

1. Introduction
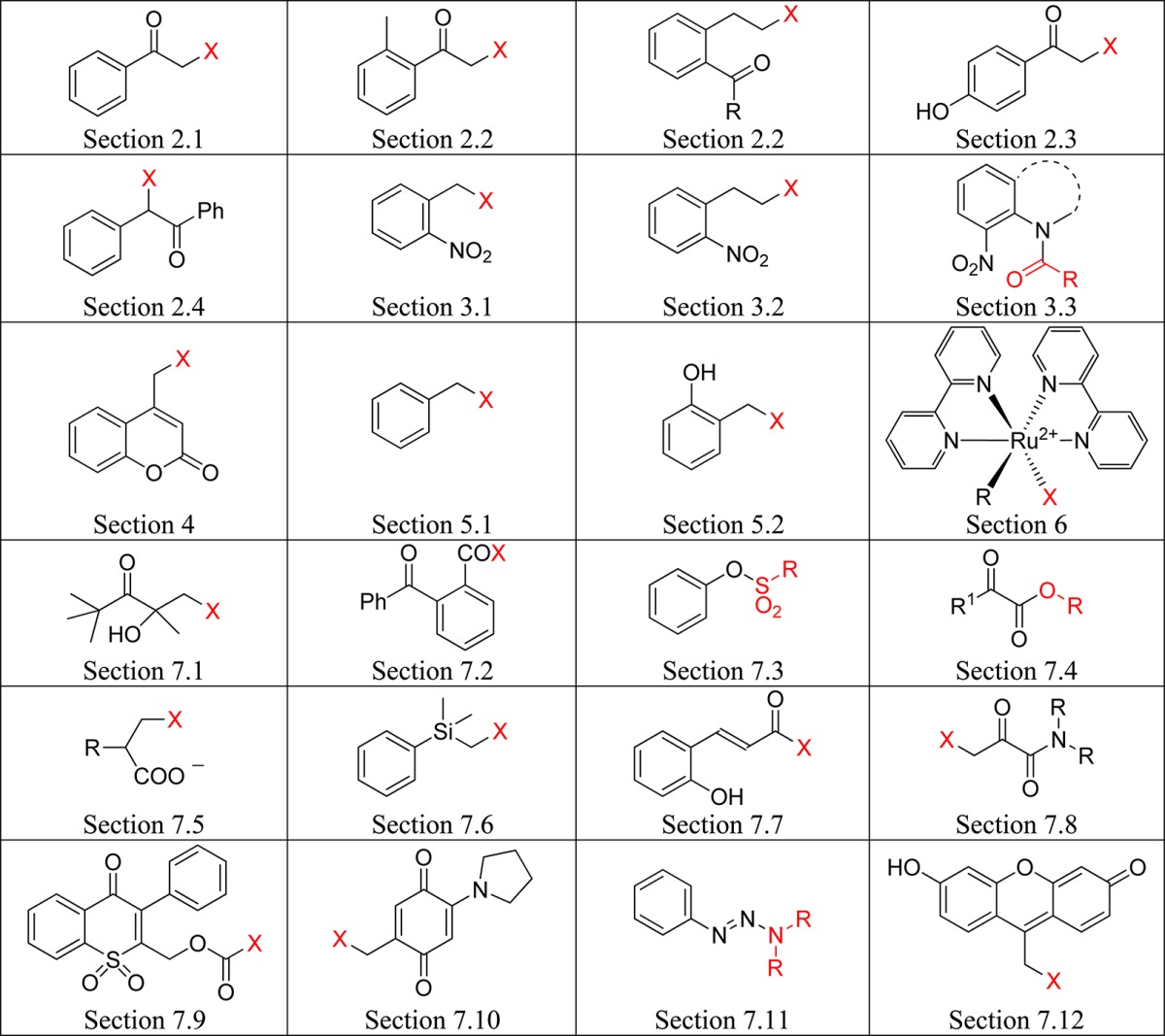
Photoremovable (sometimes called photoreleasable, photocleavable or photoactivatable) protecting groups (PPGs) provide spatial and temporal control over the release of various chemicals such as bioagents (neurotransmitters and cell-signaling molecules), acids, bases, Ca2+ ions, oxidants, insecticides, pheromones, fragrances, etc. Following early reports on PPGs for use in organic synthesis by Barltrop,1 Barton,2 Woodward,3 Sheehan4 and their co-workers, applications to biology were sparked off by Engels and Schlaeger5 and Kaplan and co-workers,6 who first achieved the photorelease of cyclic adenosine monophosphate (cAMP) and ATP, respectively. The latter authors introduced the convenient, if somewhat misleading, term “caged” to designate a compound protected by a PPG. Two general perspectives7 and many more specialized reviews covering applications of PPGs in synthesis,8 biochemistry and neurobiology,9 biomedicine,10 volatiles release,11 polymerization,12 and fluorescence activation13 have been published during the past decade, and a journal issue themed on these topics has recently been published.14 The present review covers recent developments in the field, focusing on the scope, limitations, and applications of PPGs, which are used to release organic molecules. Photoactivation of small inorganic species and ions, such as NO,15 CO,16 Ca2+,9h,17 Zn2+,18 Cd2+,19 or Cu+,20 is not covered. Simplified basic structures of the photoremovable protecting groups discussed in this review are listed in Table 1 (the leaving groups are shown in red).
Table 1. Photoremovable Protecting Groups.
The criteria for the design of a good PPG will depend on the application. No single system needs to fulfill all of the following requirements:
-
(i)
In general, the PPG should have strong absorption at wavelengths well above 300 nm, where irradiation is less likely to be absorbed by (and possibly cause damage to) the biological entity.21 Moreover, the photoreaction should be clean and should occur with a high quantum yield or efficiency for release, Φrel. The quantum yield Φrel is equal to the amount of released substrate, nrel/mol, divided by the amount of photons at the irradiation wavelength λ, np/mol = np/einstein, that were absorbed by the caged compound: Φrel = nrel/np. An important measure for the efficacy of a PPG is the product of the quantum yield and the molar decadic absorption coefficient ε of the PPG, Φrelε(λirr), which is proportional to the amount of release at the given excitation wavelength.22
-
(ii)
Sensitive detection of the response under study often depends not only on the product Φrelε(λirr) but also on the background level of activity of the caged compound prior to irradiation. Hence, the PPGs must be pure, exhibit low intrinsic activity, and be stable in the media prior to and during photolysis.
-
(iii)
The PPGs should be soluble in the targeted media; they may further be required to pass through biological barriers such as cell membranes and show affinity to specific target components, for example, binding sites on cancer cells or the active site of an enzyme.
-
(iv)
The photochemical byproducts accompanying the released bioactive reagent should ideally be transparent at the irradiation wavelength to avoid competitive absorption of the excitation wavelengths. Moreover, they must be biocompatible, i.e., they should not react with the system investigated.
-
(v)
To study the kinetics of rapid responses to a released agent in samples such as brain tissue or single live cells, the PPG must be excited by a short light pulse and the appearance rate constant kapp of the desired free substrate must exceed the rate constant of the response under investigation. Commonly, there are several reaction steps involving ground- and excited-state intermediates that precede the actual release of the free substrate. Therefore, detailed knowledge of the reaction mechanism is needed; in particular, the rate-determining step in the reaction path and its lifetime τrd or kapp must be known, unless the appearance of the free substrate (kapp) can be monitored directly by time-resolved techniques.
Nitrobenzyl, nitrophenethyl compounds, and their dimethoxy derivatives (nitroveratryl) (section 3) are by far the most commonly used PPGs. The decay of their primary quinonoid intermediates on the microsecond time scale does not generally correspond to the rate-determining step of the overall reaction, and the release of the free substrate may be orders of magnitude slower. Moreover, photolysis of these compounds forms potentially toxic and strongly absorbing byproducts such as o-nitrosobenzaldehyde. Quite a number of alternative PPGs have been developed that do not suffer from these disadvantages.
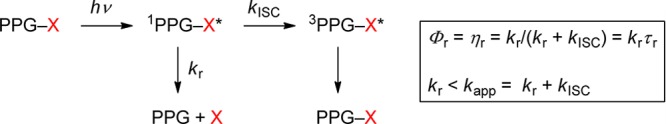
The appearance rate constant kapp of the desired product is equal to the inverse of the rate-determining intermediate’s lifetime τrd, kapp = 1/τrd, which often depends on the solvent as well as on the concentrations of acids and bases including those of the general acids and bases contained in buffers. Release rate constants, kr = ηr/τr, are sometimes quoted, where ηr is the efficiency of the releasing reaction step, ηr = kr /Σk, and τr = 1/Σk is the lifetime of the intermediate that is assumed to release the substrate; Σk includes kr and the rate constant of all competing reactions occurring from that intermediate. Note that the release rate constant kr may be higher or lower than the more relevant appearance rate constant kapp of the desired substrate. Case (a): kr < kapp if Σk > kr, i.e., if reactions other than substrate release contribute to the decay rate of the releasing intermediate. A trivial example of case (a) is shown in Scheme 1. Case (b): kr > kapp if the actual release is preceded by a slower, rate-determining step of the reaction sequence.
Scheme 1. Simple Case Where the Release Rate Constant of the Free Substrate (Leaving Group) X, kr, Is Smaller than Its Appearance Rate Constant, kapp.
The speed of release is an ambivalent expression; it may refer to the efficiency of a PPG, Φrelε(λirr), the amount released by a given irradiation dose, or to the appearance rate constant in time-resolved work. The absorption spectra of a number of chromophores frequently encountered as PPGs are shown in Figure 1.
Figure 1.

UV spectra of selected chromophores.23 (a) Benzophenone (ethanol; solid, red),24 acetophenone (ethanol; dashed, blue);24 (b) 1-acetyl-5-bromo-7-nitroindoline (acetonitrile; solid, red), 2-nitrotoluene (dashed, blue), (3,4-dimethoxy-6-nitrophenyl)methyl (nitroveratryl) derivative (acetonitrile, dashed, green);25 (c) coumarin (acetonitrile, solid, red), p-hydroxyacetophenone (acetonitrile, dashed, blue), and benzoin (acetonitrile, dotted, black); (d) tris(bipyridyl)ruthenium(II) chloride (water).26
2. Arylcarbonylmethyl Groups
Aromatic ketones are thermally stable and synthetically readily accessible compounds; their photophysical and photochemical properties are well understood. The lowest energy transition of simple carbonyl compounds is typically a weak n,π* band (ε ≈ 10–100 M–1 cm–1).27 The higher-energy π,π* absorption bands are strong, and internal conversion to the S1 state is very fast (e.g., ∼100–260 fs for acetophenone).28 The electronic transitions of aromatic ketones are sensitive to solvent polarity and to substitution on the phenyl ring. Hydrogen bonding of protic solvents to the carbonyl oxygen stabilizes the oxygen nonbonding orbital, giving rise to a hypsochromic shift of the n,π* absorption band. Both electron-donating groups and polar solvents tend to stabilize the π,π* states. The strong bathochromic shift induced by para-amino substituents is attributed to a CT interaction29 (for example, λmax for p-aminobenzaldehyde is ∼325 nm (π,π*) in cyclohexane).24 Aromatic ketones are highly phosphorescent and only weakly fluorescent30 due to their fast (>1010 s–1), very efficient intersystem crossing to the triplet state, for which two energetically close lying states (n,π* and π,π*) seem to play a crucial role, possibly due to a S1/T2/T1 intersection.31 The singlet–triplet energy gap is much larger for π,π* than for n,π* states. The lowest π,π* and n,π* triplet states are nearly degenerate, and substitution on the phenyl ring as well as polar solvents may lead to triplet-state inversion.27,32 Some of the important photophysical properties of acetophenone, the parent aryl ketone, are summarized in Table 2. Examples of the absorption spectra of other representative PPG aryl ketones are provided in Figure 1.
Table 2. Photophysical Properties of Acetophenonea.
| solvent | ES/kJ mol–1b | τS/psc | Φfd | ΦTe | ET/kJ mol–1f | Φpg |
|---|---|---|---|---|---|---|
| nonpolar | 33030b | 2533 | <1 × 10–6 34 | 135 | 31036 | 4 × 10–4 37 |
| polar | 33836 | 3933 | 135 | 31136 |
Photophysical properties of many other aromatic ketones can be found in the Handbook of Photochemistry.38
Lowest excited singlet state (S1) energy.
The lifetime of S1.
Fluorescence quantum yield.
Intersystem crossing (ISC) quantum yield.
Lowest triplet state (T1) energy.
Phosphorescence quantum yield (23 °C, isooctane).
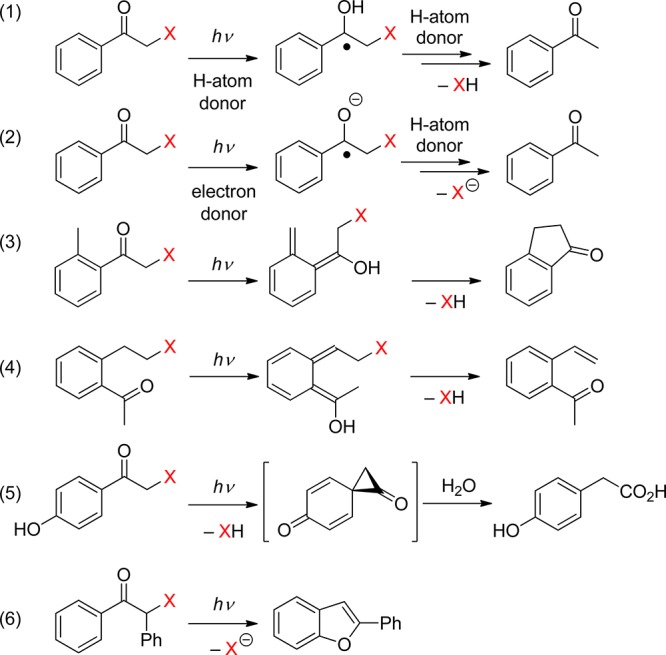
The carbonyl group of aromatic ketones is usually the center of the photochemical reactivity. Scheme 2 shows the most important photoreactions that lead to the liberation of a leaving group (X) and are discussed in the following paragraphs. Ketones with n,π* lowest triplets, possessing a half vacant n orbital localized on the carbonyl oxygen, are far more reactive than those with π,π* lowest triplets with spins delocalized on the aromatic ring. The singlet or triplet n,π* states thus readily abstract hydrogen atoms from suitable donors (entry 1), whereas both n,π* and π,π* states can be reduced in the presence of good electron donors (entry 2). The reaction intermediates hereby formed may subsequently release X– from the α-position. Intramolecular H-transfer reactions in o-alkylacetophenones result in the formation of ground-state photoenols that liberate X– from the α- (entry 3) or o-ethyl (entry 4) positions. Entry 5 shows the p-hydroxyphenacyl moiety, which undergoes a photo-Favorskii rearrangement to release X–. Finally, the benzoin derivative in entry 6 releases X– to form 2-phenylbenzofuran.
Scheme 2. Photochemistry of Aromatic Ketones that Release a Leaving Group (X).
2.1. Phenacyl and Other Related Arylcarbonylmethyl Groups
Using phenacyl compounds as PPGs has been a subject of interest for several decades.39 α-Substituted esters of the phenacyl chromophore are typical of the PPG framework for release of carboxylic acids, for example. Homolytic scission of the ester C–O bond, which would result in the formation of phenacyl and acyloxy radicals, has not been confirmed. Instead, a mechanism that involves hydrogen abstraction from a hydrogen-atom donor by the excited carbonyl group (photoreduction27) of phenacyl ester via a ketyl ester intermediate (entry 1, Scheme 2) has been established by laser flash photolysis.40
Excited phenacyl and 3-pyridacyl esters of benzoic acid were reported to react with an excess of aliphatic alcohols in a chain reaction process to give benzoic acid in addition to acetophenone and 3-acetylpyridine, respectively, as the byproducts.41 Singh and co-workers have reported that arylcarbonylmethyl groups, i.e., naphth-2-ylcarbonylmethyl42 and pyren-1-ylcarbonylmethyl,43 can release various carboxylic acids upon irradiation. The photochemistry of the 4-methoxyphenacyl moiety is discussed in section 2.3.
