

Abstract
Voltage-activated ion channels open and close in response to changes in membrane voltage, a property that is fundamental to the roles of these channels in electrical signaling. Protein toxins from venomous organisms commonly target the S1–S4 voltage-sensing domains in these channels and modify their gating properties. Studies on the interaction of hanatoxin with the Kv2.1 channel show that this tarantula toxin interacts with the S1–S4 domain and inhibits opening by stabilizing a closed state. Here we investigated the interaction of hanatoxin with the Shaker Kv channel, a voltage-activated channel that has been extensively studied with biophysical approaches. In contrast to what is observed in the Kv2.1 channel, we find that hanatoxin shifts the conductance–voltage relation to negative voltages, making it easier to open the channel with membrane depolarization. Although these actions of the toxin are subtle in the wild-type channel, strengthening the toxin–channel interaction with mutations in the S3b helix of the S1-S4 domain enhances toxin affinity and causes large shifts in the conductance–voltage relationship. Using a range of previously characterized mutants of the Shaker Kv channel, we find that hanatoxin stabilizes an activated conformation of the voltage sensors, in addition to promoting opening through an effect on the final opening transition. Chimeras in which S3b–S4 paddle motifs are transferred between Kv2.1 and Shaker Kv channels, as well as experiments with the related tarantula toxin GxTx-1E, lead us to conclude that the actions of tarantula toxins are not simply a product of where they bind to the channel, but that fine structural details of the toxin–channel interface determine whether a toxin is an inhibitor or opener.
INTRODUCTION
Voltage-activated ion channels play a wide range of critical roles in electrical and chemical signaling within biological systems. These ion channels have a modular architecture, consisting of a central pore domain that determines whether the channel is selective for K+, Ca2+, or Na+ ions (Kv, Cav, and Nav channels, respectively), and four surrounding voltage-sensing domains that drive opening or closing of the ion conduction pore (Kubo et al., 1993; Li-Smerin and Swartz, 1998; Lu et al., 2001, 2002; Long et al., 2007; Payandeh et al., 2011). In tetrameric Kv channels, each of the four subunits contain six transmembrane helices (S1–S6), with the S5–S6 forming the centrally located pore domain, and the S1–S4 helices forming each of four voltage-sensing domains.
The mechanism of voltage activation has been extensively studied in the Shaker Kv channel, which has been established to involve multiple conformational rearrangements in the S1–S4 voltage-sensing domains as they move between resting states (R1 and R2) at negative membrane voltages to activated states (A) at depolarized voltages, as conceptualized in Scheme 1 (Bezanilla et al., 1994; Hoshi et al., 1994; Stefani et al., 1994; Zagotta et al., 1994a,b; Aggarwal and MacKinnon, 1996; Mannuzzu et al., 1996; Seoh et al., 1996; Cha and Bezanilla, 1998; Schoppa and Sigworth, 1998a,b,c; Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999; Villalba-Galea et al., 2008; Tao et al., 2010; Lacroix and Bezanilla, 2011).
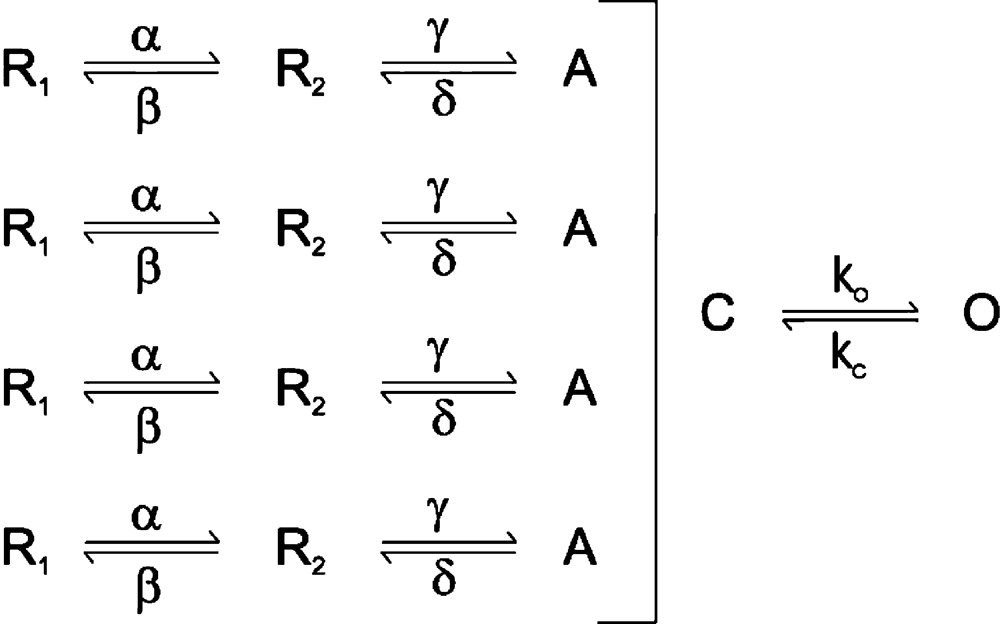 |
(SCHEME 1) |
These conformational rearrangements are thought to occur relatively independently in the four voltage sensors, but are followed by a final opening transition that is highly cooperative or concerted, as illustrated by the single closed (C) to open (O) transition in Scheme 1 (Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999; Pathak et al., 2005; Phillips and Swartz, 2010). Opening of the pore in Shaker has been extensively studied, and involves a detectable movement of the voltage sensors (Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999; del Camino et al., 2005; Pathak et al., 2005; Phillips and Swartz, 2010) along with a substantial expansion of the S6 helical bundle crossing toward the intracellular side of the membrane (Armstrong, 1974; Liu et al., 1997; Holmgren et al., 1998; del Camino and Yellen, 2001; Jiang et al., 2002; Webster et al., 2004; del Camino et al., 2005). In Nav and Cav channels, the four voltage-sensing domains are contained within a single α subunit, such that the four voltage-sensing domains are not identical (Catterall, 2012). Although our understanding of the gating mechanisms of Nav and Ca channels is less advanced than for the Shaker Kv channel, there is evidence for distinct roles of the four voltage sensors in activation and inactivation, and for cooperative interactions between voltage sensors extending to conformational changes related to the early independent transitions in the Shaker Kv channel (Yang and Horn, 1995; Sheets et al., 1999; Horn et al., 2000; Sheets et al., 2000; Chanda and Bezanilla, 2002; Chanda et al., 2004; Campos et al., 2007, 2008).
