

Abstract
To synthesize past DNA damaged by chemicals or radiation, cells have lesion bypass DNA polymerases (DNAPs), most of which are in the Y-Family. One class of Y-Family DNAPs includes DNAP η in eukaryotes and DNAP V in bacteria, which have low fidelity when replicating undamaged DNA. In E. coli, DNAP V is carefully regulated to insure it is active for lesion bypass only, and one mode of regulation involves interaction of the polymerase subunit (UmuC) and two regulatory subunits (UmuD′) with a RecA-filament bound to ss-DNA. Taking a docking approach, ~150,000 unique orientations involving UmuC, UmuD′ and RecA were evaluated to generate models, one of which was judged best able to rationalize the following published findings. (1) In the UmuD′2C/RecA-filament model, R64-UmuC interacts with S117-RecA, which is known to be at the UmuC/RecA interface. (2) At the model’s UmuC/RecA interface, UmuC has three basic amino acids (K59/R63/R64) that anchor it to RecA. No other Y-Family DNAP has three basic amino acids clustered in this region, making it a plausible site for UmuC to form its unique interaction with RecA. (3) In the model, residues N32/N33/D34 of UmuC form a second interface with RecA, which is consistent with published findings. (4) Active UmuD′ is generated when 24 amino acids in the N-terminal tail of UmuD are proteolyzed, which occurs when UmuD2C binds the RecA-filament. When UmuD is included in an UmuD2C/RecA-filament model, plausible UmuD/RecA contacts guide the UmuD cleavage site (C24/G25) into the UmuD proteolysis active site (S60/K97). One contact involves E11-UmuD interacting with R243-RecA, where the latter is known to be important for UmuD cleavage. (5) The UmuD2C/RecA-filament model rationalizes published findings that at least some UmuD-to-UmuD′ cleavage occurs intermolecularly. (6) Active DNAP V is known to be the heterotetramer UmuD′2C/RecA, a model of which can be generated by a simple rearrangement of the RecA monomer at the 3′-end of the RecA-filament. The rearranged UmuD′2C/RecA model rationalizes published findings about UmuD′ residues in proximity to RecA. In summary, docking and molecular simulations are used to develop an UmuD′2C/RecA model, whose structure rationalizes much of the known properties of the active form of DNA polymerase V.
Keywords: Cancer, Y-Family DNA polymerases, Mutations, Genotoxins, Adducts, Lesion-bypass
1. INTRODUCTION
Cells possess many DNA polymerases (DNAPs); e.g., humans, yeast (S. cerevisae) and E. coli have at least fifteen, eight and five, respectively [1–3], which serve many functions. For example, replicative DNAPs are often blocked by the DNA damage caused by chemicals and radiation, and to avoid such lethal blockage, cells possess lesion-bypass DNAPs [1–17], which conduct translesion DNA synthesis (TLS). Most lesion-bypass DNAPs are in the Y-family [1–17], where human cells have three that are template-directed (DNAPs η, ι and κ), while yeast (S. cerevisae) has one (DNAPs η) and E. coli has two (DNAPs IV and V).
Y-Family DNAPs have a conserved ~350aa core, which includes the polymerase active site [representative references: 18–35]. As with all DNA polymerases, Y-Family members resemble a right-hand with thumb, palm and fingers domains, although their “stubby” fingers and thumb result in more solvent accessible surface around the template/dNTP-binding pocket [8], which can accommodate the bypass of bulky and/or deforming DNA adducts/lesions that typically protrude into these open spaces. Y-Family DNAPs grip DNA with an additional domain [18–20], usually called the “little finger domain” or “polymerase associated domain” (PAD). Steps in the mechanism of Y-Family DNAPs have been proposed for both protein structural changes [15, 22, 24, 25, 31, 36] and for chemical catalysis [37].
The study of E. coli Y-Family DNAPs has provided many insights about Y-Family DNAPs in general. For example, human DNAP κ was originally discovered because its sequence closely resembles E. coli DNAP IV [38–40], and dNTP insertion opposite a variety of adducts/lesions is remarkably similar for the DNAP IV/κ pair suggesting they are functional orthologs [discussed in reference 41]. DNAPs IV and κ have been shown to accurately bypass a variety of N2-dG-adducts [42–49], including from endogenous sources, such as reactive derivatives of cellular trioses [47] or adducts formed from lipid peroxidation processes [49], which may be the main cellular rationale for the genesis of the IV/κ-class. E. coli DNAP V and human DNAP η are also functional orthologs, based on their similarity of dNTP insertion opposite a variety of adducts/lesions [41]. A case has been made that the main cellular rationale for the DNAP V/η-class is TLS of UV-damage (discussed in reference 41).
Our work has focused on benzo[a]pyrene (B[a]P), which is a well-studied DNA damaging agent that is a potent mutagen and carcinogen, and an example of a polycyclic aromatic hydrocarbon (PAH), a class of ubiquitous environmental substances produced by incomplete combustion [50]. PAHs in general and B[a]P in particular induce the kinds of mutations that are thought to be relevant to carcinogenesis and may be important in human cancer (51, and references therein).
Based on genetic studies, DNAPs IV and V are both involved in the non-mutagenic translesion synthesis (TLS) pathway with +BP in E. coli [44, 45, 46, 52], where DNAP IV is almost certainly responsible for correct dCTP insertion, while DNAP V is most likely involved in a subsequent extension step. In the dominant G->T mutagenic pathway, DNAP V is most likely responsible for dATP misinsertion opposite +BP, based on studies in vitro and in cells [44, 45, 52, 53]. Considering other lesions, DNAP V principally does correct dNTP insertion in some cases, such as with TT-CPDs [54] and AAF-C8-dG in 5′-GCGC sequences [45, 53], but DNAP V does incorrect or mixed correct/incorrect insertion with other lesions, such as AAF-C8-dG in 5′-GGG sequences [45, 53], and TT(6-4) photoproducts [45, 53, 54], where the latter is probably linked to DNAP V’s role in causing UV-light mutagenesis [55, 56]. DNAP V also readily inserts dATP opposite non-coding AP-sites [54], which would be the first step in a mutagenic pathway given that AP-sites are most often formed following depurination. DNAP V is E. coli’s lowest fidelity DNAP on undamaged DNA [54].
Recent studies have shown that the active form of DNAP V consists of one polymerase subunit (UmuC), two regulatory subunits (UmuD′) and one RecA monomer with ATP bound [57, 58]. This complex has been named the “DNA polymerase V mutasome” (or “pol V mut”), although herein we refer to it based on its structure (“UmuD′2C/RecA”).
Why has DNAP V evolved such a complex structure? As noted above, DNAP V is the most mutagenic of E. coli’s five DNAPs when copying undamaged DNA, so it makes sense that it would be carefully regulated to insure it only becomes active in dire circumstances when DNA damage persists and bocks replication, which might cause cell death. Based on literature findings, we count eight mechanisms that E. coli has evolved to regulate UmuD′2C/RecA such that it is principally active only on damaged DNA that has not been resolved by other less mutagenic pathways. (1) The level of the UmuC protein is normally extremely low in E. coli, and it is only turned on as part of the “SOS Response” [59], which leads to the induction of >50 genes to help E. coli survive DNA damage [4, 10, 11, 58–61]. SOS induction also increases UmuD protein levels [4, 10, 11, 58–61]. (2) Some SOS response genes help E. coli avoid the potential mutagenic consequences of DNA damage, either via lesion removal (e.g., nucleotide excision repair) or via non-mutagenic damage avoidance mechanisms (e.g., recombinational repair); these genes tend to be turned on early in the SOS response [4, 10, 11, 58–61]. In contrast, DNAP V is turned on late (~30 minutes after DNA damage first occurs), and only if cells have been unable to overcome potentially lethal DNA damage by other less mutagenic mechanisms. (3) Although UmuD binds UmuC, this gives an inactive form of DNAP V, while the active form of DNAP V contains UmuD′, which has 24aa removed from the N-terminus of UmuD [4, 10, 11, 58–61]. UmuD cleavage is regulated and requires RecA participation, probably via the following mechanism [4, 10, 11, 58–61]. When DNA replication forks are blocked by DNA damage, ss-gaps are formed downstream of the point of blockage. RecA forms filaments on these ss-gaps in order to initiate DNA homology searches during recombinational repair. If recombinational repair fails to resolve a replication blockage, then UmuD2C binds, and the RecA monomer at the 3′-end of the RecA-filament facilitates UmuD auto-proteolysis to give active UmuD′. [This requires conversion of RecA to an activated form RecA*, which also facilitates cleavage of LexA, the repressor of the SOS response.] (4) A RecA monomer at the 3′-end of a RecA-filament associates with UmuD′2C to give active DNAP V [4, 10, 11, 58–61]. The RecA-filament presents a roadblock, so at least one RecA monomer must be moved from its ss-DNA binding site such that UmuC can then move to insert a dNTP opposite the DNA damage responsible for the blocked replication fork. Thus, RecA plays two roles to ensure that UmuD′2C only becomes activated at the site of a replication fork that has been stalled by DNA damage for a prolonged period of time. (We note that evidence shows that active UmuD′2C/RecA can receive its RecA from a RecA-filament either in cis or in trans [57, 58]. In cells, others have argued [62] and we believe that addition from a cis-RecA-filament makes more sense for reasons discussed herein, though this point is not settled.) (5) Once one or a few dNTPs have been added, the rest of the RecA-filament impedes ongoing DNA synthesis by UmuD′2C/RecA, which is released, thus insuring that DNA synthesis is not extensive [57, 58]. (6) When UmuD′2C/RecA is released, it becomes inactive as a polymerase and can only become reactivated following RecA dissociation and the subsequent addition of another RecA from a RecA-filament at a blocked replication fork [57, 58]. (7) Once blocked replication forks are resolved, E. coli turns off the SOS response, because RecA* is lost, thus stopping cleavage of LexA, which again becomes an active repressor of SOS genes, including umuC and umuD [4, 10, 11, 58–61]. (8) The UmuC, UmuD and UmuD′ proteins have short half-lives, as they are rapidly degraded by the Lon and C1pXP proteases [4, 10, 11, 58–66].
