MV X-ray for radiotherapy with DT 30Gy/15F/3W in left pelvis and both femurs.
While the patient suffered from left thigh pain, bilateral periorbital xanthomas emerged. Both eyes began to bulge slightly. The exophthalmos rapidly deteriorated in the past 3 years. Redness was common and the eyelids failed to close. In addition, the patient suffered from periodic fevers as high as 39°C to 40°C in the past year. The fever usually lasted for 24 hours and vanished without any medications. The frequency of fever increased gradually from once a month to once a week.
Seven months ago, the woman, now 41 years old, presented to the endocrinology service again with severe exophthalmos (Fig. 1D, 1E). Exophthalmometry reading was 29 mm in the left eye and 24 mm in the right eye. Thyroid function tests showed primary hypothyroidism evidenced by the decrease of FT3 and FT4 as well as an increase of thyroid-stimulating hormone. However, negative autoantibodies and normal thyroid ultrasound appearance indicated the hypothyroidism was most likely not due to Hashimoto thyroiditis.
Ophthalmic ultrasound found one hypoechoic area measuring 30×22 mm in the left retrobulbar space and one of 14×14 mm in the right retrobulbar space with blood flow signals and undefined boundary. Bilateral thickening of the rectus muscles and vitreous opacities were also observed. Orbit computed tomography (CT) scan found similar results (Fig. 1B). Diffuse bone destruction and hyperosteogeny appeared in the skull CT image (Fig. 1C). Eroded edge, diffuse bone destruction, osteosclerosis, and ground-glass-like hyperosteogeny were viewed clearly on radiograph (Fig. 2A), computer reconstructed sagittal CT (Fig. 2B), and three-dimensional CT (Fig. 2C) images of the right femur. Chest radiograph (Fig. 2D) and CT (Fig. 2E) revealed extensive pulmonary fibrosis and thickening of bilateral pleura including right interlobar fissure.
Figure 2.

Eroded edge, diffuse bone destruction, osteosclerosis, and ground-glass-like hyperosteogeny were viewed clearly on plain radiograph (A), computer reconstructed sagittal computed tomography (CT) (B), and three-dimensional CT (C) images of right femur. Chest radiograph (D) and CT (E) revealed extensive pulmonary fibrosis and thickening of bilateral pleura, including right interlobar fissure.
Because osteosclerosis and hyperosteogeny are not characteristics of LCH, further investigation was conducted. Biopsy of the left retrobulbar mass was performed and the result of immunohistochemistry confirmed ECD. Radiotherapy was delivered by 6MVX linear accelerator with DT 2880cGy/16Fx+/3w+ in left retrobulbar mass. At the end of radiotherapy, the mass decreased in size. Currently, the patient is being treated with interferon-α, which has proven to be effective. Her chest CT images indicate that the lung has improved after interferon-α treatment.
Immunohistochemistry
Multiple immunohistochemistry examinations were performed. Initially, microscopic examination of the scalp mass showed the cells of the infiltrate exhibiting the characteristic morphologic features of Langerhans cells with indistinct cytoplasmic borders, indented vesicular nuclei, and a histiocytic appearance (Fig. 3A). Numerous eosinophils were also seen. Immunostaining revealed many strongly positive Langerhans cells for the CD1α (Fig. 3B), Langerin (Fig. 3C), CD68, S-100, and PGM-1 antibodies. Thus, LCH was diagnosed and the pathological findings were confirmed later in the postoperative histopathological examination of left femur tumor.
Figure 3.

Immunohistochemistry. (A): Hematoxylin and eosin (HE) staining of the scalp mass showed Langerhans cells (yellow arrows) were distributed in clusters with eosinophils (blue arrows) infiltration. Immunostaining revealed CD1α (B) and Langerin positive Langerhans cells (brown color) (C). HE staining of the retrobulbar mass showed foamy histiocytes nested in fibrosis (D). Immunostaining revealed CD68+ (E) and Lyso+ (F).