Protein toxins from venomous organisms have been invaluable tools for probing ion channel structure and mechanisms. A diversity of venom toxins interact with voltage-activated ion channels, with the external vestibule of the pore (Miller, 1995; French and Terlau, 2004; French and Zamponi, 2005), and with the S1–S4 voltage-sensing domains serving as particularly common targets (Rogers et al., 1996; Swartz and MacKinnon, 1997b; Alabi et al., 2007; Swartz, 2007; Bosmans et al., 2008). In the case of Nav channels, toxins targeting S1–S4 voltage-sensing domains can inhibit or facilitate opening (Rogers et al., 1996; Cestèle et al., 1998, 2006; French and Zamponi, 2005; Bosmans et al., 2008; Bosmans and Swartz, 2010; Wang et al., 2011; Leipold et al., 2012; Zhang et al., 2012); however, in the case of both Kv and Cav channels, only inhibitors have thus far been described (French and Zamponi, 2005; Swartz, 2007). One of the better studied examples of a toxin targeting voltage-sensing domains in Kv channels is hanatoxin, a tarantula toxin that was originally reported to produce weak inhibition of the Shaker Kv channel and robust inhibition of the Kv2.1 and Kv4.2 channels (Swartz and MacKinnon, 1995). Subsequent studies on the interaction of hanatoxin with the Kv2.1 channel showed that toxin-bound channels can open, but with a conductance–voltage (G-V) relationship that is shifted to more positive membrane voltages (Swartz and MacKinnon, 1997a; Lee et al., 2003; Phillips et al., 2005). Experiments using Kv2.1 channel mutants that exhibit weaker binding and rapid dissociation of hanatoxin demonstrate that the toxin unbinds more rapidly from open channels (Phillips et al., 2005), which supports the notion that the toxin inhibits the Kv2.1 channel by stabilizing a closed state. Mutagenesis and chimera studies suggest that hanatoxin binds to the S3b–S4 paddle motif within the S1–S4 domain of Kv2.1 to inhibit channel opening (Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 2000, 2001; Alabi et al., 2007; Bosmans et al., 2008). In addition, tarantula toxins such as hanatoxin are thought to partition into the lipid membrane and bind to the paddle motif from within the bilayer (Lee and MacKinnon, 2004; Jung et al., 2005; Phillips et al., 2005; Milescu et al., 2007, 2009).
In the present study, we set out to investigate the interaction of hanatoxin with the Shaker Kv channel to better understand the mechanisms by which tarantula toxins alter the gating mechanism of Kv channels. Although relatively little is known about the interaction of hanatoxin with the Shaker Kv channel, the toxin was reported to produce a slowing of current activation in response to depolarizing voltage steps to 0 mV (Swartz and MacKinnon, 1995), a finding that could be interpreted as rapid unbinding of the toxin from open channels, as observed with mutant Kv2.1 channels (Phillips et al., 2005). In the present experiments, we show that hanatoxin interacts with the voltage-sensing domain of the Shaker Kv channel, but that the toxin actually facilitates opening of the channel by stabilizing the activated state of the voltage sensors and the open state of the pore.
MATERIALS AND METHODS
Chimeras and point mutations were generated using sequential PCR with Kv2.1Δ7 channel (Frech et al., 1989) and Shaker-IR Kv channel (Kamb et al., 1988) as templates. The Kv2.1Δ7 construct contains seven point mutations in the outer vestibule (Li-Smerin and Swartz, 1998), rendering the channel sensitive to agitoxin-2 (AgTx-2), a pore-blocking toxin from scorpion venom (Garcia et al., 1994). The Shaker-IR construct has a deletion of residues 6–46 to remove rapid inactivation (Hoshi et al., 1990; Zagotta et al., 1990), and in this paper is referred to as wild-type Shaker. The DNA sequence of all constructs and mutants was confirmed by automated DNA sequencing, and complementary RNA (cRNA) was synthesized using T7/SP6 polymerase (mMessage mMachine kit; Ambion) after linearizing the DNA with appropriate restriction enzymes.
Hanatoxin was purified from Grammostola spatulata venom (Spider Pharm) as described previously (Swartz and MacKinnon, 1995). GxTx-1E was synthesized by solid phase methodology using Fmoc chemistry, folded in vitro, and purified as described previously (Lee et al., 2010). Linear agitoxin-2 was purchase from American Peptide, folded in vitro in a solution containing 25 µM linear peptide, 100 mM Tris-Cl, 1 mM EDTA, 2.5 mM GSH, and 0.25 mM GSSH, pH 7.8, overnight at room temperature and purified using reversed-phase HPLC.
All channel constructs were expressed in Xenopus laevis oocytes and studied after 1–4 d of incubation after cRNA injection (incubated at 17°C in 96 mM NaCl, 2 mM KCl, 5 mM Hepes, 1 mM MgCl2, 1.8 mM CaCl2, and 50 µg/ml gentamicin, pH 7.6, with NaOH) using two-electrode voltage-clamp recording techniques (OC-725C; Warner Instruments) with a 150-µl recording chamber. Data were filtered at 1–3 kHz and digitized at 20 kHz using pClamp software (Axon; Molecular Devices). Microelectrode resistances were 0.1–1 MΩ when filled with 3 M KCl. For recording macroscopic Kv channel currents, the external recording solution contained (in mM): 50 RbCl, 50 NaCl, 5 Hepes, 1 MgCl2, and 0.3 CaCl2, pH 7.6, with NaOH. In the experiments shown in Fig. 4, 50 mM KCl was used in place of RbCl. All experiments were performed at room temperature (∼22°C). Leak and background conductances were identified by blocking the channel with agitoxin-2, and in most instances have been subtracted for the Kv channel currents shown. The one exception is for data shown in Fig. 6, where the ILT construct was studied at positive voltages where full toxin occupancy of the channel could not be readily achieved, and thus unsubtracted traces are shown. G-V relationships were obtained by measuring tail currents and a single Boltzmann function was fitted to the data according to: , where I/Imax is the normalized tail-current amplitude, z is the equivalent charge, V1/2 is the half-activation voltage, F is Faraday’s constant, R is the gas constant, and T is temperature in degrees Kelvin. Gating current experiments were performed using an external recording solution containing (in mM): 100 NaCl, 5 Hepes, 1 MgCl2, and 0.3 CaCl2, pH 7.6 with NaOH. Q-V relations were obtained by integrating gating currents and an equivalent Boltzmann function fit to plots of Q/Qmax versus V.
Figure 4.

Influence of hanatoxin on the kinetics of activation and deactivation for the Shaker Δ3 Kv channel. (A) Kinetics of channel opening in the absence (black traces) or presence (red traces) of 2 µM hanatoxin. (B) Mean time constant (τ) from single-exponential fits to channel activation in the absence (black) or presence of 100 nM (red squares), 200 nM (red triangles), and 2 µM (red circles) hanatoxin. (C) Kinetics of channel closing in the absence (black traces) or presence (red traces) of 2 µM hanatoxin. (D) Mean time constant (τ) from single-exponential fits to channel deactivation in the absence (black circles) or presence of 2 µM hanatoxin (red circles). The external solution contained 50 mM Rb+, and error bars indicate SEM (n = 3).
Figure 6.

Hanatoxin facilitates opening in the Shaker Δ3/ILT channel. (A) Opening of the Shaker Δ3/ILT channel with a depolarizing step from 0 mV to +100 mV in the absence (black) and presence of 2 µM hanatoxin (red). The gray line indicates the level of zero current. (B) Normalized I-V relation in the absence (black circles) and presence of 2 µM hanatoxin (red circles). The external solution contained 50 mM Rb+. Error bars indicate SEM (n = 5).