Because of its role in mutagenic pathways with +BP, AAF, AP sites, UV light and other lesions, we have an interest in developing a DNAP V model to guide in the design of experiments to understand structure/function relationships. No X-ray structure exists for DNAP V, which prompted us to take a homology modeling approach to generate a model for UmuC, which is the polymerase subunit of DNAP V [67, and references therein]. Herein we report on the generation of two models for DNAP V based on extensive docking studies between our model of UmuC and X-ray structures for UmuD [68] and RecA [69], along with molecular dynamics simulations of ensembles of these proteins. One structure is consistent with much of what is known about how UmuD′2C initially binds to the 3′-end of a RecA-filament. In a second structure, the 3-terminal RecA in contact with UmuD′2C rearranges to give a model for active DNAP V (UmuD′2C/RecA) and in the process frees up 3-to-4 nucleotides of template-ss-DNA, which would allow a short stretch of lesion-bypass DNA synthesis.
2. METHODOLOGY
Many methods were identical to those described previously [67]. Molecular dynamics (MD) was done with CHARMM 30 using Boston University’s IBM p690 or IBM BlueGene. When water was added, it followed our previous protocol [67]. Structures were visualized using InsightII or Discovery Suite (Accelrys).
The steps in the docking protocol are described in Results and Discussion, and primarily used ZDOCK [70, http://zdock.umassmed.edu] and ClusPro [71, http://cluspro.bu.edu]. ZDOCK undertakes this search using extensive Fast Fourier transform-based docking. The docking entities are treated as rigid bodies, the assumption being that they are native structures and energetically at the global minimum. An extensive search of the translational and rotational space ensues, during which the docking entities are placed in many orientations with respect to each other. Fourier correlation, with each docking entity being transformed to a digital signal, speeds up the calculations on large data sets. Evaluation of the structures in ZDOCK is done by an energy scoring function, which takes into account electrostatics, desolvation and shape complementarity. Docking was also done with ClusPro, which also also undertakes energy evaluation (after minimization of docked structures) and clustering, where a large number of energetically favorable docked structures are in the neighborhood of the selected ‘best’ structure, pointing to the local minimum with the broadest well.
After rigid RecA was docked to rigid UmuC, the primer/template DNA (13/17 nucleotides) was added back to UmuC. [DNA had to be removed from the protein during docking for two reasons. First, the docking programs can only do searches involving structures made up of amino acids. Second, even if DNA could be included, it would have been a bad idea, because DNA is a polyanion, and it would be a strong beacon for positively charged regions in proteins, which would not allow optimal searching out of protein-protein interaction surfaces.] The best six structures (see Results and Discussion) were prepared for simulation, with DNA being added back to UmuC, along with three Mg++ (two chelating dCTP) and enough Na+ to bring net charge to zero, as well as five layers of water molecules to soak the complex. The ensemble was minimized (adopted basis Newton-Raphson, 200 steps) and heated to 300K over 40psec with DNA restraints (5.0kcal/mol per atom), followed by molecular dynamics for 50psec at 300K with DNA restraints reduced to 0.5 kcal/mol.
Thereafter the first monomer of rigid UmuD′ was docked to each of the six UmuC/RecA structures, which had been artificially turned into a single rigid protein entity. The best twelve UmuD′C/RecA structures were subjected to the same minimization/heating/molecular dynamics protocol, and a second rigid UmuD′ was docked. The best eight UmuD′2C/RecA structures (see Results and Discussion) were chosen and a new protocol was followed. By a procedure described below, three additional RecAs were added (RecA2-RecA4) to simulate the RecA-filament, including the 12 nucleotides of RecA-associated ss-DNA. Finally, the minimum number of nucleotides (i.e., three) between template-G:dCTP in the active site of UmuC were added, along with two nucleotides on the 5′-side of RecA4 to give a final length of 32 nucleotides in the ss-DNA strand, which includes the UmuC template strand. The final DNA sequence was:

The best eight structures, when prepared for simulations, contained three Mg++, 52 Na+ and ~9000 molecules of H2O, and were minimized (adopted basis Newton-Raphson, 200 steps) and heated to 300K over 40psec with DNA restraints (5.0kcal/mol per atom), followed by molecular dynamics for 50psec at 300K with DNA restraints reduced to 0.5 kcal/mol. Thereafter, molecular dynamics was continued at 300 K for 150psec and 100 psec with, respectively, a 0.5 kcal/mol and 0.1 kcal/mol of restraint on DNA. Finally, each ensemble was subjected to 600psec of unrestrained molecular dynamics at 300K.
A number of variations to this approach were also followed, though they all led to virtually the same final structures. (1) In addition to ZDOCK, another docking method, ClusPro, was also used [71]. All methods gave similar results. (2) When DNA was removed from UmuC, a large number of positively charged, DNA binding residues were exposed, and both RecA and UmuD′ frequently docked to regions containing positively charged residues. To diminish the fraction of such unlikely docking structures, relevant DNA-interacting lysines and arginines (K45, K57, K143, K147, K154, K155, R190, K194, R224, K244, R256, R273, K277, R286, K311 and K342) were mutated to methionine to remove the charge but retain bulk. This approach approximately tripled the number of viable structures that emerged from any particular docking protocol. The methionines were changed back to lysine and arginine before adding DNA and carrying out molecular dynamics. (3) Three other orders of docking were also employed: [UmuC + RecA + UmuD′2], [UmuC + UmuD′ + RecA + UmuD′], and [UmuC + UmuD′2 + RecA]. We note that when UmuD′ was docked as a dimer, most of the resulting structures interacted with UmuC/RecA via the UmuD′ N-terminal region, which is unlikely and in fact unrealistic, since N-terminal region must point into solution. Thus, we found that practically we had to dock one monomer at a time. (4) A variety of RecA mutants [72] were studied, including S117F-RecA (recA1730), E38K-RecA (recA730), F217Y-RecA (RecA4142), E38K/I298V-RecA (recA441), E38K/I298V/D32G-RecA (recA629), V37M-RecA (recA803), E38K/I298V-RecA (recA441), L277N-RecA (recA2277), G278P-RecA (recA2278), L283P-RecA (recA2283), L283E-RecA (recA2283E), I284D-RecA (recA2284), and V275F-RecA (recA2278-5). While interesting, these studies were not revealing enough to include with a few exceptions (Results and Discussion). (5) UmuD contains 139aa and the N-terminal 24aa are proteolyzed to give UmuD′. The X-ray structure of UmuD used in this study (1AY9 [68]) only contains aa32-139, where aa50-139 form a globular core and aa32-49 appear as an extended tail. UmuD′ docking was conducted with four variations: aa32-139, aa41-139, aa43-139 and aa50-139. Figure 1 shows only the core-UmuD′, as the tail has no obvious structure. (6) The little finger domain (LFD) of Y-family DNAPs is attached to the rest of the protein by a long amino acid tether (approximately aa235-250), and various X-ray structures show that the LFD can potentially swing out of the way. UmuC structures with the LFD in various orientations were also used for docking.