Abbreviation: HE, hematoxylin and eosin.
Histopathological examination of the left retrobulbar mass showed xanthogranulomatous infiltration by foamy histiocytes and multinucleated giant cells nested in fibrosis (Fig. 3D). Immunostaining revealed CD68+ (Fig. 3E), Lyso+ (Fig. 3F), PGM-1+, CD163+, CD1α-, Langerin-, CD21-, CD35-, which were consistent with ECD.
An 8-Year Follow-Up Study
In the past 8 years, 54 patients were diagnosed with LCH and 6 with ECD in our hospital, respectively. All cases have been followed up until now. Among those with LCH, 9.3% (5 cases) were HSC. Part of the information and laboratory results are presented in Table 1.
Table 1.
Langerhans cell histiocytosis and Erdheim-Chester disease
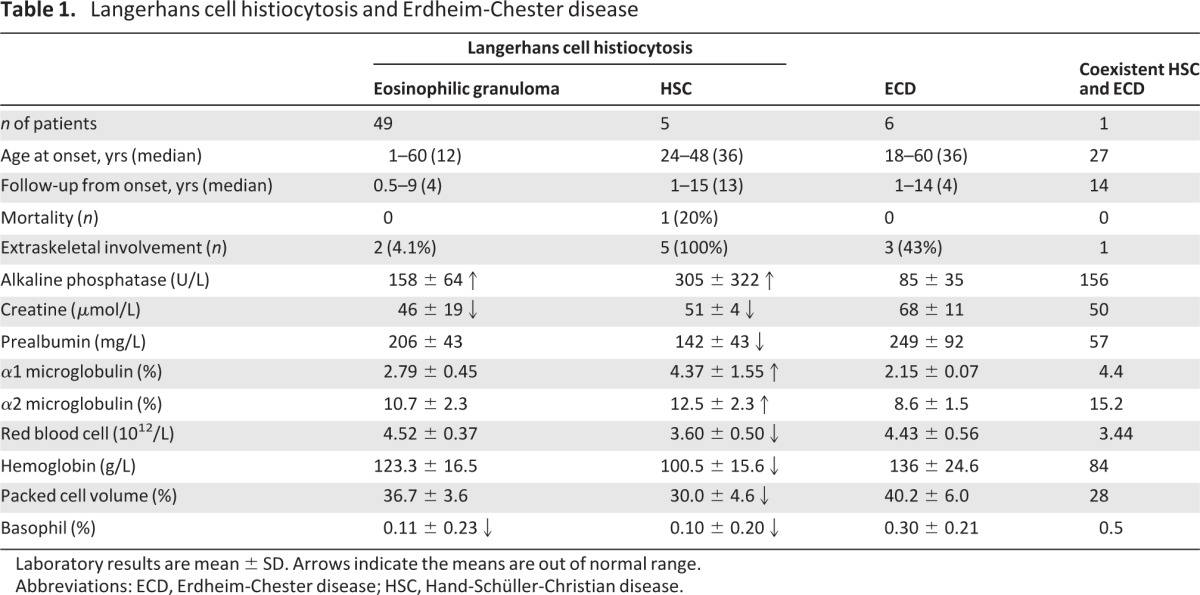
Laboratory results are mean ± SD. Arrows indicate the means are out of normal range.
Abbreviations: ECD, Erdheim-Chester disease; HSC, Hand-Schüller-Christian disease.
Except for two patients with lung involvement, the rest with eosinophilic granuloma had only bone involvement. Among them, 71.4% (35 cases) were unifocal and cured with surgery without relapse; 22.4% (11 cases) were multifocal. In addition to limbs, which were the most commonly affected sites (24 cases; 49%), almost every part of the skeleton was involved, except hands and feet: spine (9; 18.4%), skull including tympana (9; 18.4%), pelvis (7; 14.3%), and chest including rib, sternum, clavicle, and scapula (8; 16.3%).