RESULTS
Interaction of hanatoxin with the Shaker Kv channel
To begin our studies on the interaction of hanatoxin with the Shaker Kv channel, we expressed the Shaker Kv channel with rapid inactivation removed (Hoshi et al., 1990; Zagotta et al., 1990) and recorded macroscopic Kv currents in the presence of 50 mM external Rb+, a manipulation that slows channel closure so that the process of deactivation can be well-resolved (Fig. 1, A and B). When hanatoxin is applied externally at micromolar concentrations, the toxin causes a modest inhibition of outward K+ currents that gradually declines during the test depolarization (Fig. 1 A), producing a slow phase of activation that is consistent with previous observations (Swartz and MacKinnon, 1995). However, when tail currents are elicited at the end of the depolarizing pulse, it is evident that the toxin also produces a readily detectable slowing of channel deactivation. This observation contrasts with what is observed in mutant Kv2.1 channels where deactivation of the channel is faster for toxin-bound channels and is restored to control values once the toxin dissociates (Phillips et al., 2005). Thus, the results for the Shaker Kv channel suggest that the slow phase of current activation does not represent toxin unbinding, but that the toxin remains bound as the channels open and close. Interestingly, when the G-V relation of the Shaker Kv channel is examined by measuring tail current amplitudes at the end of test depolarizations, it becomes evident that hanatoxin actually produces a shift of the voltage-activation relationship to negative voltages (Fig. 1 D), opposite to what is observed for the Kv2.1 channel (Swartz and MacKinnon, 1997a; Lee et al., 2003; Phillips et al., 2005). Although the shift in the midpoint of the G-V is small (∼3 mV), it is statistically significant (P = 0.02; paired t test), and quite robust increases in macroscopic current can be readily observed when activating the channel with relatively weak depolarizations (Fig. 1, C and D). These data suggest that unlike what has been observed in the Kv2.1 channel, hanatoxin facilitates opening of the Shaker Kv. The rapid recovery of the channel after removal of the toxin from the recording chamber (see Fig. 3), as well as the relatively small effects of the toxin (Fig. 1), would be consistent with the Shaker Kv channel having relatively low affinity binding sites for hanatoxin.
Figure 1.

Hanatoxin modified the gating of the Shaker Kv channel. (A) Voltage-activated Kv channel currents in response to strong depolarization in the absence (black) and presence (red) of 2 µM hanatoxin in the external recording solution. The gray line indicates the zero current level. (B) Expanded view of tail currents from A. (C) Voltage-activated Kv channel currents in response to weak depolarization in the absence (black) and presence (red) of 2 µM hanatoxin. (D) Normalized G-V relation in the absence (black circles) and presence (red circles) of 2 µM hanatoxin. Smooth curves are Boltzmann fits to the data with the following V1/2 and z values: −33 mV and 4 for control; −36 mV and 3.8 for toxin. Conductance was measured using tail currents and the external solution contained 50 mM Rb+. Error bars indicate SEM (n = 3).
Figure 3.

Recovery of Shaker Kv channels after removal of hanatoxin from the external solution. (A) Voltage-activated Rb+ currents recorded for the Shaker Δ3 Kv channel in control solution before applying toxin (gray trace), after applying 2 µM hanatoxin to the external recording solution and allowing currents to reach equilibrium (red trace), or after removal of the toxin from the recording chamber (black traces). (B) Time course for recovery after removing hanatoxin from the external recording solution for the wild-type Shaker Kv channel (open black symbols) or for the Shaker Δ3 Kv channel (closed black symbols). Tail current amplitude was measured and normalized to the value obtained when the channel was in equilibrium with hanatoxin (red symbol).
Engineering enhanced hanatoxin receptors in the Shaker Kv channel
Our next objective was to see if we could enhance the affinity of the Shaker Kv channel for hanatoxin to facilitate studies with the channel at higher toxin occupancy. Although the very different actions of hanatoxin on Shaker and Kv2.1 channels raises the possibility that the toxin binds to distinct regions in the two types of Kv channels, a wide range of toxins have been shown to interact with the S3b–S4 paddle motif in different types of voltage-activated ion channels, and we therefore focused our attention in that region (Rogers et al., 1996; Swartz and MacKinnon, 1997b; Cestèle et al., 1998; Cestèle and Catterall, 2000; Li-Smerin and Swartz, 2000, 2001; Phillips et al., 2005; Alabi et al., 2007; Bosmans et al., 2008, 2011a,b; Wang et al., 2011; Zhang et al., 2011, 2012).
Studies of the Kv2.1 channel have identified three residues (I273, F274, and E277) within the S3b helix that are particularly important determinants for hanatoxin interaction with that channel (Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 2000, 2001; Phillips et al., 2005; Alabi et al., 2007). Fig. 2 A shows an amino acid sequence alignment for the S3b helices of the voltage sensor paddle motifs for both the Kv2.1 and Shaker Kv channels. To enhance the affinity of the Shaker Kv channel for hanatoxin, we mutated the corresponding residues (L327, A328, and V331) in the Shaker channel to those found in Kv2.1, and investigated the properties of the resulting Shaker Δ3 channel (L327I, A328F, and V331E). In contrast to what we observed for the wild-type Shaker Kv channel, hanatoxin produces dramatic facilitation of opening for the Shaker Δ3 channel (Fig. 2, B–D). In the presence of 2 µM hanatoxin, the midpoint of the G-V shifts by ∼−25 mV, and the G-V reaches a maximum that is ∼50% higher compared with that in the absence of the toxin. The concentration dependence for shifting the G-V relation (Fig. 2, D and E) indicates that the effect of hanatoxin approaches saturation at a concentration of 2 µM, which suggests that the affinity of hanatoxin for the Shaker Δ3 channel is higher than the wild-type channel. We also observed a significantly slower recovery after removal of the toxin from the recording chamber when comparing Shaker Δ3 to the wild-type channel (Fig. 3), which suggests that the unbinding rate for the toxin is decreased in the Δ3 mutant. Even several minutes after removal of the toxin, the amplitude and kinetics of tail currents are still altered, which suggests that the toxin remains tightly bound as the channel opens and closes. We also investigated the extent to which hanatoxin alters the kinetics of channel opening and closing by fitting single-exponential functions to the change in macroscopic current after voltage steps (Fig. 4). Although hanatoxin detectably slows opening of the Shaker Δ3 channel (Fig. 4, A and B), the most dramatic effect of the toxin is to produce a pronounced slowing of channel closing (Fig. 4, C and D). Collectively, these results suggest that hanatoxin interacts with the S3b helix of the S1–S4 voltage-sensing domain in the Shaker Kv channel to stabilize an open state of the channel.
Figure 2.

Generating enhanced hanatoxin receptors in the Shaker Kv channel. (A) Sequence alignment for S3 helices in Kv2.1 (blue) and Shaker (black) Kv channels. Conserved residues are shown in bold lettering. In the Shaker Δ3 construct, three residues (asterisks) were mutated to the corresponding residues in Kv2.1 (L327I, A328F, and V331E). (B and C) Voltage-activated Kv channel currents in the absence (black) and presence (red) of 2 µM hanatoxin. Recordings in B and C are from the same cell. The gray line indicates the level of zero current. (D) Normalized G-V relations in the absence (black circles) and presence of 200 nM (red triangles) or 2 µM (red circles) hanatoxin. Conductance was measured using tail currents. Smooth curves are Boltzmann fits to the data normalized to the maximal conductance in control external solution, with the following V1/2 and z values: −15 mV and 3 for control, −36 mV and 2.9 for 200 nM, and −39 mV and 2.8 for 2 µM hanatoxin. Error bars indicate SEM. (n = 5). (E) Concentration dependence of shifting the V1/2 for activation of the wild-type Shaker Kv channel (black circles) and the Shaker Δ3 Kv channel (red circles). The external solution contained 50 mM Rb+. Error bars indicate SEM (n = 3).