Figure 1.

UmuD′2C/RecA Models. (A) UmuD′2C is bound to the 3′-end of a RecA-filament, with RecA1 (yellow) primarily in contact with UmuC (Mode 1). From right-to-left, ribbons represent distal UmuD′ (dark green), proximal UmuD′ (green), UmuC (pink), RecA1 (yellow), RecA2 (turquoise), RecA3 (tan) and RecA4 (blue). The N-terminal 38 amino acids of RecA1 are highlighted in scarlet. DNA is red. (B) Structure for active UmuD′2C/RecA, in which RecA1 is rearranged to become primarily associated with UmuD′2 (Mode 2), thus freeing up three template nucleotides such that UmuC (pink) could initiate DNA synthesis. The N-terminal tail of UmuD′ (aa25-49), which is extended, is not shown. The C-terminus of UmuC (aa354-422), for which there is no model, is not present. The positioning of the N-terminus and C-terminus of RecA1, UmuC and both UmuD′s are indicated.
The RecA1 coordinates for the UmuD′2C/RecA structure were from the B-subunit of the X-ray tetramer 3CMX [69], which is the only complete monomer. In independent docking studies using ClusPro, the A-subunit (receptor) was interacted with the B-subunit (ligand), and the best dimer structure had an orientation virtually identical to the A/B structure in the 3CMX X-ray structure. (Prior to this docking, the B-subunit coordinates were used as a guide to replace missing residues in the A-subunit.) The A-subunit/B-subunit structure created using ClusPro was superimposed on the refined UmuD′2C/RecA structure to generate UmuD′2C/RecA1/RecA2, and this method was repeated until a total of four RecA subunits were present (UmuD′2C/RecA1/RecA2/RecA3/RecA4). The four RecA subunits had a structure that was virtually identical to the four RecA subunits in the 3CMX X-ray structure, and this fact suggested that the docking protocol was working effectively.
Starting coordinates for UmuC(V) were derived from refined structures in our previous work [67], using the DNA sequence we have been focusing on lately [73]. UmuC has 422aa, while our UmuC model only contains the core 353aa found in all Y-Family DNAPs, since UmuC was built by homology modeling [41, 67]. We have been unable to find a suitable model for the remaining C-terminal 69 amino acids of UmuC, so they have not been included in the UmuD′2C/RecA models.
UmuD′ proximal to RecA1 was docked using the A subunit of the 1AY9 X-ray dimer [68], and the distal UmuD′ was the B-subunit. Though a variety of UmuD′/UmuD′ dimerization orientations emerged, all of the lowest energy structures adopted virtually the same symmetrical dimer interface as present in the X-ray structure (as shown in Figure 2a of reference 68), notably with hydrogen bonding between both sets of K55/E93 residues and with F94/F94 face-stacking. The following procedure was followed to add the N-terminal 49aa tail and make full-length UmuD, which was done to investigate how RecA1 might facilitate UmuD cleavage at C24-G25 to give UmuD′. The portion of the N-terminus present in the UmuD X-ray structure (aa32-49) was added back, and the tail was extended to G24 (using insightII). NOE restraint potentials in Charmm (300kcal/mol) were added between C24 and K97 to bring the cleavage site (C24/G25) toward the active site (S60/K97) in UmuD, and the structure was heated to 300K over 40psec, after which 50 psec of molecular dynamics at 300K was performed. Finally the structure was minimized. Thereafter, the tail was extended to Methionine 1. As described in Results and Discussion, R243 in RecA is crucial for UmuD cleavage; a search for interactions to explain this fact revealed a plausible structure with Coulombic interactions between UmuD/RecA1 (E11/R243 and D8/K245), and between UmuD/RecA2 (K5/E235). NOE constraints (300kcal/mol) were placed on these three interactions (E11/R243, D8/K245 and K5/E235), and the structure was heated to 300K over 40psec, after which 50 psec of molecular dynamics at 300K was performed. Finally the structure was minimized. Loop refinement was performed with Swiss-Model [74, http://swissmodel.expasy.org/].
In summary, structures and their refinement were as follows. (1) Input RecA and UmuD structures were derived from X-ray structures, which should be accurate and reliable. (2) The input UmuC structure was taken from our previous work, in which it was equilibrated for >1ns [67]. (3) The final ensemble structure went through a process of heating and gradual release of harmonic constraints over 340 ps, followed by 600 ps of unrestrained molecular dynamics. (4) Intermediate structures were subjected to short molecular dynamics heating (40ps) and equilibration (50ps at 300K), whose objective was (a) to check if any spurious contacts had been created and to resolve them, and (b) to ensure that the hydrogen atoms on all interfacial residues attained proper geometries. Given the large number of intermediate structures generated, it would have been impractical to do more molecular dynamics on intermediates.
3. RESULTS AND DISCUSSION
3.1 Docking Steps Leading to the UmuD′2C/RecA4 Model
A systematic molecular docking approach was taken to generate a UmuD′2C/RecA4 model (details in Methods). In toto, ~150,000 structures were generated and evaluated. In addition to the docking approach described below, variations were also pursued (Methodology), though all variations led to the same final outcome.
After removal of all DNA, 60 different rotational faces of a rigid RecA monomer were docked to 60 different rotational faces of a rigid UmuC; this was accomplished using ZDOCK with the Euler angle of the rotation axis set to 15 degrees. [DNA had to be removed for reasons discussed in Methodology.] DNA was added back to the resulting 3600 structures, and the 24 lowest energy examples were viewed. Impossible structures were eliminated (e.g., those in which DNA collided with protein) and similar structures were coalesced; six unique and viable structures emerged, which were each refined by molecular dynamics and minimization. The DNA was removed from each of these six UmuC/RecA structures, and a rigid UmuD′ monomer was docked using ZDOCK, again setting the Euler angle of the rotation axis to 15 degrees, thus giving [6 × 3600] UmuD′1C/RecA structures. The 24 lowest energy structures from each of the six sets were viewed after adding back DNA. Again based on the dual criteria of eliminating impossible structures and coalescing similar structures, the total number of UmuD′1C/RecA structures was reduced to twelve. These twelve structures were refined by minimization and molecular dynamics, and then viewed again. Four structures were chosen for the next step based on the following criteria: (1) the 5′->3′ orientation of ss-DNA in RecA had to be compatible with the 5′->3′ orientation of the template strand in UmuC; (2) the UmuD′1C/RecA structure had to leave enough room to dock a second molecule of UmuD′; and (3) the structure must have significant and meaningful contour contacts between each of the interacting monomers. After removal of DNA, a second UmuD′ monomer was docked using ZDOCK, again setting the Euler angle of the rotation axis to 15 degrees, which gave [4 × 3600] UmuD′2C/RecA structures. The 24 lowest energy structures in each of the four sets were viewed after DNA was added back, and after implausible structures were excluded and similar structures were coalesced, eight reasonable UmuD′2C/RecA structures emerged, each of which was refined by molecular dynamics and minimization. The coordinates for each of these eight structures are available upon request.
These eight structures were evaluated. One structure stood out, since it could rationalize much of the available data about UmuC/UmuD′/RecA interactions, while the other seven structures could not. Using this preferred structure, three more RecA monomers were added (Methodology) to approximate the 3′-end of a RecA-filament. Figure 1A shows the resulting UmuD′2C/RecA4 structure.
3.2 The UmuC Interface with RecA
The UmuD′2C/RecA4 model shows extensive contacts between RecA1 and two regions of UmuC, namely aa32-34 and aa59-64 (Figure 2). RecA1 is primarily yellow in Figure 2A (with some amino acids highlighted in blue), while RecA2 is primarily turquoise (with some amino acids highlighted in green and brown). Only a few UmuC amino acids are shown in Figure 2; R64, R63 and K59 are shown in red in Figures 2A-2C, while N32, N33 and D34 are shown in brown Figure 2C. The former set is discussed first. To simplify viewing in Figure 2, all RecA residues are shown as van der Waals radii, while all UmuC residues are shown as sticks.
