

Abstract
The cardiovascular manifestations of Chagas disease are well known. However, the contribution of the vasculature and specifically the microvasculature has received little attention. This chapter reviews the evidence supporting the notion that alterations in the microvasculature especially in the heart contribute to the pathogenesis of chagasic cardiomyopathy. These data may also be important in understanding the contributions of the microvasculature in the aetiologies of other cardiomyopathies. The role of endothelin-1 and of thromboxane A2 vascular spasm and platelet aggregation is also discussed. Further, these observations may provide target(s) for intervention.
4.1. HISTORICAL ASPECTS
Chagas disease, caused by infection with Trypanosoma cruzi, is a cause of acute myocarditis and chronic cardiomyopathy and often associated with a vasculitis. The involvement of the vasculature in the pathogenesis of Chagas disease has not been generally appreciated. Although its involvement was described in the early years following the initial description of the parasite and the disease it caused, it remained for others to suggest an aetiologic role for the vasculature in the development of chagasic heart disease. The understanding of the contribution of vascular and, in particular, microvascular dysfunction in the pathogenesis of chagasic heart disease is important in understanding not only chagasic disease but also cardiomyopathies of other infectious and non-infectious aetiologies.
Vianna (1911) was the first to detail the pathology of Chagas disease. From the earliest description of the disease, there was a fascination with the heart so that Chagas disease became almost synonymous for chagasic heart disease or chronic chagasic cardiomyopathy. Vianna stated that the “heart is one of the viscera for which the Schizotrypanosome shows predilection both in man and in animals”. In addition, Vianna first reported vascular involvement in Chagas disease stating that “perivascular inflammations exist, some of them quite pronounced, and others barely incipient … (in the myocardium) … in many of the arterioles that irrigate the nervous substance, overt phenomena of periarteritis are found … (in the cerebellum)”. Subsequently, in autopsy specimens, Torres described alterations in the heart and considered them as unrelated to tissue parasitism but rather a result of the disruption of the coronary circulation. These alterations were also observed in experimental T. cruzi infection (Torres, 1917). In 1941, Torres also observed cardiovascular involvement and he defined these lesions as “the inflammatory cell infiltrate in the interstitial tissue of the myocardium starts at the level of and around the capillaries, and not near S. cruzi, whether the latter is exudative myocarditis related to early vascular lesions”. He also described similar lesions in the coronary arterioles of T. cruzi-infected monkeys that he considered to be ischaemic alterations in the myocardium resulting from occlusion of vessels. In 1960, Torres (1960) examined chagasic and non-chagasic human hearts and, in the former, identified marked, constriction-type irregularities with extensive myocytolysis in the intramyocardial arterioles. He suggested that the diffuse myocytolysis was caused by metabolic changes in the myocytes resulting from circulatory disorders of low intensity or short duration. Further, he suggested that the requirement for arterial blood supply was reduced resulting in areas of marked diffuse myocytolysis and extensive destruction of myocardial cells. However, this does not completely account for collapse of small arterial branches occasionally observed as these changes could also be the result of a cycle that includes passive hyperaemia, local anaemia, metabolic disturbances in myocardial fibres, myocytolysis. Several other investigators of that era described vascular lesions in Chagas disease. For example, Mazza and Benitez (1937) demonstrated amastigotes in cells of the perivascular adventitia in the conjunctiva of patients during acute infection, and Couceiro (1943) reported vascular lesions in the sciatic nerve of infected dogs and Coelho (1944) observed coronary arteriole lesions in 19 cases of chronic Chagas disease. These lesions were also detected at autopsy in coronary circulation by Ramos and Tibiriça (1945), Dias et al. (1956) and Koberle (1958).
Andrade and Andrade (1955) observed that the inflammation observed in chronic chagasic cardiomyopathy could be “allergic”, provoking ischaemic lesions of myocardium by capillary involvement. The microscopic infarctions could be responsible for alterations in the conduction system, mainly in the right branch of bundle of His, due to their preferential intramyocardial localization. They also suggested that the fibrotic lesions frequently detected at apex of the left ventricle could originate from vascular obstructions due to subendocardial parietal thrombosis. Subsequently, the “allergic phenomena” became less emphasized and the vascular changes were considered to be only congestion and marked dilatation of venules and capillaries. Brito and Vasconcelos (1959), in a study of 19 cardiac biopsies from patients with megaesophagus, detected necrotizing arteritis in nine and identified the inflammation as an “allergic phenomenon”. Vascular lesions in hearts of infected mice were also observed by Macclure and Poche (1963) using electron microscopy, and by Lucena et al. (1962) and Alencar et al. (1968) using light microscopy. Okumura et al. (1962) observed necrotizing arteritis in the myocardium and digestive tract to which they attributed an “allergic” origin. This concept was expanded when these investigators detected a parasitized endothelial cell (EC). They reported that “during the acute phase the trypanosomes may cause a focal lesion with sensitization of the vessels by an allergic mechanism, triggering hypersensitivity phenomena reflected by necrotizing arteritis”.
Jörg (1974) compared histological images obtained from a healthy heart after vascular injection of an opaque substance with those obtained in an injected heart of a patient who died of chagasic cardiomyopathy. “Decapillarization” was observed in those zones where the mesenchymal reaction was more intense. He postulated that the “angioarchitectonic” anarchy was a result of intense mesenchymal reaction secondary to the parasitic infection which led to a progressive decapillarization and a destructive loss of many meshes of the capillary net resulting in myocytolysis and destruction of cardiac ganglia. Subsequently, Jörg (1991) described vascular lesions characterized by endothelial oedema, denudation, cell accumulation and platelet–fibrin aggregation in a collecting vein of the left ventricle in a pig model of Chagas disease.
The observation that in chronic chagasic heart disease there was chronic inflammation and fibrosis and a dearth of parasites led investigators to search for a cause of the progressive pathological changes. In an effort to explain the pathology, several avenues of research were developed. Microvascular lesions in chagasic heart disease were described in the 1980s by Rossi et al. (1984) and Factor et al. (1985). The involvement of microvasculature in the pathogenesis of chronic chagasic heart disease was further underscored by Rossi (1990). It should be noted that at this time, autoimmunity and disturbances in the cardiac anatomic nervous system were being intensely investigated (Acosta and Santos-Buch, 1985; Koberle, 1968; Oliveira, 1985; Ribeiro-dos-Santos and Rossi, 1985). In this regard, a relationship between cardiac autonomic nervous system abnormalities and sudden cardiac death has been demonstrated (Rossi and Bestetti, 1995). A relationship between cardiac autonomic nervous system abnormities and sudden death has been demonstrated. Malignant ventricular tachyarrhythmias such as ventricular tachycardia and fibrillation are major causes of sudden death among patients with chronic chagasic cardiomyopathy. We are now aware that parasite persistence is present in the cardiovascular system and in other organs even though it is not obvious by histological examination (Combs et al., 2005; Zhang and Tarleton, 1999) and that this is a major contributor to the chronic disease.