In the presence of an electron donor, a mechanism involving electron transfer from the donor to the carbonyl group, followed by release of the leaving group, can also be accommodated (entry 2, Scheme 2). This PPG strategy will be discussed in section 8.2.
When a relatively stable radical can be released from the α-carbon of the phenacyl group, phenacyl radicals are produced in the primary homolytic step. This has been demonstrated in the reactions of phenacyl halogenides44 or azides.45 An alternative mechanism, formation of the phenacylium cation from phenacyl ammonium salts, which are used as photoinitiators for cationic polymerization reactions, upon irradiation via a heterolytic cleavage of the C–N bond, has been proposed.46 Recently Klán and co-workers demonstrated that readily accessible S-phenacyl xanthates undergo photoinitiated homolytic scission of the C–S bond in the primary step, opening their use as PPGs for alcohols in the presence of H-atom-donating solvents, where the xanthate moiety represents a photolabile linker.47
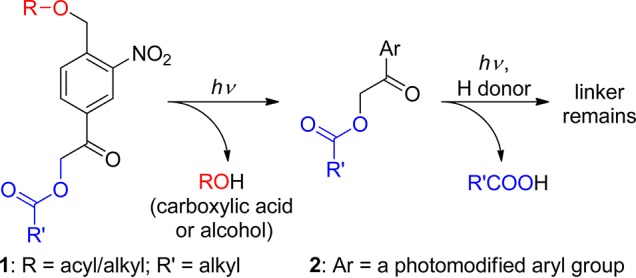
The 4-acetyl-2-nitrobenzyl (ANB, 1) moiety, substituted in both the benzylic and the phenacyl positions with leaving groups, has recently been proposed as a monochromophoric photocleavable linker (Scheme 3).48 This linker thus combines the properties of two well-known photoremovable groups, 2-nitrobenzyl (section 3.1) and phenacyl moieties, in a single chromophore. Liberation of R′CO2H from the intermediate 2 requires the presence of an H-atom donor. Depending on the presence or absence of H-atom donors, the attached groups can be disconnected selectively and orthogonally upon irradiation in high chemical yields (88–97%).
Scheme 3. Monochromophoric Photocleavable Linker48.
2.2. o-Alkylphenacyl Groups
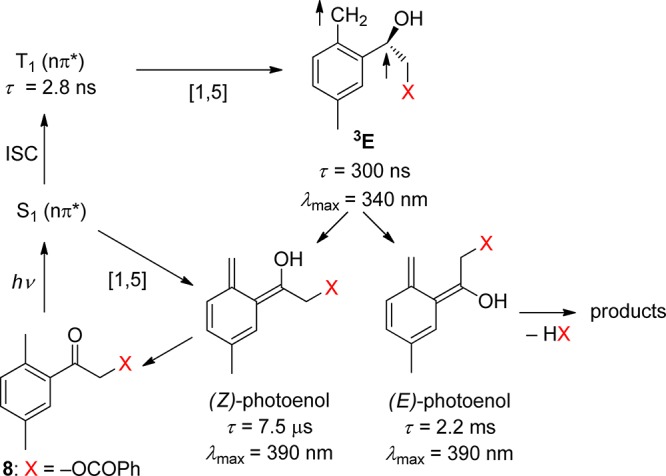
2-Alkylphenyl ketones readily photoenolize to the corresponding dienols (photoenols, o-xylylenols). For example, 2-methylacetophenone (3) undergoes intramolecular 1,5-hydrogen abstraction via the triplet state to form a triplet 1,4-biradical (enol, 3E) that yields two isomeric, (E)- and (Z)-, photoenols, whereas fast direct enolization from the lowest excited singlet state produces only the (Z)-isomer (Scheme 4).49 This scheme may serve as a blueprint for the reactions of related 2-alkylphenacyl compounds. The (Z)-isomer, having a lifetime similar to that of the triplet biradical, is generally converted efficiently back to the starting molecule via a 1,5-sigmatropic hydrogen transfer. Its lifetime is solvent-dependent because hydrogen bonding of the hydroxyl group to a polar solvent strongly retards intramolecular hydrogen back-transfer.49a In contrast, reketonization of the (E)-dienols requires intermolecular proton transfer that may occur either by protonation of the methylene group by a general acid or by proton transfer from the enol by the solvent or a general base, followed by carbon protonation of the dienol anion.49a The resulting long lifetime of the (E)-isomers in dry solvents allows for thermal conrotatory ring closure to give benzocyclobutenols, or trapping by diverse dienophiles such as alkenes, alkynes, or carbonyl compounds in a stereospecific [4 + 2]-cycloaddition reaction.50 However, they may persist up to seconds in the absence of trapping agents. Photoenolization reactions have been thoroughly reviewed by Sammes in the 1970s,51 recently by Klán et al.,52 and, to a modest extent, in several other reviews and book chapters.8d,27,32,50,53
Scheme 4. Photoenolization of 2-Methylacetophenone49.

When leaving groups are present on the α-carbon of 2-alkylphenacyl derivatives (4), they are released from the photoenol intermediates (Scheme 5). In general, the indanone54 (5) and benzocyclobutenol55 (6) side-products are formed in non-nucleophilic solvents, whereas acetophenone derivatives substituted on the o-methyl group 7 are produced in the presence of a nucleophile, such as methanol.
Scheme 5. Photochemistry of 2-Alkylphenacyl Compounds54.
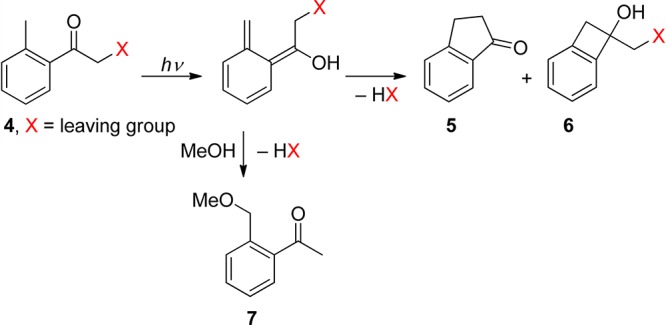
This reaction, reported for the first time on phenacyl chlorides and bromides by Bergmark,56 was shown by Klán and co-workers to be useful for PPG applications.54b Klán and Wirz later demonstrated that 2,5-dimethylphenacyl (DMP) can serve as a PPG for carboxylic acids,57 phosphates, sulfonates,54a alcohols (as carbonates),58 and amines (as carbamates).59 It was recognized that only moderately good or excellent leaving groups are released efficiently within the photoenol lifetime. Studies by laser flash photolysis showed that photolysis produces the anticipated reaction intermediates, the short-lived triplet enol 3E, and two longer-lived, ground-state photoenols assigned to the corresponding (Z)- and (E)-photoenols.54a,57b,58 For example, the (E)-photoenol was found to have a sufficient lifetime (1–100 ms) to release carboxylic acids and carbonates, while the (Z)-photoenol (τ = 0.5–10 μs) regenerated the starting ketone.57b,58 Irradiation of DMP esters in methanol efficiently releases the corresponding free acid (HX) along with indanone and 2-(methoxymethyl)-5-methylacetophenone as the major coproducts, as shown in Scheme 5. The mechanism of DMP benzoate (8) photolysis, determined by laser flash spectroscopy (LFP) in degassed methanol, is displayed in Scheme 6.57b Three intermediates, a short-lived one, λmax ≈ 340 nm (triplet enol 3E), and two longer-lived ones, λmax ≈ 390 nm (photoenols), were formed. In this case, only the longer-lived (E)-photoenol released benzoic acid via the triplet pathway with an appearance rate constant for benzoate of kapp = 1/τ(E-enol) = 4.5 × 102 s–1.
Scheme 6. Photochemistry of DMP Esters57b.
Structurally constrained phenyl ketones, such as 1-oxoindan-2-yl and 1,3-dioxoindan-2-yl derivatives, which can form only the short-lived (Z)-xylylenols, do not release carboxylic acids upon irradiation.60 Only the chloride anion was found to be eliminated from the (Z)-xylylenol (τ = 23 μs in methanol) obtained from 2,5-dimethylphenacyl chloride via the singlet pathway.61 Poor leaving groups such as alcohols and amines that are not efficiently eliminated from the short-lived photoenol intermediates have been attached through a carbonate58 or carbamate59 linkage, respectively, which have similar leaving group properties to that of a carboxylate. For example, the galactopyranosyl carbonate 9 releases a carbonate monoester in a high chemical yield that disintegrates thermally into the corresponding alcohol 10 and CO2 (Scheme 7)58 on the millisecond time scale.62 Table 3 summarizes the photochemical data for the DMP chromophore substituted by various leaving groups.
Scheme 7. Photochemistry of a DMP Galactopyranosyl Carbonate58.
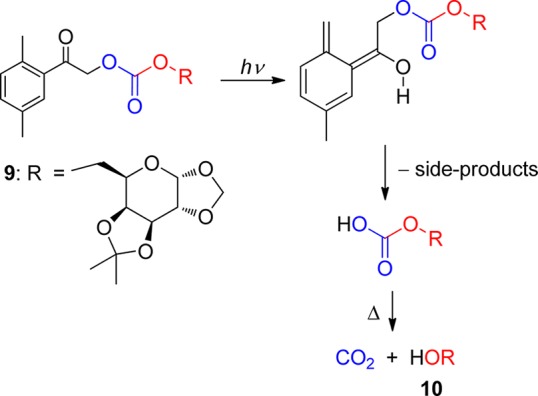
Table 3. 2,5-Dimethylphenacyl (8, DMP) Photoremovable Group.
| leaving group, X (protected species) | solvent | quantum yield, Φ | chemical yield of HX release (%) | kapp/s–1 a |
|---|---|---|---|---|
| Cl | benzene | 0.1156a (0.12)54a | 4.4 × 106 61 | |
| methanol | 0.7656a (0.78)54a | 4.4 × 104 61 | ||
| OC(=O)R (carboxylic acids) | benzene | 0.18–0.2554b,57b | 85–9554b | ∼257b |
| methanol | 0.09–0.1457b | 9254b | 4.5 × 102 57b | |
| OP(=O)(OR)2 (phosphates) | benzene | 0.0954a | ∼254a | |
| methanol | 0.7154a | 9454a | 5 × 104 54a | |
| OS(=O)2R (sulfonic acids) | benzene | 0.16–0.1954a | ||
| methanol | 0.6854a | 90–9354a | 4 × 104 54a | |
| OC(=O)OR (alcohols) | cyclohexane | 0.36–0.5158 | >7058 | b |
| methanol | 0.09–0.2058 | b | ||
| OC(=O)NR2 (amines) | cyclohexane | 0.054–0.08959 | b | |
| acetonitrile | 0.035–0.07059 | 9759 | b | |
| methanol | 0.027–0.06159 | b |
Appearance rate constant of the leaving group, calculated as kapp = 1/τenol.
Slow, presumably <1 ms–1.
Until now, only a few applications of the o-methylphenacyl moiety as a photoremovable protecting group have been reported. Wang and co-workers used the DMP photoremovable group in polymer-supported synthesis,63 and Park and Lee showed that this moiety can be part of new photoresponsive polymers.64
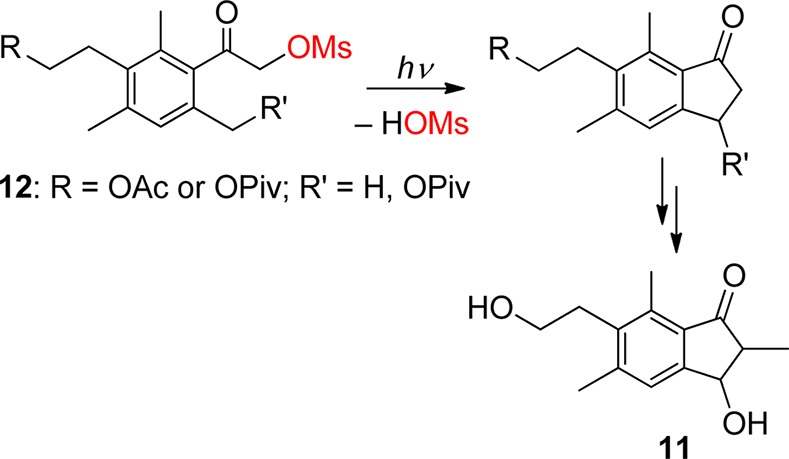
On the other hand, the photochemistry of α-substituted o-alkylphenacyl derivatives was utilized for the photochemical synthesis of interesting functionalized indan-1-ones. In such cases, releasing the leaving group is not of primary interest; it is designed to be a good leaving group and not to interfere with the course of the synthesis. Wessig and co-workers used this concept to prepare various synthetically interesting 1-indanone model derivatives65 and later two sesquiterpene indane derivatives, pterosine B or C (e.g., 11, Scheme 8), in which the key step is the photoenolization reaction of 12.66 Klán and co-workers showed that photolysis of 4,5-dimethoxy-2-methylphenacyl benzoate can lead to the corresponding indanone derivative that is a precursor for the subsequent synthesis of donepezil, a centrally acting reversible acetylcholinesterase inhibitor used to treat Alzheimer’s disease.67 Park and collaborators have recently shown that photolysis of 2,4,6-trialkylphenacyl benzoates can also lead to the corresponding benzocyclobutenols (6 in Scheme 5) in addition to indanones,55 whereas irradiation of α-dichloro-2-acetophenone yields a mixture of various photoproducts.68 Berkessel and co-workers used the photoenolization reaction as a tool to study the cyclization of 4′-benzophenone-substituted nucleoside derivatives as models for ribonucleotide reductases.69
Scheme 8. Photochemical Synthesis of Pterosines66.
Klán and co-workers reported that the photolysis of 2-(alkoxymethyl)-5-methyl-α-chloroacetophenones (13) is very sensitive to traces of water in the solvent (Scheme 9).70 Whereas 3-methoxy-6-methylindan-1-one (14) was a major product in dry, non-nucleophilic solvents, the isobenzofuran-1(3H)-one 15 was obtained in the presence of trace amounts of water. The authors demonstrated that the photoenols produced by photolysis of 13 add water as a nucleophile to yield 2-acetyl-4-methylbenzaldehyde (16), which subsequently forms 17 via a second, singlet-state photoenolization reaction. The same research group also reported that irradiation of the 2,5-dimethylbenzoyl oxiranes 18 results in a relatively efficient and high-yield formation of β-hydroxy-functionalized indanones that structurally resemble biologically active pterosines (Scheme 10).71 In this case, a ring-opening process, rather than release of a leaving group, follows the photoenolization step. An electronic excited-state switching strategy has been utilized to control the selectivity of this reaction in the total synthesis of indanorine.72 The excited-state character of the parent compound was changed to create a productive 3n,π* state by a temporary structural modification selected on the basis of quantum chemical calculations prior to the synthesis. In addition, competition of a triplet-state photoenolization reaction with a photo-Favorskii rearrangement for (o/p)-hydroxy-o-methylphenacyl esters was shown to depend on the water content of the solvent.73
Scheme 9. Photochemistry of 2-(Alkoxymethyl)-5-methyl-α-chloroacetophenones70.
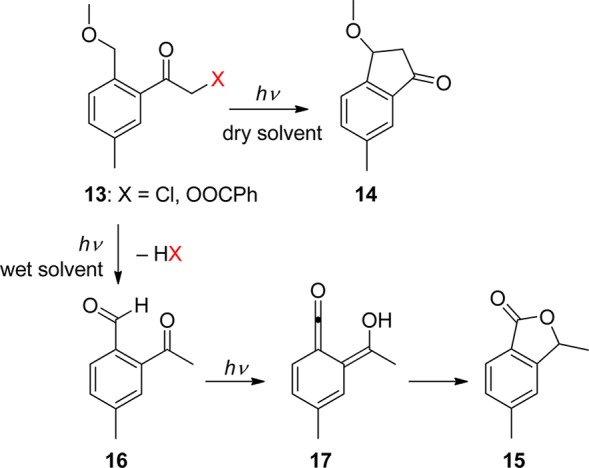
Scheme 10. Photochemistry of 2,5-Dimethylbenzoyl Oxiranes71.
