

Abstract
Bat influenza virus H17N10 represents a distinct lineage of influenza A viruses with gene segments coding for proteins that are homologs of the surface antigens, hemagglutinin (HA) and neuraminidase (NA). Our recent study of the N10 NA homolog revealed an NA-like structure, but with a highly divergent putative active site exhibiting little or no NA activity, and provided strong motivation for performing equivalent structural and functional analyses of the H17 HA protein. The overall structure of the H17 HA homolog from A/little yellow-shouldered bat/Guatemala/060/2010 at 3.18 Å resolution is very similar to other influenza HAs, with a putative receptor-binding site containing some conserved aromatic residues that form the base of the sialic acid binding site. However, the rest of the H17 receptor-binding site differs substantially from the other HA subtypes, including substitution of other conserved residues associated with receptor binding. Significantly, electrostatic potential analyses reveal that this putative receptor-binding site is highly acidic, making it unfavorable to bind any negatively charged sialylated receptors, consistent with the recombinant H17 protein exhibiting no detectable binding to sialylated glycans. Furthermore, the fusion mechanism is also distinct; trypsin digestion with recombinant H17 protein, when exposed to pH 4.0, did not degrade the HA1 and HA2, in contrast to other HAs. These distinct structural features and functional differences suggest that the H17 HA behaves very differently compared with other influenza HAs.
Keywords: crystal structure, evolution, infection, protease susceptibility, viral entry
Influenza is an infectious disease of birds and mammals caused by influenza virus, an RNA virus of the Orthomyxoviridae family. Three types of influenza virus, A, B, and C, are known. Influenza A virus infects a wide range of avian and mammalian species and is the major cause of annual human epidemics and occasional pandemics. Based on the antigenic properties of the two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), type A viruses can be classified into multiple subtypes. With the recent discovery of the H17N10 subtype of bat influenza A viruses (1), 17 subtypes of HA (H1–17) and 10 subtypes of NA (N1–10) have been found to circulate in avian and/or mammalian hosts. The H17N10 virus expands the species that can be infected by influenza virus to bats, which account for approximately one-fourth of all mammalian species and have been shown to be a reservoir for multiple emerging viruses (2).
In influenza virus infection, the HA is responsible for binding of virus to sialic acid-containing receptors on host cell surface glycoproteins and glycolipids, internalization of the virus, and subsequently membrane fusion within the endosome of the infected cell. Following virus replication, the receptor-cleavage enzyme, NA, removes sialic acid from glycans on target cell surfaces as well as from newly formed budding virions so that the progeny viruses can be released and infect other cells. However, conservation of this mechanism has been called into question for the H17N10 virus, because we and others recently found that the N10 NA-like (NAL) protein has a highly diverged putative active site and possesses no or extremely low sialic acid hydrolysis activity (3, 4). However, because the virus has retained both the HA and NA proteins, it is of considerable interest to elucidate the structure and function of the H17 HA protein and to determine whether the HA and NA retain the functions of viral entry and release, respectively.
Based on their sequences, influenza A HAs can be divided into two groups: group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, and H17) and group 2 (H3, H4, H7, H10, H14, and H15) (1). Each influenza A or influenza B HA monomer contains a receptor binding site (RBS) at its membrane-distal tip that binds sialic acid similarly in all examined HAs (5). The H17 HA shares considerable amino acid sequence identity with the other 16 HA subtypes, with an average of 50% identity with group 1 HAs and an average of 38% with group 2 HAs, compared with 49% mean pairwise identity among the 16 HA subtypes (1).
To provide insights into the structure and function of this distinct H17N10, we expressed the H17 HA protein from the H17N10 bat influenza virus, A/little yellow-shouldered bat/Guatemala/060/2010 (GU10-060) in a baculovirus system and determined its crystal structure to 3.18 Å resolution. As expected from protein sequence identity, the overall structure of the H17 HA is similar to other influenza A or influenza B HAs, and the overall shape of the H17 putative RBS is also similar to other HAs. However, the putative RBS of H17 HA is structurally and functionally divergent from other HAs. Our glycan array binding data show no binding of H17 HA to glycans containing sialic acid. Moreover, trypsin digestion of the recombinant H17 protein at pH 4.0 did not degrade the HA1 and HA2, raising questions about the pH-induced membrane fusion mechanism in H17N10 viruses.