4.2. SMALL ANIMAL STUDIES OF THE MICROCIRCULATION IN TRYPANOSOMA CRUZI INFECTION
BALB/c mice immunized with epimastigotes of the avirulent PF strain of T. cruzi and challenged with trypomastigotes of the virulent Colombian strain developed a cardiomyopathy similar to that observed in human chronic chagasic cardiomyopathy including the development of an apical aneurysm (Rossi et al., 1984). Histological examination revealed focal areas of myocytolysis, necrosis and myocardial degeneration associated with a lymphomononuclear inflammatory infiltrate accompanied by interstitial fibrosis and occasional parasite pseudocysts. Additionally, platelet aggregates forming transient occlusive thrombi were observed in small epicardial and intramyocardial vessels. The focal nature of the myocardial lesion and the type of myonecrosis indicated involvement of the microcirculation.
A/J mice infected withthe Brazil strain and perfused with silicone rubber (Microfil) 15–17 days post-infection revealed numerous areas of focal vascular constriction, microaneurysm formation, vascular dilatation and proliferation of microvessel (Factor et al., 1985) which is similar to the observations in the Syrian cardiomyopathic hamster and in human cardiomyopathies of other aetiologies (Sonnenblick et al., 1985). In that model, the administration of verapamil ameliorated the microvascular alterations and the myocardial pathology. Similarly, in the Brazil strain-infected CD-1 mouse, verapamil ameliorated the myocardial pathology when verapamil was administered early but not late infection (Chandra et al., 2002; De Souza et al., 2004; Morris et al., 1989). Verapamil increases coronary blood flow, inhibits platelet aggregation and contributes to the amelioration of the pathology. These observations were corroborated by direct in vivo visualization utilizing a surrogate murine model, that is, the cremaster microvascular bed (Tanowitz et al., 1996). Direct observation of the effects of T. cruzi infection on microcirculatory flow in vivo and quantitative measurement of parameters such as the velocity of red blood cell flow (Vrbc) and vessel diameter were provided. When the cremaster model was examined 20–25 days post-infection in male CD-1 mice infected with the Brazil strain, a significant decrease in Vrbc, reversed by verapamil treatment, was observed in the first- and third-order arterioles and venules accompanied by an attenuation of the inflammation. The arterioles of the infected mice exhibited segmental areas of vasospasm and dilatation, possibly the initiating event in microaneurysm formation (Tanowitz et al., 1996; Fig. 4.1).
FIGURE 4.1.
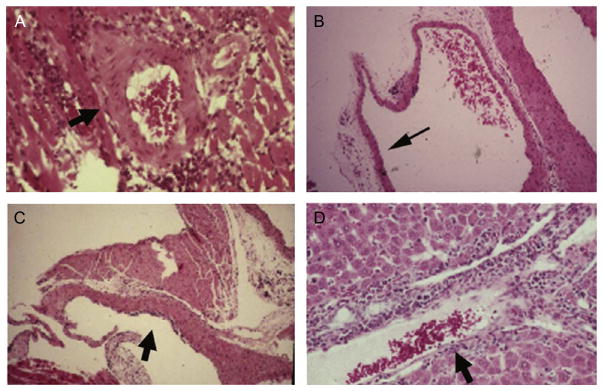
(A) Images obtained from T. cruzi-infected mouse. Perivascular inflammation. (B) Vasculitis of the pulmonary vasculature. (C) Endothelialitis of the subendocardium. (D) Vasculitis of a blood vessel in the liver (images from Petkova et al., 2001).
The infection of mice with T. cruzi caused a vasculitis. There was a gradual reduction in coronary flow in infected mice over time giving further credence to the notion that there was vascular dysfunction in experimental Chagas disease (Tanowitz, 1992b). Importantly, amastigotes are evident in the coronary microvascular ECs early in infection before parasitaemia can be detected, suggesting that the coronary endothelium could be an initial target of infection (Factor et al., 1985). Acutely infected rats developed changes in the endothelial layer characterized by EC swelling and a few points of cytoplasmic discontinuity that appeared as holes exposing the subendothelial collagen that is usually associated with platelet–fibrin aggregates, which might affect the generation of vasoactive substances, and impairs the equilibrium between opposing forces (Rossi, 1997). In vitro and in vivo studies indicate that infection of the endothelium results in expression of both pro-inflammatory cytokines and vascular adhesion molecules, which are important components of the inflammatory response (Huang et al., 1999a,b; Tanowitz et al., 1992a,b). Infection of ECs activates NF-κB and likely contributing to the induction of cytokine and adhesion molecular expression in the endothelium (Huang et al., 1999a). Further, in the myocardium obtained from T. cruzi-infected humans and experimental animals, increased expression of cytokines, nitric oxide synthases and adhesion molecules has been reported (Huang et al., 1999a; Laucella et al., 1996; Reis et al., 1993; Fig. 4.2).
FIGURE 4.2.
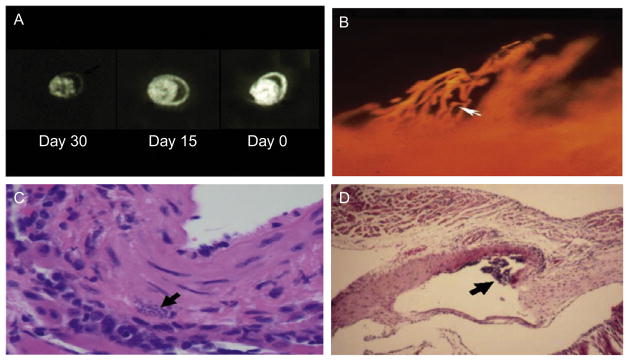
(A) Coronary perfusion of mouse hearts as determined by autoradiographic imaging utilizing the fatty acid analog 19-iodo-3,3,-dimethyl-18 nonadecenoic acid (DMIVM). A: uninfected normal mouse with normal perfusion. B: perfusion in a mouse infected for 15 days. Note the reduced perfusion C: perfusion in a mouse infected for 30 days demonstrating a marked reduction in perfusion (taken from Tanowitz, 1992). (B) Microfil injection of the coronary vasculature of an A/J mice 15 days post-infection with the Tulahuen strain of T. cruzi demonstrating a section through the subendocardium of the atrium showing saccular microaneurysms and vasospasm (Rossi et al., 2010). (C) Pseudocyst in the wall of a blood vessel (Tanowitz et al., 2009). (D) Vasculitis of a large blood vessel obtained from an infected mouse (Tanowitz et al., 2009).
