

Abstract
Purpose
Human immunodeficiency virus-1 (HIV)-associated neurocognitive disorder (HAND) is a neurodegenerative disease for which there is no available neuroprotective therapy. Viral proteins, such as Tat, have been implicated as agents of neurotoxicity via multiple mechanisms, including effects by directly binding to the NMDA receptor. We evaluated ability of the immune response against Tat to modulate neurotoxicity at glutamate receptors.
Methods
Neurotoxicity was measured in primary neuronal-glial cultures and in hippocampal slice cultures. We used immunoprecipitation experiments to demonstrate interaction between Tat, NMDA receptor, and anti-Tat antibody. Using known structures of Tat and NMDA receptors, we developed a model of their interactions.
Results
Antibodies to Tat attenuated Tat-mediated neurotoxicity. Interestingly, Tat immune complexes also blocked neurotoxicity caused by NMDA receptor agonists but not kainate/AMPA receptor agonists. Neither Tat nor antibody alone blocked the excitotoxic effect, nor did an unrelated antigen-antibody complex. The protective effect of the Tat immune complexes was also lost when Tat was modified by nitrosylation or by using a deletion mutant of Tat.
Conclusions
The ability of viral immune complexes to interact with NMDA receptors and prevent excitotoxicity represents a novel host defense mechanism. Host immune responses may influence host susceptibility to various effects of viral proteins, modulating HIV complications, such as onset of HAND. These observations provide rationale for development of vaccine therapies targeting Tat for prevention of HAND.
Keywords: neurotoxicity, neurovirology, dementia, neuroprotection, glutamate, neutralizing antibodies
INTRODUCTION
Highly active antiretroviral therapy (HAART) has significantly reduced the incidence of severe human immunodeficiency virus (HIV)-associated neurocognitive disorder (HAND). However, mild forms persist and nearly 50% of patients have neurocognitive dysfunction despite adequate viral suppression (Heaton et al., 2011). HIV encephalitis is still seen on autopsy in HAART-treated patients, suggesting that HAART is ineffective in eliminating central nervous system (CNS) infection (Anthony et al., 2005; Langford et al., 2003).
Viral proteins, such as Tat and gp120, have been implicated in the neurotoxicity of HAND by multiple mechanisms, prominent among which is production of glutamate excitotoxicity. Tat’s production is not impacted by available antiretroviral drugs once proviral DNA has been formed. Tat is a potent excitotoxin and is known to stimulate NMDA receptors via direct cysteine-cysteine interactions with the extracellular domains of the receptor (Li et al., 2008; Prendergast et al., 2002). Tat also promotes the phosphorylation of the NMDA receptor leading to its further stimulation (Haughey et al., 2001). Tat sensitizes neurons so that normally physiological levels of glutamate cause significant excitotoxicity and massive derangement in intracellular calcium (Nath et al., 2000) and intra-hippocampal Tat administration in rats promotes learning deficits following alcohol withdrawal that is NMDA receptor-dependent (Self et al., 2009).
While investigating the interactions between Tat and the NMDA receptor, we discovered that some anti-Tat antibodies not only block the neurotoxicity of Tat but also block toxicity of other excitotoxins.
MATERIALS AND METHODS
Tat
Recombinant Tat1–72, Tat1–101, and mutant TatΔ31–61 was produced in our laboratory. Details of Tat production and purification have been published (Ma and Nath, 1997; Turchan et al., 2001). It was greater than 99% pure as analyzed by HPLC and silver stained gel electrophoresis. Each batch is monitored for purity by Western blot analysis, for endotoxin contamination by Litmus amebocyte lysate assay (Associates of Cape Cod, Inc.), and for bioactivity by LTR-CAT assay. Tat was nitrosylated as described (Jaffrey and Snyder, 2001).
Anti-Tat Antibodies
Mouse monoclonal antibody3 against the N-terminal of Tat was obtained from the NIBSC Centre for AIDS Reagents and also produced in our own laboratory (cat#ARP352, hybridoma code TR1)(Ruckwardt et al., 2004; Tikhonov et al., 2003). The mAb against the C-terminal of Tat was produced in our laboratory with a hybridoma obtained from the NIH AIDS Repository (cat#7383). Affinity-purified rabbit anti-Tat polyclonal antibodies4 to the C-terminus, N-terminus, and whole Tat were generated in our laboratory.
Other Reagents
NMDA, kainate, and kynurenic acid were from Sigma. p24 was obtained from NIH AIDS Repository. Anti-p24 was from NIBSC Centre for AIDS Reagents. Anti-NR1 was obtained from BD Biosciences, anti-NR2A from Molecular Probes, anti-NR2B from Upstate, anti-GluR1–4 from Chemicon or R&D.