Results
Evaluation of Sialoside Binding of GU10-060 HA.
The H17 HA ectodomain from bat influenza A virus GU10-060 was overexpressed in a baculovirus expression system, as previously described (6) (Materials and Methods). After thrombin cleavage to remove the foldon trimerization domain and His-tag, the purified GU10-060 HA protein was recovered in mostly monomeric forms. However, we found that mutating HA2 Ala47 (in H3 numbering) in the trimer interface that interacts with a γ-turn at HA1 position 30 from a neighboring monomer to Gly47 that lacks a side chain (A47G, Fig. 1A) enabled the purified HA mutant protein to be recovered mainly as trimers, even after thrombin cleavage. To evaluate the HA receptor-binding affinity, these two forms of the H17 HA proteins with their foldon domain and His-tag still attached were studied extensively by glycan microarray analysis using different batches of protein to evaluate sialic acid binding (7, 8). Even with conservation of some key residues at the base of the RBS (1), no glycan receptor-binding activity for GU10-060 HA was observed on a glycan microarray against diverse sialosides as well as other glycans, such as galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), or N-glycolylneuraminic acid (NeuGc) (SI Appendix, Figs. S1 and S2).
Fig. 1.

Crystal structure of GU10-060 HA2-47G HA. (A) Overview of the H17 HA trimer. For clarity, one of the monomers is highlighted in magenta (HA1) and cyan (HA2). The receptor-binding site (RBS) in influenza A and B HAs is highlighted in blue and designated here as the HA putative RBS because no binding activity has yet been found for H17 HA. The HA1/HA2 single Arg cleavage site is highlighted in green. Carbohydrate observed at HA1 Asn117 in the electron density map is colored yellow, and other asparagines, 23 and 289 of HA1, and 145 and 154 of HA2, that code for potential N-glycosylation sites are also labeled. The GU10-060 HA2 A47G mutation was made to stabilize an ectodomain trimer during baculovirus expression; the HA2 47 position is on the trimer interface and far from the RBS. (B) Putative RBS of the H17 HA with key side chains shown in sticks. The four residues that are conserved in the H17 HA and other HAs are colored with green carbon atoms, whereas other nonconserved putative RBS residues are colored with yellow carbon atoms.
Structural Overview of GU10-060 HA2-47G HA.
The overall structure of GU10-060 HA2-47G HA (Fig. 1A) is very similar to other published influenza A HAs (5), as well as influenza B HAs (9), as expected from sequence comparisons (1). The HA homotrimer comprises a membrane-distal, globular head that includes the RBS and the vestigial esterase domain, and a membrane-proximal domain composed of a long, extended stem region and HA1/HA2 cleavage site (Fig. 1A). Each H17 HA monomer has six disulfides that are conserved in other influenza A virus HAs, five of which are conserved with influenza B virus HAs, which also have two unique disulfide bonds (10). Consistent with phylogenetic analysis that positions GU10-060 H17 in influenza A group 1 HAs (1), superimposing GU10-060 HA on other HA structures by individual domains reveals that the H17 HA is slightly more closely related to group 1 H1 (PDB ID code 3UBQ), H2 (3KU6), H5 (2FK0), and H9 (1JSD) with Cα rmsd values of 1.9–2.2 Å (HA1), 0.9–1.2 Å (HA2), and 1.8–2.2 Å (HA monomer) than to group 2 H3 (2HMG), H7(1TI8), and H14 (3EYJ) with rmsd values of 2.3–2.4 Å (HA1), 1.4 Å (HA2), and 2.4–2.5 Å (HA monomer) (SI Appendix, Table S1). As expected, the H17 HA is structurally more distant from influenza B HA with Cα rmsd values of 3.0 Å (HA1), 2.8 Å (HA2), and 4.0 Å (HA monomer) (SI Appendix, Table S1). Interestingly, the RBS subdomain of H17 HA (residues 117–265) (11) is similar to all influenza A HAs with Cα rmsd values of 1.3–1.7 Å, in contrast to 2.5 Å with influenza B HA (SI Appendix, Table S1).