Taken together, all of aforementioned studies in experimental animals strongly suggest that the vasospasm of the branches of the coronary microcirculation leads to a reduction in blood flow and ischaemia to a small area of the myocardium subserved by that microvessel which resulted in a microinfarct. When this process is repeated over a period of time in different areas of the heart, these areas may coalesce and lead to falling out of cardiac myocytes and replacement by fibrous tissue. The focal but widespread nature of the pathology supports, in part, this hypothesis.
4.3. STUDIES IN DOGS
Dogs have been used in investigations of Chagas disease because they are a larger animal than the standard mouse model and may better recapitulate the human disease. Hearts obtained from dogs sacrificed 18–26 days after intraperitoneal inoculation with the 12SF strain of T. cruzi demonstrated myocarditis characterized by small focal areas of lesion and myocytic necrosis associated with interstitial mononuclear infiltration. Electron microscopic studies revealed degenerative changes in the ECs in contact with T lymphocytes, as well as platelet aggregates and fibrin thrombi in the intramyocardial capillaries. These alterations suggested that a possible interaction between ECs and effector immune cells might play an important role in the pathogenesis of the myocellular lesion and of the observed microangiopathy (Andrade et al., 1994). More recently, Melo et al. (2011) demonstrated that the administration of simvastatin ameliorated the cardiac remodelling in a canine model of chronic chagasic heart disease by histological and functional criteria. Importantly, statins have been demonstrated to inhibit platelet aggregation (Lee et al., 2010) and reduce the inflammation in the vasculature (Liu et al., 2009), thus increasing coronary blood flow in some studies (Brands et al., 1991).
4.4. VASOACTIVE PEPTIDES AND EICOSANOIDS
Endothelin-1 (ET-1), a 21-amino acid peptide (Yanagisawa et al., 1988), was originally described as a powerful vasoconstrictor secreted by endothelial cells (ECs). T. cruzi infection of ECs results in a dramatic increase in biologically active ET-1. However, other cell types have found to be sources of ET-1 such as cardiac myocytes, fibroblasts, astrocytes and macrophages (Kedzierski and Yanagisawa, 2001). The synthesis of ET-1 is mediated by endothelin-converting enzyme (ECE) which converts Big ET-1 (31 amino acids) to ET-1. The actions of ET-1 are mediated by the G-protein-coupled endothelin receptors ETA and ETB. Although ET-1 is constitutively expressed in many cells, increased synthesis has been associated with many disease states such as malignant hypertension, primary pulmonary hypertension, CHF, sepsis, meningitis, eclampsia and subarachnoid haemorrhage (Kedzierski and Yanagisawa, 2001). Increased expression/synthesis of ET-1 has been implicated in the pathogenesis of cerebral malaria (Machado et al., 2006) and chagasic cardiomyopathy (Petkova et al., 2000, 2001; Tanowitz et al., 2005).
Eicosanoids are lipid mediators that participate in many biological activities including vascular tone, inflammation, ischaemia and tissue homeostasis (Haeggstrom et al., 2010). The biosynthetic pathways in mammals for these important biological mediators are dependent upon liberation of arachidonic acid for the inner leaflet of the plasma membrane. Thromboxane A2 (TXA2), an eicosanoid generated during arachidonic acid metabolism, is the most potent vasoconstrictor known and acts via its receptors TPα and its splice variant TPβ, both of which are expressed on human ECs. Several parasitic organisms produce eicosanoids which may modulate host response and the progress of an infection (Belley and Chadee, 1995; Kubata et al., 1998, 2000; Liu and Weller, 1990; Noverr et al., 2003).
Thus, the observation in experimental animals and humans regarding vasospasm and platelet aggregation and thrombi in the coronary microcirculation was reminiscent of the actions of TXA2. Tanowitz et al. (1990) observed that there was increased platelet aggregation in infected mice accompanied by an increase in plasma TXA2. The increased levels of TXA2 could explain the vascular spasm and the platelet aggregation (Tanowitz et al., 1990). Ashton et al. (2007), 17 years later, demonstrated that T. cruzi was capable of synthesizing TXA2. It was further demonstrated that the majority of TXA2 detected in the blood of infected mice is parasite derived. These observations suggest that TXA2 could contribute to the pathogenesis of chronic chagasic cardiomyopathy and its clinical manifestations. More recently, on the basis of these observations, Mukherjee et al. (2011) administered aspirin (ASA) to T. cruzi (Brazil strain)-infected mice. There was a reduction in the plasma levels of TXA2. ASA inhibits the mammalian COX-1 enzyme thus reducing the levels of PGH2 available for the synthesis of TXA2. Thus, we believe that ASA treatment of the infected host decreases the ability of the parasite to scavenge PGH2 from the host to synthesize TXA2. In addition, ASA-treated infected mice suffer a high parasitaemia and mortality. This effect of ASA is a result of “off-target” factors unrelated to TXA2. It may suggest that caution should be used in the treatment of fever and pain with ASA during acute infection.
TXA2 and ET-1 share several important properties important in the pathogenesis of Chagas disease. They both cause vasoconstriction and platelet aggregation. Additionally, they are both pro-inflammatory. Mice infected with T. cruzi display an increased expression of ET-1 protein and mRNA in the myocardium and an increase in plasma ET-1 levels (Petkova et al., 2000). Treatment of infected mice with phosphoramidon, an inhibitor of ECE, reduced T. cruzi-infection-induced right ventricular dilation (Tanowitz et al., 2005). T. cruzi infection of mice in which the gene for ET-1 is deleted either in cardiac myocytes or in ECs ameliorated cardiac remodelling as demonstrated by histopathology, echocardiography and cardiac MRI (Tanowitz et al., 2005). Elevated plasma levels of ET-1 have been demonstrated in patients with chronic chagasic cardiomyopathy (Salomone et al., 2001). However, it is unclear if this is a result of congestive heart failure in general or chagasic cardiomyopathy in particular. It is important to note that Hassan et al. (2006) found increased expression of ET-1 in the carotid arteries of infected mice. This observation clearly demonstrated the importance of ET-1 in the vasculature of infected mice and by implication in infected humans. The release of platelet-activating factor by macrophages in this infection causes transient ischaemia and myocytolytic necrosis (Talvani et al., 2003; see Chapter 1 for a discussion of eicosanoids and Chapter 5 for a discussion of the role of bradykinin and bradykinin receptors).