Influence of hanatoxin on voltage sensor activation in the Shaker Kv channel
To explore the influence of hanatoxin on conformational changes in the voltage-sensing domains, we recorded gating currents from the Shaker Δ3 channel in the background of the W434F nonconducting mutant (Perozo et al., 1993). Our initial objective was to obtain gating charge versus voltage (Q-V) relations from individual cells both in the absence and presence of hanatoxin. However, hanatoxin reaches equilibrium with the channel rather slowly at the concentrations we used (200 nM; τ = ∼150 s), and cells expressing high levels of Shaker Δ3 W434F tend to become leaky and unstable when maintained under a voltage clamp for prolonged periods of time. (We also observed that higher toxin concentrations made cells leaky and difficult to study.) We therefore compared cells with and without preincubation with 200 nM hanatoxin for 5 min before recording gating currents. Comparison of the gating currents obtained in this fashion shows that hanatoxin produces a pronounced slowing of the “off” gating charge elicited by repolarization to a holding voltage of −120 mV (Fig. 5 A). Integration of the gating currents and plotting of the Q-V relations reveals that the toxin produces a shift of charge movement to negative voltages (Fig. 5 B). The Q-V relation is also shallower in the presence of hanatoxin, although this should be interpreted cautiously because it could be simply explained by heterogeneity in toxin occupancy of the channel given that the concentration of toxin used in these experiments is not saturating and up to four toxins can bind to each channel (Swartz and MacKinnon, 1997a). To investigate the effects of hanatoxin on the kinetics of charge movement, we fit the decay of gating currents elicited by depolarization (“on” gating currents) with single exponential functions and plotted those values against membrane voltage (Fig. 5, A and C). The resulting time constants are not influenced by hanatoxin at positive voltages, but are dramatically slowed at negative voltages, which suggests that the toxin stabilizes the activated state of the voltage sensors by slowing their return to the resting state populated at negative voltages.
Figure 5.

Influence of hanatoxin on gating charge movement in the Shaker Δ3/W434F channel. (A) Families of gating currents recorded from one cell bathed in control external solution (black) and another preequilibrated with 200 nM hanatoxin (red). Leak, background, and capacitive currents were subtracted using a P/−4 protocol. (B) Normalized Q-V relation in the absence (black circles) and presence of 200 nM hanatoxin (red circles). In both cases, on and off gating currents were integrated, and their average was normalized to the maximal charge measured for depolarizations to +50 mV. Smooth curves are fits of Boltzmann functions to the data with the following V1/2 and z values: −40 mV and 3.7 for control, and −53 mV and 1.3 for 200 nM hanatoxin. Error bars indicate SEM (n = 4). (C) Mean time constants (τ) for charge movement obtained by fitting single-exponential functions to the decay of on gating currents in the absence (black circles) and presence (red circles) of 200 nM hanatoxin. Error bars indicate SEM (n = 6).
Influence of hanatoxin on the final opening transition in the Shaker Kv channel
To investigate whether hanatoxin also influences the final opening transition, we recorded macroscopic ionic currents from the Shaker Δ3 channel in the background of the ILT mutant (V369I, I372L, S376T), a well-characterized triple mutant of the Shaker Kv channel that assists in isolating the final concerted opening transition (Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999; del Camino et al., 2005; Pathak et al., 2005; Phillips and Swartz, 2010). In this ILT mutant, voltage sensor activation occurs at voltages negative to −50 mV, whereas the final opening transition occurs at voltages positive to 0 mV. When the Shaker Δ3/ILT channels are activated by depolarization to +100 mV from a holding voltage of 0 mV, application of hanatoxin causes a pronounced increase in macroscopic current (Fig. 6 A). It was not possible to determine the G-V relation for the Shaker Δ3 ILT mutant because saturation occurs at extremely depolarized voltages (Smith-Maxwell et al., 1998a,b; Ledwell and Aldrich, 1999); however, we observed larger facilitation by hanatoxin for less positive membrane depolarizations (Fig. 6 B), which suggests that the toxin likely produces a shift in the G-V relationship. Collectively, these results demonstrate that hanatoxin promotes opening of the Shaker Kv channel by stabilizing the activated state of the S1–S4 voltage-sensing domain and by altering the final opening transition to stabilize the open state of the channel.
Transferring paddle motifs between Shaker and Kv2.1 channels
The S3b–S4 paddle motif contains the most important determinants for hanatoxin interaction with Kv2.1 channels (Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 1998, 2000, 2001; Winterfield and Swartz, 2000; Phillips et al., 2005), and the enhanced affinity of hanatoxin for the Shaker Δ3 construct suggests that the paddle motif is also important for hanatoxin interaction with the Shaker Kv channel (Fig. 2). The paddle motif can also be transferred between different types of voltage-activated channels and conveys sensitivity to a wide variety of tarantula and scorpion toxins (Alabi et al., 2007; Bosmans et al., 2008, 2011b; Milescu et al., 2009). We therefore wondered whether transferring the paddle motif between Kv2.1 and Shaker channels might also influence whether hanatoxin facilitates or inhibits opening. To explore this possibility, we constructed chimeras in which the S3b–S4 paddle motifs were swapped between Shaker Δ3 and Kv2.1 channels (Fig. 7 A). Both chimeras were robustly activated by membrane depolarization (Fig. 7 B,C), were K+ selective (not depicted), and were sensitive to the pore-blocking toxin agitoxin-2 (toxin-sensitive currents are those shown in Fig. 7 B). Interestingly, hanatoxin facilitates opening of the Kv2.1 channel containing the Shaker Δ3 paddle motif, and the toxin inhibits the Shaker Kv channel containing the paddle motif from Kv2.1 (Fig. 7, B and C), which is qualitatively similar to the effects observed for hanatoxin on the channels donating each paddle motif. These results suggest that interactions between hanatoxin and S3b–S4 paddle motifs play an important role in determining whether the tarantula toxin facilitates or inhibits opening of Kv channels.
Figure 7.

Influence of hanatoxin on paddle chimeras between Kv2.1 and Shaker Kv channels. (A) Sequence alignments for the S3-S4 regions of Kv2.1 (blue), Shaker Δ3 (black), and two chimeras wherein the S3b-S4 paddle motifs were swapped between the two Kv channels. Conserved residues are shown using bold lettering. (B) Voltage-activated Kv channel currents for two chimeras in the absence (black) and presence (red) of hanatoxin. The top sets of traces are for depolarization to 0 mV, whereas the lower are for depolarization to +60 mV. The gray line indicates the level of zero current. (C) Normalized G-V relations in the absence (black circles) and presence of (red circles) 2 µM hanatoxin. The external solution contained 50 mM K+. Error bars indicate SEM (n = 5).