The amino acid sequence of GU10-060 HA predicts five possible glycosylation sites per monomer, with three on HA1 (Asn23, Asn117, and Asn289) and two on HA2 (Asn145 and Asn154) (Fig. 1A). Interpretable electron density is only observed for HA1 Asn117, which is between the RBS subdomain and the vestigial esterase subdomain (E′ subdomain) (11) and is the glycosylation site closest to the putative RBS. The HA1 289 site is located in the fusion subdomain, whereas HA1 23 is close to the HA0 cleavage site. For the N-linked glycosylation sites in HA2, Asn154 is conserved in all influenza A HAs, but Asn145 is only found in three group 1 HA subtypes (H13, H16, and H17).
Putative Receptor Binding Site of GU10-060 HA.
The putative RBS of H17 HA has diverged from other HAs. For other HA serotypes, the RBS at the membrane-distal end (HA1) of each HA monomer (Fig. 1A) binds to sialic acid-containing receptors with weak millimolar affinity (12). All HAs, including H17 HA, have similar RBSs, which are shallow cavities surrounded by a rim of three HA1 structural elements: 190-helix (HA1 190–198), 130-loop (HA1 134–138), and 220-loop (HA1 221–228), as well as HA1 155 (Fig. 1B). A number of conserved aromatic residues, including Trp153, His183, and Tyr195, form the base of the RBS (Fig. 1B and SI Appendix, Table S2). Residues on the rim of the RBS site tend to be more variable than those at the base (Fig. 1B and SI Appendix, Table S3) because they accommodate variations in the glycan receptor structures, especially between birds and humans.
Comparison of ligand-bound structures of HAs with α2–3 linked “avian-type” or α2–6 linked “human-type” sialylated receptors or their analogs show that the structure and orientation of the sialic acid moiety is very similar (5, 13). Sialic acid is bound by hydrophobic interactions and hydrogen bonds with the 130-, 190-, and 220-loops and conserved aromatic residues at the base of the RBS; its carboxylate hydrogen bonds with the side chain of HA1 136 and other polar side chains or main chain, whereas the acetamido-nitrogen and glycerol moiety insert into the site and hydrogen bond with surrounding RBS residues (5, 13). In the putative RBS of GU10-060 HA, Trp153, His183, and Tyr195, as well as Leu194, are conserved in the base of the site (Fig. 1B and SI Appendix, Table S3). However, all other H17 HA putative RBS residues have diverged from the conserved amino acids in other HAs (Fig. 1B and SI Appendix, Tables S2 and S3). Residue 136 is mainly Thr/Ser in all other HA subtypes, and hydrogen bonds with the sialic acid carboxyl group. A change to Asp136 in H17HA is inconsistent with binding a sialic acid receptor. The highly conserved Tyr98 is substituted by Phe98 in H17 HA, abolishing the potential for hydrogen bonding to the sialic acid glycerol moiety (carbons C7–C9). In this regard, it is notable that a Y98F mutation in some H3 HAs has severely reduced receptor binding (13, 14). Four RBS residues, 190, 225, 226, and 228, which are important determinants of receptor-binding specificity, such as in H1, H2, or H3 HAs, are not conserved, but instead are replaced by Gln190, Ala225, His226, and Asp228 in H17 HA. Asp228 is mostly Gly and Ser in other influenza A HAs. In H17 HAs, Asn134 replaces the highly conserved Gly134, and Gln155 is also unique among other influenza A HAs. To compare the RBS structures of GU10-060 HA with other HA structures, the HA RBS subdomain was superimposed with a group 1 H1 HA (3UBQ) (Fig. 2) with Cα rmsd values of 1.3 Å (SI Appendix, Table S1). The three RBS 130-, 190-, and 220-loops are superimposable, and the conserved base residues, Trp153, His183, and Tyr195, as well as Leu194, overlap with the H1 HA. All other key RBS residues are substituted in H17 HA compared with H1, including Y98F, T136D, and G228D (Fig. 2). Interestingly, two deletions at 157 and 158 result in a truncated 150-loop in H17 HA near the RBS.
Fig. 2.

Stereoview of structural comparison of the putative RBS of H17 HA (in green side chains and yellow Cα carbons) with the H1 HA from A/California/04/2009 (H1N1) (in gray; PDB ID code 3UBQ) after superimposing the RBS subdomain. The overall structures are all superimposable, including Trp153, His183, and Tyr195, as well as Leu194, in the base of the RBS. Other key residues are substituted between H17 HA and H1 HA, and are labeled in green for H17 HA and in gray for H1 HA.