4.5. IN VITRO STUDIES
Direct infection of human ECs in culture with T. cruzi resulted in the alteration of various critical biochemical processes responsible for the maintenance of microvascular perfusion, such as calcium homeostasis and generation of inositol trisphosphate (IP3), ET-1, TXA2 and prostacyclin (PG12) which is a vasodilator and inhibits platelet aggregation (Morris et al., 1988). EC infection also resulted in alterations of cyclic AMP metabolism, which plays a protective role against the direct and/or indirect lesion caused by the adhesion and aggregation of circulating platelets to ECs (Morris et al., 1992).
Inflammatory cells contribute to microvascular hypoperfusion by secreting cytokines and other factors known to affect platelets and ECs. Infection of cultured ECs results in increased synthesis of interleukin-1β (IL-1β), IL-6 and colony-stimulating factor 1 (CSF-1) which may result in altered function (Tanowitz et al., 1992a). IL-1β is elaborated by activated macrophages and by peripheral blood mononuclear cells, including those infected with T. cruzi, and by a variety of other cell types, such as ECs (Van Voorhis, 1992). The antithrombotic properties of ECs may be altered by IL-1β. This cytokine may reduce tissue production of the plasminogen activator and increase production of the inhibitor of this activator, which may result in thrombus formation (Bevilacqua et al., 1984; Nachman et al., 1986). CSF-1 is an important growth factor needed for the proliferation and maturation of cells of the mononuclear lineage (Mantovani et al., 1990). It is also important in recruitment, possibly acting in conjunction with IL-1β. High CSF-1 levels have been detected in infected cultured ECs. These observations may reflect the growth of the monocyte population in the microvasculature resulting in the synthesis of pro-inflammatory cytokines (Mantovani et al., 1990; Tanowitz et al., 1992a). In addition, trypomastigotes have been demonstrated to produce neuraminidase (trans-sialidase) that may be involved in the removal of sialic acid from the surface of mammalian myocardial cells and ECs, facilitating thrombin binding. The loss of this endothelial surface protector molecule could contribute to platelet aggregation and thrombosis within the small coronary vessels (Libby et al., 1986). These factors acting together may ultimately result in spasm and thrombosis in the small coronary vessels, inducing focal myocardial damage.
Mukherjee et al. (2004) examined infected human ECs which resulted in activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) but not c-Jun N-terminal kinase or p38 MAPK. Treatment of these cells with the MAPK kinase inhibitor PD98059 prior to infection blocked the increase in phosphorylated ERK1/2 observed with infection. Transfection with dominant-negative Raf(301) or Ras(N17) constructs reduced the infection-associated levels of phospho-ERK1/2, indicating that the activation of ERK1/2 involved the Ras–Raf–ERK pathway. Infection also resulted in an increase in activator protein 1 (AP-1) activity, which was inhibited by transfection with a dominant-negative Raf(301) construct. Infected ECs were found to synthesize ET-1 and IL-1β, which activated ERK1/2 and induced cyclin D1 expression in uninfected smooth muscle cells. More recently, Tonelli et al. (2010) demonstrated that T. cruzi gp85/trans-sialidase surface protein family is important in the attachment of the parasite to the host cells.
Taken together, these data suggest a possible molecular paradigm for the pathogenesis of the vasculopathy in this infection.
4.6. STUDIES IN HUMANS
Anatomical studies have shown structural derangement and rarefied microvasculature in the left ventricular myocardium. A histotopographical study comparing the microcirculatory system after injection of an opaque medium into chagasic and control human hearts demonstrated focal decapillarization in chronic Chagas disease due to extraluminal compression, suggesting that this might be the cause of focal myocytolytic necrosis ( Jörg, 1974). Similarly, a post-mortem radiological study of chagasic hearts revealed vascular changes at the heart apex characterized by distorted and/or scarce vessels associated with decreased arterial density, presumably related to the pathogenesis of apical aneurysm (Ferreira et al., 1980).
Patients with Chagas disease may exhibit symptoms that are atypical for classic angina pectoris. Although symptoms suggestive of myocardial ischaemia are present, coronary angiographic studies show normal or nearly normal coronary arteries in more than 90% of patients studied (Marin-Neto et al., 1992). Patients specifically selected on the basis of chest pain did show perfusion abnormalities detectable by thallium-201 scintigraphy, suggesting that myocardial ischaemia may be due to alterations in the microvasculature. Abnormal perfusion in different groups of chagasic patients has been confirmed using isonitrile-99m-technetium (Castro et al., 1988) or thallium-201 (Hagar and Rahimtoola, 1991; Marin-Neto et al., 1992). Myocardial capillary blood flow in chronic chagasic patients with no significant clinical or electrocardiographic manifestations proved to be markedly reduced when evaluated with rubidium-86, while the major coronary vessels appeared normal. The reduction observed is comparable to that exhibited by a group of non-chagasic patients with obstructive coronary disease (Kuschnir et al., 1974a,b). Vasospasm has been proposed in the genesis of myocardial ischaemia in patients with chronic chagasic cardiomyopathy (Vianna et al., 1979). For example, it was demonstrated that in patients with chagasic cardiomyopathy, there is an abnormal, endothelium-dependent, coronary-vasodilating mechanism as demonstrated by acetylcholine and adenosine infusion into the left coronary artery, suggesting that epicardial and microvascular coronary reactivity may be altered in these patients. The clinical importance of this alteration awaits elucidation. However, this abnormality of the coronary microvasculature may contribute to the genesis of the symptoms related to the ischaemic processes observed in chronic chagasic patients and to acute myocardial infarction in the absence of significant coronary damage (Torres et al., 1995).
Biopsies of chronic chagasic hearts revealed a marked thickening of the basement membrane in most myocytes and capillaries (Ferrans et al., 1988). These alterations are similar to the thickening reported for the basement membranes of myocardial capillaries in other cardiomyopathies (Factor et al., 1983). A very well developed capillary network has been observed in chagasic human hearts using a cell-maceration scanning electron microscopic method (Higuchi et al., 1999). This network may result in reduced flow of blood thus contributing to the hypoxic changes observed in chronic chagasic cardiomyopathy. Significant dilatations of arterioles and capillaries in ventricular areas of chagasic hearts compared to hearts with dilated cardiomyopathy were described. These microcirculatory dilatations could be responsible for a reduction in blood flow distribution in the watershed area lying between the two main coronary flow sources (the anterior- and posterior-descending arteries, and the right and circumflex coronary arteries). These findings could result in ischaemia and extensive fibrosis within the left ventricle apical and posterior regions (Higuchi et al., 1999).