Interaction of the tarantula toxin GxTx-1E with the Shaker Kv channel
We were also interested in exploring the extent to which variations in tarantula toxins might influence whether they facilitate or inhibit opening of Kv channels. A growing number of tarantula toxins have been shown to target paddle motifs in Kv channels (Swartz, 2007), and we focused our attention on GxTx-1E because the toxin has several intriguing similarities and differences when compared with hanatoxin. Like hanatoxin, GxTx-1E interacts with the paddle motif in Kv2.1 channels and inhibits the channel by shifting the G-V relationship to depolarized voltages (Herrington et al., 2006, 2007; Milescu et al., 2009; Schmalhofer et al., 2009; Lee et al., 2010). Although the solution NMR structures of hanatoxin and GxTx-1E have similar backbone folds and amphipathic surfaces, the detailed structure of the putative active faces of the two toxins show many differences (Takahashi et al., 2000; Wang et al., 2004; Lee et al., 2010). In addition, alanine-scanning mutagenesis of the paddle motif in Kv2.1 shows that a larger number of mutations influence GxTx-1E interaction with the channel compared with hanatoxin, although there are also common determinants (Li-Smerin and Swartz, 2000, 2001; Milescu et al., 2009). If the protein–protein interactions between toxin and paddle determine whether a toxin promotes or inhibits opening, we might expect that hanatoxin and GxTx-1E would have distinct actions. At a concentration as high as 2 µM, however, GxTx-1E does not affect the gating of wild-type or Shaker Δ3 channels (not depicted). The three residues mutated in the S3b helix of Shaker Δ3 are important determinants for GxTx-1E interaction with Kv2.1, though several other positions are also critical (Milescu et al., 2009). In an attempt to engineer sensitivity to GxTx-1E, we therefore mutated two additional residues (V330 and A332) within the Shaker Δ3 channel that correspond to residues in Kv2.1 where mutations dramatically weaken GxTx-1E interaction with the channel (Milescu et al., 2009). The resulting Shaker Δ5 channel (L327I, A328F, V330T, V331E, and A332S; Fig. 8 A) retains sensitivity to hanatoxin, and much like what is seen in Shaker Δ3, the toxin facilitates opening of the channel by shifting activation by 29 mV to more negative voltages and by increasing the maximal conductance (Fig. 8 B). The Shaker Δ5 channel also displays robust sensitivity to GxTx-1E, but in this case the tarantula toxin produces robust inhibition (Fig. 8 C). These stark differences in the activity of hanatoxin and GxTx-1E on the Shaker Δ5 channel suggest that the influence of tarantula toxins on the gating properties of Kv channels is determined by specific molecular interactions between the toxin and the voltage sensor paddle motif.
Figure 8.

Engineering sensitivity to the tarantula toxin GxTx-1E into the Shaker Kv channel. (A) Sequence alignment for S3 helices in Kv2.1 (blue) and Shaker (black) Kv channels. Conserved residues are shown in bold lettering. In the Shaker Δ5 construct, five residues (asterisks) were mutated to the corresponding residues in Kv2.1 (L327I, A328F, V330T, V331E, and A332S). (B and C) Normalized G-V relations in the absence (black circles) and presence (red circles) of hanatoxin or GxTx-1E, both applied to the external solution at a concentration of 2 µM. Conductance was measured using tail currents and the external solution contained 50 mM Rb+. Error bars indicate SEM (n = 6).
DISCUSSION
In this paper, we investigated the interaction of hanatoxin with the Shaker Kv channel. In contrast to the inhibitory actions of the toxin on Kv2.1 channels investigated in previous studies (Swartz and MacKinnon, 1997a,b; Li-Smerin and Swartz, 1998, 2000, 2001; Lee et al., 2003; Wang et al., 2004; Phillips et al., 2005; Alabi et al., 2007; Bosmans et al., 2008), our results show that hanatoxin facilitates opening of the Shaker Kv channel by interacting with the paddle motif (Figs. 1–3), stabilizing the voltage sensor in the activated state (Fig. 5), and influencing the final opening transition to stabilize the open state of the pore (Figs. 4 and 6). Although the effects of hanatoxin on the G-V relations (Figs. 1 and 2), the kinetics of channel closure (Figs. 1 and 4), and the ILT channel (Fig. 6) could in part be explained by effects on the final opening transition in Shaker, the pronounced effects of the toxin on gating currents (Fig. 5) suggest that early transitions in the voltage sensors are also affected. Our experiments with paddle chimeras between Shaker and Kv2.1 suggest that the paddle motif determines whether hanatoxin inhibits or facilitates opening of these two Kv channels (Fig. 7). We also succeeded in making the Shaker Kv channel sensitive to GxTx-1E, a toxin that also inhibits the Kv2.1 channel (Herrington et al., 2006; Milescu et al., 2009; Schmalhofer et al., 2009; Lee et al., 2010), and found that in the Shaker Δ5 construct, hanatoxin facilitates opening while GxTx-1E inhibits (Fig. 7). Collectively, these rather striking results demonstrate that the actions of tarantula toxins are not solely a product of where they bind to the channel, but that fine structural details at the interface between toxin and channel determine whether a toxin acts to inhibit or facilitate channel opening.
It is interesting to consider the mechanism by which the Δ3 mutations enhance the sensitivity of the Shaker Kv channel to hanatoxin. Because hanatoxin does not increase the single channel conductance of this construct (not depicted), the relatively large 50% increase in maximal conductance the toxin produces (Fig. 2) might suggest that this construct has a maximal open probability in the absence of toxin that is lower than the value of ∼0.8 observed for the Shaker Kv channel (Hoshi et al., 1994). When considered together with evidence that hanatoxin stabilizes the open state (Figs. 4 and 6), the enhanced sensitivity of Shaker Δ3 to hanatoxin could in part be explained by the lower maximal open probability of that construct. However, this mechanism cannot readily explain the observed concentration dependence for shifting the midpoint of voltage activation (Fig. 2 E), which shows no indication of saturation for the Shaker Kv channel and approaches saturation in the low micromolar range for the Shaker Δ3 channel. In addition, the return of channels to control conditions after removal of hanatoxin from the external solution is far slower for the Δ3 Shaker Kv channel (Fig. 3), which is consistent with a slower dissociation rate for the toxin (Phillips et al., 2005). For these reasons, we think that our engineering of the S3b helix in Shaker actually enhances the binding affinity of hanatoxin. This mechanism would also explain the sensitivity of the Δ5 Shaker Kv channel to not only hanatoxin, but also to GxTx-1E, a toxin that inhibits channel activation by stabilizing a closed state and whose effects would not be expected to be enhanced by decreases in maximal open probability.
One intriguing feature of the interaction of hanatoxin with the Shaker Kv channel is the pronounced slow phase of channel opening that is observed in the presence of the toxin (Fig. 1 A). Indeed, the kinetics of channel opening have two components in the presence of hanatoxin, one that is similar to that observed in the absence of toxin, which we attribute to unbound channels, and a second that is much slower, which we attribute to slow opening of toxin bound channels. We did not investigate this feature in detail because it is less pronounced in the Shaker Δ3 Kv channel exhibiting enhanced toxin sensitivity, which suggests that the kinetic properties of this phenomenon are likely influenced by the S3b mutations themselves. Nevertheless, this slow phase of channel opening brings to mind the slow process by which the voltage sensors in the Shaker Kv channel are stabilized in a “relaxed” state upon prolonged depolarization (Labro et al., 2012). In this state, voltage sensors exhibit Q-V relations that are shifted to more negative voltages and charge movement upon hyperpolarization that occurs more slowly, both of which are similar to the actions of hanatoxin. It is tempting to speculate that the slow phase of channel opening in the presence of hanatoxin may be related to toxin stabilization of a relaxed state of the voltage sensor.