Despite these amino acid changes in the putative RBS in GU10-060 HA, the overall shape of the putative RBS pocket is still similar to other HAs (Fig. 3). However, mainly due to the substitutions of larger side chains, docking simulations with a modeled canonical sialic acid indicate a clash with the pocket around its carboxylate group and glycerol side chain. Significantly, electrostatic surface representations of the RBS of all known HA structures reveal major differences between H17 HA and other influenza HAs that bind sialic acid. For most influenza A and B HAs, the RBS is slightly basic or neutral around HA1 136 that normally interacts with the sialic acid carboxylate. In contrast, H17 HA is strongly acidic in its entire putative RBS (Fig. 3), which renders it unfavorable for the binding of sialic acid-containing receptors, consistent with the fact that the H17 HA exhibits no binding affinity for sialylated glycans in the glycan arrays (SI Appendix, Fig. S1). In fact, the whole molecular surface of the H17 HA trimer is generally acidic, in contrast to other influenza A and influenza B HAs (SI Appendix, Fig. S3).
Fig. 3.

Electrostatic potential surface around putative RBS of different HAs. Electrostatic surface potentials were calculated using the APBS program (28). Negatively charged regions are red, positively charged regions are blue, and neutral regions are white (−10–10 KbT/ec potential range). The coordinates used in this figure are as follows. Group 1 HAs: H17, GU10-060 HA2-47G HA; H1 (PDB ID code 3UBQ), H2 (PDB ID code 3KU6), H5 (PDB ID code 2FK0), and H9 (PDB ID code 1JSD). Group 2 HAs: H3 (PDB ID code 2HMG), H7 (PDB ID code 1TI8), and H14 (PDB ID code 3EYJ), as well as influenza B HA (PDB ID code 3BT6). The putative RBS of the H17 HA is unusual in its strong negative charge.
HA1/HA2 Cleavage Site and Putative Fusion Peptide of GU10-060 HA.
During viral infection, each monomer of the homotrimer is synthesized as a single peptide chain precursor (HA0) that is cleaved into HA1/HA2 by host proteases. So far, HA0 structures have been determined for the R329Q mutant of H3 HA (15) and 1918 H1 HA0 of H1 (6), as well as a very recent report of H16 HA0 (16). The cleavage site is a loop in H3 HA0 and H1 HA0, with the unique exception of a recently reported α-helical element in H16 HA0. In the baculovirus expression system, HAs with monobasic cleavage sequences (a single Arg) are usually expressed as HA0 (6), whereas HAs with polybasic cleavage sequences are expressed mostly in the cleaved HA1/HA2 form (8). Accordingly, GU10-060 HA and its HA2-A47G mutant were expressed as HA0 due to the single monobasic E-G-R cleavage site (Fig. 4). As expected, trypsin digestion of the H17 HA at pH 8.0 gave HA1 and HA2. Significantly, trypsin digestion of H17 HA exposed to pH 4.9, and even as low as pH 4.0, did not degrade H17 HA (Fig. 4), in contrast to other HAs, which are cleaved into small oligopeptides (17). In the HA0 structure of GU10-060 HA2-47G HA, electron density for HA1 325–329 and HA2 1–5 was not observed due to flexibility of the cleavage loop and, therefore, not modeled (Fig. 5A).
Fig. 4.