The relation of regional sympathetic denervation and myocardial perfusion disturbance to wall motion impairment was described in patients with chronic chagasic cardiomyopathy. Global left ventricular function, segmental wall motion analysis and myocardial perfusion were evaluated in 58 patients. There were myocardial perfusion defects in the absence of epicardial coronary artery disease, and the extension and severity of perfusion abnormalities paralleled the progression of myocardial damage. These observations support the notion that perfusion disturbances in chronic chagasic cardiomyopathy may be caused by transient disturbances of coronary blood flow regulation at the microvascular level (Simoes et al., 2000). The same group correlated the clinical, electrocardiographic, angiographic, electrophysiologic and wall motion/myocardial perfusion disturbances in chronic chagasic patients with either sustained or non-sustained ventricular tachycardia. The fact that both fixed perfusion defects (which reflect local fibrosis) and reversible and paradoxical defects predominate in the arrhythmias in the left ventricular region is also compatible with the hypothesis that microvascular ischaemia is aetiologic. Thus, several observations suggest that in human chagasic heart disease, transient disturbances of coronary blood flow regulation at the level of the microvasculature may result in regional myocardial degeneration, with a consequent reparative fibrosis that ultimately constitutes the substrate for re-entrant circuits and the appearance of both sustained and non-sustained ventricular tachycardia (Sarabanda et al., 2005).
4.7. CONCLUSIONS
Abnormalities in the coronary circulations were observed since the earliest studies by Vianna and Torres conducted soon after the discovery by Carlos Chagas of the disease that bears his name. Since then, much information has accumulated from attempts to define the physiopathology of chagasic heart disease. The changes observed both on humans and in experimental models of T. cruzi infection suggest that myocardial lesions are multifactorial including parasite persistence, autoimmunity and microvascular involvement. Importantly, they are not mutually exclusive.
Acknowledgments
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (M. A. R.; FAPESP 09/17787-8; 10/19216-5) and National Institutes of Health (NIH) Grants AI-076248 (H. B. T.) and CA-123334 (L. A. J.). C. M. P. was supported in part by a grant from the Fogarty International Center–NIH (D43-TW007129). M. A. R. is senior investigator of the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
References
- Acosta AM, Santos-Buch CA. Autoimmune myocarditis induced by Trypanosoma cruzi. Circulation. 1985;71:1255–1261. doi: 10.1161/01.cir.71.6.1255. [DOI] [PubMed] [Google Scholar]
- Alencar A, de Karsten MRQ, Cerqueira MC. Estudo do sistema nervoso autônomo do aparelho digestivo em camundongos albinos cronicamente infectados pelo Schizotrypanum cruzi. Hospital (Rio J) 1968;73:165–176. [PubMed] [Google Scholar]
- Andrade ZA, Andrade SG. Pathogenesis of Chagas’ chronic myocarditis; importance of ischemic lesions. Arq Bras Med. 1955;45:279–288. [PubMed] [Google Scholar]
- Andrade ZA, Andrade SG, Correa R, Sadigursky M, Ferrans VJ. Myocardial changes in acute Trypanosoma cruzi infection. Ultrastructural evidence of immune damage and the role of microangiopathy. Am J Pathol. 1994;144:1403–1411. [PMC free article] [PubMed] [Google Scholar]
- Ashton AW, Mukherjee S, Nagajyothi FN, Huang H, Braunstein VL, Desruisseaux MS, et al. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J Exp Med. 2007;204:929–940. doi: 10.1084/jem.20062432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belley A, Chadee K. Eicosanoid production by parasites: from pathogenesis to immunomodulation? Parasitol Today. 1995;11:327–334. doi: 10.1016/0169-4758(95)80185-5. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA., Jr Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med. 1984;160:618–623. doi: 10.1084/jem.160.2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands MW, Hildebrandt DA, Mizelle HL, Hall JE. Sustained hyperinsulinemia increases arterial pressure in conscious rats. Am J Physiol. 1991;260:R764–R768. doi: 10.1152/ajpregu.1991.260.4.R764. [DOI] [PubMed] [Google Scholar]
- Brito T, Vasconcelos E. Necrotizing arteritis in megaesophagus. Histopathology of ninety-one biopsies taken from the cardiac. Rev Inst Med Trop São Paulo. 1959;1:195–206. [Google Scholar]
- Castro R, Kuschnir E, Sgammini H. Evaluacion de la performance cardiaca y perfusion miocardica con radiotrazadores en la cardiopatia chagasica cronica. Rev Argent Cardiol. 1988;17:226–231. [Google Scholar]
- Chandra M, Shirani J, Shtutin V, Weiss LM, Factor SM, Petkova SB, et al. Cardioprotective effects of verapamil on myocardial structure and function in a murine model of chronic Trypanosoma cruzi infection (Brazil Strain): an echocardiographic study. Int J Parasitol. 2002;32:207–215. doi: 10.1016/s0020-7519(01)00320-4. [DOI] [PubMed] [Google Scholar]
- Coelho NA. Aspecto anatomopatológico do coração na moléstia de Chagas. Rev Med Chirurg S Paulo. 1944;4:209–211. [Google Scholar]
- Combs TP, Nagajyothi F, Mukherjee S, de Almeida CJ, Jelicks LA, Schubert W, et al. The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem. 2005;280:24085–24094. doi: 10.1074/jbc.M412802200. [DOI] [PubMed] [Google Scholar]
- Couceiro A. Lesões do ciático na infecção experimental de cães pelo Schizotrypanum cruzi (nota prévia) Mem Inst Oswaldo Cruz (Rio J) 1943;39:435–439. [Google Scholar]