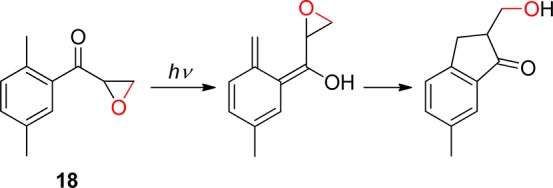
In the 1970s, Tseng and Ullman proposed a new PPG based on (2-hydroxyethyl)benzophenone derivatives (19, R = Ph, Scheme 11), having a leaving group attached in the benzophenone ortho position via an ethylene linker.74 A recent methodical investigation by Pirrung and his co-workers was carried out to elucidate the scope and limitations of the deprotection reaction.75 Alternatively, Wirz7a and later Banerjee and their co-workers76 proposed a similar photoremovable protecting group based on the 1-[2-(2-hydroxyalkyl)phenyl]ethanone 19 (R = alkyl, Scheme 11). The leaving group was reported to be released with a low photochemical efficiency.76 Interestingly, irradiation of 2-acetylphenyl- or 2-benzoylphenylacetic acid results in efficient release of CO2.77
Scheme 11. Photochemistry of 1-[2-(2-Hydroxyalkyl)phenyl]ethanones74.
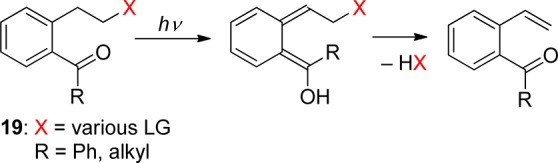
The same mechanism, photoenolization followed by heterolytic elimination of HX, was shown to operate in substituted 5-(ethylen-2-yl)-1,4-naphthoquinones (20, X = Br, dialkyl phosphate, carboxylate), a photoremovable protecting group that absorbs up to 405 nm and provides fast and efficient release of bromide or diethyl phosphate (Φ = 0.7 in aqueous solution) (Scheme 12).78 The blue photoenol is formed in the ground state within 2 ps of excitation and with a quantum yield of unity.79
Scheme 12. Photochemistry of 5-(Ethylen-2-yl)-1,4-naphthoquinones78.
Photoenolization reactions can also be used for releasing protected alcohols through intramolecular lactonization. Gudmundsdottir and her collaborators reported that the corresponding (Z)- and (E)-photoenols are produced by irradiation of the 2-(2-isopropylbenzoyl)benzoate ester 21 via the triplet excited state (Scheme 13).8d,80 An alcohol, such as geraniol (in up to 90% chemical yield), and the side-product 22 are formed in various solvents as well as in thin films. 2-(2-Methylbenzoyl)benzoate esters are not reactive under the same conditions.80a In addition, the 4-oxo-4-o-tolylbutanoate 23 releases methanol by a photoenolization-induced lactonization process (Scheme 14).81
Scheme 13. Photochemistry of 2-(2-Isopropylbenzoyl)benzoate Esters8d,80.
Scheme 14. Photochemistry of 4-Oxo-4-o-tolylbutanoate81.
2.3. p-Hydroxyphenacyl Groups
Among the known photoremovable protecting groups, p-hydroxyphenacyl (24, pHP, Scheme 2 entry 5 and Scheme 15) has emerged as a promising candidate.82 Since debuting a little over a decade ago, the pHP chromophore has found application as a photoremovable protecting group in neurobiology,7,83 enzyme catalysis,7b,9u,83b,83c and synthetic organic chemistry.84 The intriguing features of this protecting group are the skeletal rearrangement that accompanies the release of a substrate, the quantitative chemical yield of released product, and the necessary role of water.7,9u,82,83,85 Advantageous properties are the hydrophilicity of the pHP ligand, the high quantum yields, and the unusually clean reaction that yields only one significant byproduct.
Scheme 15. Photochemistry of pHP as a Protecting Group82.
The absorption spectrum changes drastically as the reaction progresses from a conjugated phenyl ketone (Figure 1) to a nonconjugated phenol, 4-hydroxyphenyl acetate (25, R = H, Figure 2). The purported intermediate (26) shown in Scheme 15 is reminiscent of the cyclopropanone intermediates proposed for the Favorskii rearrangement;86 thus this transformation has been termed the photo-Favorskii rearrangement.87
Figure 2.
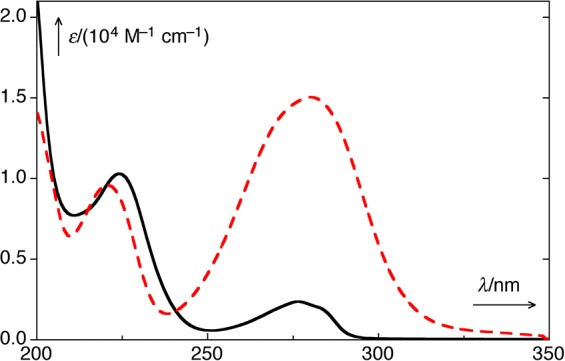
UV–vis absorption spectra of p-hydroxyphenacyl diethyl phosphate (24, X = OPO(OEt)2; pHP DEP; dashed, red) and p-hydroxyphenylacetic acid (25, R = H; black) in H2O/MeCN (1:1).88
p-Hydroxyacetophenone (pHA, 24, X = H) serves as a model for the pHP chromophore. Figure 3 displays the absorption spectra of pHA in neutral water, of pHA– in aqueous NaOH, and of protonated HpHA+ in aqueous HClO4.89
Figure 3.
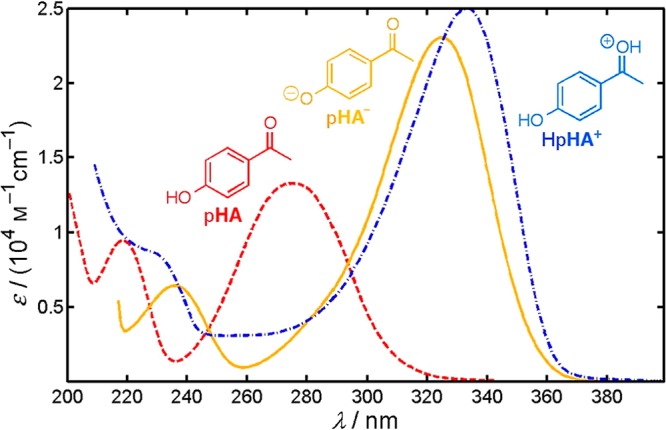
Absorption spectra of pHA (24, X = H; dashed, in red) in neutral water (λmax = 278 nm), of pHA– in 0.05 M aqueous NaOH (λmax = 325 nm; solid, in yellow), and of HpHA+ in 70% aqueous HClO4 (λmax = 333 nm; dash–dot, in blue). The triplet excited state equilibria are shown in Scheme 18. The pKa of ground state pHA is 7.9 ± 0.1 (the concentration quotient at ionic strength I = 0.1 M, 25 °C). Adapted with permission from ref (89). Copyright 2012 American Chemical Society.
pHA (24, X = H) is also the basic framework for the synthesis of the parent pHP protecting group that accommodates an expanding number of leaving groups (HX).7,9u,82,83,85d,85e Most of the leaving groups have been introduced through a sequence of bromination of pHA followed by its SN2 displacement with the conjugate base of the leaving group (X–) under basic conditions82,83c,85a−85c (Scheme 16a–c). In some instances, protection of the phenol group by benzylation, silylation, or acetylation is required.
Scheme 16. Nucleophilic Substitution Routes for pHP (24) Functional Group Protection.

More complex syntheses are required for more reactive or highly functionalized leaving groups, such as protected nucleotides, i.e., pHP ATP (24, X = ATP),82,83c,84,90 pHP GTP,91 and 18O-labeled isotopomers of pHP GTP.91,92p-Hydroxyphenacyl monophosphates are available either through displacement of pHP Br (24, X = Br) or through esterification of 2,4′-dihydroxyacetophenone.85b Dibenzyl, diphenyl, and diethyl phosphates, for example, are sufficiently nucleophilic to undergo SN2 replacement when the reagents and solvents are rigorously dried. The benzyl groups can be removed by hydrogenolysis with H2/Pd after ketal protection of the phenacyl carbonyl.82,83cp-Hydroxyphenacyl phosphoric acid then can be coupled with ADP or GDP through their imidazolium salts to provide the protected nucleotides pHP ATP82,83c,90 and pHP GTP,90,91 respectively (Scheme 16d). These protected nucleosides have found several applications in studies on enzyme catalysis. An advantage of this sequence is the ability to introduce site-specific 18O-labeled isotopomers of GTP90,91 that are used as probes for functional group assignment and dynamic changes in time-resolved Fourier transform infrared (TR-FTIR) studies. Of the leaving groups thus far explored, sulfonates,9u,85d,93 phosphates,7,9u,82,83,85a−85d,90−93 and carboxylates,7,9u,83,85a−85c,93 are the most efficacious and therefore most commonly encountered.
Another, less frequently encountered synthetic method uses addition of α-diazo-p-hydroxyacetophenone (27) to the conjugate acid of the leaving group (HX) under acidic conditions (Scheme 17).93 This approach is particularly useful for protection of highly reactive or base-sensitive leaving groups. Advantages of the diazoketone approach include the ease of synthesis of a variety of substituted diazoacetophenones and the mild conditions for the coupling reaction. The yields are generally good, and the only byproduct is N2. Furthermore, protection of the phenolic OH group or other, less acidic functional groups on the leaving group is normally unnecessary. When the phenolic OH does require protection, the acetate ester is either retained or otherwise readily prepared and is later removed by mild hydrolysis.
Scheme 17. Strategies for α-Diazo-p-Hydroxyacetophenone Coupling to Protect Acidic Leaving Groups93.

The excited-state equilibria of p-hydroxyacetophenone (24, X = H; pHA) reflect the important nonproductive reactions and the photophysical properties of pHP. A recent, detailed study89 of the primary photophysical processes of pHA and the ensuing proton transfer reactions in aqueous solution by picosecond pump–probe spectroscopy and nanosecond laser flash photolysis has provided a comprehensive reaction scheme (Scheme 18): Following fast and quantitative ISC of excited pHA, τ(1pHA*) = 3.4 ps, to the triplet state, 3pHA*, spontaneous adiabatic ionization of 3pHA* in aqueous solution occurs with a rate constant kH+ ≈ 1 × 108 s–1, yielding the triplet of the conjugate base anion 3pHA–* and, simultaneously, the quinoid triplet enol tautomer 3pQ*. The latter is formed by in-cage capture of a proton at the more basic carbonyl oxygen of 3pHA–*. The equilibrium 3pQ* ⇆ 3pHA–* + H+ is established subsequently by diffusional processes on the nanosecond time scale. The formation of 3pQ* from 3pHA* is accelerated by strong acids (via the protonated species 3HpHA+*) and is suppressed by buffer bases, which form 3pHA–* upon encounter with 3pHA*. The triplet-state proton-transfer equilibria of 3pHA* are summarized in Scheme 18.89
Scheme 18. Triplet-State Proton Transfer Equilibria of pHA (24, X = H) in Aqueous Solution (the Experimental Values for the pKa's and Absorption Maxima Are in Black and Calculated Values Are in Red). Adapted with Permission from Ref (89). Copyright 2012 American Chemical Society.
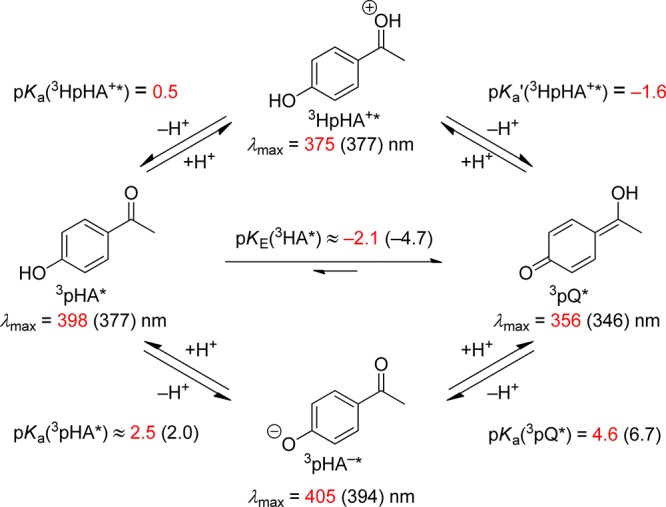
It has been suggested89 that similar proton-transfer processes may account for the lower-than-unity quantum yields found for most pHP PPGs, especially those carrying poor leaving groups, as formation of the less-reactive pHP triplet anion and the nonreactive triplet quinoid enol represent energy-wasting pathways.85d,85e,94 Furthermore, earlier studies on pHP phosphate and carboxylate esters had documented the importance of aqueous solvents for the photochemical release of the leaving group, the rearrangement of the chromophore, and the role of the triplet state as the reactive excited state, i.e., a short-lived, quenchable triplet (ET = 71.2 kcal mol–1).82,83c Subsequent work by Wan and Corrie,95 Phillips85a,85b,96 and Givens and Wirz7,9u,83b,83c,85d,94,97 and their co-workers added a rich compilation of spectroscopic and kinetic information. Recently, the effect of ring size on the photo-Favorskii-induced ring-contraction reaction of various hydroxybenzocycloalkanonyl acetate and mesylate esters has provided new insight into the mechanism of the rearrangement.98
The Phillips group assigned electronic configurations of the key excited states, confirming the triplet state as the reactive excited state, using a combination of time-resolved transient absorption, fluorescence, and resonance Raman spectroscopy, as well as femtosecond and picosecond Kerr-gated resonance Raman spectroscopy (KTRF). An examination of the weak fluorescence from p-hydroxyphenacyl acetate (pHP OAc; 24, X = OAc) in anhydrous CH3CN revealed that the excited singlet manifold of the pHP chromophore is composed of two fluorescing states, a 1π,π* (334 nm) state and a lower lying 1n,π* (427 nm).96a The positions of the two emission bands are influenced by the solvent: in more polar, aqueous media (e.g., 90% aq. CH3CN), the two bands are shifted toward one another to 356 and 392 nm, respectively, or to an energy difference between the 1π,π* and 1n,π* states of 7.4 kcal mol–1 from an energy difference of 18.7 kcal mol–1 in anhydrous CH3CN. The shift enhances the overlap of the two states, resulting in increased vibronic coupling and consequently more efficient internal conversion to the lower 1n,π* state. Density functional theory (DFT) calculations of the electronic states of pHP OAc further suggest that the 3π,π* state (ET = 72.9 kcal mol–1) lies just below the 1n,π* state (ES = 75 kcal mol–1) and is sandwiched between the 1n,π* singlet and the nearby 3n,π* state (71.5 kcal mol–1).85a,95,96,99 The authors suggest that the surfaces of these three states31 merge, resulting in enhanced intersystem crossing (ΦST = 1.0) with a rate of kisc = 5 × 1011 s–1 to a nearly degenerate, “mixed 3n,π*–3π,π*” state. Their findings reaffirmed the important role of water on the photophysical and photochemical processes of pHP.96a
For instance, the picosecond (ps)-KTRF studies showed that added water made only a small difference in the growth rate of the triplet (from 7 to 12 ps) but greatly influenced its decay rate, resulting in second-order quenching of the triplet.96a A solvent change from anhydrous to 50% aqueous CH3CN caused a 100-fold diminution in the 3π,π* triplet lifetime. Phillips and co-workers attributed the large decrease in the lifetime to a leaving group effect: pHP OAc, with the poorer leaving group, had nearly the same triplet rate constant in neat, air-saturated CH3CN as that of pHP diethyl phosphate (pHP DEP, 150 ns). In the aqueous media, both triplet lifetimes (3τ ∼ 150 ns) decreased, but the pHP OAc lifetime (3τ = 2.13 ns) was five times longer than the lifetime of the more reactive pHP DEP (3τ ≈ 420 ps; 70% CH3CN).96a
The most important mechanistic information obtained by Phillips’ group was from the picosecond time-resolved resonance Raman (ps-TR-RR) results of the 600–1600 cm–1 spectral region measured during photolysis of pHP DEP (Figure 4). Scans taken in the first few ps show only diffuse, weak absorption signals attributable to the excited singlet and triplet states of pHP DEP. At ∼300 ps, the scans show the emergence of four new bands that become prominent after 0.7–1.0 ns and by 6 ns are the only bands remaining. These four peaks precisely match those obtained with an authentic sample of the photoproduct, p-hydroxyphenylacetic acid (25, R = H). This TR-RR profile sets the reaction time-constant for the rearrangement and, therefore, encompasses the period for both the release of the leaving group and the rearrangement of the chromophore. In fact, the rearrangement product is in full bloom within just 1 or 2 ns, demonstrating both that the leaving group has departed and, more strikingly, that the complex rearrangement including any intermediates that may intervene between the excited triplet state and 25 had silently formed and then expired completely, escaping ps-TR-RR detection. On the basis of a kinetic analysis of the appearance of 25, Phillips and co-workers showed that such a silent intermediate or intermediates were necessary. He assigned a candidate for the intermediate to “M” to a p-quinone methide cation that was formed by direct heterolysis of the leaving group from triplet pHP DEP. This assignment was corrected in a later study (vide infra).100
Figure 4.