Figure 2.

A portion of the UmuD′2C/RecA4 model from Figure 1A, showing how two regions of UmuC interact with RecA to form the UmuC-RecA1 interface. Panel A shows van der Waals radii both for RecA1 (yellow, blue and dark blue) and for RecA2 (turquoise, green, dark green and brown), while three UmuC residues (K59, R63 and R64) are shown as sticks along with a ribbon (purple) of a portion of the UmuC structure. The three basic amino acids R64/R63/K59 (red) on the surface of UmuC interact with seven amino acids of RecA1 (blue: E96, P119, S117, D120, Q124, A147 and A148). Panel B shows a close-up of amino acids R64/R63/K59 in UmuC (red) interacting with the RecA1 amino acids P119/S117/D120/Q124/A147/A148 (blue) and E96 (turquoise). Panel C shows a different orientation of the same interactions as in panel B, along with UmuC amino acids N32/N33/D34 (brown) interacting with RecA1 amino acids E68/Q194/R196 (blue) and E96 (turquoise).
S117F-RecA (recA1730) is deficient in lesion bypass and has been shown to bind less tightly to UmuD′2C, which has been taken as evidence that S117-RecA is in contact with UmuD′2C [57, 75–79]. S117, along with several other amino acids (blue in the otherwise yellow RecA1 in Figure 2A), interact with three basic amino acids on the surface of UmuC: R64, R63 and K59 (red). Figure 2B shows a close-up of these interactions. The hydroxyl-oxygen of S117-RecA1 forms a hydrogen bond with a hydrogen on the guanidinium moiety of R64-UmuC, which also forms hydrogen bonds with two other RecA1 residues: D120 (carboxylate-oxygen) and N124 (amide-oxygen). The guanidinium moiety of R63-UmuC forms hydrogen bonds with the RecA amino acids P119 (carbonyl-oxygen), D120 (carbonyl-oxygen) and D120 (carboxylate-oxygen). The terminal amino group on K59-UmuC forms hydrogen bonds with the RecA amino acids E96 (carbonyl-oxygen), A147 (carbonyl-oxygen) and A148 (carbonyl-oxygen).
An interface between the UmuC/aa59-64 region and the RecA/aa96-148 region is sensible for several reasons. Arginines R63 and R64 and lysine K59 must be on the UmuC protein surface, since the amino acid sequence in this region of UmuC aligns with a region that is known to be on the surface of other Y-Family DNAPs based on their X-ray structures [41]. The uniqueness of the K59-R64 region stands out as a plausible place for UmuC to form a unique interaction with RecA for the following reasons (refer to Figure 3): (1) Other Y-Family DNAPs have at most one basic amino acid projecting from the protein surface in this region; (2) UmuC has two extra amino acids in this region based on amino acid alignment; and (3) UmuC conforms least to a consensus AKxxCP sequence in this region.
Figure 3.

Amino acid alignment of six Y-family DNAPs, for which X-ray structures exist, along with EcDNAP IV and EcUmuC, for which modeled structures exist, in a region that includes the B-α-helix (turquoise), C-α-helix (turquoise) and 4-β-strand (yellow). Conserved amino acids are boxed. Note that EcUmuC has an additional two amino acids (R63 and R64) and conforms least to the conserved AKxxCP sequence in this region.
Recently, alanine-scanning mutagenesis was conducted in two UmuC loops (aa31-38 and aa50-54), and based on a UV hypersensitivity screen, the residues N32, N33 and D34 of UmuC were judged likely to be in contact with RecA [80], which is consistent with our model. Figure 2C shows the aa32-34 region of UmuC (brown). Amide hydrogens on N32-UmuC interact with E68-RecA1 (carboxylate-oxygen). Amide hydrogens on N33-UmuC interact with E96-RecA1 (carboxylate-oxygen) and with Q194-RecA1 (amide-oxygen). Carboxylate oxygens on D34-UmuC interact with hydrogens on the guanidinium moiety of R196-RecA1. Amino acids N32/N33/D34 are at the tip of the aa31-38 loop in UmuC, and no other residues in this loop are in contact with RecA1. In the UmuD′2C/RecA4 model, none of the residues in the UmuC-aa50-54 loop are in contact with RecA1. Thus, our UmuD′2C/RecA4 model shows contacts with RecA1 that are consistent with the findings in the alanine scanning/UV hypersensitivity screen [80]. Note that E96-RecA1 (turquoise in Figures 2B and 2C) serves as a bridge, in that E96 interacts both with N33-UmuC in the aa31-38 loop and with K59-UmuC in the aa59-64 region.
To identify amino acids in RecA that might interact with UmuD′2C, the following screen was developed [81]. Constitutive UmuD′2C overexpression in E. coli causes UV-sensitivity by inhibiting recombinational repair. Revertants that overcome the UV-sensitivity to UmuD′2C overexpression are named “RecA[UmuR]”, which could arise if the RecA allele were to bind less tightly to UmuD′2C. Though S117F was originally isolated by other means [79], it was also isolated in the screen for RecA[UmuR] mutants [81]. Near S117 are a cluster of other RecA[UmuR] mutants (i.e., C116T, L114V, N113K and D112G). In our UmuD′2C/RecA4 model, these amino acids are not part of the RecA-UmuC interface. Rather, based on the RecA X-ray structure [69], aa112-116 are at the RecA-RecA interface as shown in Figure 2A, where aa112-116 of RecA2 are highlighted in brown and lie under a segment of RecA1 (aa26-30). The developers of the RecA[UmuR] screen pointed out that mutants with stronger RecA-RecA interactions might also be isolated in their screen [81], and this seems a likely explanation for why C116T, L114V, N113K and D112G were isolated, though this notion has not been investigated experimentally. Based on the X-ray structure, S117-RecA is not part of the RecA-RecA interface. Most of the other RecA residues at the UmuC/RecA interface (shown in blue/turquoise in Figure 2C) also do not participate in the RecA-RecA interface. (The exceptions are E68-RecA and E96-RecA, which participate in both RecA-RecA and UmuC-RecA interactions.) The RecA[UmuR] genetic screen also isolated the mutants S44L and V274M. In fact, S44 and V274 contact each other in the interior of a RecA monomer, where they cannot possibly interact with either UmuC or another RecA monomer. This finding demonstrates that the location of RecA[UmuR] mutants does not necessarily reveal definitive information about RecA residues that are involved in either UmuC-RecA or RecA-RecA contacts.
In the UmuD′2C/RecA4 model, a few other UmuC residues also interact with RecA1, including K25 with D100-RecA, C65 with Q118-RecA, G66 with D100-RecA, W55 with E96-RecA, and F56 with both A148- and R196-RecA.
The UmuC/RecA interface in Figure 2 makes sense for other reasons. RecA has binding sites for three strands of DNA; one site is thought to be for ss-DNA, while the other two are thought to be for ds-DNA [72]. Our UmuD′2C/RecA4 model (Figure 1A) allows the ss-DNA tail leaving RecA1 in a 5′->3′ orientation to smoothly connect to the ss-DNA primer entering UmuC. In fact, the model requires as few as three nucleotides of ss-DNA to get from the template:dNTP base pair in the UmuC active site to the ss-DNA bound to RecA1. (More than three nucleotides can also be accommodated.)
3.3 UmuD and UmuD′ Interactions with RecA
Several UmuD′ structures have been published [68, 82, 83]. During docking, we used the monomeric X-ray coordinates, because they best orient S60 and K97 for their roles as catalytically active residues that cleave UmuD to give UmuD′ [66]. A variety of UmuD′ dimerization orientations have been observed [68, 82, 83] or computed [84]. In the UmuD′2C/RecA4 model, the computed lowest energy UmuD′ dimerization orientation adopted a structure with virtually the same symmetrical dimer interface as present in the X-ray structure (as shown in Figure 2a of reference 68), notably with hydrogen bonding between both sets of K55/E93 residues and with F94/F94 face-stacking.