- De Souza AP, Tanowitz HB, Chandra M, Shtutin V, Weiss LM, Morris SA, et al. Effects of early and late verapamil administration on the development of cardiomyopathy in experimental chronic Trypanosoma cruzi (Brazil strain) infection. Parasitol Res. 2004;92:496–501. doi: 10.1007/s00436-004-1080-1. [DOI] [PubMed] [Google Scholar]
- Dias E, Laranja FS, Miranda A, Nobrega G. Chagas’ disease; a clinical, epidemiologic, and pathologic study. Circulation. 1956;14:1035–1060. doi: 10.1161/01.cir.14.6.1035. [DOI] [PubMed] [Google Scholar]
- Factor SM, Minase T, Bhan R, Wolinsky H, Sonnenblick EH. Hypertensive diabetic cardiomyopathy in the rat: ultrastructural features. Virchows Arch A Pathol Pathol Anat. 1983;398:305–317. doi: 10.1007/BF00583587. [DOI] [PubMed] [Google Scholar]
- Factor SM, Cho S, Wittner M, Tanowitz H. Abnormalities of the coronary micro-circulation in acute murine Chagas’ disease. Am J Trop Med Hyg. 1985;34:246–253. doi: 10.4269/ajtmh.1985.34.246. [DOI] [PubMed] [Google Scholar]
- Ferrans VJ, Milei J, Tomita Y, Storino RA. Basement membrane thickening in cardiac myocytes and capillaries in chronic Chagas’ disease. Am J Cardiol. 1988;61:1137–1140. doi: 10.1016/0002-9149(88)90148-8. [DOI] [PubMed] [Google Scholar]
- Ferreira CS, Lopes ER, Chapadeiro E, de Almeida HO, de Souza WF, de Silva Neto IJ. Coronariografia post mortem na cardite chagàsica crônica: correlação anátomo-radiológica. Arq Bras Cardiol (Sao Paulo) 1980;34:81–86. [PubMed] [Google Scholar]
- Haeggstrom JZ, Rinaldo-Matthis A, Wheelock CE, Wetterholm A. Advances in eicosanoid research, novel therapeutic implications. Biochem Biophys Res Commun. 2010;396:135–139. doi: 10.1016/j.bbrc.2010.03.140. [DOI] [PubMed] [Google Scholar]
- Hagar JM, Rahimtoola SH. Chagas’ heart disease in the United States. N Engl J Med. 1991;325:763–768. doi: 10.1056/NEJM199109123251103. [DOI] [PubMed] [Google Scholar]
- Hassan GS, Mukherjee S, Nagajyothi F, Weiss LM, Petkova SB, de Almeida CJ, et al. Trypanosoma cruzi infection induces proliferation of vascular smooth muscle cells. Infect Immun. 2006;74:152–159. doi: 10.1128/IAI.74.1.152-159.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi ML, Fukasawa S, De Brito T, Parzianello LC, Bellotti G, Ramires JA. Different microcirculatory and interstitial matrix patterns in idiopathic dilated cardiomyopathy and Chagas’ disease: a three dimensional confocal microscopy study. Heart. 1999;82:279–285. doi: 10.1136/hrt.82.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, Wittner M, et al. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun. 1999a;67:5434–5440. doi: 10.1128/iai.67.10.5434-5440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chan J, Wittner M, Jelicks LA, Morris SA, Factor SM, et al. Expression of cardiac cytokines and inducible form of nitric oxide synthase (NOS2) in Trypanosoma cruzi-infected mice. J Mol Cell Cardiol. 1999b;31:75–88. doi: 10.1006/jmcc.1998.0848. [DOI] [PubMed] [Google Scholar]
- Jörg ME. Tripanosomiasis cruzi; anarquía angiotopográfica por descapilarización mesenquimorreactiva: cofactor patogénico de la miocardiopatia crónica. Prensa Méd Argent. 1974;61:94–106. [Google Scholar]
- Jörg ME. Reactividad de endotelios vasculares frente a antigenos del Trypanosoma cruzi. C M Publ Cient (Mar del Plata) 1991;4:134–143. [Google Scholar]
- Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- Koberle F. Strength conditions of the left and right halves of the heart. Cardiologia. 1958;33:384–394. [PubMed] [Google Scholar]
- Koberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol. 1968;6:63–116. doi: 10.1016/s0065-308x(08)60472-8. [DOI] [PubMed] [Google Scholar]
- Kubata BK, Eguchi N, Urade Y, Yamashita K, Mitamura T, Tai K, et al. Plasmodium falciparum produces prostaglandins that are pyrogenic, somnogenic, and immunosuppressive substances in humans. J Exp Med. 1998;188:1197–1202. doi: 10.1084/jem.188.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubata BK, Duszenko M, Kabututu Z, Rawer M, Szallies A, Fujimori K, et al. Identification of a novel prostaglandin f(2alpha) synthase in Trypanosoma brucei. J Exp Med. 2000;192:1327–1338. doi: 10.1084/jem.192.9.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschnir E, Kustich F, Epelman M, Santamarina N, Podio RB. Valoración del flujo miocárdico con Rb 86, en pacientes con cardiopatia chagasica, con insuficiencia coronaria y en controles normales. Parte 1: estudios basales. Arq Bras Cardiol (Sao Paulo) 1974a;27:187–196. [Google Scholar]
- Kuschnir E, Kustich F, Epelman M, Santamarina N, Podio RB. Valoración del flujo miocárdico con Rb 86, en pacientes con cardiopatia chagasica, con insuficiencia coronaria y en controles normales. Parte 2: respuesta al ejercicio y la cardiotonificación aguda. Arq Bras Cardiol (Sao Paulo) 1974b;27:721–732. [Google Scholar]
- Laucella S, De Titto EH, Segura EL, Orn A, Rottenberg ME. Soluble cell adhesion molecules in human Chagas’ disease: association with disease severity and stage of infection. Am J Trop Med Hyg. 1996;55:629–634. doi: 10.4269/ajtmh.1996.55.629. [DOI] [PubMed] [Google Scholar]
- Lee YM, Chen WF, Chou DS, Jayakumar T, Hou SY, Lee JJ, et al. Cyclic nucleotides and mitogen-activated protein kinases: regulation of simvastatin in platelet activation. J Biomed Sci. 2010;17:45. doi: 10.1186/1423-0127-17-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Alroy J, Pereira ME. A neuraminidase from Trypanosoma cruzi removes sialic acid from the surface of mammalian myocardial and endothelial cells. J Clin Invest. 1986;77:127–135. doi: 10.1172/JCI112266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LX, Weller PF. Arachidonic acid metabolism in filarial parasites. Exp Parasitol. 1990;71:496–501. doi: 10.1016/0014-4894(90)90076-o. [DOI] [PubMed] [Google Scholar]