Neuronal Cultures
Mixed neuronal-glial cultures were prepared, as described (Magnuson et al., 1995), from human fetal brain specimens of 12–15 weeks’ gestational age, with consent from women undergoing elective termination of pregnancy, as approved by Johns Hopkins University Institutional Review Board. Cells were maintained in tissue culture flasks for at least four weeks then plated in 96 well plates for 3–7 days before use in neurotoxicity assays. These cells contained approximately 70% neurons as determined by immunostaining for microtubule associated protein-2, 30% astrocytes by immunostaining for glial fibrillary acidic protein, and 1% microglia by immunostaining for CD68.
Cortical neuronal cultures were prepared from embryonic day 17 or 18 Sprague Dawley rats, as described (Caporello et al., 2006). Immunofluorescent staining for MAP-2 shows that these cultures are >98% neurons; the remainder of cells are predominantly astrocytes.
Rat hippocampal slice cultures were prepared as described (Stoppini et al., 1991), from eight-day old male and female Sprague Dawley rat pups (Harlan Laboratories; Indianapolis, IN). Slices were allowed five days to attach to the insert membrane before any experiments were conducted.
Care of animals was in accordance with NIH Guide for the Care and Use of Laboratory Animals, as well as University of Kentucky’s and Johns Hopkins University’s Institutional Animal Care and Use Committees, and in accordance with EU Directive 2010/63/EU.
Cytotoxicity Assays
Mitochondrial assay
At the time of performing the experiment, culture medium was replaced by fresh OptiMEM (without phenol red). Mitochondrial potential was monitored in the cultures by using 5,5′6,6-tetrachloro-1,1′3,3′-tetraethylbenzimidazole carbocyanide iodide5 dye (Molecular Probes) as we have described(Turchan et al., 2001). We performed this assay at 15–18 hours after exposure to experimental agents, a time period we have found to have optimal kinetics for consistent measurements. Results were normalized so that control mean values were equal to 100. The means +/− standard error of means were calculated and data analyzed by one way ANOVA and Neumann-Keuls post-test.
Propidium Iodide Staining of Hippocampal Slice Cultures
At 5 days in vitro, male and female rat hippocampal slice cultures were transferred into new culture plates containing 1 ml of culture medium on the bottom with the addition of the Tat1–72 (200nM) or Tat plus the monoclonal antibody against the N-terminal of Tat (1:200 dilution, 5ng/ul) in culture medium. For experiments using NMDA, cultures were exposed to Tat+anti-Tat for a 30 minute pre-incubation period, after which they were put into fresh media containing Tat+anti-Tat+20μM NMDA. Additional cultures were exposed only to 20μM NMDA. Culture medium contained the fluorescent marker propidium iodide6, which binds to nucleic acids when cell membrane integrity has been compromised. Fluorescent images to visualize cell death as indicated by PI uptake were taken at 24 hours of exposure to experimental agents. All experiments were replicated with separate rat litters.
Images were taken using SPOT Advanced version 4.0.2 software for Windows with a 5× objective on an inverted Leica DMIRB microscope fitted for fluorescence detection (mercury-arc lamp) and connected to a personal computer via a SPOT 7.2 color mosaic camera (W. Nuhsbaum Inc., McHenry, IL). Fluorescent intensity was analyzed by densitometry using Image J software (National Institutes of Health, Bethesda, MD) in the granule cell layer of DG and the pyramidal cell layers of CA3 and CA1. Background fluorescent intensity was subtracted prior to statistical analysis. All measurements were converted to percent control values. One-way analysis of variance7 was conducted within CA1, CA3, and DG. When appropriate, Fisher’s LSD post-hoc analyses were interpreted with significance level set at p<0.05.
RESULTS
Anti-Tat antibodies neutralize Tat neurotoxicity
We coincubated Tat with anti-Tat, and exposed neuronal cultures to the immune complexes. Antibodies against either C- or N-terminal of Tat attenuated neurotoxicity caused by Tat alone (p<0.05) (figure 1). Other antibodies against Tat, particularly rabbit polyclonal antibodies made against whole Tat, did not protect. An unrelated antibody (anti-p24) also showed no protection.
Figure 1. Attenuation of Tat neurotoxicity by a monoclonal antibody.

Mixed rat neuronal cultures were exposed to Tat1–72 and/or C-terminal anti-Tat monoclonal antibody as indicated. Mitochondrial membrane potential was measured 15–18 hours later. Tat alone caused toxicity versus untreated controls (p<0.001). When the antibody was incubated with Tat, it provided significant protection versus the toxicity seen with Tat alone (p<0.05). Anti-p24 provided no neuroprotection. Concentrations: Tat 200nM, antibody 5ng/μl. Data represents mean ± SEM of at least six independent experiments, analyzed by ANOVA with Neumann-Keuls post-test.