Because cleavage of UmuD to UmuD′ is stimulated by RecA when bound to ss-DNA (see Introduction), it is generally believed that cleavage of UmuD to UmuD′ occurs when UmuD2C is bound to a RecA-filament. Thus, our model should provide a reasonable mechanism by which the cleavage site in UmuD (C24-G25) can lie in the proteolysis active site of UmuD (S60/K97). UmuD has a globular core (aa50-139) and a long N-terminal tail (aa1-49), which, if fully extended, could stretch up to ~125Å. In our UmuD′2C/RecA4 model, the orientation of UmuD′2 is such that the N-terminal tails point away from RecA. Figure 4 shows a portion of the model with the full-length N-terminal tail added to one monomer to give full-length proximal-UmuD (see Methods). If the N-terminal tail is folded back over the surface of core-UmuD, for which there is evidence [84, 85], Figure 4 shows the following plausible interactions: E11-UmuD with R243-RecA1; D8-UmuD with K245-RecA1; and K5-UmuD with E235-RecA2. [If the N-terminal tail were included in Figure 1A, it would be wrapped behind UmuD′.] The E11-UmuD/R243-RecA1 interaction is of particular interest, because several lines of evidence suggest that UmuD forms a key interaction with R243-RecA1, which is crucial for UmuD cleavage [86]. This positioning allows the C24-G25 cleavage site to bind in the S60/K97 (dark green) proteolysis active site (red oval in Figure 4). Thus, the positioning of UmuD in the UmuD2C/RecA4 model provides a plausible way of rationalizing RecA stimulated cleavage of UmuD to UmuD′.
Figure 4.
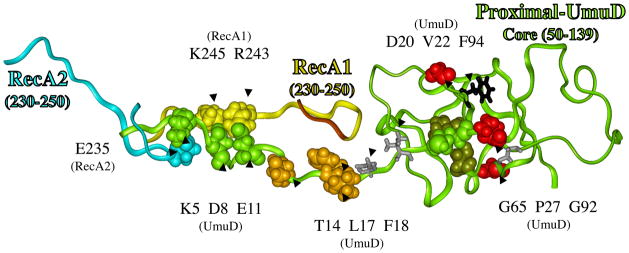
Structural model for how RecA might facilitate the cleavage of UmuD in its active site (red circle), where the catalytically important residue (S60/K97, dark green) cleave between amino acids C24 and G25 (green) to give UmuD′. A portion of RecA1 (aa230-250, yellow ribbon) and RecA2 (aa230-250, turquoise ribbon) are also shown. The N-terminal 49 amino acids of UmuD form a long tail, and evidence suggests it is in contact with core-UmuD (aa50-139) [84, 85]. Evidence also suggests that some portion of UmuD interacts with R243 in RecA to facilitate UmuD cleavage [86]. The N-terminal tail of UmuD is long enough and the spacing is reasonable to allow interactions involving: E11-UmuD with R243-RecA1; D8-UmuD with K245-RecA1; and K5-UmuD with E235-RecA2 E11. When these interactions are made, the cleavage site in UmuD (C24-G25, green) can sit in the proteolysis active site of UmuD (S60/K97, dark green), as indicated by the red oval. The figure also shows P27, G65 and G92 (red), which when mutated each give non-cleavable UmuD (see text). A UmuD triple mutant involving T14/L17/F18 (brown) is also non-cleavable (see text). The significance of D20 and V22 (gray sticks), along with F94 (black sticks) are discussed in the text.
Figure 4 shows the tail of proximal-UmuD in the proximal-UmuD active site (red oval). If distal-UmuD were included in Figure 4, it would be to the right of proximal-UmuD. A related distal-UmuD structure was also generated. When the distal-UmuD tail is directed into its own active site, the tail points away from RecA1 and, thus, its cleavage could not be facilitated by interacting with RecA. In fact, the distal-UmuD tail can only interact with RecA1 if it is directed into the proximal-UmuD active site. Thus, the structure in Figure 4 predicts that one tail cleavage would be intramolecular (i.e. the proximal-UmuD tail in the proximal-UmuD active site) and one cleavage would be intermolecular (i.e., the distal-UmuD tail in the proximal-UmuD active site). Evidence shows that UmuD cleavage can occur intermolecularly [64], though this evidence does not speak to whether intramolecular cleavage is also possible.
A variety of residues in UmuD and UmuD′ have been changed to cysteine and disulfide bond formation studied to map potential sites of protein-protein interactions [84]. For example, this approach was used to show that each of the residues D20C, V22C, C24 and S28C must be close to F94C, which led to a model of how the N-terminus of UmuD must drape over its core. None of these residues (i.e., D20, V22, C24 or S28) are close to F94 in the model shown in Figure 4. This may indicate a problem with the model; alternatively, the disulfide study was done in the absence of RecA, and the structure of UmuD′s N-terminal tail might be different in the presence of RecA. In fact, the proposed knotted fold of UmuD′s N-terminal tail based on the disulfide study (i.e., Figure 6B in reference 84) precludes it from being long enough to interact with RecA, at least based on any of our lowest energy structures for the UmuD/RecA interface. Finally, a cryo-EM study visualized UmuD′2C interacting with the terminus of RecA-filament [87], but the resolution of this structure was not sufficient for it to provide insights relevant to our proposed structure.
In the UmuD′2C/RecA4 model, interactions between UmuD′ and RecA are modest. The proximal UmuD′ monomer has several interactions with RecA. S71-UmuD′ interacts with E235-RecA (carbonyl oxygen) and with N236-RecA (amide-hydrogen). The methyl group on T69-UmuD′ forms hydrophobic interactions with methylenes in the E235-RecA and N236-RecA. The distal UmuD′ monomer does not interact with RecA. [The contacts noted above between E11, D8 and K5 of UmuD and R243,K245, and E235 of RecA, respectively, are not present once the first 24 amino acids of UmuD are proteolyzed to give UmuD′.]
3.4 A RecA1 Conformational Change from Mode 1 to Mode 2
One study [88] showed that when a cross-linking azido moiety (p-azidoiodoacetanilide, AIA) was attached to UmuD at residues V34, S56, S67, S81 or S112, UmuD became crosslinked to RecA, which was taken as evidence that these sites in UmuD are likely to be in proximity to RecA [88]. [The AIA moiety was attached after these amino acids were converted to cysteine.] In contrast, AIA attached to UmuD at S19, C24, L44 and S60 did not yield significant crosslinking with RecA.
In the UmuD′2C/RecA4 model shown in Figure 1A, RecA is neither near the sites that crosslink (V34, S56, S67, S81 and S112) nor near the sites that do not cross-link (S19, C24, V34, L44 and C60). This suggests either that the model is wrong or that something else must be invoked. Evidence suggests that RecA interacts with UmuD′2C in two modes: in “Mode 1” RecA is primarily in contact with UmuC, while in “Mode 2” RecA is primarily in contact with UmuD′2 [89]. The UmuD′2C/RecA4 model depicted in Figures 1A and 2 is more consistent with Mode 1. The cross-linking data, however, might be revealing about interactions in Mode 2.
Given the UmuD′2C/RecA4 model (Figure 1A), DNA synthesis seems impossible, because the entire RecA-filament would have to slide, unless RecA1 rearranged in order to free up template-ss-DNA for DNA synthesis. A RecA1 rearrangement is an attractive possibility, given that active DNAP V is known to include one RecA monomer bound to UmuD′2C [57, 58], which suggests a rearrangement might occur. Finally, a RecA1 rearrangement might explain the existence of two RecA binding modes. We know of no explicit evidence for a RecA rearrangement, though some kind of rearrangement must occur if Modes 1 and 2 are both biologically relevant.
In the docking phase of our work, different docking combinations were tried, including RecA with UmuD′2C and UmuD′2 alone. Four potential low-energy structures emerged in which RecA1 was more associated with UmuD′2 and not in contact with RecA2-RecA4. [The coordinates for all four structures are available upon request.] We favor the structure in Figure 1B for reasons discussed in the next several paragraphs.
Of the options for the Mode 2 structure, the positioning of RecA1 in Figure 1B is closest to its original positioning in Figure 1A, and RecA1 could rearrange via the following simple mechanism. In Figure 1A, RecA1’s N-terminal tail (aa1-38, scarlet ribbon) contacts RecA2 (as revealed in the X-ray structure), while in Figure 1B the N-terminal tail interacts with UmuD′2. One could imagine RecA1’s N-terminal tail releasing from RecA2 and attaching to UmuD′2, with core-RecA1 turning over into its new position either concurrently or subsequently. The RecA1 residues K6, F21, I26, L29 and M35 interact prominently with RecA2 in the putative Mode 1 structure in Figure 1A, and the same residues interact with UmuD′2 in the putative Mode 2 structure in Figure 1B. Figure 5 shows a simple model for the steps leading from RecA1 in the putative Mode 1 structure (Figure 1A) to the putative Mode 2 structure (Figure 1B). The “backside” orientation of the UmuD N-terminal tail would tend to inhibit the ability of RecA1 to rearrange; however, once cleavage occurs, the contacts between UmuD/RecA would be eliminated and RecA1 could turn over to the Mode 2 structure and become active DNAP V.