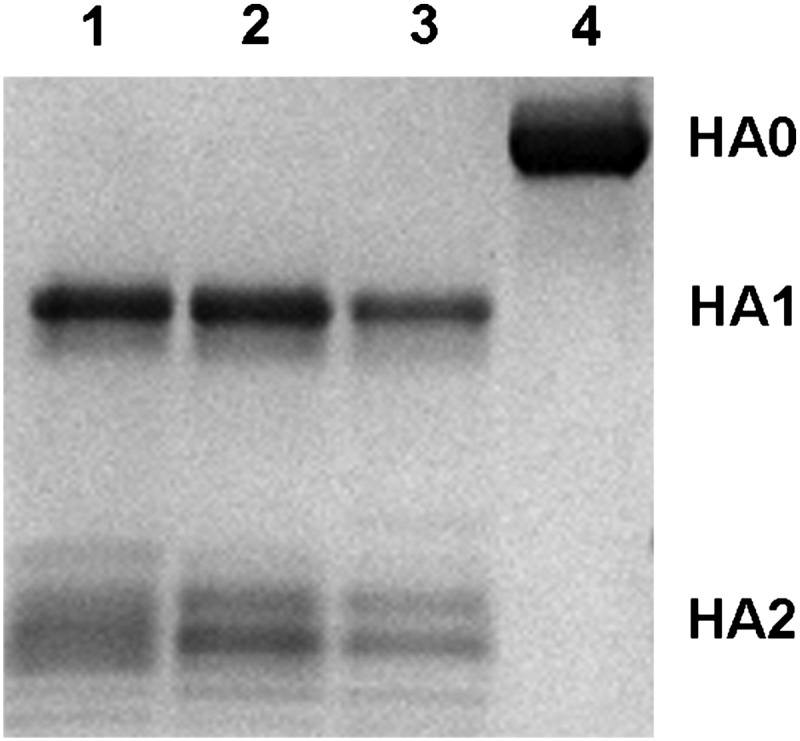
Reducing SDS/PAGE of trypsin-digested GU10-060 HA2-47G HA0 to HA1 and HA2 subdomains at different pHs. Lanes 1, 2, and 3 show GU10-060 HA0 trypsin-digested after exposure to pH 4.0, 4.9, and 8.0, respectively, which cleaves HA0 only to HA1 and HA2 (note: HA2 has different glycan forms). Trypsin did not degrade the HA exposed to low pH, as observed in other HA subtypes. Lane 4 shows uncleaved H17 HA2-47G HA0. These results suggest that GU10-060 HA requires processing to the HA1/HA2 subunits to generate the putative fusion peptide, but low-pH–induced conformational changes, which render HA susceptible to extensive degradation, are not apparent.
Fig. 5.

Structural comparison around the HA1/HA2 cleavage site of H17 HA with other HAs. (A) GU10-060 HA HA0 cleavage site with HA1 colored green and HA2 colored cyan. Due to flexibility of the cleavage loop, the residues corresponding to HA1 325–329 and HA2 1–5 are not modeled. Overlay around the cleavage loops of H17 HA0 (in green) with H3 HA0 (in brown; PDB ID code 1HA0) (B) and H1 HA0 (in gray; PDB ID code 1RD8) (C). The conformation of modeled putative fusion peptide of H17 is more similar to H3 HA0 than to H1 HA0. For comparison, Fig. 5 A–C are generated in the same orientation.
The putative fusion peptide sequence (18) of H17 HA is conserved compared with other influenza A HAs (SI Appendix, Table S2). Superimposing the HA2 domain of GU10-060 HA with H3 HA0 (Fig. 5B) and 1918 H1 HA0 (Fig. 5C) shows that the conformation of the putative fusion peptide of GU10-060 H17 is more similar to H3 HA0 (group 2 HA) than to 1918 H1 HA0 (group 1 HA). In H3 and H17, the HA2 Trp21 indole points toward the HA distal end and the HA2 Phe9 residues are in virtually identical positions (Fig. 5B). In H1, the Trp21 indole points in the opposite orientation and HA2 Phe9 Cα is shifted about 15 Å from that in H3 and H17 (Fig. 5C). It has been proposed that the different HA0 cleavage loop conformations in H3 and H1, as well as H16, may be influenced by nearby glycosylation sites (6). In H1 HA0 (6) and H16 HA0 (16), the HA1 Asn20 glycosylation site may be partially responsible for causing the loop to abut the HA surface rather than protrude from it, such as in H3 HA0, in which the equivalent HA1 Asn22 site is further distanced from the cleavage loop. Interestingly, in H17 HA, similar to H3 HA0, the equivalent HA1 Asn23 site is even further from the cleavage loop (Fig. 5A).
Comparison of GU10-060 HA with Other H17 HAs.
In addition to GU10-060 H17N10 virus, we reported two other subtype H17N10 bat viruses A/little yellow-shouldered bat/Guatemala/164/2009 (GU09-164) and A/little yellow-shouldered bat/Guatemala/153/2009 (GU09-153), in which the HA and NAL gene sequences are identical (1). The GU10-060 and GU09-164/GU09-153 HAs have very similar sequences, including an identical putative RBS, HA1/HA2 cleavage site, and entire HA2. Only four substitutions are found in HA1 at N78S, N92S, D216E, and I223V (from GU10-060 to GU09-164) (SI Appendix, Table S2), and one substitution in the signal peptide (SI Appendix, Table S2) and another in the transmembrane domain (1). HA1 78 and 92 are in the vestigial esterase subdomain, and HA1 216 and 223 are in the trimer interface of the HA receptor-binding domain. The structural and functional role of these substitutions, if any, between the two H17 HAs is not known.