- Liu M, Wang F, Wang Y, Jin R. Atorvastatin improves endothelial function and cardiac performance in patients with dilated cardiomyopathy: the role of inflammation. Cardiovasc Drugs Ther. 2009;23:369–376. doi: 10.1007/s10557-009-6186-3. [DOI] [PubMed] [Google Scholar]
- Lucena DT, Carvalho JAM, Abath EC, Amorim N. Terapêutica experimental da doença de Chagas. I Ação de uma 8-aminoquinoleina em ratos albinos. Hospital (Rio J) 1962;62:1278–1296. [Google Scholar]
- Macclure E, Poche R. Microscopia eletrônica da miocardite chagásica experimental em camundongos albinos. Int Cong Trop Med Malar. 1963;2:249–250. [Google Scholar]
- Machado FS, Desruisseaux MS, Nagajyothi F, Kennan RP, Hetherington HP, Wittner M, et al. Endothelin in a murine model of cerebral malaria. Exp Biol Med. 2006;231:1176–1181. [PubMed] [Google Scholar]
- Mantovani A, Sica A, Colotta F, Dejana E. The role of cytokines as communication signals between leukocytes and endothelial cells. Prog Clin Biol Res. 1990;349:343–353. [PubMed] [Google Scholar]
- Marin-Neto JA, Marzullo P, Marcassa C, Gallo Junior L, Maciel BC, Bellina CR, et al. Myocardial perfusion abnormalities in chronic Chagas’ disease as detected by thallium-201 scintigraphy. Am J Cardiol. 1992;69:780–784. doi: 10.1016/0002-9149(92)90505-s. [DOI] [PubMed] [Google Scholar]
- Mazza S, Benitez C. Comprobación de la natureza esquizotripanósica y frecuencia de la dacrioretinitis en la enfermedad de Chagas. MEPRA. 1937;31:3–31. [Google Scholar]
- Melo L, Caldas IS, Azevedo MA, Goncalves KR, da Silva do Nascimento AF, Figueiredo VP, et al. Low doses of simvastatin therapy ameliorate cardiac inflammatory remodeling in Trypanosoma cruzi-infected dogs. Am J Trop Med Hyg. 2011;84:325–331. doi: 10.4269/ajtmh.2011.10-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Tanowitz H, Hatcher V, Bilezikian JP, Wittner M. Alterations in intracellular calcium following infection of human endothelial cells with Trypanosoma cruzi. Mol Biochem Parasitol. 1988;29:213–221. doi: 10.1016/0166-6851(88)90076-x. [DOI] [PubMed] [Google Scholar]
- Morris SA, Weiss LM, Factor S, Bilezikian JP, Tanowitz H, Wittner M. Verapamil ameliorates clinical, pathologic and biochemical manifestations of experimental chagasic cardiomyopathy in mice. J Am Coll Cardiol. 1989;14:782–789. doi: 10.1016/0735-1097(89)90126-5. [DOI] [PubMed] [Google Scholar]
- Morris SA, Tanowitz H, Makman M, Hatcher VB, Bilezikian JP, Wittner M. Trypanosoma cruzi: alteration of cAMP metabolism following infection of human endothelial cells. Exp Parasitol. 1992;74:69–76. doi: 10.1016/0014-4894(92)90140-6. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Huang H, Petkova SB, Albanese C, Pestell RG, Braunstein VL, et al. Trypanosoma cruzi infection activates extracellular signal-regulated kinase in cultured endothelial and smooth muscle cells. Infect Immun. 2004;72:5274–5282. doi: 10.1128/IAI.72.9.5274-5282.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Machado FS, Huang H, Oz HS, Jelicks LA, Prado CM, et al. Aspirin treatment of mice infected with Trypanosoma cruzi and implications for the pathogenesis of Chagas disease. PLoS One. 2011;6:e16959. doi: 10.1371/journal.pone.0016959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman RL, Hajjar KA, Silverstein RL, Dinarello CA. Interleukin 1 induces endothelial cell synthesis of plasminogen activator inhibitor. J Exp Med. 1986;163:1595–1600. doi: 10.1084/jem.163.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noverr MC, Erb-Downward JR, Huffnagle GB. Production of eicosanoids and other oxylipins by pathogenic eukaryotic microbes. Clin Microbiol Rev. 2003;16:517–533. doi: 10.1128/CMR.16.3.517-533.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura M, Silva AC, Correa Neto A. Contribuição para o estudo da patogenia das lesões vasculares na doença de Chagas experimental em camundongos brancos. Rev Paulista Med (Sao Paulo) 1962;61:265–266. [Google Scholar]
- Oliveira JS. A natural human model of intrinsic heart nervous system denervation: Chagas’ cardiopathy. Am Heart J. 1985;110:1092–1098. doi: 10.1016/0002-8703(85)90222-4. [DOI] [PubMed] [Google Scholar]
- Petkova SB, Tanowitz HB, Magazine HI, Factor SM, Chan J, Pestell RG, et al. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc Pathol. 2000;9:257–265. doi: 10.1016/s1054-8807(00)00045-4. [DOI] [PubMed] [Google Scholar]
- Petkova SB, Huang H, Factor SM, Pestell RG, Bouzahzah B, Jelicks LA, et al. The role of endothelin in the pathogenesis of Chagas’ disease. Int J Parasitol. 2001;31:499–511. doi: 10.1016/s0020-7519(01)00168-0. [DOI] [PubMed] [Google Scholar]
- Ramos JJ, Tibiriça PQT. Miocardite crônica na moléstia de Chagas. Rev Bras Med. 1945;2:1–9. [Google Scholar]
- Reis DD, Jones EM, Tostes S, Lopes ER, Chapadeiro E, Gazzinelli G, et al. Expression of major histocompatibility complex antigens and adhesion molecules in hearts of patients with chronic Chagas’ disease. Am J Trop Med Hyg. 1993;49:192–200. doi: 10.4269/ajtmh.1993.49.192. [DOI] [PubMed] [Google Scholar]
- Ribeiro-dos-Santos R, Rossi MA. Imunopatologia. Fundação Carlos Chagas; Belo Horizonte, Brazil: 1985. [Google Scholar]
- Rossi MA. Microvascular changes as a cause of chronic cardiomyopathy in Chagas’ disease. Am Heart J. 1990;120:233–236. doi: 10.1016/0002-8703(90)90191-y. [DOI] [PubMed] [Google Scholar]
- Rossi MA. Aortic endothelial cell changes in the acute septicemic phase of experimental Trypanosoma cruzi infection in rats: scanning and transmission electron microscopic study. Am J Trop Med Hyg. 1997;57:321–327. doi: 10.4269/ajtmh.1997.57.321. [DOI] [PubMed] [Google Scholar]