As far as we are aware, hanatoxin is the first example of a protein toxin that can facilitate opening of a Kv channel. In contrast, a large class of toxins from scorpion venom facilitate opening of Nav channels (Catterall et al., 2007). A primary target for these β-scorpion toxins is the voltage sensor paddle motif in domain II of Nav channels (Rogers et al., 1996; Cestèle et al., 1998, 2006; Cestèle and Catterall, 2000; Bosmans et al., 2008; Zhang et al., 2011), and these toxins are thought to stabilize the domain II voltage sensor in an activated state (Cestèle et al., 1998; Leipold et al., 2012), similar to what we have observed here for the Shaker Kv channel. Examples of tarantula toxins that facilitate opening of Nav channels have also been described (Corzo et al., 2003, 2007; Bosmans et al., 2011b). Although the voltage-sensing domains they target to influence channel activity remain to be further defined, Magi5 and ProTx-I facilitate opening of Nav1.2 and Nav1.9, respectively. In the case of Magi5, the toxin likely interacts with the voltage sensor paddle in domain II, as it has shown to compete with a β-scorpion toxin (Corzo et al., 2007; Bosmans and Swartz, 2010). However, in the case of ProTx-I, the toxin has been shown to interact with paddle motifs from multiple domains when transplanted into Kv channels, but the most important domains targeted in the intact Nav1.9 channel have not been explored because this noncanonical Nav channel cannot be expressed in heterologous expression systems (Bosmans et al., 2011b).
The parallel between the present work on the interaction of hanatoxin with the Shaker Kv channel and β-scorpion toxins with Nav channels raises an interesting question about whether these two classes of toxins bind to voltage sensors within the membrane or directly from aqueous solution. In the case of tarantula toxins, the prevailing view is that they interact with voltage sensors within the membrane. Hanatoxin and related tarantula toxins partition into membranes (Lee and MacKinnon, 2004; Jung et al., 2005, 2010; Phillips et al., 2005; Milescu et al., 2007, 2009) and do not bind tightly to detergent solubilized channels (Lee and MacKinnon, 2004; Ruta and MacKinnon, 2004), and toxin mutations have been identified where correlated effects on the strength of membrane partitioning and apparent affinity for the channel are observed (Milescu et al., 2007). In addition, mutations on the voltage sensor paddle that weaken toxin affinity have been identified that can be rescued by modification of the lipid sphingomyelin, as if there is an intimate interaction between lipids, toxins, and voltage sensor paddle motifs (Milescu et al., 2009). In contrast, models of β-scorpion toxin binding to Nav channel voltage sensors have depicted toxin binding from aqueous solution (Wang et al., 2011; Zhang et al., 2011, 2012), and there have been reports that some scorpion toxins can partition into membranes whereas others cannot (Smith et al., 2005; Cohen et al., 2006). It will be interesting to revisit these issues for scorpion toxins to clarify the environment in which they interact with voltage-sensing domains.
One fascinating and relatively unexplored aspect of the interaction of protein toxins with S1–S4 voltage-sensing domains is the mechanism by which specific conformations of the voltage sensor are stabilized. The results presented here suggest that both the toxin and the S3b–S4 paddle motif within the voltage-sensing domain of the channel can play decisive roles in determining whether a toxin facilitates or inhibits opening. When only considering hanatoxin, the paddle chimeras we studied (Fig. 7) lead us to conclude that variations in the paddle motifs between Kv2.1 and Shaker Kv channels play a crucial role in determining whether the toxin inhibits or promotes opening. However, the interactions of hanatoxin and GxTx-1E with the Shaker Δ5 channel (Fig. 8) clearly demonstrate that two related tarantula toxins can have opposite effects on a single Kv channel. These results seem to indicate that both the toxin and the paddle motif are critical. One mechanism by which toxins might stabilize different states of the voltage sensors in these Kv channels would be that the protein–protein interface between the toxin and paddle motif is largely responsible, and that the toxin simply binds tighter to the conformation it stabilizes because the complementarity between the two proteins is optimized in that state. However, another possibility would be to suppose that the shape of the toxin-binding surface doesn’t actually change substantially as the voltage sensors move, but that it is the interaction between the toxin and membrane that dynamically rearranges as the paddle motif moves relative to the surrounding membrane. Tarantula toxins interact strongly with the S3b helix and to some extent the S4 helix of the paddle motif (Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 2000; Alabi et al., 2007; Bosmans et al., 2008; Milescu et al., 2009), and there is agreement that the S4 helix moves roughly perpendicular to the plane of the membrane in response to changes in membrane voltage (Bezanilla, 2008; Swartz, 2008; Vargas et al., 2011). It will be fascinating to explore whether the interaction between the toxin and the membrane plays an important role in determining which state is stabilized by a toxin. Some of the constructs characterized here may be particularly useful tools for distinguishing between these two types of mechanisms. The Shaker Δ5 construct, for example, is sensitive to structurally related tarantula toxins that have opposite effects on activation of the channel by membrane voltage.
Acknowledgments
We thank the National Institute of Neurological Disorders and Stroke (NINDS) DNA sequencing facility for DNA sequencing, and Miguel Holmgren, Mark Mayer, Joe Mindell, Jeet Kalia, and Dmitriy Krepkiy for helpful discussions.
This work was supported by the Intramural Research Program of the NINDS, National Institutes of Health, to K.J. Swartz (grant no. NS002945-16).
Richard W. Aldrich served as guest editor.
References
- Aggarwal S.K., MacKinnon R. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177 10.1016/S0896-6273(00)80143-9 [DOI] [PubMed] [Google Scholar]
- Alabi A.A., Bahamonde M.I., Jung H.J., Kim J.I., Swartz K.J. 2007. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 450:370–375 10.1038/nature06266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M. 1974. Ionic pores, gates, and gating currents. Q. Rev. Biophys. 7:179–210 10.1017/S0033583500001402 [DOI] [PubMed] [Google Scholar]
- Bezanilla F. 2008. How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 9:323–332 10.1038/nrm2376 [DOI] [PubMed] [Google Scholar]
- Bezanilla F., Perozo E., Stefani E. 1994. Gating of Shaker K+ channels: II. The components of gating currents and a model of channel activation. Biophys. J. 66:1011–1021 10.1016/S0006-3495(94)80882-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F., Swartz K.J. 2010. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 31:175–182 10.1016/j.tips.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F., Martin-Eauclaire M.F., Swartz K.J. 2008. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature. 456:202–208 10.1038/nature07473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F., Milescu M., Swartz K.J. 2011a. Palmitoylation influences the function and pharmacology of sodium channels. Proc. Natl. Acad. Sci. USA. 108:20213–20218 10.1073/pnas.1108497108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F., Puopolo M., Martin-Eauclaire M.F., Bean B.P., Swartz K.J. 2011b. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 138:59–72 10.1085/jgp.201110614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F.V., Chanda B., Beirão P.S., Bezanilla F. 2007. β-Scorpion toxin modifies gating transitions in all four voltage sensors of the sodium channel. J. Gen. Physiol. 130:257–268 10.1085/jgp.200609719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos F.V., Chanda B., Beirão P.S., Bezanilla F. 2008. α-Scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J. Gen. Physiol. 132:251–263 10.1085/jgp.200809995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. 2012. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590:2577–2589 10.1113/jphysiol.2011.224204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A., Cestèle S., Yarov-Yarovoy V., Yu F.H., Konoki K., Scheuer T. 2007. Voltage-gated ion channels and gating modifier toxins. Toxicon. 49:124–141 10.1016/j.toxicon.2006.09.022 [DOI] [PubMed] [Google Scholar]