Picosecond time-resolved resonance Raman spectra of pHP DEP (24, X = diethyl phosphate) obtained with a 267 nm pump and 200 nm probe wavelengths in a H2O/CH3CN (1:1) mixed solvent. The resonance Raman spectrum of an authentic sample of p-hydroxyphenylacetic acid recorded with 200 nm excitation is displayed at the top. Reprinted with permission from ref (96a). Copyright 2005 American Chemical Society.
Another significant result came from the analyses of the photolysis products from a series of pHP-substituted acetate esters in the study by Corrie, Wan, and co-workers.95 The acetates were chosen for their increasing propensity toward decarboxylation when converted to carboxy radicals by photoinduced homolysis of the corresponding arylmethyl esters. Photolysis of the pHP esters, i.e., acetate, phenylacetate, pivalate, and diphenylhydroxyacetate, however, produced only carboxylic acids in >90% yield, free of any radical-derived decarboxylation products.101 This confirmed the heterolytic pathway suggested by the groups of Givens,85d,85e Falvey,40 and Phillips.99,100
The next layer of evidence on the photo-Favorskii mechanism arose from time-resolved transient absorption (TR-TA) studies,85d which revealed two additional reactive intermediates: an early, very short-lived transient appearing on the tail of the triplet decay (Figure 5) and a later, long-lived species. The critical evidence for the first transient was obtained using pHP OTs (24, X = OTs), OMs, and DEP, all excellent leaving groups that depart efficiently and rapidly. For example, the transient formed from 3pHP DEP (lifetime, 3τ = 63 ps; in water) appears as a weak set of absorptions on the tail of the pHP triplet. These three maxima were assigned to an allyloxy–phenoxy triplet biradical (328, Scheme 19): the decay profile of 324 (3τ = 100 ps in 87% aqueous CH3CN) transforms into the profile of the slightly longer-lived transient 328 (τbirad = 500 ps).
Figure 5.
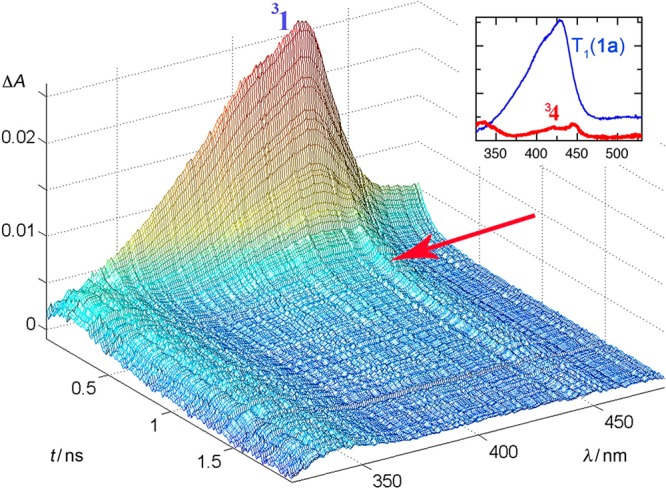
Pump–probe spectra of pHP DEP (24, X = diethyl phosphate) in 87% aqueous CH3CN. The sample was excited with a pulse from a Ti/Sa–NOPA laser system (266 nm, 150 fs pulse width, pulse energy 1 μJ). The inset shows the species spectra of 3pHP DEP and biradical 328 that were determined by global analysis of the spectra taken with delays of 10–1800 ps using a biexponential fit. Reprinted with permission from ref (85d). Copyright 2008 American Chemical Society.
Scheme 19. Refined Mechanism Based on Time-Resolved Transient Absorption Analysis85d.

The three weak absorptions of 28 were detected85d only with the best leaving groups. The bands at 340, 430, and 440 nm were taken as evidence of a phenoxy radical intermediate and thus assigned to the biradical 28. As noted earlier, kinetic analysis of the ps-TR-RR spectra by Phillips and co-workers85a,85b had suggested the intervention of an intermediate “M”, formed from the triplet state that proceeded to the final product 25. The intermediate “M” is now attributed85d to the triplet biradical 328 that is assumed to be formed adiabatically. The formation of 328 can be viewed as being extruded from 324, leaving behind the leaving group X– and a proton in the ground state, thus obeying the Wigner spin rule. The fate of 328 is ISC and closure to an as yet undetected spirodienedione 26. The resulting traditional Favorskii-like intermediate very rapidly hydrolyzes to p-hydroxyphenylacetic acid, completing the formal ground state events normally proposed for the Favorskii rearrangement.102
A further, long-lived intermediate was identified as the known p-quinone methide10329 (τ = 0.3 s) that hydrates yielding p-hydroxybenzyl alcohol (30).85d The formation of small amounts of 30 is also a signature of the elusive spirodione intermediate 26, the lifetime of which appears to be shorter than its rate of formation under the reaction conditions. Thus, the validation of 26 is based solely on a requirement for the carbon skeleton reorganization and a very facile CO extrusion from 26 due to its strained bicyclic structure, and is complemented by DFT calculations.
A summary of the photo-Favorskii mechanism, as currently understood, is outlined in Scheme 19.104 The rearrangement proceeds from the chromophore’s triplet excited state (324) by a concerted departure of the leaving group and the phenolic proton generating the triplet biradical 328. Intersystem crossing of 328 gives an intermediate common to the ground state Favorskii rearrangement,102a−102c the putative cyclopropanone,86,98,102 that either hydrolyzes or decarbonylates on its pathway to the final products. The evidence provided, however, requires involvement of another intermediate, presumably the singlet allyloxy-phenoxy species 128,98,99,104b to account for the complete racemization of a p-hydroxypropiophenone analogue where the leaving group is affixed to a stereogenic α-carbon.104b This mechanism also provides a pathway for the minor photohydrolysis byproduct 24 (X = OH) that becomes predominant for the ring-contraction photoreactions of hydroxybenzocycloalkanonyl esters when ring strain discourages or prevents cyclopropanone formation.98
Position and Requirement for a p-Hydroxy Group
Unsubstituted phenacyl, which is also a PPG (section 2.1), does not undergo a photo-Favorskii rearrangement but rather reacts through a photoreduction mechanism. The p-OH modification of the phenacyl chromophore causes a profound change in the photochemical behavior. The search for alternative functional groups or other locations of the OH group on the phenacyl chromophore that would accommodate a Favorskii rearrangement pathway has met with very little success. Only 2-hydroxyphenacyl esters were shown to release carboxylic acids.73m-Hydroxyphenacyl acetate and a 5-hydroxy-1-naphthacyl analog were unreactive under photo-Favorskii conditions.105
Electron donors such as p-methoxy and o-methoxyphenacyl have been tested as early as the seminal report of the photo-Favorskii rearrangement by Anderson and Reese.87 For these examples, the photo-Favorskii rearrangement competes with photoreduction, forming mixtures of the corresponding methoxyacetophenones and phenyl acetates. In the early 1970s Sheehan and Umezawa developed the p-methoxyphenacyl derivatives as a PPG for photolysis in dioxane or ethanol, producing reduction products.39,106 Givens and co-workers later showed that the reaction in methanol or t-butanol (hydroxylic solvents) did undergo the Favorskii rearrangement as the major pathway, yielding p-methoxyphenylacetates. The competing photoreduction pathway was also evident from the significant proportion of reduction to p-methoxyacetophenone.107 Phillips and co-workers showed that p-methoxyphenacyl diethyl phosphate undergoes a rapid heterolytic cleavage that results in deprotection and formation of a solvolytic rearrangement product.108
Other electron-donating substituents have met with even less success toward the rearrangement of the chromophore.82 Although several p-methoxy and other p-alkoxy analogues have been successfully employed as PPGs for the release of carboxylates,39,106,109 phosphates,82,107 carbonates, and carbamates,110 they do not lead to rearrangement of the chromophore.
Nature of the Leaving Group
The most efficacious leaving groups are conjugate bases of moderate to strong acids, e.g., sulfates,85d,93 phosphates7,9u,82−85,99 (thiophosphate),100,111 carboxylates,7b,9u,85a−85c,93−95,97b,97c,112 phenolates,9u,93 and thiolates.111,113 In general, quantum yields monotonically decrease with an increase in the acid leaving group’s pKa (Table 4), conforming to a Brønsted leaving group relationship (βLG), which correlates the pHP release rate constant with the Ka of the leaving group. A correlation of the log of the rate constants (log kr), derived from the quantum yields and the triplet lifetimes as kr = Φ/3τ (see Table 4 footnote b for details), with the pKa of the leaving groups gave βLG = −0.24 ± 0.03.9u,114
Table 4. Disappearance Quantum Yields, pKa’s, and Rates of Release for Different Leaving Groups (X) for pHP X (24) Arranged According to the pKa of HX.
| X (released substrate) | λmax (log ε)a (pHP X) | pKa (HX) | Φ–Xb | kr/108 s–1c | ref |
|---|---|---|---|---|---|
| mesylate | –1.54 | 0.93 | 50 | (85d, 93, 114) | |
| tosylate | –0.43 | 1.00 | 100 | (85d, 93, 114) | |
| OPO3Et2 | 271 (4.18) | 2.12 | 0.4 | 12. | (82, 85a−85c, 96a, 99, 114) |
| Glu | 273 (3.94) | 4.33 | 0.14 | 1.9 | (83c, 97b, 97c) |
| Ala·Ala | 282 (4.12) | 3.4 | 0.27 | 1.8 | (83c, 112a) |
| bradykinin | 282 (4.07) | 3.4 | 0.22 | 1.8e | (83c, 112a) |
| p-CF3C6H4CO2– | 3.69 | 0.2 | 3.2 | (9u, 93, 114) | |
| formate | 3.75 | 0.94 | 14 | (9u) | |
| benzoate | 4.21 | 0.32 | 2.8 | (9u, 73, 85c, 93, 114) | |
| acetate | 279 (4.09) | 4.76 | 0.4 | N/Af | (85a−85c, 95, 96) |
| GABA | 282 (4.16); 325 | 4.76 | 0.2 | 6.2 | (83c, 85e, 94, 97b, 97c) |
| RPO3S–d | 5.3 | 0.21 | N/A | (111) | |
| OPO3–2 | 280 (4.48) | 7.19 | 0.38 | N/A | (82) |
| GTP | 7.4 | N/A | N/A | (91, 92) | |
| ATP | 253 (4.3); 320 (2.70) | 7.4 | 0.37 | 68 | (82, 83c, 90) |
| p-CNC6H4O– | 7.8 | 0.11 | 0.76 | (9u, 93) | |
| RS–d | 8.4 | 0.085 | N/A | (111, 113, 115) | |
| C6H5O– | 9.89 | 0.04 | 1.0 | (9u, 93) | |
| HO– | 15.7 | <0.01 | N/A | (95) |
In H2O unless otherwise noted.
Appearance efficiencies were identical within experimental error (±5%). See Table 5 for examples.
The rates are derived from several sources and conditions vary. The H2O content was between 10% and 50%, causing small variations in the quantum yields/rates (see text).
Decaging of the catalytic subunit C199A/C343A of PKA (protein kinase A) at Thr-197.111
Assumed to be the same as the model, Ala·Ala.
N/A = not available.
The good quantum efficiencies and high appearance rate constants of the free substrates (kapp = 1/3τ) make the pHP protecting group attractive for quantitative and mechanistic studies in biology and physiology.9u Other beneficial features include good aqueous solubility and stability, the ease of synthesis, the biologically benign quality of the pHP group and its photoproducts, and the lack of quenching by adventitious O2 in aqueous solvents.
ATP and GTP release from the pHP-protected nucleotides has been extensively investigated, resulting in pHP becoming the “phototrigger” of choice for fast kinetic studies of the enzyme-catalyzed hydrolysis by Ras and Rap GTPase activating proteins (GAP proteins).90−92 The phototrigger methodology for activating hydrolysis by photodeprotection of GTP or ATP is rapid (an appearance rate constant kapp = 1/3τ = 1.6 × 1010 s–1 as measured for pHP diethyl phosphate in water99) and was assumed by the authors to be sufficient for measuring the kinetic rate constants for most subsequent binding and hydrolysis steps for the nucleotide.116 Thus, the pHP protecting group provides researchers with a powerful arsenal for fast kinetic mechanistic investigations.
Kötting, Gerwert, and co-workers, for example, compared the release rates for pHP versus NPE (1-(2-nitrophenyl)ethyl; section 3.2) GTP esters (Figure 6).116 The rise time for photorelease of GTP from pHP GTP was too fast to record by their TR-FTIR instrument (τrise = 10 ms), whereas they were able to monitor the GTP appearance rate constant (kapp = 2 ± 1 s–1, Figure 6). They then exploited the rate advantage of pHP GTP to study the catalytic GTP hydrolysis by Ras GTPase and other GAP-based catalytic hydrolysis mechanisms.
Figure 6.
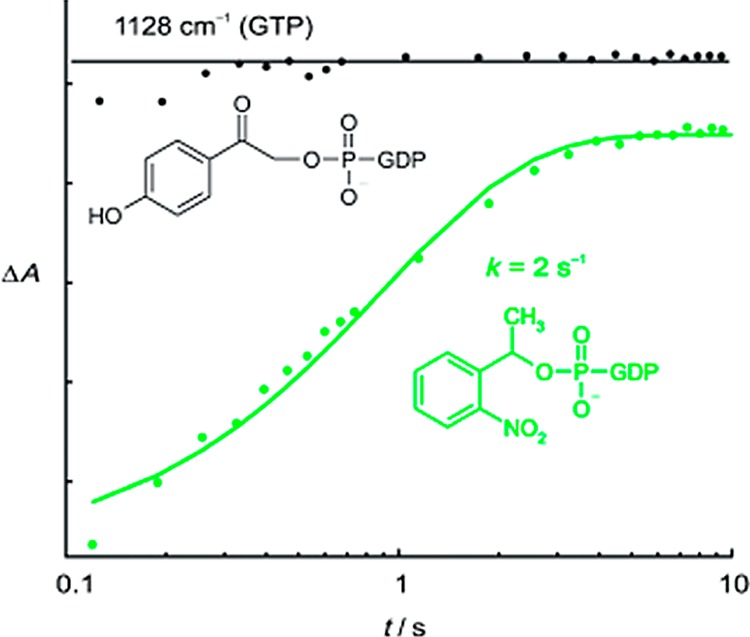
Formation of GTP measured as its Mg2+ complex at 1128 cm–1 from pHP-caged GTP (black) is already complete at the first data point. Formation of GTP from NPE-caged GTP (green) takes place more slowly with a rate constant of 2 s–1 because the rate-limiting step is the release from a ground-state hemiacetal intermediate (section 3.2), an inherently slow process on the time scale necessary for the kinetic measurements reported here.116 Reprinted with permission from ref (116). Copyright 2007 Wiley and Sons.
In pursuing the mechanistic pathway for Ras GTPase catalysis, comparisons of the FTIR spectra of individual α-, β-, and γ-18O-labeled and unlabeled phosphates of GTP, inorganic phosphate (Pi), and GDP hydrolysis products as well as 13C and 14N-labeled site-specific amino acids provided detailed information on both bonding and environmental changes at the enzyme active site. TR-FTIR was then employed to monitor the changes in binding and the evolution and decay of the intermediates during hydrolysis as well as the product-release step and to determine the rate constants.92 Figure 7 illustrates the power of TR-FTIR to resolve the changes in structure and binding at the labeled sites as a function of reaction time. Scheme 20 summarizes the key steps for the hydrolysis, beginning with initial binding of free GTP and ending with the release of inorganic phosphate (Pi) from the enzyme “pocket”, in the rate-limiting step that controls signal transduction. In contrast, with NPE GTP as the phototrigger, only the (last) rate-limiting step could be determined.92
Figure 7.