Hippocampal slice cultures have the advantage that the synaptic interactions of neurons and neuro-glial interactions are intact (Mulholland and Prendergast, 2003; Prendergast et al., 2004). Thus, to determine if the antibodies have similar protective properties in the setting of this more intact network, rat hippocampal slice cultures were used. We coincubated slice cultures with Tat and mAb against N-terminal of Tat, and measured neurotoxicity with PI staining after 24 hours of exposure. (Figure 2) depicts representative images, while (figure 3) quantitates results. In CA1, a main effect of treatment was present [F(2,157)=10.905, p<0.001]. Specifically, Tat produced 17% greater PI uptake compared to control cultures (p<0.001), which was attenuated by co-exposure to Tat and anti-Tat (p<0.05). A main effect was also present in CA3 [F(2,157)=8.870, p<0.001] and DG [F(2,157)=5.723, p<0.01], such that Tat produced significant toxicity above control (p<0.01), and co-exposure of Tat with anti-Tat did not produce PI uptake greater than control (p>0.2). Exposure to antibody alone produced no significant changes in PI uptake compared to controls.
Figure 2. Representative images of propidium iodide uptake in organotypic hippocampal slice cultures.
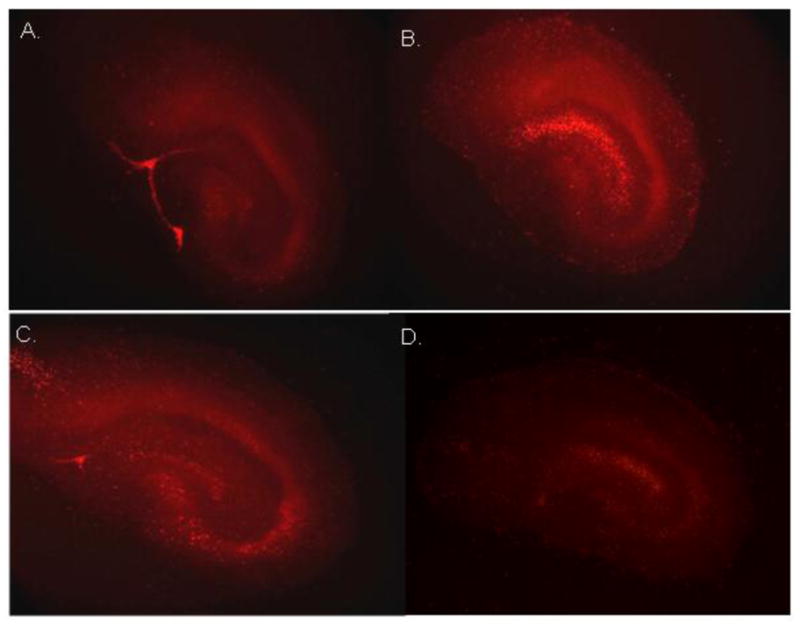
following 24 hr of exposure to: (A) control cell culture medium; (B) Tat 1–72 (200nM); and (C) Tat 1–72+anti-Tat antibody (5ng/μl); and (D) anti-Tat antibody (5 ng/μl)
Figure 3. Twenty-four hours of exposure to Tat 1–72 (200nM) produced significant injury (increased uptake of propidium iodide) in dentate gyrus granule cells, as well as, pyramidal cells of the CA3 and CA1 regions.

Co-exposure of slices to Tat 1–72 with C-terminal anti-Tat monoclonal antibody significantly attenuated Tat toxicity in the pyramidal cell layer of the CA1 hippocampal region, and reduced PI uptake compared to Tat alone in all three regions. All values were converted to percent control before analysis and graphing, and the dashed line indicates control values.*p<0.05 vs. control cultures; #p<0.05 vs. 200nM Tat.
It is important to determine if the Tat-immune complexes have the observed functions only in dissociated neuronal cultures or have the ability to penetrate tight extracellular spaces in vivo. These experiments thus provide in vivo relevance to these observations. Although the effect size is small in the hippocampal slice cultures, this is not unexpected because the Tat-immune complexes are large and would not penetrate the tissue as well as relatively small molecule toxins like Tat or NMDA. The consistent results with both primary dissociated neuronal cultures and hippocampal slice cultures supports the biological relevance of the neuroprotective effect of Tat-immune complexes.
Tat immune complexes attenuate NMDA-excitotoxity
Since peptides from N-terminal of Tat are non-toxic (Nath et al., 1996), and since antibodies against both N- and C-terminal were protective, we explored a more indirect neuroprotective function. We found the immune complex formed by Tat and C-terminal anti-Tat attenuated NMDA mediated excitotoxicity (p<0.01) (figure 4a). The protective effect of the immune complex was similar to that of kynurenic acid, a glutamate receptor antagonist (supplementary figure S1). However, the Tat-anti-Tat immune complex did not protect against excitotoxicity mediated by the AMPA agonist, kainate (figure 4b). Immune complexes formed by Tat and N-terminal mAb were also protective against NMDA (p<0.05) (supplementary figure S2). An unrelated immune complex (p24 and anti-p24) showed no protection (supplementary figure S3). Immune complexes made with rabbit polyclonal antibodies against whole Tat did not protect. Neither Tat nor antibody alone blocked NMDA excitotoxicity.