Discussion
The two viral surface glycoproteins of the influenza virus, HA and NA, both engage sialic acid-containing receptors on host cells, and their interplay has important implications for viral replication, tissue tropism, transmission, and host range (19). HA is responsible for sialic acid receptor binding and subsequently membrane fusion and is the primary target for infectivity-neutralizing antibodies. NA is a sialic acid receptor-destroying enzyme that is responsible for viral progeny release. However, the recently reported N10 NAL structures, including GU10-060 NAL and GU09-164 NAL, from bat H17N10 viruses revealed a highly diverged putative active site that is wider than other NAs and in which most of the residues required for NA activity are substituted. These structural features are consistent with the recombinant N10 proteins exhibiting no, or extremely low, NA activity (3, 4). Thus, these observations have raised further interest in the structure and function of the HA and NA proteins of bat H17N10 viruses. Here, we characterized the H17 HA protein (1) by determining its crystal structure and analyzing its receptor-binding and pH-induced fusion properties.
The overall structure of H17 HA is similar to other HAs and closer to influenza A group 1 HAs, consistent with the phylogenetic analysis that H17 is more closely related to the group 1 HAs than to group 2 HAs; H17 HA shares ancestry with H1 clade HAs that contain H1, H2, H5, and H6 subtypes (1). For the RBS, the overall architecture is similar to other influenza A HAs, and the three RBS structural elements, the 190-helix, the 130-loop, and the 220-loop, are comparable to other HAs. A number of conserved aromatic residues at the base of the RBS, including Trp153, His183, and Tyr195, as well as Leu194, are identical with most influenza A HAs (SI Appendix, Table S3). However, due to numerous unique or nearly unique substitutions of other key residues around the RBS, the structure and properties of the H17 putative RBS have significantly diverged from other HAs. Moreover, in contrast to other influenza HAs, the electrostatic surface potential analysis revealed a strong acidic RBS for the H17 HA, especially around residue 136, which normally interacts with the carboxylate group of sialic acid. Based on the unfavorable properties for sialic acid binding, and the fact that there was no measurable binding to diverse sialosides on the glycan microarray, it is likely that the bat H17 HA protein recognizes a receptor other than sialic acid.
These results and those reported previously with the N10 NAL protein strongly support the idea that neither the putative HA nor the NA of H17N10 viruses interact with sialic acid receptors (3). However, it is possible, and even likely, that they still play complementary roles in virus entry and release, but interact with a completely different receptor(s). Influenza C virus is exemplary for interaction with an alternative receptor in that it uses a unique sialic acid, 9-O-acetyl-N-acetylneuraminic acid, for virus entry, and 9-O-acetyl esterase activity instead of NA activity to destroy receptors for virus release (20). However, the H17N10 virus may bind a radically different receptor, such as a protein receptor (21), as seen for other bat viruses that have converted sialic acid binding sites into protein binding sites (22). To date, all attempts to propagate the bat H17N10 viruses in embryonated chicken eggs and several mammalian cells have been unsuccessful. The eventual identification of the receptors on bat cells or tissues for H17 HA and/or N10 NAL will undoubtedly reveal the roles of the H17 HA and N10 NAL proteins and their role in the mechanism of H17N10 influenza virus infection.
In summary, the H17 HA protein is similar to other influenza A HAs in its 3D structure but lacks sialic acid receptor-binding activity and is insensitive to low pH. Together with the previous studies on the N10 NAL, which also has an overall structure similar to other influenza NAs but exhibits little or no NA activity, these observations strongly suggest that the H17N10 bat influenza virus has a unique mechanism for viral entry and release. Given the large numbers of bat species, many of which harbor other emerging disease viruses, such as Hendra and Nipah viruses, among others (2), it is essential to find out whether bats are another natural reservoir for influenza viruses. In this regard, identification of any additional influenza viruses in bats would be extremely helpful.
Materials and Methods
The methods are briefly summarized here, and a more detailed description is provided in SI Appendix, SI Materials and Methods.
Cloning, Expression, and Purification of the H17 HA Proteins.