Figure 5.

Steps leading to the putative Mode 1 to Mode 2 rearrangement. In Mode 1, the N-terminal tail of one UmuD (green) can potentially bind to the backside of RecA1 (yellow) and inhibit the RecA1 rearrangement, until UmuD is proteolyzed to UmuD′, which yields a shorter N-terminal tail that can no longer interact with RecA1 (see text) and might allow rearrangement to Mode 2.
Let us reconsider the AIA crosslinking data in light of the structure in Figure 1B, where a close-up of UmuD′2C/RecA is shown in Figure 6. Though many factors might affect the quantitative level of crosslinking, as pointed out by the authors [88], certainly proximity would be important, given that the AIA moiety is only ~9Å long when fully extended. Attachment of AIA to S81-UmuD led to the greatest amount of crosslinking to RecA. In the UmuD′2C/RecA model (Figure 6), there are no obstructions between RecA1 and the R-group of S81 in either UmuD′ monomer, and S81 in the proximal UmuD′ monomer is closer than 9Å to RecA amino acids R60 and K250, while S81 in distal UmuD′ is closer than 9Å to RecA1 amino acids A1 and I2. S67-UmuD′ had the second greatest level of crosslinking [88]; there are no obstructions between Reac A1 and S67 on proximal UmuD′, and S67 is closer than 9Å to RecA1 residues R176 and Y218. S112-UmuD′ showed the third greatest level of crosslinking [88]; there are no obstructions between RecA1 and S112 on proximal UmuD′, and S112 is closer than 9Å to RecA1 residues S172, R176, G212, A214, L215 and Y217. S57-UmuD showed the least crosslinking with RecA [88]; an AIA-moiety on S57 would be obstructed from reaching all but a small portion of the N-terminal tail of RecA, and the closest RecA1 amino acid (K6) is ~19Å away. S60-UmuD′ does not crosslink [88]; S60 is completely obstructed, since it is on the face opposite RecA1 and is buried in the active site crevice. Thus, the UmuD′2C/RecA model in Figures 1B and 5 seems qualitatively consistent with the AIA crosslinking findings. [In the UmuD′2C/RecA model, UmuD′ amino acids C24, V34 and L44 would be in the disordered N-terminal tail, making them hard to evaluate.]
Figure 6.

Close up of UmuD′2C/RecA, which is the active form of DNAP V, as derived from the model in Figure 1B (Mode 2). The N-terminal tail of RecA1 (aa1-38, scarlet) is shown to be in contact with UmuD′ and is close to S56, S67, S81 and S112 (red), as suggested by published findings [88]. RecA1 residues, which are closer than 9Å to S56, S67, S81 and S112 of UmuD′, are shown as sticks in a color-coordinated fashion (see text).
In the UmuD′2C/RecA model in Figure 6, the two UmuD′ monomers are decidedly asymmetric, with proximal UmuD′ (lighter green) having much more contact with RecA1. Recent work suggests that UmuD′ can be active as a monomer [90]. It is not unreasonable to imagine that an UmuD′C/RecA complex lacking distal UmuD′ (darker green in Figure 6) might still retain activity.
Our model in Figure 4, which shows how UmuD might interact with RecA in order to facilitate UmuD-to-UmuD′ proteolysis, must be evaluated in terms of known non-cleavable UmuD mutants [91, 92], which fall into two categories: (1) inactive for lesion bypass and (2) active for lesion bypass. As depicted in Figure 5, the “backside” orientation of the UmuD N-terminal tail would tend to inhibit the ability of RecA1 to rearrange; however, once cleavage occurs, the contacts between UmuD/RecA would be eliminated and RecA1 could turn over to the Mode 2 structure and become active DNAP V. Non-cleavable UmuD mutants might lock RecA1 in the inactive Mode 1 structure. Three non-cleavable UmuD mutants (P27S, G65R and G92D) result in inactive DNAP V [84, 91], and viewing these residues in the X-ray structure suggests why each is non-cleavable. P27 (red in Figure 4) enforces a protein backbone kink, which appears to facilitate the ability of the cleavage site (C24-G25) to enter the active site. G65 (red in Figure 4) has φ/ψ angles that are unique to glycines, which allows a tight loop to form in order to properly orient S60 as part of the UmuD active site. Similarly, G92 (red in Figure 4) has φ/ψ angles unique to glycine that allow the proper orientation of a loop near active site residue K97. Thus, while the non-cleavability of the P27S, G65R and G92D mutants can be rationalized, these rationale do not provide insight about the plausibility of the UmuD2C/RecA model in Figure 4. One non-cleavable UmuD triple mutant (T14A/L17A/F18A) behaves like UmuD′, in that it is always active [92]. Residues T14, L17 and F18 (brown in Figure 4) are close to aa-230-233 in RecA1 (dark brown ribbon), and if this interaction were lost in the T14A/L17A/F18A-UmuD triple mutant then perhaps the affinity of UmuD for RecA1 would be low enough to permit the RecA1 rearrangement from Mode 1 to Mode 2 without UmuD cleavage.
3.5 The UmuD′2 Interface with UmuC
Due to the difficulty of purifying active UmuD′2C, only limited studies have been attempted on its structure, and we are not aware of any published work that reveals potential contact points between UmuD′ and UmuC. In our UmuD′2C/RecA model, the UmuD′ proximal to RecA1 interacts with UmuC amino acids 82, 90, and 126-132, while the UmuD′ distal to RecA1 interacts with UmuC amino acids 89, 93, 94 and 239.
3.6 Implications of the UmuD′2C/RecA Models for Lesion Bypass
The UmuD′2C/RecA4 structure in Figures 1A and 2 can rationalize why S117F-RecA (recA1730) might have decreased affinity for UmuD′2C [57, 75–79], since it could no longer effectively interact with R64-UmuC. S117-RecA is in a region of RecA1 (aa96-148) that shows a plausible interface with aa59-66 of UmuC. Figure 2C shows RecA1 interacting with another region involving residues N32/N33/D34 of UmuC, which is consistent with published findings [80]. As illustrated in Figure 4, the model also provides a plausible structure, whereby amino acids at the N-terminus of UmuD (i.e., E11, D8 and K5) could interact with RecA (i.e., with R243, K245 and E235, respectively), in such a way that the cleavage site in proximal-UmuD (C24/G25) is brought into the proximal-UmuD active site (S60/K97). The putative E11-UmuD/R243-RecA1 interaction provides a sensible rationale for the observation that R243 is important to the cleavage of UmuD to give UmuD′ [86]. The tail of distal-UmuD could only interact with RecA1 if it were cleaved in proximal-UmuD′s active site, which provides a plausible structural explanation for the observation that at least some UmuD cleavage can occur intermolecularly [64]. Once cleavage occurs, RecA1 could move from being primarily associated with UmuC (Mode 1, Figures 1A an 2) to being primarily associated with UmuD′2 (Mode 2, Figures 1B and 6), which is a structure that can rationalize how RecA could become cross-linked when an AIA moiety was attached at UmuD residues S56, S67, S81 and S112 [88]. In going from the structure in Figure 1A to the structure in Figure 1B, RecA1 is removed from template-ss-DNA, thus freeing up three nucleotides previously covered by the RecA1 monomer, which would allow UmuC to insert a dNTP opposite the lesion in its active site, followed by several dNTP extension steps. At that point, UmuD′2C/RecA would come into contact again with the RecA-filament. Since RecA1 is occupying the RecA binding site on UmuD′2/C, it seems unlikely that RecA2 could be removed from the end of the RecA-filament, and UmuD′2C/RecA might ultimately be released, thus limiting its role in DNA synthesis. Regarding the latter point, it is known that DNAP III cannot resume normal DNA synthesis until a lesion-bypass polymerase does insertion opposite a lesion, along with 3-to-5 extension steps [93–95], which seems consistent with the number of total incorporations that would be possible following the RecA1 rearrangement.
Highlights.