Figure 4. Modulation of NMDA excitotoxicity by a Tat immune complex.

Mixed rat neuronal cultures were exposed to Tat1–72, anti-Tat antibody, NMDA, and/or kainic acid. Mitochondrial membrane potential was measured 15–18 hours later. (A) NMDA alone caused toxicity versus untreated controls (p<0.001). Antibody alone demonstrated no protective effect against NMDA excitotoxicity, but the combination of Tat and anti-Tat antibody was neuroprotective against NMDA (p<0.01). (B) Kainic acid alone caused toxicity versus untreated controls (p<0.001), but neither the anti-Tat antibody alone nor the Tat immune complex demonstrated a protective effect against kainate-mediated excitotoxicity. Concentrations: Tat 200nM, antibody 5ng/μl, NMDA 125 μM, kainic acid 50 μM. Data represents mean ± SEM of at least four independent experiments, analyzed by ANOVA with Neumann-Keuls post-test.
We confirmed these results with slice cultures. (Figure 5) quantitates results from DG, CA1, and CA3. In all three areas (DG [F(2, 111)=19.590, p<0.001], CA3 [F(2, 111)=26.870, p<0.001], CA1 [F(2,110)=120.515, p<0.001]), NMDA produced significant toxicity compared to control cultures (p<0.001), and 30 minute pre-incubation with Tat+antiTat significantly attenuated NMDA toxicity (p<0.001). Tat immunocomplex may be less efficient in reverting NMDA-induced propidium uptake in CA1 when compared to DG or CA3. This may be due to differences in subtypes of NMDA receptors in these regions.
Figure 5. The Tat 1–72+anti-Tat antibody complex blocks NMDA toxicity in organotypic hippocampal cultures.

Cultures were pre-incubated for 30 minutes in culture media containing Tat 1–72 (200nM)+anti-Tat antibody before being transferred into media containing Tat 1–72+anti-Tat antibody+20μM NMDA. Cultures were imaged 24 hours after the initial exposure to NMDA. All values were converted to percent control before analysis and graphing, and the dashed line indicates control values. **p<0.001 vs. 20μM NMDA; *p<0.001 vs. control cultures.
Tat immune complexes physically interact with NMDA receptors
Next, we conducted a series of immunoprecipitation experiments (figure 6a). Using antibody to NR1 subunit of NMDA receptor, we were able to immunoprecipitate the Tat-NMDA receptor complex. Co-incubation of Tat immune complexes with NMDA receptor expressing cells also allowed immunoprecipitation of Tat-NMDA receptor complex with antibody to NR1. The formation of these complexes was not affected by the presence of NMDA. In reverse immunoprecipation experiments Tat was incubated with cells expressing the NMDA receptor and immunoprecipitated with anti-Tat antibody. Complexes were detected by western blot analysis using antibodies to NR1a (figure 6b). However, cells expressing AMPA receptors did not bind to Tat when similarly immunoprecipated using Tat antibody and Western blots probed with GluR-1 antibody. This is consistent with experiments above demonstrating that Tat-immune complex did not attenuate kainate-toxicity.
Figure 6. Interaction of Tat immune complexes with the NMDA receptor, but not kainate receptors.

(A) Lanes 1–6: Protein G sepharose beads were prepared with a monoclonal anti-NR1 antibody. HEK 293 cells expressing NR1A and NR2A were incubated for 1 hour in conditioned media with the indicated agent(s): untreated (lane 1), Tat protein (lane 2), nitrosylated Tat (lane 3), TatΔ31–61 (lane 4), Tat and anti-Tat antibody (lane 5), Tat and anti-Tat antibody and NMDA (lane 6). The cells were then harvested for immunoprecipitation over the anti-NR1 loaded protein G sepharose. Eluted proteins were separated by SDS-PAGE and immunoblotted with anti-Tat antibody. As controls, NR1 (lane 7) and Tat (lane 8) were run on the gel. (B) Top panel: Tat protein or vehicle was incubated with HEK 293 cells which had been transfected with NMDA receptor proteins, NR1A and NR2A. Immunoprecipitation was performed with anti-Tat antibody. Eluted proteins were immunoblotted with anti-NR1A antibody. Bottom panel: Tat protein or vehicle was incubated with HEK 293 cells which had been transfected with an AMPA receptor-GFP fusion protein, GluR1-GFP. Immunoprecipitation was performed with anti-Tat antibody. Eluted proteins were immunoblotted with an antibody against the GluR1-GFP. As a control, the third lane, shows cell lysate from HEK 293 cells transfected with NR1A and NR2A (top panel) and with GluR1-GFP (bottom panel). The arrows indicate the monomeric forms of NR1A (top) and GluR1 (bottom). (C) Mixed rat neuronal cultures were exposed to NMDA and/or anti-NR2b antibody. Mitochondrial membrane potential was measured 18 hours later. NMDA alone caused toxicity versus untreated controls (p<0.001). When the cells were pre-treated with anti-NR2b antibody for 30 minutes prior to addition of NMDA, it provided significant protection versus the toxicity seen with NMDA alone (*p<0.05, **p<0.01). Concentrations: NMDA 125 μM, ab1 5ng/μl, ab2 10ng/μl. Data represents mean ± SEM of at least three independent experiments, analyzed by ANOVA with Neumann-Keuls post-test.