HSC can be quite easily distinguished from other types of LCH in terms of clinical presentation. CDI was the first manifestation in all five cases of HSC. In the early stage, patients presented with polydipsia, polyuria, and pituitary stalk thickening on MRI. In addition, the prolactin level was significantly elevated with the onset of HSC, and an initial triad of CDI, hyperprolactinemia and pituitary stalk thickening on MRI preceded typical bone lesions of HSC by 4–9 years. One case was diagnosed with CDI originally but MRI investigation revealed pituitary stalk thickening and elevated plasma prolactin was observed. We speculated the patient probably suffered from HSC, so a bone scan was performed. The images showed increased radionuclide uptake in the right zygoma, temporal bone, mandible, thoracic vertebra, and ribs. Eventually, he was diagnosed with HSC. This case suggests the initial triad as described previously is very useful in diagnosis of HSC in the absence of bone pain.
The incidence of ECD is comparable to that of HSC in this study. Based on our observation, there are few differences between LCH and ECD with regard to clinical manifestations and outcomes. Zero deaths from ECD were recorded in the past 8 years. All of the six ECD cases in our hospital were diagnosed based on histoimmunostaining after enucleation of bone tumor in the lower limb. Among them, unifocal involvement was observed in three patients, who were cured after surgery. Although multisystem involvement was observed in the other three patients, the progress is very slow and so far all of them have a normal life after enucleation of bone tumor.
The fatality rates from LCH and ECD were quite low in our cases. Only a 43-year-old woman with HSC died after chemotherapy. She suffered from severe oral lesions and followed only a liquid diet in the last year of her life. Currently, most of the other patients are living well. Interestingly, although symptoms and outcomes are very similar between LCH and ECD, routine laboratory tests turned out differently (Table 1). Typical ECD is associated with normal laboratory results. In contrast, patients with LCH (especially HSC) tend to have elevated alkaline phosphatase, α1 and α2 microglobulin levels, and mild anemia.
Discussion
In general, LCH is considered to be a pediatric disease [10]. However, it also can occur in adults. In this study, 15 (30.6%) patients with eosinophilic granuloma were more than 18 years old at the onset, with the oldest being 60 years of age. For HSC cases, all five patients were adults.
It is known that HSC features a triad of CDI, exophthalmos, and lytic bone lesions [11, 12]. Among the triad, CDI may be the most important component because it was the first manifestation in our five cases of HSC and preceded other manifestations of HSC by years. Other groups also had similar observations of CDI as the initial presentation [13–17]. With isolated CDI, it is usually hard to trace the etiology; however, it is important for a clinician determine whether a CDI is part of HSC. In this study, we found hyperprolactinemia to be another sign of HSC. For a patient presenting with CDI, pituitary MRI and a blood prolactin test may be used for differential diagnosis. If pituitary stalk thickening and hyperprolactinemia are observed in the patient, HSC should be considered and a bone scan is necessary. In early stages of HSC, radionuclide uptake of multiple sites already increases in bone scintigraphy despite the lack of pain, which is useful to establish the diagnosis of HSC.
Pituitary stalk thickening is one of the important signs of HSC. Thickening of the pituitary stalk is often seen on MRI of brain tumors such as craniopharyngioma, germinoma, and metastases (including lymphoma) [13]. The imaging of germinoma sometimes mimics that of HSC. In a case report, a thickened pituitary stalk suggested LCH, which later was revealed to be a germinoma [14]. Although HSC and germinoma share similar clinical features, such as CDI and pituitary stalk thickening, skull images are different. HSC is characterized as osteolysis, which is absent in germinoma.
Osteolysis is a term used to define bone loss associated with bone resorption and osteopenia. Bone erosions of different shapes are obvious on radiograph. In contrast, osteosclerosis indicates an elevation in bone density. Radiograph shows an area of increased opacity, which results from more mineral in the bone absorbing or deflecting the x-ray beam. Osteolysis and osteosclerosis are common characteristics of LCH and ECD, respectively.