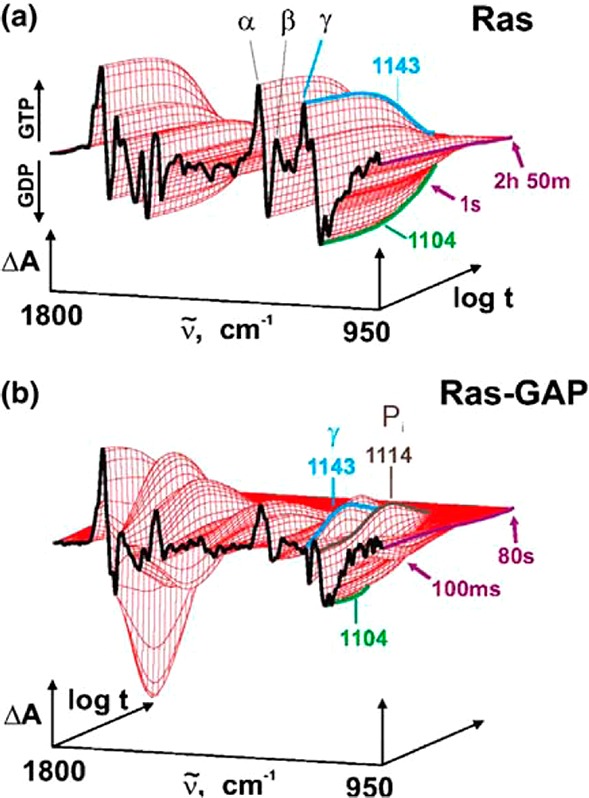
(a) Difference spectra by TR-IR absorption of the intrinsic Ras-catalyzed GTPase reaction. A single exponential function by global fit analysis shows the change from Ras GTP to Ras GDP at 1143 cm–1. (b) TR-IR absorbance difference spectra for the GAP-catalyzed GTPase by Ras. Two intermediates are seen by the fit of three exponential functions at 1143 and 1114 cm–1. The appearance of GTP at 1143 cm–1 arises from pHP-caged GTP followed by GTP hydrolysis. Protein bound Pi appears at 1114 cm–1, which is subsequently released as the rate-limiting step (Scheme 20). Reprinted with permission from ref (92a). Copyright 2004 Elsevier B. V.
Scheme 20. Rate Constants and Mechanism for Ras GTPase (GAP) Hydrolysis of GTP Derived from the Initial Photorelease of GTP from pHP-Caged GTP (the Nonsignaling “OFF” to the signaling “ON” States Are Shown)92b.

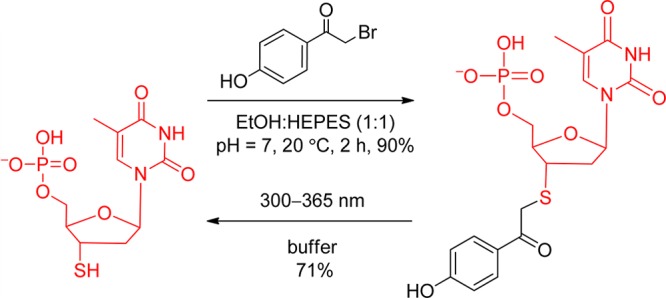
For thiolates, the nucleophilicity of the leaving group is especially noteworthy because leaving groups can readily be protected through in situ derivatization. pHP Br can be added directly to thiols and thiophosphates, even in the presence of other nucleophilic groups on the substrate or in the media. Direct derivatization of thiols and thiophosphates has been exploited for peptides and proteins that possess exposed cysteine and thiophosphate residues, an especially useful feature when the thiol group is an integral part of the catalytic center.115 Model reactions where pHP Br was reacted directly with 3′-thiodeoxythymidine, cysteine, and glutathione produced the corresponding pHP thioethers in 80–90% yields in buffered solutions (Scheme 21). Deprotection by irradiation at 300–365 nm releases the thiol in 60–70% yield, performing essentially as a protection–deprotection switch.
Scheme 21. Reversible Protection–Deprotection of a Thiol on 3′-Thiodeoxythymidine with pHP Br115.
The switch sequence was employed by Pei and co-workers113 and by Bayley and co-workers.111 Pei and co-workers inhibited the phosphorylation of a cysteine located at the active site of protein tyrosine phosphatases (PTK) by direct addition of pHP Br. The phosphorylated cysteine turned “OFF” PTK, a common type of suicide inhibition used with other phenacyl halides. However, photolysis of the pHP thiolates freed the catalytic cysteine unit, turning PTK back “ON”. Interestingly, the protection step was regioselective for blocking only Cys 453, the cysteine at the catalytic site, and none of the other three available cysteine residues.
The protection–deprotection sequence was also reported for the C subunit of protein kinase A (PKA) at Thr-197 and for a thiophosphorylated tyrosine (Y) on a model 11-aa peptide, EPQYEEIPILG, by Bayley’s group.111 Two PPGs were compared: reacting thiophosphate with o-nitrobenzyl bromide (75%) and protection with pHP Br (90%). Deprotection proved more difficult with the o-nitrobenzyl (oNB, section 3.1) thioether because the nitrosobenzaldehyde as a side-product reacted with the newly exposed thiol, causing inhibition. The quantum yield for the reaction was modest (0.37). pHP deprotection was more efficient (Φ = 0.56 to 0.65) and the 70% recovery of the activity was higher because there were no competing reactions of the byproducts with the exposed thiophosphate. This methodology was transferrable into in vitro cell machinery by simply importing the pHP Br into human B cells.111
Substituent Effects on the Chromophore
Very recently, a number of new ortho- and meta-substituted p-hydroxyphenacyl PPGs were introduced to extend the versatility by absorbing at longer wavelengths and by altering the solubility properties. The influence of substituents on the chromophore’s physical, spectral, and mechanistic capabilities, by necessity, became the target of several studies. GABA was selected as the common leaving group because it imparts good aqueous solubility and is biologically significant. Quantum yields of a representative collection of 2- and 3-substituted pHP GABA (31, Table 5) vary only modestly for these substituents. meta-Electron donors such as 3-OCH3 generally display lowered quantum yields whereas electron-withdrawing groups such as 3-CF3 and 3-CN often give rise to slightly increased yields. The rate constants for release are consistently high, in the range of 109 s–1.94
Table 5. Effects of Substituents and pKa on Quantum Yieldsa for the Substituted pHP GABA 31 in Unbuffered H2O;b Entries Are Arranged in the Order of Decreasing pKa of the Substituted pHP Chromophore94.
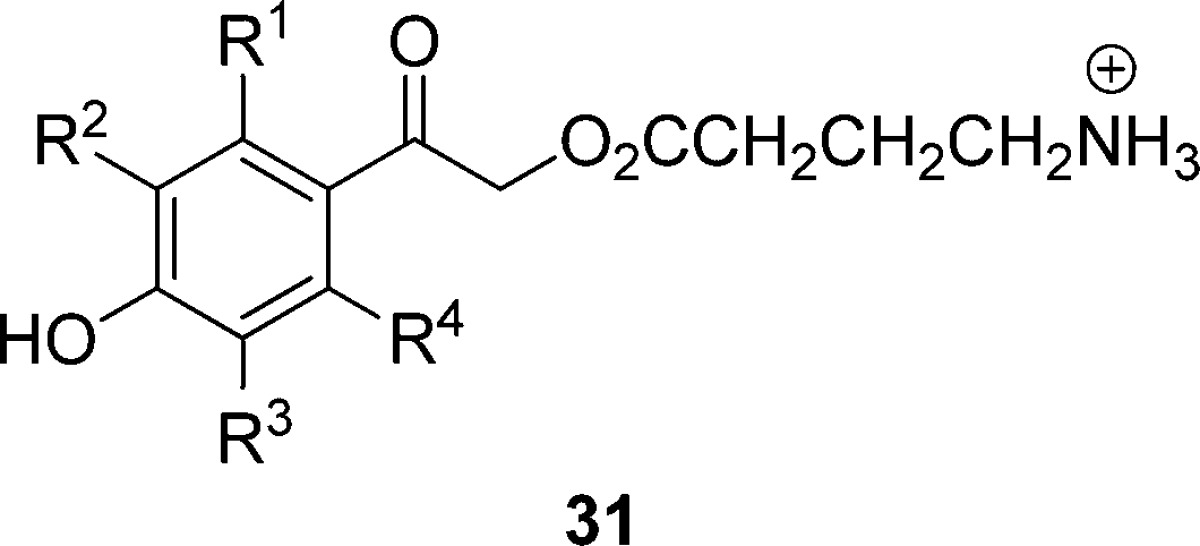
| 31 | pKa | Φdisc | ΦGABA | Φ (25)d | Φdis (Ac or DEP)d |
|---|---|---|---|---|---|
| 3,5-CH3 | 8.2 | 0.15 | 0.14 | 0.13 | |
| 3-CH3 | 8.1 | 0.15 | 0.14 | 0.13 | |
| 2-CH3 | 8.0 | 0.11 | 0.1 | 0.1 | |
| 3-OCH3 | 7.9 | 0.07 | 0.06 | NDe | 0.39 (DEP) |
| R1–R4 = H | 7.8 | 0.20 | 0.19 | 0.16 | 0.30 (Ac) |
| 0.40 (DEP) | |||||
| 3,5-OCH3 | 7.8 | 0.03 | 0.03 | ND | 0.44 (DEP) |
| 2-F | 7.2 | 0.28 | 0.27 | 0.26 | |
| 2,6-F | 6.8 | 0.16 | 0.16 | 0.15 | |
| 3-F | 6.7 | 0.16 | 0.15 | 0.15 | |
| 3-OCF3 | 6.5 | 0.09 | 0.09 | 0.07 | |
| 2,3-diF | 5.9 | 0.24 | 0.24 | 0.22 | |
| 2,5-diF | 5.7 | 0.22 | 0.21 | 0.2 | |
| 3-CF3 | 5.5 | 0.17 | 0.16 | 0.14 | |
| 3,5-F | 5.3 | 0.11 | 0.11 | 0.1 | |
| 3-CN | 5.2 | 0.42 | 0.35 | 0.39 | 0.17 (Ac) |
| 2,3,5-triF | 4.5 | 0.08 | 0.07 | 0.06 | |
| tetra-F | 3.9 | 0.11 | 0.1 | 0.1 |
All runs were low conversions to products (<5%); standard deviations were < ±0.02.
Unbuffered 18 MΩ ultrapure H2O.
Disappearance quantum yield when GABA is the leaving group.
Quantum yield for the substituted phenylacetic acid (25).
Disappearance quantum yield when Ac (acetate) or DEP (diethyl phosphate) is the leaving group.
ND = not determined.94
Certain groups, m-nitro, m-OH, and m-acetyl, when present on the chromophore completely quench the photorearrangement reaction.94
In addition to GABA, there are many other small-molecule and amino acid neuroactive agonists and antagonist with carboxylic acid end-groups. Although carboxylate release is generally less efficient than those of phosphates and tosylates, caged carboxylates find useful applications in neurobiology and neurophysiology, taking advantage of the rapid rate of release and the unreactive, benign qualities of 25 photoproduct vis-à-vis o-nitrobenzyl-based PPGs (section 3.1).92,97b,97c,117 Substituent modification of the pHP derivative 32, such as the 3-CF3 and 3-OCH3 pHP GABA compounds shown in Scheme 22, expands and extends the versatility of the PPG methodology. Here, the relative efficacy of modified pHP GABA to stimulate the GABAA receptor is documented in Figure 8.
Scheme 22. m-Electron-Donor and -Acceptor Group Compatibility for Photorelease of GABA from m-Substituted pHP GABA97b.
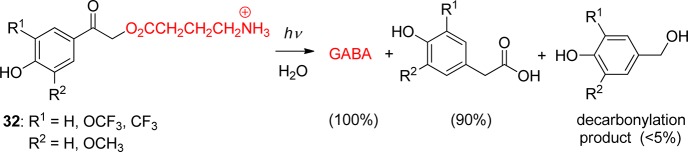
Figure 8.
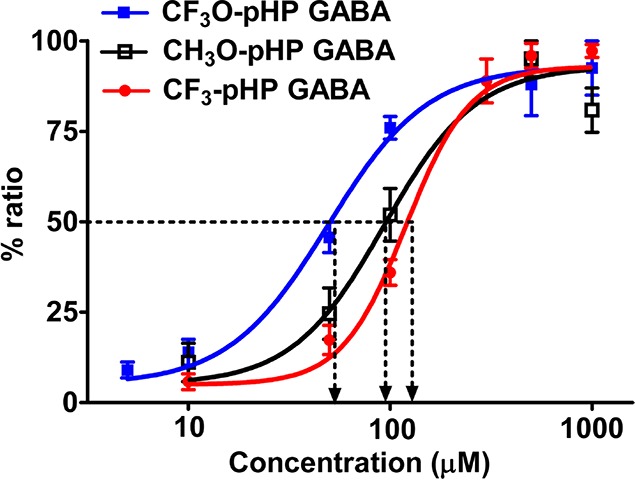
Comparison of EC50’s for GABAA receptor activation by rapid photolysis of pHP (24) GABA. Dose–response curves for 3-CF3O-pHP GABA (blue, n = 7 neurons), 3-CF3-pHP GABA (red, n = 6 neurons), and 3-CH3O-pHP GABA (black, n = 6 neurons) with population data of peak currents normalized to the maximum peak response. EC50 and Hill’s coefficient values were as follows: 3-CF3O-pHP GABA, 49.2 μM, 1.8, n = 7 neurons; 3-CH3O-pHP GABA, 93.4 μM, 1.9, n = 6; and 3-CF3-pHP GABA 119.8 μM, 2.73, n = 6. Reprinted with permission from ref (97b). Copyright 2009 American Chemical Society.
The effect of m-methoxy, trifluoromethoxy, and trifluoromethyl groups on pHP-caged GABAs were tested for their efficacy to release GABA in whole-cell patch-clamp studies on neurons in cortical slices. Local photolysis with short UV light pulses (10–50 ms) delivered through a small-diameter optical fiber produced transient whole-cell inward currents from released GABA.97b
Effect of Media pH and pHP pKa
Because the ionization of substituted pHP derivatives to their conjugate bases changes during irradiation in unbuffered media (Figure 3), the pH effects on the pHP photochemistry were examined (Table 6). The extent of quinone methide formation is altered also by the pH and the substituents on the chromophore (Scheme 18).
Table 6. Substituent Effects on the Quantum Yieldsa As a Function of pH for GABA Release from 31 at 300 nm in Buffered CH3CN–H2O; Entries Are Arranged According to Decreasing pKa of the Substituted pHP GABA94.
| pHP GABA | pKa | Φdis pH 5.0b | Φdis pH 7.3c | Φdis pH 8.2c |
|---|---|---|---|---|
| 3,5-(CH3)2 | 8.2 | N/A | 0.17 | 0.11 |
| 3-CH3 | 8.1 | N/A | 0.15 | 0.08 |
| parent | 8.0 | 0.21 | 0.21 | 0.09 |
| 2-F | 7.2 | 0.24 | 0.21 | 0.06 |
| 3-F | 6.7 | 0.15 | 0.12 | 0.02 |
| 3-OCF3 | 6.5 | 0.07 | 0.06 | 0.02 |
| 3-CF3 | 5.5 | 0.24 | 0.12 | 0.08 |
| 3,5-F2 | 5.3 | 0.08 | 0.05 | 0.02 |
| 3-CN | 5.2 | 0.21 | 0.33d | 0.19e |
| 2,3,5,6-F4 | 3.9 | 0.08 | 0.10 | 0.09 |
Standard deviations were < ±0.02.