Amino acids 31–61 of Tat are necessary to cause neurotoxicity (Nath et al., 1996), and cysteine residues of Tat are critical in direct interactions with the NMDA receptor (Li et al., 2008). Hence, we used a mutant Tat protein, TatΔ31–61, made by deleting the domain that is known to bind to the NMDA receptor and is critical for mediating its neurotoxic properties. The antibody to Tat binds to the C terminal region hence is capable of binding to the mutant Tat. We also used Tat in which we had nitrosylated the cysteine residues. When nitrosylated Tat or TatΔ31–61 was incubated with NMDA receptor expressing cells, Tat-NMDA receptor complexes could not be immunoprecipated. The anti-Tat antibodies still bind the mutant Tat or nitrosylated Tat, but these forms of Tat do not bind to the NMDA receptor. Immune complexes of this mutant Tat and N-terminal anti-Tat, failed to attenuate NMDA excitotoxicity (figure 7a). Since the protective effect of the antibody occurred in the presence of wild-type Tat, but not with this mutant Tat, it suggested that protection requires interaction of Tat with the NMDA receptor even when the immune complexes are formed.
Figure 7. Tat immune complexes which do not interact with the NMDA receptor do not neuroprotect.

Mixed rat neuronal cultures were exposed to TatΔ31–61, nitrosylated Tat, anti-Tat antibody, and/or NMDA. Mitochondrial membrane potential was measured 15–18 hours later. (A) NMDA caused toxicity versus untreated controls (p<0.01), which the mutant Tat-antiTat immune complex could not attenuate. (B) Nitrosylated Tat-antiTat immune complex could not attenuate toxicity caused by NMDA. Concentrations: Tat 200nM, antibody 5ng/μl, NMDA 125 μM. Data represents mean ± SEM of at least five independent experiments, analyzed by ANOVA with Neumann-Keuls post-test.
We further found that immune complexes of nitrosylated Tat with N-terminal mAb failed to protect against NMDA mediated neurotoxicity (figure 7b). Similar lack of protection was seen with immune complexes formed with heat denatured Tat and N-terminal mAb (not shown).
Collectively, these experiments suggested that direct interaction between Tat and NMDA receptor is necessary for the immune complexes to protect against NMDA agonists. The observation that immune complexes of the mutant Tat protein and nitrosylated Tat do not protect against NMDA toxicity, suggests that the domain of Tat that binds to the NMDA receptor is critical for mediating the protection. Since Tat is known to be an agonist of the receptor, resulting in excitotoxicity, this also suggests that the antibody must change the tertiary configuration of the molecule, thus preventing the toxic effects without altering its NMDA receptor binding properties.
Anti-NR2b antibodies attentuate NMDA-excitotoxity
We wanted to determine if the ability of Tat-immune complexes to block NMDA excitotoxicity was specific for these antibodies or if other antibodies directed against the NMDA receptor can also block NMDA effects. This is particularly important since in patients with NMDA receptor encephalitis, the antibodies against the receptor have agonist properties. Thus, we coincubated NMDA with an anti-NR2b mAb, and found that the mAb attenuated neurotoxicity caused by NMDA alone (p<0.05) (figure 6c). Protection was not observed with an anti-NR1a mAb (supplementary figure S4), suggesting that only some antibody-receptor interactions can block NMDA access to or stimulation of the receptor. This experiment clearly shows that blocking antibodies against this receptor can be generated and the effects are not specific to Tat-immune complexes.