- Cestèle S., Catterall W.A. 2000. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 82:883–892 10.1016/S0300-9084(00)01174-3 [DOI] [PubMed] [Google Scholar]
- Cestèle S., Qu Y., Rogers J.C., Rochat H., Scheuer T., Catterall W.A. 1998. Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 21:919–931 10.1016/S0896-6273(00)80606-6 [DOI] [PubMed] [Google Scholar]
- Cestèle S., Yarov-Yarovoy V., Qu Y., Sampieri F., Scheuer T., Catterall W.A. 2006. Structure and function of the voltage sensor of sodium channels probed by a beta-scorpion toxin. J. Biol. Chem. 281:21332–21344 10.1074/jbc.M603814200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A., Bezanilla F. 1998. Structural implications of fluorescence quenching in the Shaker K+ channel. J. Gen. Physiol. 112:391–408 10.1085/jgp.112.4.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B., Bezanilla F. 2002. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120:629–645 10.1085/jgp.20028679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B., Asamoah O.K., Bezanilla F. 2004. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J. Gen. Physiol. 123:217–230 10.1085/jgp.200308971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L., Gilles N., Karbat I., Ilan N., Gordon D., Gurevitz M. 2006. Direct evidence that receptor site-4 of sodium channel gating modifiers is not dipped in the phospholipid bilayer of neuronal membranes. J. Biol. Chem. 281:20673–20679 10.1074/jbc.M603212200 [DOI] [PubMed] [Google Scholar]
- Corzo G., Gilles N., Satake H., Villegas E., Dai L., Nakajima T., Haupt J. 2003. Distinct primary structures of the major peptide toxins from the venom of the spider Macrothele gigas that bind to sites 3 and 4 in the sodium channel. FEBS Lett. 547:43–50 10.1016/S0014-5793(03)00666-5 [DOI] [PubMed] [Google Scholar]
- Corzo G., Sabo J.K., Bosmans F., Billen B., Villegas E., Tytgat J., Norton R.S. 2007. Solution structure and alanine scan of a spider toxin that affects the activation of mammalian voltage-gated sodium channels. J. Biol. Chem. 282:4643–4652 10.1074/jbc.M605403200 [DOI] [PubMed] [Google Scholar]
- del Camino D., Yellen G. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K(+) channel. Neuron. 32:649–656 10.1016/S0896-6273(01)00487-1 [DOI] [PubMed] [Google Scholar]
- del Camino D., Kanevsky M., Yellen G. 2005. Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J. Gen. Physiol. 126:419–428 10.1085/jgp.200509385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech G.C., VanDongen A.M., Schuster G., Brown A.M., Joho R.H. 1989. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 340:642–645 10.1038/340642a0 [DOI] [PubMed] [Google Scholar]
- French R.J., Terlau H. 2004. Sodium channel toxins—receptor targeting and therapeutic potential. Curr. Med. Chem. 11:3053–3064 10.2174/0929867043363866 [DOI] [PubMed] [Google Scholar]
- French R.J., Zamponi G.W. 2005. Voltage-gated sodium and calcium channels in nerve, muscle, and heart. IEEE Trans. Nanobioscience. 4:58–69 10.1109/TNB.2004.842500 [DOI] [PubMed] [Google Scholar]
- Garcia M.L., Garcia-Calvo M., Hidalgo P., Lee A., MacKinnon R. 1994. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 33:6834–6839 10.1021/bi00188a012 [DOI] [PubMed] [Google Scholar]
- Herrington J. 2007. Gating modifier peptides as probes of pancreatic beta-cell physiology. Toxicon. 49:231–238 10.1016/j.toxicon.2006.09.012 [DOI] [PubMed] [Google Scholar]
- Herrington J., Zhou Y.P., Bugianesi R.M., Dulski P.M., Feng Y., Warren V.A., Smith M.M., Kohler M.G., Garsky V.M., Sanchez M., et al. 2006. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 55:1034–1042 10.2337/diabetes.55.04.06.db05-0788 [DOI] [PubMed] [Google Scholar]
- Holmgren M., Shin K.S., Yellen G. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621 10.1016/S0896-6273(00)80571-1 [DOI] [PubMed] [Google Scholar]
- Horn R., Ding S., Gruber H.J. 2000. Immobilizing the moving parts of voltage-gated ion channels. J. Gen. Physiol. 116:461–476 10.1085/jgp.116.3.461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538 10.1126/science.2122519 [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1994. Shaker potassium channel gating. I: Transitions near the open state. J. Gen. Physiol. 103:249–278 10.1085/jgp.103.2.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Lee A., Chen J., Cadene M., Chait B.T., MacKinnon R. 2002. The open pore conformation of potassium channels. Nature. 417:523–526 10.1038/417523a [DOI] [PubMed] [Google Scholar]
- Jung H.J., Lee J.Y., Kim S.H., Eu Y.J., Shin S.Y., Milescu M., Swartz K.J., Kim J.I. 2005. Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry. 44:6015–6023 10.1021/bi0477034 [DOI] [PubMed] [Google Scholar]
- Jung H.H., Jung H.J., Milescu M., Lee C.W., Lee S., Lee J.Y., Eu Y.J., Kim H.H., Swartz K.J., Kim J.I. 2010. Structure and orientation of a voltage-sensor toxin in lipid membranes. Biophys. J. 99:638–646 10.1016/j.bpj.2010.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A., Tseng-Crank J., Tanouye M.A. 1988. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1:421–430 10.1016/0896-6273(88)90192-4 [DOI] [PubMed] [Google Scholar]
- Kubo Y., Baldwin T.J., Jan Y.N., Jan L.Y. 1993. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 362:127–133 10.1038/362127a0 [DOI] [PubMed] [Google Scholar]
- Labro A.J., Lacroix J.J., Villalba-Galea C.A., Snyders D.J., Bezanilla F. 2012. Molecular mechanism for depolarization-induced modulation of Kv channel closure. J. Gen. Physiol. 140:481–493 10.1085/jgp.201210817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix J.J., Bezanilla F. 2011. Control of a final gating charge transition by a hydrophobic residue in the S2 segment of a K+ channel voltage sensor. Proc. Natl. Acad. Sci. USA. 108:6444–6449 10.1073/pnas.1103397108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledwell J.L., Aldrich R.W. 1999. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113:389–414 10.1085/jgp.113.3.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.Y., MacKinnon R. 2004. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature. 430:232–235 10.1038/nature02632 [DOI] [PubMed] [Google Scholar]
- Lee H.C., Wang J.M., Swartz K.J. 2003. Interaction between extracellular Hanatoxin and the resting conformation of the voltage-sensor paddle in Kv channels. Neuron. 40:527–536 10.1016/S0896-6273(03)00636-6 [DOI] [PubMed] [Google Scholar]
- Lee S., Milescu M., Jung H.H., Lee J.Y., Bae C.H., Lee C.W., Kim H.H., Swartz K.J., Kim J.I. 2010. Solution structure of GxTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in Kv2.1 potassium channels. Biochemistry. 49:5134–5142 10.1021/bi100246u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold E., Borges A., Heinemann S.H. 2012. Scorpion β-toxin interference with NaV channel voltage sensor gives rise to excitatory and depressant modes. J. Gen. Physiol. 139:305–319 10.1085/jgp.201110720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Swartz K.J. 1998. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl. Acad. Sci. USA. 95:8585–8589 10.1073/pnas.95.15.8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Swartz K.J. 2000. Localization and molecular determinants of the Hanatoxin receptors on the voltage-sensing domains of a K(+) channel. J. Gen. Physiol. 115:673–684 10.1085/jgp.115.6.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Swartz K.J. 2001. Helical structure of the COOH terminus of S3 and its contribution to the gating modifier toxin receptor in voltage-gated ion channels. J. Gen. Physiol. 117:205–218 10.1085/jgp.117.3.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Holmgren M., Jurman M.E., Yellen G. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184 10.1016/S0896-6273(00)80357-8 [DOI] [PubMed] [Google Scholar]
- Long S.B., Tao X., Campbell E.B., MacKinnon R. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 450:376–382 10.1038/nature06265 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A.M., Ramu Y. 2001. Ion conduction pore is conserved among potassium channels. Nature. 413:809–813 10.1038/35101535 [DOI] [PubMed] [Google Scholar]