The ectodomain (residues 30–527, equivalent to 11–329 of HA1 and 1–174 of HA2 in H3 numbering) of HA from GU10-060 H17N10 bat influenza virus (GenBank accession no. CY103892) and its HA2-A47G mutant were expressed in a baculovirus expression system using Hi5 insect cells with an N-terminal gp67 signal peptide, a C-terminal thrombin cleavage site, a foldon trimerization sequence, and a His6-tag and expressed as described previously (6).
HA Glycan Microarray Receptor Binding Assay.
Protocols for microarray HA analysis and the glycan list (SI Appendix, Fig. S2) were as previously described (7, 23). Briefly, HA–antibody complexes were prepared by mixing HA, mouse anti-His Alexa Fluor 488, and goat anti-mouse IgG Alexa Fluor, and the complex mixture was then added directly to the surface of the array and allowed to incubate for 1 h at room temperature (∼22 °C). After washing with 1× PBS and Tween, the array slides were dried and scanned for fluorescence signal.
Crystal Structure Determination.
Crystallization experiments were set up using the sitting drop vapor diffusion method. The GU10-060 HA2-47G HA at 10 mg/mL in 20 mM Tris (pH 8.0), 100 mM NaCl, and 0.02% (vol/vol) NaN3 was crystallized in 0.1 M sodium citrate (pH 5.5), 1 M LiCl2, and 15% (wt/vol) polyethylene glycol (PEG) 6000. Diffraction data were collected at beamline 08ID-1 at the Canadian Light Source (Table 1). The GU10-060 HA structure was determined by molecular replacement using the program Phaser (24) with the starting model from A/California/04/2009 (H1N1) H1 HA structure (PDB ID code 3UBQ). The refinement was performed in Refmac5 (25), and model building was carried out with the program Coot (26). Final statistics for both structures are represented in Table 1. Figs. 1, 2 and 5 were generated with Bobscript (27), and Fig. 3 was generated with PyMol (www.pymol.org).
Table 1.
Data collection and refinement statistics for GU10-060 HA2-47G HA
| Dataset | GU10-060 HA2-47G HA |
| Space group | R32 |
| Unit cell (Å) | a = b = 100.8, c = 760.5 |
| α = β = 90°, γ = 120° | |
| Resolution (Å)* | 50.0–3.18 (3.27–3.18) |
| X-ray source | Canadian Light Source 08ID-1 |
| Unique reflections | 24,997 |
| Redundancy* | 4.5 (3.8) |
| Average I/σ(I)* | 10.9 (2.0) |
| Completeness* | 97.6 (91.2) |
| Rsym*,† | 0.16 (0.79) |
| Rpim*,† | 0.09 (0.47) |
| Monomers in asymmetric unit | 2 |
| Vm (Å3/Da) | 3.4 |
| Reflections used in refinement | 23,738 |
| Refined residues | 978 |
| Refined waters | — |
| Rcryst‡ | 0.203 |
| Rfree§ | 0.258 |
| B-values (Å2) protein | 64.4 |
| Wilson B-value (Å2) | 53.7 |
| Ramachandran plot (%)¶ | 95.2, 0.5 |
| rmsd bond (Å) | 0.012 |
| rmsd angle (°) | 1.7 |
| PDB ID code | 4I78 |
*Values in parentheses denote outer-shell statistics.
†Rsym = ∑hkl∑i |Ihkl,i − <Ihkl>| /∑hkl∑i Ihkl,i and Rpim = ∑hkl[1/(N − 1)]1/2∑i |Ihkl,i − <Ihkl>| /∑hkl∑i Ihkl,i, where Ihkl,i is the scaled intensity of the ith measurement of reflection h, k, l, < Ihkl> is the average intensity for that reflection, and N is the redundancy.
‡Rcryst = ∑hkl |Fo − Fc|/∑hkl |Fo|, where Fo and Fc are the observed and calculated structure factors.
§Rfree was calculated as for Rcryst, but on 5% of data excluded before refinement.
¶The values are percentage of residues in the favored and outliers regions analyzed by MolProbity (29).
Protease Susceptibility Assay.
Protocols for trypsin susceptibility analysis were as previously described (17). For GU10-060 HA2-47G HA, each reaction contained ∼5.0 μg of HA0 incubated at 37 °C for 1 h under pH values of 4.0, 4.9, and 8.0. After incubation, the reaction pH was neutralized to pH 8.4. Trypsin was then added to all samples except controls, at a final ratio of 1:10 (wt/wt) of trypsin to the HA, and reactions were incubated overnight at 22 °C. Samples were then analyzed by SDS/PAGE.