0.01 M ammonium acetate.
0.01 M HEPES, 0.1 M LiClO4, pH 7.3.
0.01 M ammonium acetate, pH 7.
0.01 M ammonium acetate, pH 9.
Raising the pH above 8 lowers the quantum yields, reflecting the lower reactivities of the conjugate bases (Table 6). In all cases, the quantum yields were maximal in neutral or slightly acidic conditions but dropped at higher pH. As shown for the pKa’s of the corresponding acetophenones, there is a substantial substituent effect on the pKa, which is manifested in a pronounced UV–vis spectral change (see Figure 3). The prominent π,π* transition at 260–280 nm for neutral p-hydroxyacetophenone is shifted to 320–340 nm, the π,π* transition for the conjugate base, and the absorptivity nearly doubles.
The resultant interplay of pKa of the substituted pHP derivative and the pH of the solution influence the quantum yield as illustrated with 3-CF3 pHP GABA. The quantum yields at pH 5 (Φ = 0.24) decrease to half of their value when the pH is 7.3 (Φ = 0.12) and a third at pH 9.2 (Φ = 0.08). Product yields remain the same at all three pH values, demonstrating that the photo-Favorskii rearrangement is still the major reaction pathway for the conjugate base. These initial results on substituted pHP protecting groups show promise for extending their use in chemistry, physiology, and biochemistry.
2.4. Benzoin Groups
In the course of their ground-breaking studies on benzoin (desyl alcohol; Figure 1; Scheme 2, entry 6) acetates, Sheehan and Wilson determined that the 3′,5′-dimethoxybenzoin (DMB, 33, X = OAc) derivative performed best as a PPG of acetate.4,118 The reaction proceeded in an extraordinarily smooth fashion as illustrated by the spectra shown in Figure 9. The expected product 2-phenyl-5,7-dimethoxybenzofuran (34, DMBF, Scheme 23) was formed in quantitative yield, and the quantum yield was determined as 0.64 ± 0.03. The authors noted that the photocyclization of DMB acetate was not quenched by either naphthalene or neat piperylene, and they concluded that the reaction proceeds from the excited singlet state or an extremely short-lived triplet state.
Figure 9.

Course of the photolysis of DMB (33) acetate to DMBF (34) in acetonitrile (Scheme 23); irradiated in a photochemical reactor at 360 nm. Reprinted with permission from ref (118). Copyright 1971 American Chemical Society.
Scheme 23. Photocyclization of DMB Acetate4,118.
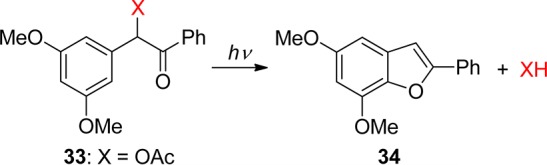
It has taken substantial efforts to elucidate the detailed mechanism of this reaction, and a clear picture (Scheme 24) has emerged only in recent years. Time-resolved work on DMB derivatives was performed by the groups of Trentham,119 Wan,120 Simon,121 Wirz,122 and Phillips.123 By ns-LFP of several DMB carboxylate esters (33, X = OCOR) in dry acetonitrile, Shi, Corrie, and Wan120 observed a strong transient absorption at 485 nm that was formed within the lifetime of their laser pulse (∼10 ns) and decayed with a lifetime of 1 μs. This transient was assigned to the cyclohexadienyl cation (35, Scheme 24). An additional transient absorption with μs lifetime was observed at λmax = 330 and 420 nm. Introduction of air resulted in a faster decay of this transient but did not affect the lifetime of the 485-nm transient or the yield of the final product 34 (DMBF). The (330, 420)-nm transient was therefore assigned to the (nonreactive) triplet state of the DMB esters (333). These assignments have stood the test of time. However, subsequent studies with better time resolution showed that heterolytic cleavage of the excited singlet state is not, as claimed,120 the primary photochemical step of DMB esters, and that the cyclohexadienyl cation is not even an intermediate along the predominant reaction path releasing the substrate HX.122,123b
Scheme 24. Mechanism of the Photocyclization of 3′,5′-Dimethoxybenzoin (DMB) Derivatives (X = OCOR, OPO(OEt)2, F)122,123b.
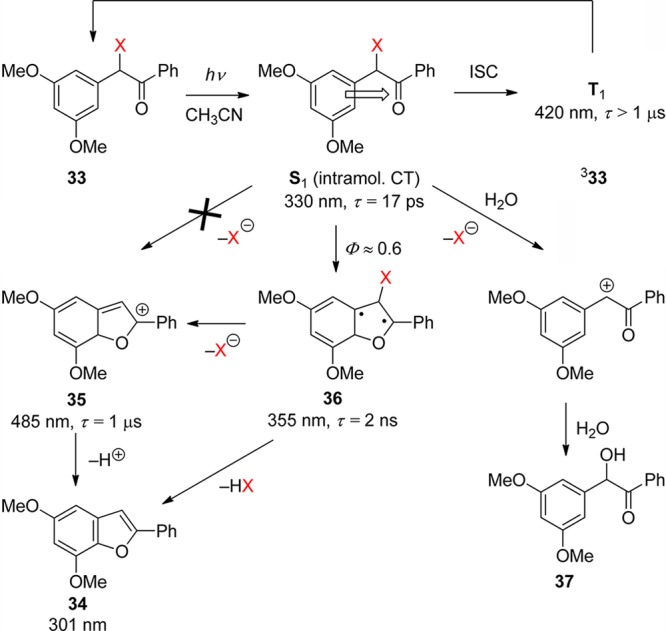
Pump–probe experiments of DMB acetate and fluoride (33, X = OCOMe, F) with picosecond time resolution revealed a preoxetane biradical intermediate 36, λmax = 355 nm, that was formed from the singlet state, τ = 17 ps, and decayed with a lifetime of 1–2 ns.122 The biradical 36 was shown to be the precursor of the cyclohexadienyl cation 35, λmax = 485 nm, the lifetime of which was found to be strongly reduced by the addition of water to the acetonitrile solution, kw = 6 × 106 M–1 s–1. This work proved that the reactive intermediate is the biradical 36, which releases the DMB-caged ligands HX on a time scale of 1–2 ns, largely independent of the leaving group ability of X. Thus, DMB is an excellent PPG for the kinetic investigation of fast processes such as protein folding.124 The existence of a biradical intermediate 36 had been proposed earlier on the basis of preparative work by Rock and Chan, who also showed that a benzoin side-product (37) is formed in aqueous solution.125 Otherwise, the reaction proceeds cleanly and efficiently in both polar and apolar solvents.
In the most recent time-resolved study of DMB phosphate (X = OPO(OEt2), Phillips and co-workers123b investigated the ultrafast primary photophysical processes by absorption spectroscopy and provided evidence for the formation of an intramolecular charge-transfer singlet state (S1, τ = 14 ps), as had been proposed earlier.120 Moreover, using nanosecond time-resolved resonance Raman spectroscopy, they proved that the final product 34 (DMBF) is largely formed within the time resolution (∼10 ns) of their setup. It was postulated that DMBF is formed by concerted HX elimination directly from the biradical 36, largely independent of the solvent, and that the cyclohexadienyl cation is not a precursor of DMBF. Nevertheless, earlier ns-LFP experiments had shown that a second, minor wave of DMBF is also released from the cyclohexadienyl cation on a time scale of 1 μs in dry acetonitrile.122,126 Finally, Phillips and co-workers127 reported extensive CASPT2 calculations in support of the ultrafast primary processes that they had observed by transient absorption.123b
The photoreaction of parent benzoin derivatives follows an entirely different course (Scheme 25). Givens and Matuszewski reported that irradiation of benzoin diethyl phosphate (38, X = OPO(OEt)2; BDP) in acetonitrile proceeds cleanly and quantitatively with a quantum yield of 0.28.128 Stern–Volmer quenching studies with either piperylene or naphthalene indicated that the reaction proceeds via the triplet state with a lifetime of a few nanoseconds. A study using ps-pump–probe and ns-LFP of BDP was reported by Rajesh et al.129 The assignment of the observed transient intermediates was assisted by DFT calculations. Two competing reaction paths of diethyl phosphate elimination proceed from the triplet state of BDP: Reaction via path (a) yielding 39 (R = H or CH2CF3) predominates in water and 2,2,2-trifluoroethanol (TFE), while exclusively path (b) yielding 2-phenylbenzofuran (40) is followed in acetonitrile. Path (a) proceeds via a transient intermediate, λmax = 570 nm, that was attributed to the triplet state of the carbocation formed adiabatically from 338 by heterolytic release of the phosphate anion. A triplet multiplicity of the cation 341 was indicated by the observation of oxygen quenching, kq ≈ 1 × 109 M–1 s–1, and by the fact that its lifetime in water (430 ns) is similar to that in the much less nucleophilic solvent TFE (660 ns). This indicated that the rate-determining step for hydrolysis of the triplet cation is ISC to the singlet ground state. For both pathways, the reactive intermediate releasing diethyl phosphate is thus the excited triplet state with a lifetime of about 10 ns.
Scheme 25. Mechanism of the Photorelease of Diethylphosphate from 38 (X = OPO(OEt)2) in Various Solvents129,130.
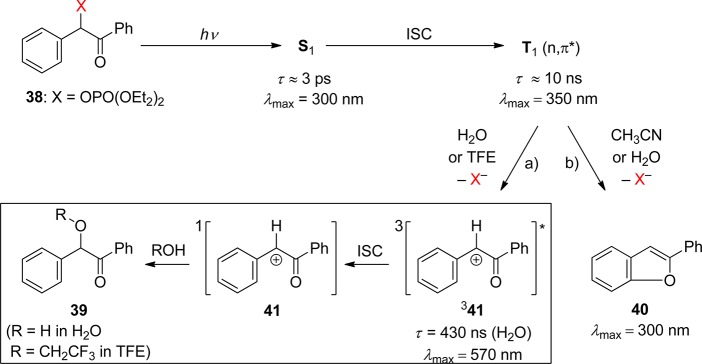
The assignments of the observed transients have been largely confirmed and corroborated by Phillips and co-workers using fs-pump–probe and ns-resonance Raman spectroscopies as well as DFT calculations.130,131 The formation and decay of the cation intermediate in 75% aqueous acetonitrile solution was measured by ns-time-resolved resonance Raman spectroscopy. An essential difference from the previous study129 is that two cationic species appeared in sequence: The first of these (main feature at 1560 cm–1), which was formed within the rise time of the instrument (∼10 ns), was attributed to the triplet cation 341 with optical absorption at 570 nm. The 1560 cm–1 signal decayed with a lifetime of 100 ns, forming another transient species (main feature 1626 cm–1), which decayed with a lifetime of 200 ns and was attributed to the singlet ground state of the cation (41). The lifetime of the short-lived species (100 ns, 1560 cm–1) was reduced by purging the solution with oxygen. It is hard to reconcile the observations by optical LFP129 and Raman spectroscopy,130 and more work may be required to settle this point.
Very soon after the initial reports on PPGs, Sheehan published the photoconversion of substituted benzoin esters into benzofurans with the concomitant loss of acetate (38, X = OAc, Scheme 23).4,132 Several years later he exploited this reaction as a PPG for carboxylic acids.118
The benzoin group remained remarkably underutilized for two decades, until Baldwin and co-workers used it for the photorelease of phosphates. Thus, phosphate derivatives of 33, 38, and 42 (X = OPO32–) were investigated, and all three released inorganic phosphate upon irradiation with a laser at 308 or 355 nm with appreciable yields (35–55% under continuous irradiation).106a
Although, as Sheehan and co-workers had already pointed out in their initial studies, the 3′,5′-dimethoxybenzoin derivative 33 was the most reactive, easier synthesis of the symmetrical benzoin derivative 38 made it equally attractive. Doubts on the purity of the substrates prompted another study, which provided a reliable preparative method for the phosphate (X = OPO32–) and esters (X = OAc), rendering 33 an ideal candidate for a fast and clean release of phosphates.119 The same PPG was used to protect the 3′-phosphate of nucleotides.133
DMB phototriggers have been extensively used for applications in drug delivery,134 muscle relaxation studies,119 lithography,135 biochip fabrication,133,136 protein folding and unfolding,124 or for masking a photochemical switch.137 cAMP derivatives of 38 were also able to release cAMP upon photolysis.107b,107c,138 Likewise, glutamate and GABA were released from 38, with the PPG at the γ-carboxyl group (Scheme 26). However, neither the α- nor the N-protected derivatives of 38 led to a clean photolysis.139
Scheme 26. Release of Glutamate and GABA from Benzoin Derivatives107b,107c,138.
Various types of leaving groups, such as amines, can be released from derivatives of 3′,5′-dimethoxybenzoin (33).140 Although there are no photochemical reasons preventing the use of a whole array of leaving groups, the sometimes delicate preparation of certain derivatives has to be considered. α-Ketol rearrangement can scramble the position of the methoxy groups on both arenes, and an activated ester precursor can react intramolecularly into an inert cyclic product. However, carbamates could be prepared by the reaction of 33 (X = OH) with cyclohexylisocyanate or by preparing the p-nitrophenyl mixed carbonate.140c,141 They were utilized for the photogeneration of bases in films.142 In the solid state, 33 released cyclohexylamine with Φ = 0.067 (254 nm), 0.08 (313 nm), 0.054 (336 nm), and 0.028 (365 nm). The liberated benzofuran side-product absorbs at longer wavelengths; this photobleaching is crucial for applications in thick films, allowing light to reach deeper layers. Again, the methoxy groups on the benzylic side were found to be important for the reactivity; other substituents on the benzoyl side also had an impact, but less significant. An alternative preparative method was devised by Pirrung and Huang, where the benzoin is allowed to react with carbonyl diimidazole (preactivated by methyl triflate), followed by the amine.140b Primary amines, however, failed to give the desired carbamate and gave instead the cyclic product mentioned above.
The activation procedure with carbonyldiimidazole proved to be efficient also for the preparation of carbonates, thus allowing 33 to release alcohols (X = OCOOR). It was used to protect the 5′ primary alcohol of nucleotides,135b various kinds of alcohols, or benzylthiol.136
The protection of chiral molecules (such as nucleotides) can be problematic with DMB, because it also bears a stereogenic center. Access to an enantiopure derivative of 33 would therefore be highly desirable. Enantioselective syntheses of 33 have been published, using enantiopure TMS-protected cyanohydrins generated either by asymmetric catalysis or enzymatic resolution (Scheme 27).143 Addition of an aryl-Grignard reagent to the nitrile, followed by acidic hydrolysis, leads to chiral unsymmetrical benzoin. When asymmetry is not required, more straightforward routes are available, in particular by using a dithiane as a benzoyl anion equivalent.144 This method was used to prepare derivatives of DMB that could release a phenol (ubiquinol),145 or as a linker for peptides.144 This dithiane route was cleverly exploited in a safety-catch strategy, where keeping the carbonyl function masked prevented any photolytic activity, whereas hydrolysis restored the initial sensitivity.146 A related strategy was recently proposed, where the carbonyl group is masked as a dimethyl ketal, which can be smoothly hydrolyzed into the photolabile benzoin derivative (3% trifluoroacetic acid (TFA) in CH2Cl2, 5 min).147
Scheme 27. Preparation of Racemic and Enantiopure 3′,5′-Dimethoxybenzoin (DMB; 33, X = OH)143,144.

As will be mentioned in the next section on nitrobenzyl derivatives (section 3.1), water-insolubility is a major issue when the release of bioactive material is sought under physiologic conditions. Thus, the water-soluble derivative 43 was prepared. The possibility of carrying out the photolysis in aqueous solution gave an additional hint in favor of the cationic mechanism (Scheme 24), as the free DMB alcohol was also observed as a side-product, in addition to the expected benzofuran.125
Attempts to improve the reactivity by further modification of the main core were proposed, such as the replacement of both aromatic groups by the 2-furyl moiety. Dipeptide esters of this “furoin” analogue 44 indeed could be deprotected, but in lower yields than the parent structure.148 On the other hand, esters of the achiral 1,2,2-triphenylethanone 45 proved to be as reactive as 33 (DMB) (Scheme 28).149 The diphenylbenzofuran side-product continues to react under irradiation to form a more conjugated heterocycle.
Scheme 28. Photolysis of 1,2,2-Triphenylethanone Esters149.