DISCUSSION
The role of anti-Tat antibodies in progression of HAND needs to be determined. In as of yet unpublished experiments, we have found that antibodies to Tat were present in CSF of HIV-infected patients. Pending further analysis, these CSF studies are consistent with the in vitro findings shown in this manuscript, suggesting that anti-Tat antibodies are neuroprotective, with the highest CSF anti-Tat antibody levels in patients who were cognitively normal. Though not previously identified in patients with HAND, immune complexes have been identified in CSF from many neurological diseases and there is clear evidence of intrathecal synthesis of antibodies in these patients (Cawley et al., 1976; Cojocaru et al., 1992; Rastogi et al., 1983; Rudick et al., 1985; Wajgt and Gorny, 1983). Additionally, antibodies may cross the blood brain barrier. For example in paraneoplastic syndromes, antibodies directed against specific CNS antigens arise in the periphery and easily cross the blood brain barrier (Toothaker and Rubin, 2009). B cells may be found in the brain of HIV-infected patients and intrathecal synthesis of antibodies may occur (Anthony et al., 2003; Fainardi et al., 2001). Similarly, antibodies to gp120 have been demonstrated in the CSF of HIV infected individuals (Trujillo et al., 1996). Therapeutic strategies using CNS vaccines hold great promise despite previous controversy, such as with the amyloid vaccine for Alzheimers disease (Pul et al., 2011).
We observed that antibodies to Tat prevent Tat mediated excitotoxicity, but do not prevent its binding to NMDA receptor. Interactions between Tat and NMDA receptor are mediated via the basic domain of Tat and cysteine-cysteine interactions between the two molecules(Li et al., 2008) (figure 8). The tertiary configuration of Tat is essential to mediate these interactions. These observations are supported by our findings that heat denaturation or nitrosylation of cysteine moieties on Tat prevented any direct interactions between Tat and NMDA receptor. Since antibodies to N- or C-terminal of Tat prevented Tat-mediated neurotoxicity but did not prevent Tat binding to NMDA receptor, it suggests that these regions are freely available for binding to antibody. It also suggests that antibody must alter the tertiary configuration of Tat such that it prevents excitation of the NMDA receptor. This can only occur if binding properties and excitatory properties of Tat are mediated via different regions of the molecule. Other anti-Tat antibodies, such as antibodies made against whole Tat as opposed to mAbs against the N- or C-terminus, did not protect. This supports the requirement that the antibody binds to Tat at an epitope which is accessible, yet does not prevent binding of Tat to the NMDA receptor.
Figure 8. Neuroprotection by Tat immune complexes at NMDA receptors.
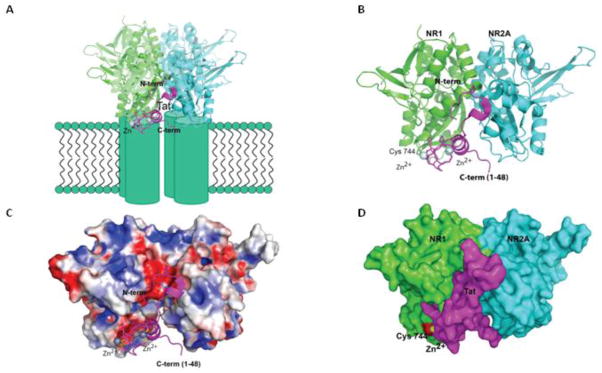
Model of the interaction between Tat and NMDA. Tat (aa 1–48, PDBid 3MIA) was manually docked to a cleft at the interface between the NR1/NR2A complex (PDBid 2AT5). The close proximity of NR1’s cysteine-744 and Tat’s cysteine-31 and Zn-site (coordinated by cysteines 25, 27 and 30) suggests the possibility of a Zn-mediated or disulfide bridge between Tat and the receptor. A similar interaction was observed between HIV-Tat and P-TEFb(Tahirov et al., 2010) (A) Tat (colored magenta) bound to the ligand binding domain of the NMDA receptor (cyan and green). N- and C-term of Tat are exposed to the solvent, and available for binding to anti-Tat antibody. The interaction between the molecules may stabilize the receptor in an active conformation, leading to excitotoxicity. When the anti-Tat antibody binds to the exposed N- or C-terminal of Tat, Tat is still able to bind to the NMDA receptor, but the receptor must then be stabilized in an inactive confirmation. Furthermore, in the presence of this Tat-antiTat immune complex, small molecule agonists are not able to stimulate the receptor either. (B) Protein cartoon of Tat bound to NR1-NR2A heterodimer (NR1 in cyan, NR2A in green, and Tat in magenta.) (C) Solvent accessible surface of the NR1/NR2A heterodimer colored by surface-charge distribution (blue positively charged, red negatively). The interaction between Tat and cysteine-744 on the NMDA receptor is also depicted. (D) Tat, represented as a solvent accessible surface colored in magenta, is docked in the NR1-NR2A interface cleft. The surface corresponding to the Zn2+ ion and cysteine-744 are colored yellow and red respectively. The molecular modeling program Pymol™(Schrodinger, LLC) was used for the modeling and figures.