In this study, few differences were observed in prognosis between LCH and ECD, which is different from previous reports indicating that ECD has a poorer outcome than LCH [15, 16]. It has been reported that approximately 60% of patients die within 32 months of ECD onset [16], despite that the survival is significantly improved with interferon-α treatment [17, 18]. However, all of our patients with ECD are in good condition after surgery. The discrepancy may be due to ethnic background; this is the first systemic report in Chinese people. Also, although symmetrical bone involvement has been noted for ECD, unifocal involvement was observed in three cases in this study; this discrepancy may be due to a lack of symptoms and therefore lack of radiography on the other side of skeleton.
Over the past 30 years, 121 cases of HSC were reported in China. The median (average) age of onset was 4.5 (11.1) years old. The ratio of male to female patients was 2.1:1. CDI was the first sign of the disease in 25% of the patients. Spontaneous recovery occurred in three cases. Compared to the abundant information in HSC, ECD was sparse in the Chinese literature. Until now, only four cases of ECD were reported, all in adults. Taking into account that we had six cases of ECD diagnosed in our hospital, it is speculated that there are patients with ECD being misdiagnosed elsewhere. Considering that the majority of physicians are unaware of the disease in China, education about ECD is essential.
We observed that plasma ALP level increased markedly in LCH, particularly the HSC. ALP is abundant in bone and elevated in many bone diseases, such as Paget disease, which features osteolytic lesions like LCH [19]. We propose the increase of ALP may result from bone destruction in addition to other proposed mechanisms of hepatomegaly or hepatic involvement [10, 20]. In contrast, ALP level is normal in ECD, probably because the characteristic of ECD is osteosclerosis rather than osteolysis.
Until now, 13 cases of coexistent LCH and ECD have been reported worldwide [21, 22]. Coexistence has occurred exclusively in adults so far. An in vitro study demonstrated that the dendritic/Langerhans progenitors in bone marrow had a capacity for both macrophage and dendritic cell differentiation [23]. The coexistence of LCH and ECD (or LCH evolving to ECD) may be due to different microenvironments or microenvironment alterations in the same patient. Interestingly, radiograph and CT results of the case highlighted here were consistent with ECD. However, the laboratory results indicated HSC (Table 1). The typical radiographic feature of ECD is characterized by symmetric sclerosis of long bones [24]. Therefore, when osteosclerosis is observed in a patient with LCH patient, as in this case, coexistence of ECD should be considered and a new biopsy is necessary.
Although the molecular mechanism of LCH and ECD remains unknown, high prevalence of BRAF V600E mutations was reported in both LCH and ECD [25–27]. This indicates potential anti-BRAF targeted therapy for these diseases. Since 2011, vemurafenib, a BRAF enzyme inhibitor (with the name coming from V600E mutated BRAF inhibition) [28], has been approved for the treatment of late-stage melanoma with the mutation by U.S. Food and Drug Administration, Health Canada, and the European Commission. Positive results from clinical trials of vemurafenib on melanoma suggest anti-BRAF targeted therapy is plausible in the treatment of LCH and ECD. New clinical trials on vemurafenib or other agents targeting V600E-mutated BRAF for LCH and ECD are necessary.
Conclusion
For patients with HSC, CDI, hyperprolactinemia, and pituitary stalk thickening on MRI usually precede skeletal symptoms. The triad can be used to screen for HSC in patients with CDI. Bone scan is a very useful examination to reveal the disease. Additionally, if osteosclerosis appears in a patient with HSC, the coexistence of ECD should be considered and further investigation is needed.
Acknowledgments
This work was supported by a grant from the Shanghai Pujiang Program (11PJ1407700 to J.Y.) and a National 973 Program of China grant (2011CB504001 to W.J.).
Footnotes
Editor's Note: See the accompanying commentary on pages 2–4 of this issue.