- Lu Z., Klem A.M., Ramu Y. 2002. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120:663–676 10.1085/jgp.20028696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuzzu L.M., Moronne M.M., Isacoff E.Y. 1996. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271:213–216 10.1126/science.271.5246.213 [DOI] [PubMed] [Google Scholar]
- Milescu M., Vobecky J., Roh S.H., Kim S.H., Jung H.J., Kim J.I., Swartz K.J. 2007. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 130:497–511 10.1085/jgp.200709869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu M., Bosmans F., Lee S., Alabi A.A., Kim J.I., Swartz K.J. 2009. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16:1080–1085 10.1038/nsmb.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. 1995. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 15:5–10 10.1016/0896-6273(95)90057-8 [DOI] [PubMed] [Google Scholar]
- Pathak M., Kurtz L., Tombola F., Isacoff E. 2005. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 125:57–69 10.1085/jgp.200409197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J., Scheuer T., Zheng N., Catterall W.A. 2011. The crystal structure of a voltage-gated sodium channel. Nature. 475:353–358 10.1038/nature10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E., MacKinnon R., Bezanilla F., Stefani E. 1993. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 11:353–358 10.1016/0896-6273(93)90190-3 [DOI] [PubMed] [Google Scholar]
- Phillips L.R., Swartz K.J. 2010. Position and motions of the S4 helix during opening of the Shaker potassium channel. J. Gen. Physiol. 136:629–644 10.1085/jgp.201010517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips L.R., Milescu M., Li-Smerin Y., Mindell J.A., Kim J.I., Swartz K.J. 2005. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 436:857–860 10.1038/nature03873 [DOI] [PubMed] [Google Scholar]
- Rogers J.C., Qu Y., Tanada T.N., Scheuer T., Catterall W.A. 1996. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 271:15950–15962 10.1074/jbc.271.27.15950 [DOI] [PubMed] [Google Scholar]
- Ruta V., MacKinnon R. 2004. Localization of the voltage-sensor toxin receptor on KvAP. Biochemistry. 43:10071–10079 10.1021/bi049463y [DOI] [PubMed] [Google Scholar]
- Schmalhofer W.A., Ratliff K.S., Weinglass A., Kaczorowski G.J., Garcia M.L., Herrington J. 2009. A KV2.1 gating modifier binding assay suitable for high throughput screening. Channels (Austin). 3:437–447 10.4161/chan.3.6.10201 [DOI] [PubMed] [Google Scholar]
- Schoppa N.E., Sigworth F.J. 1998a. Activation of shaker potassium channels. I. Characterization of voltage-dependent transitions. J. Gen. Physiol. 111:271–294 10.1085/jgp.111.2.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa N.E., Sigworth F.J. 1998b. Activation of Shaker potassium channels. II. Kinetics of the V2 mutant channel. J. Gen. Physiol. 111:295–311 10.1085/jgp.111.2.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa N.E., Sigworth F.J. 1998c. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 111:313–342 10.1085/jgp.111.2.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoh S.A., Sigg D., Papazian D.M., Bezanilla F. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167 10.1016/S0896-6273(00)80142-7 [DOI] [PubMed] [Google Scholar]
- Sheets M.F., Kyle J.W., Kallen R.G., Hanck D.A. 1999. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77:747–757 10.1016/S0006-3495(99)76929-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets M.F., Kyle J.W., Hanck D.A. 2000. The role of the putative inactivation lid in sodium channel gating current immobilization. J. Gen. Physiol. 115:609–620 10.1085/jgp.115.5.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.J., Alphy S., Seibert A.L., Blumenthal K.M. 2005. Differential phospholipid binding by site 3 and site 4 toxins. Implications for structural variability between voltage-sensitive sodium channel domains. J. Biol. Chem. 280:11127–11133 10.1074/jbc.M412552200 [DOI] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. 1998a. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J. Gen. Physiol. 111:399–420 10.1085/jgp.111.3.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. 1998b. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 111:421–439 10.1085/jgp.111.3.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani E., Toro L., Perozo E., Bezanilla F. 1994. Gating of Shaker K+ channels: I. Ionic and gating currents. Biophys. J. 66:996–1010 10.1016/S0006-3495(94)80881-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K.J. 2007. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 49:213–230 10.1016/j.toxicon.2006.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K.J. 2008. Sensing voltage across lipid membranes. Nature. 456:891–897 10.1038/nature07620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. 1995. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 15:941–949 10.1016/0896-6273(95)90184-1 [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. 1997a. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 18:665–673 10.1016/S0896-6273(00)80306-2 [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. 1997b. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 18:675–682 10.1016/S0896-6273(00)80307-4 [DOI] [PubMed] [Google Scholar]
- Takahashi H., Kim J.I., Min H.J., Sato K., Swartz K.J., Shimada I. 2000. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K(+) channels: common surface features of gating modifier toxins. J. Mol. Biol. 297:771–780 10.1006/jmbi.2000.3609 [DOI] [PubMed] [Google Scholar]
- Tao X., Lee A., Limapichat W., Dougherty D.A., MacKinnon R. 2010. A gating charge transfer center in voltage sensors. Science. 328:67–73 10.1126/science.1185954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas E., Bezanilla F., Roux B. 2011. In search of a consensus model of the resting state of a voltage-sensing domain. Neuron. 72:713–720 10.1016/j.neuron.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba-Galea C.A., Sandtner W., Starace D.M., Bezanilla F. 2008. S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. USA. 105:17600–17607 10.1073/pnas.0807387105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.M., Roh S.H., Kim S., Lee C.W., Kim J.I., Swartz K.J. 2004. Molecular surface of tarantula toxins interacting with voltage sensors in K(v) channels. J. Gen. Physiol. 123:455–467 10.1085/jgp.200309005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Yarov-Yarovoy V., Kahn R., Gordon D., Gurevitz M., Scheuer T., Catterall W.A. 2011. Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA. 108:15426–15431 10.1073/pnas.1112320108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster S.M., Del Camino D., Dekker J.P., Yellen G. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868 10.1038/nature02468 [DOI] [PubMed] [Google Scholar]
- Winterfield J.R., Swartz K.J. 2000. A hot spot for the interaction of gating modifier toxins with voltage-dependent ion channels. J. Gen. Physiol. 116:637–644 10.1085/jgp.116.5.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Horn R. 1995. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 15:213–218 10.1016/0896-6273(95)90078-0 [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Hoshi T., Aldrich R.W. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571 10.1126/science.2122520 [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Hoshi T., Aldrich R.W. 1994a. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103:321–362 10.1085/jgp.103.2.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta W.N., Hoshi T., Dittman J., Aldrich R.W. 1994b. Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol. 103:279–319 10.1085/jgp.103.2.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., Catterall W.A. 2011. Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 286:33641–33651 10.1074/jbc.M111.282509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., Catterall W.A. 2012. Mapping the interaction site for a β-scorpion toxin in the pore module of domain III of voltage-gated Na(+) channels. J. Biol. Chem. 287:30719–30728 10.1074/jbc.M112.370742 [DOI] [PMC free article] [PubMed] [Google Scholar]