Since Tat antibody blocked Tat-mediated excitotoxicity, it could be termed a neutralizing antibody. Typically, neutralizing antibodies prevent viral entry by interacting with epitopes on the virus necessary for binding to cell surface receptors. Similarly, sera that neutralize various toxins, usually do so by preventing binding to receptors by targeting epitopes on the toxin (Poignard et al., 1996). We observed that Tat immune complexes bound to NMDA receptor prevented excitation of these receptors by small molecule agonists. This suggests that the complex produces steric hindrance and prevents access to the active sites on the extracellular domain of the receptor. Alternatively, the alteration in tertiary configuration which prevents Tat from exciting the receptor must make the receptor unexcitable by other NMDA agonists. This is a novel mechanism by which neutralizing antibodies may develop broad receptor neutralizing properties by binding to antigen rather than receptor. Typically, receptor blocking antibodies have been demonstrated in autoimmune diseases such as lupus and paraneoplastic syndromes (Ardoin and Pisetsky, 2008; Toothaker and Rubin, 2009). We observed that by binding to viral antigen that in turn binds to receptor, anti-Tat can act as a receptor antagonist without causing an autoimmune syndrome.
A recent study (Rezza et al., 2005) of 252 HIV-1 positive individuals found that anti-Tat seropositivity was protective against progression to advanced HIV disease. Our work suggests one mechanism by which anti-Tat positivity could protect against progression, at least for progression to HAND.
Previous work has shown the potential for producing neuroprotection by modulating NMDA excitotoxicity via vaccine strategies. Rats immunized with NR1 subunit of NMDA receptor showed neuroprotection against effects of seizure and stroke (During et al., 2000). The ability of viral immune complexes to interact with NMDA receptors and prevent excitotoxicity represents a novel host defense mechanism. This may have important implications for our understanding of viral host relationships. These neutralizing antibodies could potentially be used therapeutically. It is also possible that this phenomenon could be exploited for development of therapeutic vaccines.
CONCLUSIONS
The ability of viral immune complexes to interact with NMDA receptors and prevent excitotoxicity represents a novel host defense mechanism. Host immune responses may influence host susceptibility to various effects of viral proteins, modulating HIV complications, such as onset of HAND. These observations provide rationale for development of vaccine therapies targeting Tat for prevention of HAND.
Supplementary Material
HIGHLIGHTS.
Antibodies to HIV Tat protein attenuate Tat-mediated neurotoxicity
Tat immune complexes also block neurotoxicity caused by NMDA receptor agonists
Vaccine therapies targeting Tat may prevent HIV-associated cognitive impairment
Acknowledgments
We gratefully acknowledge Phillip Ray for preparation of recombinant Tat protein; Tonya Malpica-Llanos for technical assistance with neuronal cultures; and Ilia Tikhonov and C. David Pauza for providing anti-Tat antibody.
This work was supported by NIH grants to J. Rumbaugh, A. Nath, and M. Prendergast.
Footnotes
mAb;
pAb;
JC-1;
PI;
ANOVA;
MSK;
EDTA;
EGTA;
RLU
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anthony IC, Crawford DH, Bell JE. B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain. 2003;126:1058–67. doi: 10.1093/brain/awg118. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–36. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- Ardoin SP, Pisetsky DS. Developments in the scientific understanding of lupus. Arthritis Res Ther. 2008;10:218. doi: 10.1186/ar2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporello E, Nath A, Slevin J, Galey D, Hamilton G, Williams L, et al. The immunophilin ligand GPI1046 protects neurons from the lethal effects of the HIV-1 proteins gp120 and Tat by modulating endoplasmic reticulum calcium load. J Neurochem. 2006;98:146–55. doi: 10.1111/j.1471-4159.2006.03863.x. [DOI] [PubMed] [Google Scholar]
- Cawley LP, Minard BJ, Tourtellotte WW, Ma BI, Chelle C. Immunofixation electrophoretic techniques applied to identification of proteins in serum and cerebrospinal fluid. Clin Chem. 1976;22:1262–8. [PubMed] [Google Scholar]
- Cojocaru M, Cojocaru MI, Steriu O, Baldescu R. Changes of circulating immune complexes in the serum and CSF of patients with multiple sclerosis. Rom J Intern Med. 1992;30:51–6. [PubMed] [Google Scholar]
- During MJ, Symes CW, Lawlor PA, Lin J, Dunning J, Fitzsimons HL, et al. An oral vaccine against NMDAR1 with efficacy in experimental stroke and epilepsy. Science. 2000;287:1453–60. doi: 10.1126/science.287.5457.1453. [DOI] [PubMed] [Google Scholar]