Y-Family DNA Polymerase V models, which explain most experimental findings
Model for DNA Polymerase V at a RecA filament prior to UmuD cleavage
Structural model for UmuD cleavage to give active DNA Polymerase V
Model for active DNA Polymerase V, which is the tetramer UmuD′2C/RecA
Acknowledgments
FUNDING
This work was supported by United States Public Health Services Grant R01ES03775.
Abbreviations
- B[a]P
benzo[a]pyrene
- +BP
[+ta]-B[a]P-N2-dG (Figure 1)
- -BP
[-ta]-B[a]P-N2-dG (Figure 1)
- TLS
translesion synthesis: the insertion of a base opposite a DNA adduct, as well as subsequent elongation
- DNAP
DNA polymerase
- S1-dNTP
dNTP active shape 1
- S2-dNTP
dNTP active shape 2
- MD
molecular dynamics
- TT-CPD
thymine-thymine cyclopyrimidine dimer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCulloc SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bebenek K, Kunkel TA. Functions of DNA Polymerases. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 3.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. American Society for Microbiology; Washington, D.C: [Google Scholar]
- 5.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci U S A. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-Family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 7.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 8.Yang W. Damage Repair DNA polymerases. Curr Opin Struct Biol. 2003;13:23–30. doi: 10.1016/s0959-440x(02)00003-9. [DOI] [PubMed] [Google Scholar]
- 9.Prakash S, Johnson RE, Prakash L. L Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 10.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-Family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs RP, Fujii S, Wagner J. Properties and Functions of Escherchia coli: Pol IV and Pol V. In: Yang W, editor. Advances in Protein Chemistry. Vol. 69. 2004. pp. 230–264. [DOI] [PubMed] [Google Scholar]
- 12.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–61. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo C, Kosarek-Stancel JN, Tang TS, Friedberg EC. Y-family DNA polymerases in mammalian cells. Cell Mol Life Sci. 2009;66:2363–2381. doi: 10.1007/s00018-009-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–54. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pata JD. Structural diversity of the Y-family DNA polymerases. Biochim Biophys Acta. 2010;1804:1124–35. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider S, Schorr S, Carell T. Crystal structure analysis of DNA lesion repair and tolerance mechanisms. Curr Opin Struct Biol. 2009;19:87–95. doi: 10.1016/j.sbi.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Washington MT, Carlson KD, Freudenthal BD, Pryor JM. Variations on a theme: eukaryotic Y-family DNA polymerases. Biochim Biophys Acta. 2010;1804:1113–23. doi: 10.1016/j.bbapap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou BL, Pata JD, Steitz TA. Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- 19.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-Family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 20.Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis-syn thymine dimer at atomic resolution. Nature. 2004;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 21.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;25:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rechkoblit O, Malinina L, Cheng Y, Kuryavyi V, Broyde S, Geacintov NE, Patel DJ. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;4:25–42. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. A structural gap in Dpo4 supports mutagenic bypass of a major benzo[a]pyrene dG adduct in DNA through template misalignment. Proc Natl Acad Sci USA. 2007;104:14905–14910. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong JH, Fiala KA, Suo Z, Ling ZH. Snapshots of a Y-family DNA polymerase in replication: substrate-induced conformational transitions and implications for fidelity of Dpo4. J Mol Biol. 2008;379:317–330. doi: 10.1016/j.jmb.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 25.Eoff RL, Sanchez-Ponce R, Guengerich FP. Conformational changes during nucleotide selection by Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2009;284:21090–21999. doi: 10.1074/jbc.M109.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Guengerich FP. Effect of N2-guanyl modifications on early steps in catalysis of polymerization by Sulfolobus solfataricus P2 DNA polymerase Dpo4 T239W. J Mol Biol. 2010;395:1007–18. doi: 10.1016/j.jmb.2009.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rechkoblit O, Kolbanovskiy A, Malinina L, Geacintov NE, Broyde S, Patel DJ. Mechanism of error-free and semitargeted mutagenic bypass of an aromatic amine lesion by Y-family polymerase Dpo4. Nat Struct Mol Biol. 2010;17:379–88. doi: 10.1038/nsmb.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 29.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 30.Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol Cell. 2007;23:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 31.Ummat A, Silverstein TD, Jain R, Buku A, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Human DNA polymerase h is pre-aligned for dNTP binding and catalysis. J Mol Biol. 2012;415:627–634. doi: 10.1016/j.jmb.2011.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Human DNA polymerase iota incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure. 2005;13:1569–77. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase iota. Nat Struct Mol Biol. 2006;13:619–25. doi: 10.1038/nsmb1118. [DOI] [PubMed] [Google Scholar]
- 34.Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, Aggarwal AK. Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature. 2010;465:1039–1043. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biertümpfel C, Zhao Y, Kondo Y, Ramón-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, Yang W. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maxwell BA, Xu C, Suo Z. DNA Lesion Alters Global Conformational Dynamics of Y-family DNA Polymerase during Catalysis. J Biol Chem. 2012;287:13040–13047. doi: 10.1074/jbc.M112.345835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Yu X, Hu P, Broyde S, Zhang Y. A water-mediated and substrate-assisted catalytic mechanism for Sulfolobus solfataricus DNA polymerase IV. J Am Chem Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson RE, Prakash S, Prakash L. The human DINB1 gene encodes the DNA polymerase Pol theta. Proc Natl Acad Sci U S A. 97(92000):3838–3843. doi: 10.1073/pnas.97.8.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee CH, Chandani S, Loechler EL. Homology modeling of four lesion-bypass DNA polymerases: structure and lesion bypass findings suggest that E. coli pol IV and human Pol k are orthologs, and E. coli pol V and human Pol h are orthologs. Journal of Molecular Graphics and Modelling. 2006;25:87–102. doi: 10.1016/j.jmgm.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 42.Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 43.Shen X, Sayer JM, Kroth H, Ponten I, O’Donnell M, Woodgate R, Jerina DM, Goodman MF. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J Biol Chem. 2002;277:5265–5674. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- 44.Lenne-Samuel N, Janel-Bintz R, Kolbanovskiy A, Geacintov NE, Fuchs RP. The processing of a benzo(a)pyrene adduct into a frameshift or a base substitution mutation requires a different set of genes in Escherichia coli. Mol Microbiol. 2000;38:299–307. doi: 10.1046/j.1365-2958.2000.02116.x. [DOI] [PubMed] [Google Scholar]
- 45.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seo KY, Nagalingam A, Miri S, Yin J, Kolbanovskiy A, Shastry A, Loechler EL. Mirror Image Stereoisomers of the Major Benzo[a]pyrene N2-dG Adduct Are Bypassed by Different Lesion-Bypass DNA Polymerases in E. coli. DNA Repair. 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 47.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci USA. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarosz DF, Cohen SE, Delaney JC, Essigmann JM, Walker GC. A DinB variant reveals diverse physiological consequences of incomplete TLS extension by a Y-family DNA polymerase. Proc Natl Acad Sci USA. 2009;106:21137–21142. doi: 10.1073/pnas.0907257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem Res Toxicol. 2009;22:759–778. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harvey RG. Polycyclic Aromatic Hydrocarbons: Chemistry and Cancer. Wiley-VCH, Inc; New York: 1997. [Google Scholar]