Author Contributions
Conception and design: Jun Yin, Feng Zhang, Weiping Jia
Provision of study materials or patients: Jun Yin, Feng Zhang, Huizhen Zhang, Qing Li, Shundong Hu, Qinghua Tian, Yuqian Bao, Weiping Jia
Collection and/or assembly of data: Jun Yin, Feng Zhang, Huizhen Zhang, Qing Li, Yuqian Bao
Data analysis and interpretation: Jun Yin, Huizhen Zhang, Li Shen, Shundong Hu, Qinghua Tian, Yuqian Bao
Manuscript writing: Jun Yin, Li Shen
Final approval of manuscript: Jun Yin, Feng Zhang, Huizhen Zhang, Li Shen, Qing Li, Shundong Hu, Qinghua Tian, Yuqian Bao, Weiping Jia
Disclosures
The authors indicated no financial relationships.
Section Editors: Fred Hirsch: coinventor of a patent for EGFR FISH as a predictive biomarker for EGFR inhibitors, licensed to Abbott; OSI/Genentech/Roche, Pfizer, Boehringer Ingelheim, BMS, Celgene, Merck Serono, Imclone/Lilly (C/A); AstraZeneca, Syndax, OSI, Genentech, Merck, Imclone (RF); Jeffrey Ross: leadership position with Foundation Medicine; stock held in Foundation Medicine and Syfr; Genentech/Roche, Bristol-Myers Squibb, Boehringer-Ingelheim (H), Foundation Medicine (RF); Foundation Medicine, Affiliated Pathology Services at Albany Medical Center (E)
Reviewer “A”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Broadbent V, Gadner H, Komp DM, et al. Histiocytosis syndromes in children: II. Approach to the clinical and laboratory evaluation of children with langerhans cell histiocytosis. Med Pediatr Oncol. 1989;17:492–495. doi: 10.1002/mpo.2950170527. [DOI] [PubMed] [Google Scholar]
- 2.Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim-Chester disease. Curr Opin Rheumatol. 2012;24:90–96. doi: 10.1097/BOR.0b013e32834db53e. [DOI] [PubMed] [Google Scholar]
- 3.Orii T, Takeda H, Kawata S, et al. Differential immunophenotypic analysis of dendritic cell tumours. J Clin Pathol. 2010;63:497–503. doi: 10.1136/jcp.2009.067819. [DOI] [PubMed] [Google Scholar]
- 4.Novak N, Gros E, Bieber T, et al. Human skin and oral mucosal dendritic cells as ‘good guys’ and ‘bad guys’ in allergic immune responses. Clin Exp Immunol. 2010;161:28–33. doi: 10.1111/j.1365-2249.2010.04162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singhi AD, Montgomery EA. Gastrointestinal tract Langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol. 2011;35:305–310. doi: 10.1097/PAS.0b013e31820654e4. [DOI] [PubMed] [Google Scholar]
- 6.Love FM, Fashena GJ. Eosinophilic granuloma of bone and hand-schuller-christian disease. J Pediatr. 1948;32:46–54. doi: 10.1016/s0022-3476(48)80129-0. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Wu X, Xi ZJ. Langerhans cell histiocytosis of bone in children: A clinicopathologic study of 108 cases. World J Pediatr. 2010;6:255–259. doi: 10.1007/s12519-010-0205-0. [DOI] [PubMed] [Google Scholar]
- 8.Kimura T, Ota K, Shoji M, et al. Hand-Schuller-Christian disease with occult diabetes insipidus, cardiac failure and renal dysfunction. Jpn J Med. 1990;29:405–410. doi: 10.2169/internalmedicine1962.29.405. [DOI] [PubMed] [Google Scholar]
- 9.Haroche J, Arnaud L, Amoura Z. Erdheim-Chester Disease. Curr Opin Rheumatol. 2012;24:53–59. doi: 10.1097/BOR.0b013e32834d861d. [DOI] [PubMed] [Google Scholar]
- 10.Bansal D, Marwaha RK, Trehan A, et al. Langerhans' cell histiocytosis: Experience from a single center. Indian Pediatr. 2008;45:685–688. [PubMed] [Google Scholar]