- Fainardi E, Contini C, Benassi N, Bedetti A, Castellazzi M, Vaghi L, et al. Assessment of HIV-intrathecal humoral immune response in AIDS-related neurological disorders. J Neuroimmunol. 2001;119:278–86. doi: 10.1016/s0165-5728(01)00386-1. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Langford TD, Letendre SL, Larrea GJ, Masliah E. Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol. 2003;13:195–210. doi: 10.1111/j.1750-3639.2003.tb00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, et al. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–8. doi: 10.1523/JNEUROSCI.3019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–2499. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37:373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Prendergast MA. Transection of intrinsic polysynaptic pathways reduces N-methyl-D-aspartate neurotoxicity in hippocampal slice cultures. Neurosci Res. 2003;46:369–76. doi: 10.1016/s0168-0102(03)00102-0. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–94. [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poignard P, Klasse PJ, Sattentau QJ. Antibody neutralization of HIV-1. Immunol Today. 1996;17:239–46. doi: 10.1016/0167-5699(96)10007-4. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Harris BR, Mullholland PJ, Blanchard JA, 2nd, Gibson DA, Holley RC, et al. Hippocampal CA1 region neurodegeneration produced by ethanol withdrawal requires activation of intrinsic polysynaptic hippocampal pathways and function of N-methyl-D-aspartate receptors. Neuroscience. 2004;124:869–77. doi: 10.1016/j.neuroscience.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH, Jr, Self RL, et al. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 2002;954:300–7. doi: 10.1016/s0006-8993(02)03360-7. [DOI] [PubMed] [Google Scholar]
- Pul R, Dodel R, Stangel M. Antibody-based therapy in Alzheimer’s disease. Expert Opin Biol Ther. 2011;11:343–57. doi: 10.1517/14712598.2011.552884. [DOI] [PubMed] [Google Scholar]
- Rastogi SC, Clausen J, Hansen HJ, Pedersen E, Tourtellotte WW. Estimation of levels of IgG to multiple sclerosis specific brain antigens in the cerebrospinal fluid of MS patients. Neurochem Res. 1983;8:1261–9. doi: 10.1007/BF00963996. [DOI] [PubMed] [Google Scholar]
- Rezza G, Fiorelli V, Dorrucci M, Ciccozzi M, Tripiciano A, Scoglio A, et al. The presence of anti-Tat antibodies is predictive of long-term nonprogression to AIDS or severe immunodeficiency: findings in a cohort of HIV-1 seroconverters. J Infect Dis. 2005;191:1321–4. doi: 10.1086/428909. [DOI] [PubMed] [Google Scholar]
- Ruckwardt TJ, Tikhonov I, Berg S, Hatfield GS, Chandra A, Chandra P, et al. Sequence variation within the dominant amino terminus epitope affects antibody binding and neutralization of human immunodeficiency virus type 1 Tat protein. J Virol. 2004;78:13190–6. doi: 10.1128/JVI.78.23.13190-13196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudick RA, Bidlack JM, Knutson DW. Multiple sclerosis. Cerebrospinal fluid immune complexes that bind C1q. Arch Neurol. 1985;42:856–8. doi: 10.1001/archneur.1985.04060080034012. [DOI] [PubMed] [Google Scholar]
- Self RL, Smith KJ, Butler TR, Pauly JR, Prendergast MA. Intra-cornu ammonis 1 administration of the human immunodeficiency virus-1 protein trans-activator of transcription exacerbates the ethanol withdrawal syndrome in rodents and activates N-methyl-D-aspartate glutamate receptors to produce persisting spatial learning deficits. Neuroscience. 2009;163:868–76. doi: 10.1016/j.neuroscience.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–82. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–51. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonov I, Ruckwardt TJ, Hatfield GS, Pauza CD. Tat-neutralizing antibodies in vaccinated macaques. J Virol. 2003;77:3157–66. doi: 10.1128/JVI.77.5.3157-3166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toothaker TB, Rubin M. Paraneoplastic neurological syndromes: a review. Neurologist. 2009;15:21–33. doi: 10.1097/NRL.0b013e3181870aa2. [DOI] [PubMed] [Google Scholar]
- Trujillo JR, Navia BA, Worth J, Lucey DR, McLane MF, Lee TH, et al. High levels of anti-HIV-1 envelope antibodies in cerebrospinal fluid as compared to serum from patients with AIDS dementia complex. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12:19–25. doi: 10.1097/00042560-199605010-00003. [DOI] [PubMed] [Google Scholar]
- Turchan J, Anderson C, Hauser KF, Sun Q, Zhang J, Liu Y, et al. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci. 2001;2:3. doi: 10.1186/1471-2202-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajgt A, Gorny M. CSF antibodies to myelin basic protein and to myelin-associated glycoprotein in multiple sclerosis. Evidence of the intrathecal production of antibodies. Acta Neurol Scand. 1983;68:337–43. doi: 10.1111/j.1600-0404.1983.tb04841.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.