- 51.Pfeifer GP, Hainaut P. On the origin of G-to-T transversions in lung cancer. Mutat Res. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 52.Yin J, Seo KY, Loechler EL. A role for DNA polymerase V in G-to-T mutagenesis from the major benzo[a]pyrene N2-dG adduct when studied in a 5′-TGT sequence in Escherichia coli. DNA Repair. 2004;3:323–334. doi: 10.1016/j.dnarep.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 53.Wagner J, Etienne H, Janel-Bintz R, Fuchs RP. Genetics of mutagenesis in E. coli: various combinations of translesion polymerases (Pol II, IV and V) deal with lesion/sequence context diversity. DNA Repair (Amst) 2002;1:159–167. doi: 10.1016/s1568-7864(01)00012-x. [DOI] [PubMed] [Google Scholar]
- 54.Tang M, Pham P, Shen X, Taylor JS, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 55.LeClerc JE, Borden A, Lawrence CW. The thymine-thymine pyrimidine-pyrimidone(6–4) ultraviolet light photoproduct is highly mutagenic and specifically induces 3′ thymine-to-cytosine transitions in Escherichia coli. Proc Natl Acad Sci U S A. 1991;88:9685–9689. doi: 10.1073/pnas.88.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szekeres ES, Jr, Woodgate R, Lawrence CW. Substitution of mucAB or rumAB for umuDC alters the relative frequencies of the two classes of mutations induced by a site-specific T-T cyclobutane dimer and the efficiency of translesion DNA synthesis. J Bacteriol. 1996;178:2559–2563. doi: 10.1128/jb.178.9.2559-2563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF. The active form of DNA polymerase V is UmuD′(2)C-RecA-ATP. Nature. 2009;460:359–363. doi: 10.1038/nature08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel M, Jiang Q, Woodgate R, Cox MM, Goodman MF. A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit Rev Biochem Mol Biol. 2010;45:171–184. doi: 10.3109/10409238.2010.480968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol Gen Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 60.Simmons LA, Foti JJ, Cohen SE, Walker GW. In: The SOS Regulatory Network, in EcoSal-Escherchia coli and Salmonella, cellular and Molecular Biology. Bock A III, Kaper RC, Karp JB, et al., editors. Chapter 5.4.3. ASM Press; Washington D.C., USA: [Google Scholar]
- 61.Ollivierre JN, Fang J, Beuning PJ. The Roles of UmuD in Regulating Mutagenesis. J Nucleic Acids. 2010 doi: 10.4061/2010/947680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fujii S, Fuchs RP. Biochemical basis for the essential genetic requirements of RecA and the beta-clamp in Pol V activation. Proc Natl Acad Sci U S A. 2009;106:14825–14830. doi: 10.1073/pnas.0905855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez M, Rasulova F, Maurizi MR, Woodgate R. Subunit-specific degradation of the UmuD/D′ heterodimer by the ClpXP protease: the role of trans recognition in UmuD′ stability. EMBO J. 2000;19:5251–5258. doi: 10.1093/emboj/19.19.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDonald JP, Frank EG, Levine AS, Woodgate R. Intermolecular cleavage by UmuD-like mutagenesis proteins. Proc Natl Acad Sci U S A. 1998;95:1478–1483. doi: 10.1073/pnas.95.4.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. Regulation of SOS mutagenesis by proteolysis. Proc Natl Acad Sci U S A. 1996;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chandani S, Loechler EL. Y-Family DNA polymerases may use two different dNTP shapes for insertion: a hypothesis and its implications. J Mol Graph Model. 2009;27:759–769. doi: 10.1016/j.jmgm.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. Structure of the UmuD′ protein and its regulation in response to DNA damage. Nature. 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- 69.Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–484. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 70.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 71.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 72.McGrew DA, Knight KL. Molecular Design and Functional Organization of the RecA Protein. Crit Rev Biochem Mol Biol. 2003;38:385–432. doi: 10.1080/10409230390242489. [DOI] [PubMed] [Google Scholar]
- 73.Seo KY, Yin J, Donthamsetti P, Chandani S, Lee CH, Loechler EL. Amino Acid Architecture that Influences dNTP Insertion Efficiency in Y-Family DNA Polymerase V of E. coli. Journal of Molecular Biology. 2009;392:270–282. doi: 10.1016/j.jmb.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 75.Boudsocq F, Campbell M, Devoret R, Bailone A. Quantitation of the inhibition of Hfr x F- recombination by the mutagenesis complex UmuD′C. J Mol Biol. 1997;270:201–211. doi: 10.1006/jmbi.1997.1098. [DOI] [PubMed] [Google Scholar]
- 76.Tang M, Bruck I, Eritja R, Turner J, Frank EG, Woodgate R, O’Donnell M, Goodman MF. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc Natl Acad Sci U S A. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frank EG, Hauser J, Levine AS, Woodgate R. Targeting of the UmuD, UmuD′, and MucA′ mutagenesis proteins to DNA by RecA protein. Proc Natl Acad Sci U S A. 1993;90:8169–8173. doi: 10.1073/pnas.90.17.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dutreix M, Burnett B, Bailone A, Radding CM, Devoret R. A partially deficient mutant, recA1730, that fails to form normal nucleoprotein filaments. Mol Gen Genet. 1992;232:489–497. doi: 10.1007/BF00266254. [DOI] [PubMed] [Google Scholar]
- 79.Dutreix M, Moreau PL, Bailone A, Galibert F, Battista JR, Walker GC, Devoret R. New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J Bacteriol. 1989;171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hawver LA, Gillooly CA, Beuning PJ. Characterization of Escherichia coli UmuC active-site loops identifies variants that confer UV hypersensitivity. J Bacteriol. 2011;193:5400–5411. doi: 10.1128/JB.05301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sommer S, Boudsocq F, Devoret R, Bailone A. Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol Microbiol. 1998;28:281–291. doi: 10.1046/j.1365-2958.1998.00803.x. [DOI] [PubMed] [Google Scholar]
- 82.Ferentz AE, Opperman T, Walker GC, Wagner G. Dimerization of the UmuD′ protein in solution and its implications for regulation of SOS mutagenesis. Nat Struct Biol. 1997;4:979–983. doi: 10.1038/nsb1297-979. [DOI] [PubMed] [Google Scholar]
- 83.Ferentz AE, Walker GC, Wagner G. Converting a DNA damage checkpoint effector (UmuD2C) into a lesion bypass polymerase (UmuD′2C) EMBO J. 2001;20:4287–4298. doi: 10.1093/emboj/20.15.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sutton MD, Guzzo A, Narumi I, Costanzo M, Altenbach C, Ferentz AE, Hubbell WL, Walker GC. A model for the structure of the Escherichia coli SOS-regulated UmuD2 protein. DNA Repair (Amst) 2002;1:77–93. doi: 10.1016/s1568-7864(01)00006-4. [DOI] [PubMed] [Google Scholar]
- 85.Beuning PJ, Simon SM, Zemla A, Barsky D, Walker GC. A non-cleavable UmuD variant that acts as a UmuD′ mimic. J Biol Chem. 2006;281:9633–9640/23296. doi: 10.1074/jbc.M511101200. [DOI] [PubMed] [Google Scholar]
- 86.Mustard JA, Little JW. Analysis of Escherichia coli RecA interactions with LexA, lambda CI, and UmuD by site-directed mutagenesis of recA. J Bacteriol. 2000;182:1659–1670. doi: 10.1128/jb.182.6.1659-1670.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee MH, Walker GC. Interactions of Escherichia coli UmuD with activated RecA analyzed by cross-linking UmuD monocysteine derivatives. J Bacteriol. 1996;178:7285–7294. doi: 10.1128/jb.178.24.7285-7294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frank EG, Cheng N, Do CC, Cerritelli ME, Bruck I, Goodman MF, Egelman EH, Woodgate R, Steven AC. Visulaization of two binding sites for the Escherchia coli UmuD′(2)C complex (DNA pol V) on RecA-ssDNA filaments. J Mol Biol. 2000;297:585–597. doi: 10.1006/jmbi.2000.3591. [DOI] [PubMed] [Google Scholar]
- 89.Schlacher K, Leslie K, Wyman C, Woodgate R, Cox MM, Goodman MF. DNA polymerase V and RecA protein, a minimal mutasome. Mol Cell. 2005;17:561–572. doi: 10.1016/j.molcel.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 90.Ollivierre JN, Sikora JL, Beuning PJ. The dimeric SOS mutagenesis protein UmuD is active as a monomer. J Biol Chem. 2011;286:3607–3617. doi: 10.1074/jbc.M110.167254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koch WH, Ennis DG, Levine AS, Woodgate R. Escherichia coli umuDC mutants: DNA sequence alterations and UmuD cleavage. Mol Gen Genet. 1992;233:443–448. doi: 10.1007/BF00265442. [DOI] [PubMed] [Google Scholar]
- 92.Beuning PJ, Simon SM, Zemla A, Barsky D, Walker GC. A non-cleavable UmuD variant that acts as a UmuD′ mimic. J Biol Chem. 2006;281:9633–9640. doi: 10.1074/jbc.M511101200. [DOI] [PubMed] [Google Scholar]
- 93.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fujii S, Fuchs RP. Interplay among replicative and specialized DNA polymerases determines failure or success of translesion synthesis pathways. J Mol Biol. 2007;372:883–893. doi: 10.1016/j.jmb.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 95.Fuchs RP, Fujii S. Translesion synthesis in Escherichia coli: lessons from the NarI mutation hot spot. DNA Repair. 2007;6:1032–1041. doi: 10.1016/j.dnarep.2007.02.021. [DOI] [PubMed] [Google Scholar]