Supplementary Material
Acknowledgments
We thank Henry Tien of the Robotics Core at the Joint Center for Structural Genomics (supported by Grant U54 GM094586) for automated crystal screening. X-ray diffraction datasets were collected at the Canadian Light Source (CLS) beamline 08ID-1 with the help of Drs. Jean-Philippe Julien and Minsun Hong. We thank Dr. Rui Xu for helpful discussions. This work was supported in part by National Institutes of Health Grant AI058113 (to J.C.P. and I.A.W.), a contract from the Centers for Disease Control (to J.C.P.), and the Skaggs Institute for Chemical Biology. Portions of this research were carried out at the CLS, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. This is publication 21983 of The Scripps Research Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 4I78).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218509110/-/DCSupplemental.
References
- 1.Tong S, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA. 2012;109(11):4269–4274. doi: 10.1073/pnas.1116200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: Important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19(3):531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X, et al. Crystal structures of two subtype N10 neuraminidase-like proteins from bat influenza A viruses reveal a diverged putative active site. Proc Natl Acad Sci USA. 2012;109(46):18903–18908. doi: 10.1073/pnas.1212579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, et al. Structural and functional characterization of neuraminidase-like molecule N10 derived from bat influenza A virus. Proc Natl Acad Sci USA. 2012;109(46):18897–18902. doi: 10.1073/pnas.1211037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gamblin SJ, Skehel JJ. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J Biol Chem. 2010;285(37):28403–28409. doi: 10.1074/jbc.R110.129809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens J, et al. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303(5665):1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 7.Blixt O, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci USA. 2004;101(49):17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens J, et al. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312(5772):404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 9.Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337(6100):1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Cheng F, Lu M, Tian X, Ma J. Crystal structure of unliganded influenza B virus hemagglutinin. J Virol. 2008;82(6):3011–3020. doi: 10.1128/JVI.02477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha Y, Stevens DJ, Skehel JJ, Wiley DC. H5 avian and H9 swine influenza virus haemagglutinin structures: Possible origin of influenza subtypes. EMBO J. 2002;21(5):865–875. doi: 10.1093/emboj/21.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauter NK, et al. Hemagglutinins from two influenza virus variants bind to sialic acid derivatives with millimolar dissociation constants: A 500-MHz proton nuclear magnetic resonance study. Biochemistry. 1989;28(21):8388–8396. doi: 10.1021/bi00447a018. [DOI] [PubMed] [Google Scholar]
- 13.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 14.Bradley KC, et al. Analysis of influenza virus hemagglutinin receptor binding mutants with limited receptor recognition properties and conditional replication characteristics. J Virol. 2011;85(23):12387–12398. doi: 10.1128/JVI.05570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, et al. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95(3):409–417. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 16.Lu X, et al. Insights into avian influenza virus pathogenicity: The hemagglutinin precursor HA0 of H16 subtype has an α-helix structure in its cleavage site with an inefficient HA1/HA2 cleavage. J Virol. 2012;86(23):12861–12870. doi: 10.1128/JVI.01606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324(5924):246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skehel JJ, Cross K, Steinhauer D, Wiley DC. Influenza fusion peptides. Biochem Soc Trans. 2001;29(Pt 4):623–626. doi: 10.1042/bst0290623. [DOI] [PubMed] [Google Scholar]
- 19.Wagner R, Matrosovich M, Klenk HD. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol. 2002;12(3):159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- 20.Rogers GN, Herrler G, Paulson JC, Klenk HD. Influenza C virus uses 9-O-acetyl-N-acetylneuraminic acid as a high affinity receptor determinant for attachment to cells. J Biol Chem. 1986;261(13):5947–5951. [PubMed] [Google Scholar]
- 21.Chang A, Dutch RE. Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses. 2012;4(4):613–636. doi: 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowden TA, et al. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat Struct Mol Biol. 2008;15(6):567–572. doi: 10.1038/nsmb.1435. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, et al. Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic acid receptors. J Virol. 2012;86(24):13371–13383. doi: 10.1128/JVI.01426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model. 1997;15(2):132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 28.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.