- Rossi MA, Bestetti RB. The challenge of chagasic cardiomyopathy. The pathologic roles of autonomic abnormalities, autoimmune mechanisms and microvascular changes, and therapeutic implications. Cardiology. 1995;86:1–7. doi: 10.1159/000176822. [DOI] [PubMed] [Google Scholar]
- Rossi MA, Goncalves S, Ribeiro-dos-Santos R. Experimental Trypanosoma cruzi cardiomyopathy in BALB/c mice. The potential role of intravascular platelet aggregation in its genesis. Am J Pathol. 1984;114:209–216. [PMC free article] [PubMed] [Google Scholar]
- Rossi MA, Tanowitz HB, Malvestio LM, Celes MR, Campos EC, Blefari V, et al. Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic mechanisms. PLoS Negl Trop Dis. 2010;4:e674. doi: 10.1371/journal.pntd.0000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomone OA, Caeiro TF, Madoery RJ, Amuchastegui M, Omelinauk M, Juri D, et al. High plasma immunoreactive endothelin levels in patients with Chagas’ cardiomyopathy. Am J Cardiol. 2001;87:1217–1220. A1217. doi: 10.1016/s0002-9149(01)01502-8. [DOI] [PubMed] [Google Scholar]
- Sarabanda AV, Sosa E, Simoes MV, Figueiredo GL, Pintya AO, Marin-Neto JA. Ventricular tachycardia in Chagas’ disease: a comparison of clinical, angiographic, electrophysiologic and myocardial perfusion disturbances between patients presenting with either sustained or nonsustained forms. Int J Cardiol. 2005;102:9–19. doi: 10.1016/j.ijcard.2004.03.087. [DOI] [PubMed] [Google Scholar]
- Simoes MV, Pintya AO, Bromberg-Marin G, Sarabanda AV, Antloga CM, Pazin-Filho A, et al. Relation of regional sympathetic denervation and myocardial perfusion disturbance to wall motion impairment in Chagas’ cardiomyopathy. Am J Cardiol. 2000;86:975–981. doi: 10.1016/s0002-9149(00)01133-4. [DOI] [PubMed] [Google Scholar]
- Sonnenblick EH, Fein F, Capasso JM, Factor SM. Microvascular spasm as a cause of cardiomyopathies and the calcium-blocking agent verapamil as potential primary therapy. Am J Cardiol. 1985;55:179B–184B. doi: 10.1016/0002-9149(85)90629-0. [DOI] [PubMed] [Google Scholar]
- Talvani A, Santana G, Barcelos LS, Ishii S, Shimizu T, Romanha AJ, et al. Experimental Trypanosoma cruzi infection in platelet-activating factor receptor-deficient mice. Microbes Infect. 2003;5:789–796. doi: 10.1016/s1286-4579(03)00146-1. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Burns ER, Sinha AK, Kahn NN, Morris SA, Factor SM, et al. Enhanced platelet adherence and aggregation in Chagas’ disease: a potential pathogenic mechanism for cardiomyopathy. Am J Trop Med Hyg. 1990;43:274–281. doi: 10.4269/ajtmh.1990.43.274. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Gumprecht JP, Spurr D, Calderon TM, Ventura MC, Raventos-Suarez C, et al. Cytokine gene expression of endothelial cells infected with Trypanosoma cruzi. J Infect Dis. 1992a;166:598–603. doi: 10.1093/infdis/166.3.598. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Morris SA, Factor SM, Weiss LM, Wittner M. Parasitic diseases of the heart I: acute and chronic Chagas’ disease. Cardiovasc Pathol. 1992b;1:7–15. doi: 10.1016/1054-8807(92)90004-8. [DOI] [PubMed] [Google Scholar]
- Tanowitz HB, Kaul DK, Chen B, Morris SA, Factor SM, Weiss LM, et al. Compromised microcirculation in acute murine Trypanosoma cruzi infection. J Parasitol. 1996;82:124–130. [PubMed] [Google Scholar]
- Tanowitz HB, Huang H, Jelicks LA, Chandra M, Loredo ML, Weiss LM, et al. Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect Immun. 2005;73:2496–2503. doi: 10.1128/IAI.73.4.2496-2503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, et al. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease) Prog Cardiovasc Dis. 2009;51:524–539. doi: 10.1016/j.pcad.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli RR, Giordano RJ, Barbu EM, Torrecilhas AC, Kobayashi GS, Langley RR, et al. Role of the gp85/trans-sialidases in Trypanosoma cruzi tissue tropism: preferential binding of a conserved peptide motif to the vasculature in vivo. PLoS Negl Trop Dis. 2010;4:e864. doi: 10.1371/journal.pntd.0000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres CM. Estudo do miocárdio na moléstia de Chagas (forma aguda) Mem Inst Oswaldo Cruz (Rio J) 1917;9:114–139. [Google Scholar]
- Torres CM. Sobre a anatomia patológica da doença de Chagas. Hospital (Rio J) 1941;36:391–409. [Google Scholar]
- Torres CM. Miocitólise e fibrose do miocárdio na doença de Chagas. Mem Inst Oswaldo Cruz (Rio J) 1960;58:161–182. doi: 10.1590/s0074-02761960000200004. [DOI] [PubMed] [Google Scholar]
- Torres FW, Acquatella H, Condado JA, Dinsmore R, Palacios IF. Coronary vascular reactivity is abnormal in patients with Chagas’ heart disease. Am Heart J. 1995;129:995–1001. doi: 10.1016/0002-8703(95)90122-1. [DOI] [PubMed] [Google Scholar]
- Van Voorhis WC. Coculture of human peripheral blood mononuclear cells with Trypanosoma cruzi leads to proliferation of lymphocytes and cytokine production. J Immunol. 1992;148:239–248. [PubMed] [Google Scholar]
- Vianna G. Contribuição para o estudo da anatomia patológica da moléstia de Carlos Chagas. Mem Inst Oswaldo Cruz (Rio J) 1911;3:199–226. [Google Scholar]
- Vianna LG, Campos GP, de Magalhaes AV. Myocardial infarct without coronary obstruction associated with chronic Chagas cardiopathy. Arq Bras Cardiol. 1979;33:41–47. [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- Zhang L, Tarleton RL. Parasite persistence correlates with disease severity and localization in chronic Chagas’ disease. J Infect Dis. 1999;180:480–486. doi: 10.1086/314889. [DOI] [PubMed] [Google Scholar]