Chirality in the PPG is not necessarily a problem and can actually be exploited. Klán and co-workers used enantiopure acrylate derivatives as a photoremovable chiral auxiliary (PCA). Thus, the acrylate 46 reacted in a highly enantioselective manner with cylclopentadiene in the presence of a Lewis acid, and the cycloadduct was photolyzed (Φ = 0.43) at 313 nm to give the Diels–Alder exo product as the main diasteroisomer, with enantiomeric excessed (ee’s) up to 96% (Scheme 29).150 The photochemistry of several benzoin derivatives is summarized in Table 7.
Scheme 29. Use of a Chiral Benzoin as a Photoremovable Chiral Auxiliary150.
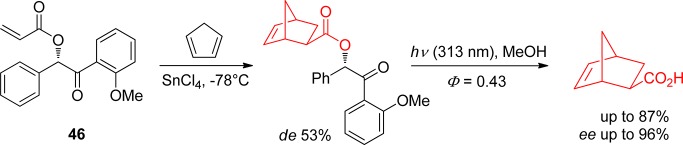
Table 7. Photolysis Quantum Yields for Benzoin Derivatives.
3. Nitroaryl Groups
3.1. o-Nitrobenzyl Groups
o-Nitrobenzylic derivatives have been widely used despite their disadvantages. They were proposed as general PPGs in 1970,3 but there were earlier reports on their photochemistry,151 including the one on the photoisomerization of o-nitrobenzaldehyde into the corresponding nitrosobenzoic acid.152
Hydrogen transfer from the o-alkyl substituent to the nitro group forming an aci-nitro tautomer in the ground state is commonly taken to be the primary photoreaction of o-alkylnitroarenes. Parent o-nitrotoluene (47, oNT, Scheme 30) and several derivatives have been studied by time-resolved spectroscopy.153 The most recent investigation of oNT and the analogous o-nitrobenzaldehyde by Gilch and co-workers,154 who used femtosecond transient absorption and stimulated Raman spectroscopy, has finally provided a convincing and complete picture of the early events. The results reported by these authors about oNT154a are summarized in Scheme 30, which bears a striking similarity to that for the photoenolization of o-methylacetophenone (Scheme 4 in section 2.2).
Scheme 30. Reaction Mechanism for the Phototautomerization of oNT in THF154a.

In aqueous solution, equilibration between the (Z)- and (E)-aci-isomers by proton exchange between the oxygen atoms through solvent water is faster than the intramolecular back-reaction (Z)-aci → oNT and the aci-decay obeys a single exponential rate law.153f A detailed investigation of the pH–rate profile provided the acidity constant of the equilibrated aci-tautomers of oNT, pKa = 3.57 ± 0.02, and kinetic isotope effects on the aci-decay kinetics indicated that the dominant rate-determing step for aci-decay switches from carbon protonation by H+ below pH 6 to carbon protonation by water above pH 6. In strongly acidic solutions, acid-catalyzed addition of water to the methylene carbon followed by dehydration of the resulting nitroso hydrate yields o-nitrosobenzyl alcohol.153f
A total quantum yield (Φaci = 0.08) for the formation of aci-nitro tautomers from oNT in tetrahydrofuran (THF) was estimated on the basis of transient absorbance intensities;154a this value is an order of magnitude larger than previous estimates that were obtained with aqueous solutions of oNT by photoinduced H/D exchange153f and by another comparison of transient absorbance intensities.155 The discrepancy was attributed to fast retautomerization of the (Z)-aci-nitro isomer to oNT.154a However, this explanation cannot hold as the (Z)- and (E)-isomers are rapidly equilibrated in aqueous solution.153f An independent value of Φaci for oNT might be obtained by measuring the quantum yield of the irreversible reaction in strongly acidic solutions.153f Fortunately, many derivatives of oNT show much higher quantum yields Φaci (vide infra).
o-Nitrobenzyl (oNB) and 1-(2-nitrophenyl)ethyl (NPE; see section 3.2) derivatives that carry a leaving group at the benzylic position release the protected substrate upon irradiation. The reaction proceeds via aci-nitro intermediates that are readily observed by flash photolysis at λmax ≈ 400 nm. The decay of these aci-transients frequently follows a biexponential rate law (due, presumably, to the formation of both geometrical isomers at the methylene group); the aci-decay rate constants are on the order of 102–104 s–1 and vary strongly with substitution, solvent, and pH in aqueous solution. A detailed mechanistic study of the release of ATP from “caged ATP”,6P3-1-(2-nitrophenyl)ethyl ester of adenosine triphosphate 48 (Scheme 31), was reported in 1988 by Trentham and co-workers.156
Scheme 31. Photochemistry of P3-1-(2-Nitrophenyl)ethyl Ester of Adenosine Triphosphate156.
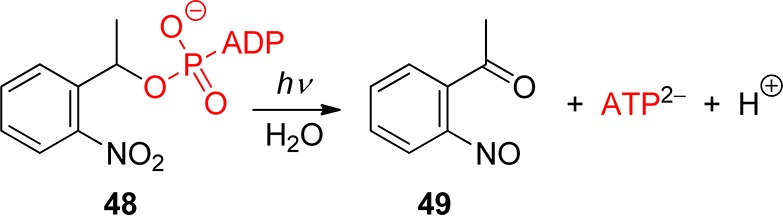
The appearance rates of the three products formed by irradiation of caged ATP, namely, ATP2–(bioassay), 2-nitrosoacetophenone (49, absorption at 740 nm), and H+ (using an indicator dye), were each monitored and were found to coincide with the decay of the aci-nitro intermediate. Later work using time-resolved infrared detection and isotopic labeling further established that the release of ATP occurs in a first-order reaction that is synchronous with the decay of the aci-anion (k = 52 s–1 at pH 7, 10 °C).157 The reaction mechanism proposed by Trentham and co-workers156 was unchallenged for many years, and it was frequently taken for granted that aci-decays were synchronous with substrate release. That assumption is not warranted in general, however, especially with more nucleophilic leaving groups.
Subsequent LFP and time-resolved IR studies were used to study the release of alcohols from oNB158 and NPE159 (section 3.2) ethers at near-neutral pH. Detailed kinetic studies covering a wide pH range (Figure 10)158 indicated that the mechanism proposed by Trentham and co-workers,156 although it had been fully consistent with their results, needed to be revised on several counts (Scheme 32): (a) Cyclization to the 1,3-dihydrobenz[c]isoxazol-1-ol intermediate (50) occurs from the neutral aci-compounds (51), as predicted by DFT calculations,160 not from the conjugate bases (51–). This accounts for the specific acid catalysis often observed for the decay rates of the aci-intermediates. (b) The cyclization 51 → 50 is irreversible. (c) At pH values below 8, hydrolysis of the hemiacetal intermediate 52 formed by ring-opening of 50 is rate-determining for the release of methanol and may be rate-determining with other poor leaving groups. The intermediates 50 and 51 have also been identified by IR analysis following irradiation of oNB methyl ether in Ar and N2 matrices at 12 K.161
Figure 10.
pH–rate profiles for the reaction steps 51/51– → 50 (• and o), 50 → 52 (+ and × ), and 52 → 53 (□) of oNB methyl ether in aqueous solution. Reprinted with permission from ref (158). Copyright 2004 American Chemical Society.
Scheme 32. Mechanism of 1-(Methoxymethyl)-2-nitrobenzene Photoreaction160.
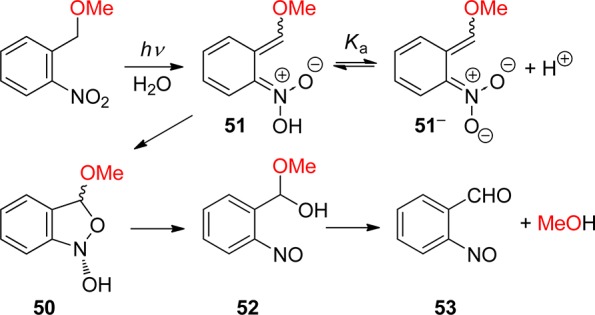
As can be seen from Figure 10, the release of methanol from oNB methyl ether in the final reaction step 52 → 53 is many orders of magnitude slower than the decay of the aci-intermediates 51/51– at pH values near neutral. A faster reaction path bypassing intermediates 50 that was proposed by Corrie et al.159 was subsequently shown not to be operative.158 The same holds for the release of glycolic acid from oNB glycolic acid ether, which also proceeds via a long-lived hemiacetal with a lifetime of 4 s at 20 °C in wholly aqueous solution at pH 7.162 Moreover, buffers were found to intercept the aci-intermediates when used in high concentrations, thereby further retarding the desired release of alcohols from oNB ethers.162
Nevertheless, Ellis-Davies and Barsotti reported fast release of Ca2+ ions from the ethylene glycol tetraacetic acid (EGTA) chelator complexes that are attached through an ether linkage to 3-nitrodibenzofuran-2-yl (54, NDBF EGTA) and several 6-nitroveratryl (55, NV EGTA)163 derivatives. Rate constants of Ca2+ ion release on the order of 104–105 s–1 were observed by monitoring the fluorescence signal arising from the Ca2+-dyes Ca-Green-5N164 or Ca-Orange-5N.163 This enables relatively fast mobilization of intracellular Ca2+ by photolysis of NV- and NDBF-caged IP3 (inositol 1,4,5-trisphosphate).165 The authors assumed that cleavage of the benzylic carbon–ether linkage is much faster in derivatives of NDBF and NV than that of oNB, possibly bypassing a hemiacetal intermediate. In our view, a more likely interpretation of these observations is that the formation of the cyclic intermediates of type 50 already reduces the binding constants of the chelator complexes sufficiently to afford substantial Ca2+ ion release with rates corresponding to the aci-decay rates. Indeed, biphasic release of Ca2+ from compound 55 (NV EGTA) with rate constants of 5 × 104 and 1.5 s–1 has been observed.163

The formation of the reactive aci-nitro intermediates may proceed from both the excited singlet and triplet states. Steiner and co-workers have shown that the 2-(2-nitrophenyl)prop-1-oxycarbonyl (NPPOC, section 3.2) PPGs used in photolithographic DNA chip synthesis can be considerably enhanced by covalently linked triplet sensitizers (section 8.1) such as thioxanthone.166 Moreover, the same group recently reported that sensitization by diffusion in a thin film can be more effective than intramolecular sensitization for sensitizer concentrations higher than 5 mM.167 On the other hand, Görner showed that triplet states of CT character are formed by direct excitation of NV derivatives and related compounds that, however, do not participate in aci-nitro formation or the release of the substrate.168
Various substituents at the benzylic position of oNB were found to increase their perfomance as PPGs. For example, Hess and co-workers discovered that addition of a carboxylate to the benzylic carbon atom of the oNB caging chromophore increased release rates.169 However, decarboxylation is a significant reaction pathway for photolabile calcium chelator derivatives of ethylenediaminetetraacetic acid (EDTA) and EGTA.170 Radical stabilization energies computed by DFT methods were shown to be useful predictors of the relative efficiency with which LGs are photoreleased from oNB protecting groups.171
The presence of a hydroxy group at the benzylic carbon in 56 opens a new pathway for the decay of aci-nitro intermediates via a nitroso hydrate (Scheme 33, path b).168a,172 pH–Rate profiles for the reaction steps involved in paths (a) and (b) in aqueous solution have been determined.172 The caged Ca2+ chelator “nitr-5” is a BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) derivative of an o-nitrobenzyl alcohol reported by Tsien and co-workers,173 who proposed that the relatively fast Ca2+ release (3000 s–1) from “nitr-5” may be attributable to proton shuffling, i.e., path b of Scheme 33.
Scheme 33. Photochemistry of the Hydroxy Derivative 56(168a,172).

The parent oNB structure 57, although not the most widely used, represents the generic form of this family of PPGs. The leaving group can be directly attached to the benzylic site, as in 57, and this is the typical mode for the release of carboxylic acids,3 thiols,174 histidine,175 and phosphates (Scheme 34, several examples shown).5,107b Although it is possible to directly attach alcohols and amines, they are most frequently linked as carbonic acid derivatives 58 (X = OCO–X′), which are better leaving groups and make the synthesis more convenient, i.e., by the direct reaction of the alcohol or amine with the readily available o-nitrobenzyl chloroformate. These strategies are, however, inappropriate when rapid release is needed (e.g., for electrophysiological applications), as the postphotolytic fragmentation is the rate-limiting step for release.140a,159 This also does not prevent other major drawbacks of this PPG for the release of alcohols, which is slow (vide supra), and of amines, which undergo condensation with the nitrosoaldehyde side-product, leading to a stable imine; however, the latter reaction can be avoided by the addition of carbonyl scavengers, such as semicarbazide hydrochloride.3 Another more serious problem is the formation of nitrosoaldehyde that yields brown-colored degradation products that absorb the incident light, thus creating an internal filter.
Scheme 34. Photolysis of Some o-Nitrobenzyl Derivatives.
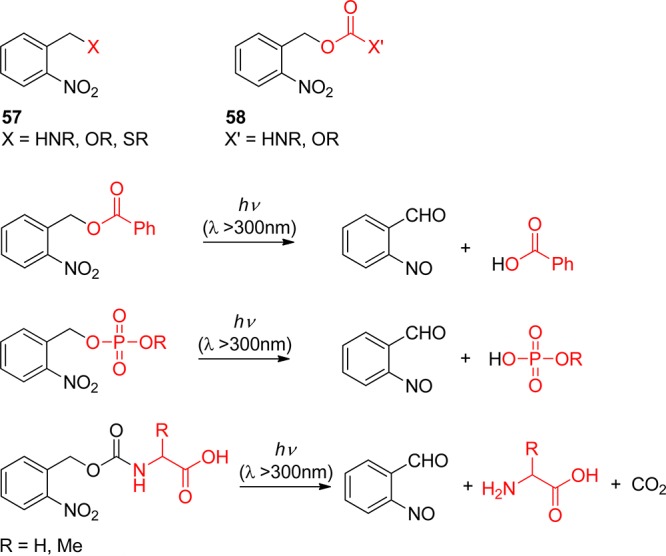
o-Nitrobenzylic PPGs have naturally led to various synthetic and biological applications, especially for caged biomolecules, and only selected examples will be discussed here as many reviews have been published on the field during the past decade.9c,9f,9l,9m,12b,176 For example, photolytic deprotection of PPGs under very mild conditions provides a great advantage for automated DNA or RNA synthesis compared to the traditionally acyl-protecting group used. In this aim, a pioneer application was developed by Stutz and Pitsch using oNB (57) as a protecting group for the 2′-oxygen of the four different bases for oligoribonucleotide synthesis (Scheme 35).177 Interestingly, whereas nucleobases were protected via the usual carbamate function, the 2′-oxygen and the 2-nitrobenzyl group were linked together via an oxymethyl acetal.
Scheme 35. Protected Phosphoramidite Building Blocks for Automated RNA Synthesis177.
2,4-Dinitrophenol was released in mitochondria, by first linking it to a cationic phosphonium targeting group via an o-nitrobenzyl ether. Incubation in the cytoplasm allowed migration of the assembly into the mitochondria and was followed by subsequent photolysis (λ = 355 nm).178 Ingenious inclusion of nitrobenzylic units in vesicles allowed a dose-controlled release of the encapsulated hydrophobic guests upon irradiation (λ > 315 nm).179 Other interesting applications were developed in the preparation of otherwise inaccessible metal–organic frameworks, by inclusion of o-nitrobenzyl ethers and subsequent photolysis.180
o-Nitrobenzylic PPGs have also been employed in natural product syntheses. One well-known and impressive application was reported by Nicolaou and co-workers in the total synthesis of the Calicheamicin γ1 where the high compatibility of 57 with other classical PGs and functions was demonstrated.181 Another smart example is the use of 57 in the synthesis of an analogue of Leukotriene C4 (LTC4, Scheme 36).182 In the final steps, deprotection by irradiation at 350 nm of the nitrobenzyl derivative affords the secondary amine in 74% yield. It is noteworthy that no isomerization of the triene part and no racemization were induced by photolysis.
Scheme 36. Final Steps in the Total Synthesis of N-Methyl LTC4182.
Over the years, multiple modifications have been developed to tune the properties, mostly toward an increase in quantum yield, an increase in the rate of release, and increasing absorbance at longer wavelengths. Substitution at the benzylic site mainly affects quantum yields, whereas modification on the aromatic moiety affects the absorbance.