- 11.Lichtenstein L. Histiocytosis X (eosinophilic granuloma of bone, letterer-siwe disease, and schueller-christian disease) Further observations of pathological and clinical importance J Bone Joint Surg Am. 1964;46:76–90. [PubMed] [Google Scholar]
- 12.Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans' cell histiocytosis (Hand-Schuller-Christian disease) in an adult: A case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261:326–330. doi: 10.1007/s00405-003-0690-z. [DOI] [PubMed] [Google Scholar]
- 13.Rupp D, Molitch M. Pituitary stalk lesions. Curr Opin Endocrinol Diabetes Obes. 2008;15:339–345. doi: 10.1097/MED.0b013e3283050844. [DOI] [PubMed] [Google Scholar]
- 14.Prosch H, Grois N, Prayer D, et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2004;43:594–599. doi: 10.1002/pbc.20102. [DOI] [PubMed] [Google Scholar]
- 15.Andrysek K. Erdheim-Chester disease: A case study and literature review. Dimens Crit Care Nurs. 2011;30:184–189. doi: 10.1097/DCC.0b013e31821b7dff. [DOI] [PubMed] [Google Scholar]
- 16.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75:157–169. doi: 10.1097/00005792-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: A multicenter survival analysis of 53 patients. Blood. 2011;117:2778–2782. doi: 10.1182/blood-2010-06-294108. [DOI] [PubMed] [Google Scholar]
- 18.Braiteh F, Boxrud C, Esmaeli B, et al. Successful treatment of Erdheim-Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood. 2005;106:2992–2994. doi: 10.1182/blood-2005-06-2238. [DOI] [PubMed] [Google Scholar]
- 19.Cortis K, Micallef K, Mizzi A. Imaging Paget's disease of bone—from head to toe. Clin Radiol. 2011;66:662–672. doi: 10.1016/j.crad.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Braier J, Ciocca M, Latella A, et al. Cholestasis, sclerosing cholangitis, and liver transplantation in langerhans cell histiocytosis. Med Pediatr Oncol. 2002;38:178–182. doi: 10.1002/mpo.1306. [DOI] [PubMed] [Google Scholar]
- 21.Brower AC, Worsham GF, Dudley AH. Erdheim-Chester disease: A distinct lipoidosis or part of the spectrum of histiocytosis? Radiology. 1984;151:35–38. doi: 10.1148/radiology.151.1.6608118. [DOI] [PubMed] [Google Scholar]
- 22.Pineles SL, Liu GT, Acebes X, et al. Presence of Erdheim-Chester disease and Langerhans cell histiocytosis in the same patient: A report of 2 cases. J Neuroophthalmol. 2011;31:217–223. doi: 10.1097/WNO.0b013e31820a204e. [DOI] [PubMed] [Google Scholar]
- 23.Reid CD, Stackpoole A, Meager A, et al. Interactions of tumor necrosis factor with granulocyte-macrophage colony-stimulating factor and other cytokines in the regulation of dendritic cell growth in vitro from early bipotent cd34+ progenitors in human bone marrow. J Immunol. 1992;149:2681–2688. [PubMed] [Google Scholar]
- 24.Balink H, Hemmelder MH, de Graaf W, et al. Scintigraphic diagnosis of Erdheim-Chester disease. J Clin Oncol. 2011;29:e470–472. doi: 10.1200/JCO.2010.34.0307. [DOI] [PubMed] [Google Scholar]
- 25.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF v600e mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–2703. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 27.Sahm F, Capper D, Preusser M, et al. BRAFv600e mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012;120:e28–e34. doi: 10.1182/blood-2012-06-429597. [DOI] [PubMed] [Google Scholar]
- 28.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a Raf inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]