Substitution at the Benzylic Position
In addition to providing an electronic effect at the benzylic site, a second hydrogen-abstracting unit could, in principle, increase the efficiency. Already in the 1970s, Patchornik, Amit, and Woodward proposed this modification, which worked up to a certain point.3 It should be kept in mind that most substitutions at this site create a chiral center, which can become a drawback if chiral molecules have to be protected (and that is the case for most of the relevant applications, such as amino acids, carbohydrates, and oligonucleotides), unless the added subsituent is identical to the aromatic core. A second o-nitrophenyl would fit in this category. However, an increase in photolysis quantum yields was not explicitly mentioned, and the increase in chemical yield might just be the consequence of the formation of a ketone instead of an aldehyde side-product, which is less prone to the parasitic imine formation. Thus, 59 was able to release carboxylic acids quantitatively and inorganic phosphate with 85% yield,106a whereas 60 released amino acids in yields between 70% and 95% in the absence of a carbonyl scavenger. Various derivatives have been prepared since, among them 61 and 62, which liberated acetic acid in good quantum yields (Φ = 0.2 for 61),168b or 63.183
Despite the problem of chirality, a phenyl group can have a beneficial effect, as had been shown much earlier by Barltrop and co-workers with esters of 64 and carbamates of 65,151 and was later used in linkers for solid-phase synthesis.184
Simpler substituents, such as a methyl group, have a beneficial impact on the quantum yield. Thus, pivalate esters of 57 have release quantum yields in a polymeric matrix of 0.04 and 0.13 in acetonitrile solution, whereas they are much higher for pivalate ester 66 (Φ = 0.09 in polymer and 0.64 in acetonitrile).185 Solution-phase quantum yields cannot be directly extrapolated to those of the solid phase, as the available conformations can be quite different.186 Derivatives of 66 have many other uses, such as the release of biologically active carboxylic acids in plant cells,187 or on solid support.188 Specht and Goeldner proposed the introduction of a strongly electron-withdrawing group at the benzylic carbon, which had a massive effect on the quantum efficiency on choline and arseniocholine ether derivatives (Φ = 0.7 for 67 and Φ = 0.43 for 68) and sugar ethers (Φ = 0.62 for 67 and Φ = 0.52 for 68). Such derivatives were, however, not widely used in organic synthesis, as they were assembled by Mistunobu reactions, which were sluggish for secondary alcohols. Nevertheless, interesting applications, such as the release of choline or arsenocholine, were used as examples.189
Jullien and co-workers prepared and evaluated the 6-nitroveratryl (NV, or 4,5-dimethoxy-2-nitrobenzyl, DMNB) derivatives 69, 70, 71, and 72 with electron-withdrawing substituents at the benzylic site; despite earlier claims, it was found that the substituents at the benzylic site, while having an unambiguously observable effect (up to a factor 3 between the most and least efficient), did not increase the quantum efficiency by orders of magnitude (Φ = 0.013 for −Br and Φ = 0.003 for −CN).190 Interestingly, the cyano compound had been independently identified by a clever combinatorial technique by Pirrung and co-workers as being quite efficient.191 Such apparent contradictions have been reported in the field of PPGs for decades and could potentially slow down future development of new or modified PPGs, as no clear trend emerges from the huge amount of sometimes unreliable data accumulated. The determination of quantum yields remains a delicate operation subject to significant experimental error; furthermore, all these different groups are frequently tested for the release of different leaving groups (phenols, carboxylic acids, amine, aliphatic alcohols, and carbamates) under different irradiation wavelengths, in different solvents and buffers, and each with a different photochemical apparatus. We have seen above the enormous importance that the effects of solvation and proton transfer play on the reaction mechanism.
As mentioned for the benzoin PPG (see section 2.4), water-solubility is a crucial point for biological applications, and this property can be achieved by the introduction of a CO2H substituent. Hess and co-workers designed the α-carboxynitrobenzyl (α-CNB) derivative 73 for the release of phenolic OH groups; there was a significant increase in the release rate, but this was observed only at shorter wavelengths.169,192 Bassani and co-workers recently reported that the α-carboxy-6-nitroveratryl (α-CNV) analogue 74 released carboxylic acids, in quantitative chemical yields with Φ = 0.17 and a rate of release 2–3 orders of magnitude larger than the parent compound,193 and capsaicin through an ether bond.194 A related strategy (based on 75) had been devised by Schaper and co-workers.195 Thiols could also be released with the derivative 76.196 A time-resolved infrared study of the photoreactivity of α-CNB 74 was performed by Corrie and co-workers.197 Related groups were studied computationally by Schaper and co-workers.198
Pirrung and co-workers designed a clever way of dealing with the nitroso side-product, responsible for the internal filter effect. It was trapped in situ by a Diels–Alder reaction with a diene function included at the benzylic site (Scheme 37).199 These pentadienylnitrobenzyl PPG esters, ethers, and carbamates 77 were used to release carboxylic acids, alcohols, and amines (Φ = 0.22, 0.41, and 0.38, respectively); an analogue with a methylenedioxy group on the aromatic ring was also discussed, with improved properties (in this case, the increase in absorbance more than compensated for the usual decrease in quantum yields).
Scheme 37. Trapping of the Nitroso Byproduct in a Diels–Alder Cycloaddition199.
Substituents on the Aromatic Ring of the o-Nitrobenzyl Chromophore
Because of the problems of chirality, the synthetic issues, and the limited impact on the absorbance, considerably less work was devoted on substitution at the benzylic site than the very numerous modifications on the aromatic ring. Such modifications bring three features: (a) the tuning of the absorbance with concomitant effects on the quantum yield, (b) the possibility to anchor the group on a solid support or on a linker (this will not be discussed here, as a book chapter reviewed the field recently),200 and (c) the possibility to modulate the solubility properties of the group.
A second electron-withdrawing nitro group was added to increase the hydrogen-abstraction capacity (reminiscent of 59 and 60), and the release of amines from carbamates 78 was much more efficient than for 58 (Φ = 0.62 versus Φ = 0.13 in the solid state).186 Interestingly, in the same study, the carbamate derivative of 66 showed a quantum yield of release of 0.11, which differs from earlier data. This was attributed to the influence of the matrix. The analogue 79 was also exploited a few years later.187
Various groups have been added to increase the hydrophilicity of the cage again to address the water-solubility issue. As an example, the carboxylic acid 80 with a one-carbon spacer was reported by Allan et al.187 Similarly, 81 and 82 were also exploited.169b,201
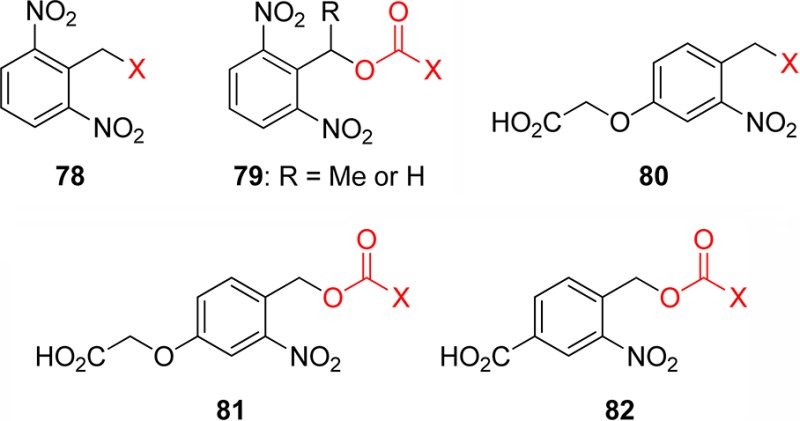
Early on, two methoxy groups were added to increase the absorbance at longer wavelengths (λ > 350 nm, and Rayonet lamps at 420 nm still provide photolysis at reasonable rates), leading to the 6-nitroveratryl (NV, 83) and 6-nitroveratryloxycarbonyl (NVOC, 84) moieties. These are undoubtedly the most frequently used groups to date both in solution3 and in the solid phase (for two recent, among many other, examples, see ref (202)). Multiple variants have been evaluated, such as the introduction of an α-methyl group at the benzylic site: the methyl-6-nitroveratryl (MeNV, 85) and methyl-6-nitroveratryloxycarbonyl (MeNVOC, 86) groups. Very close analogues, but with slightly altered properties, presumably due to a restriction in the freedom in aligning the nonbonding orbitals of the oxygen with the aromatic π system, are the methylenedioxy derivatives 87 and 88. Further addition of an α-methyl group led to α-methyl-(6-nitropiperonyloxymethyl) (MeNPOM, 89) and the now famous analogous 3,4-(methylenedioxy)-6-nitrophenylethoxycarbonyl (MeNPOC 90) groups. The latter moiety was used in the automated synthesis of DNA chips.203 A solution-phase comparison of the quantum yield of release of thymidine derivatives of NVOC (Φ = 0.0013) and MeNPOC (Φ = 0.0075) was performed.204
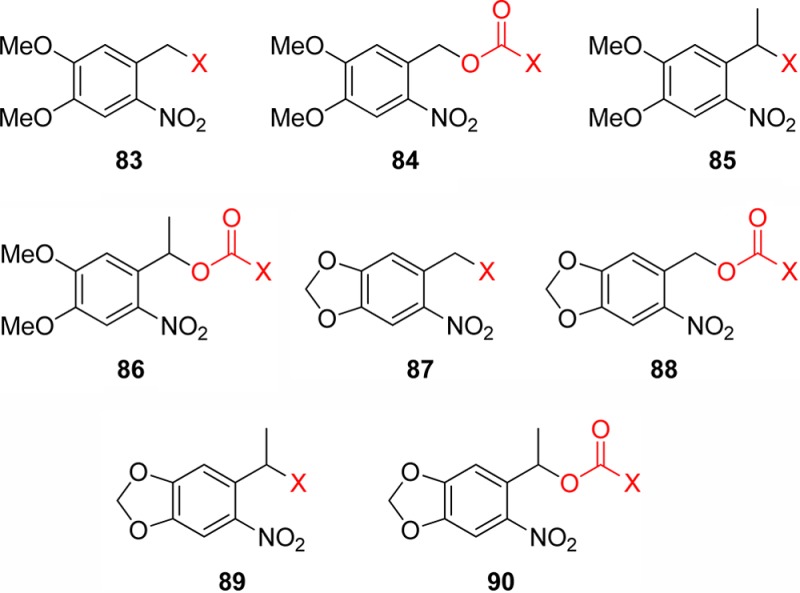
Caged nucleobases were used for the transient disruption of DNA hybridization to photochemically controlled DNAzyme activity.205 This strategy is illustrated in Scheme 38 where the 89 (MeNPOM)-protected thymidine phosphoramidite is incorporated into DNA using classical synthetic procedures. The caged-DNAzyme thus obtained can be quantitatively reactivated by irradiation at higher wavelengths. MeNPOM presents several advantages compared to the parent compounds 73, 87, and 90: the very efficient conversion upon irradiation at 365 nm, the high stability of the caged molecules in aqueous media, the less toxic acetophenone byproduct, and its easy incorporation via the chloromethyl ether derivative.206
Scheme 38. Preparation of Caged-DNAzyme and Activation by Nonphoto-Damaging UV Light205.
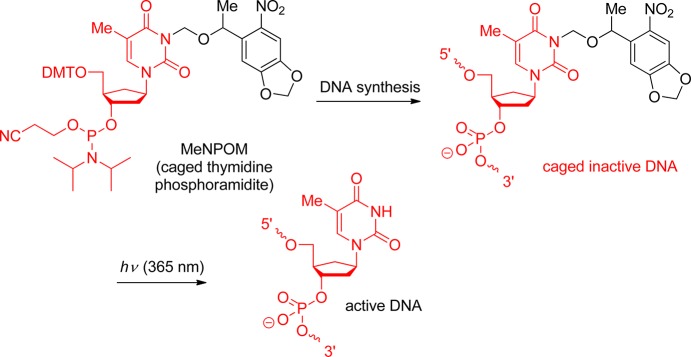
Inactivation of fluorophores (see section 11) was effected by similar caging, for example, by binding nitroveratryl groups to rhodamines and fluoresceins.207 A recent computational study198 was complemented by an experimental systematic evaluation of substituents on the aromatic ring and at the benzylic site168b and showed surprisingly low influence of these groups (except for the strongly electron-releasing amine of 91, an effect that was reported earlier). Indeed, one of us reported a very significant lowering of the deprotection efficiency by having electron-releasing groups para to the nitro group, a manifestation of the CT-character of the excitation, as mentioned earlier.208 This was exploited in a so-called safety-catch approach, where esters of 92, 93, 94, and 95 were protected against photolysis in normal conditions. Upon protonation with a strong acid, the reactivity was restored, thus allowing normal photolysis.209
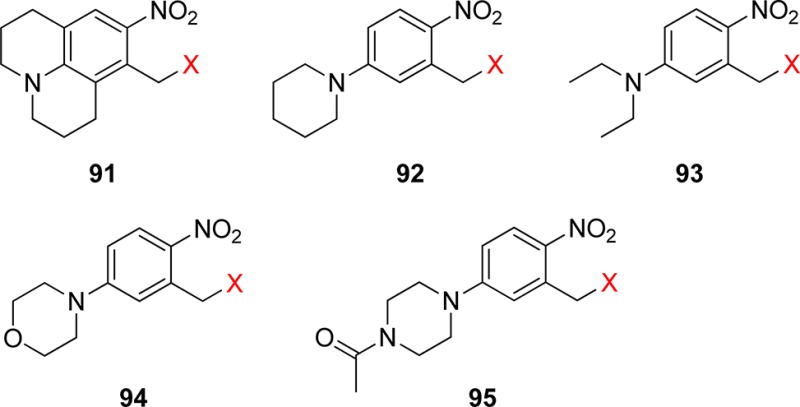
Similarly, the reactions of the compounds 96, 97, and 98, evaluated by Jullien and co-workers, were found to be less efficient than those of the nonsubstituted congeners (in fact, they were too slow to be measured in the case of 96 and 97).190
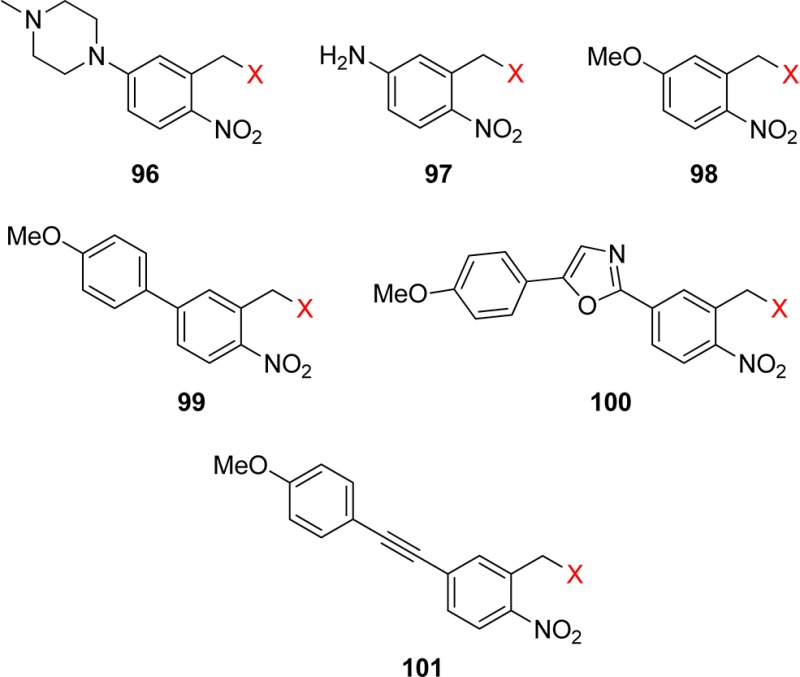
The same authors also examined the effect of other groups, in para-position to the nitro group, with or without additional conjugation (99, 100, and 101).190 As was the case for the benzylic substituent, these extra groups had a limited impact on the quantum yield, although the highly conjugated group in 101 decreased the reaction efficiency. The authors also explicitly pointed out an inverse correlation between the quantum yield and longer absorption wavelengths. Various other o-nitrobenzyl alcohol (e.g., 102 and 103) derivatives substituted in the ortho-position to the benzylic group, or meta-position to the nitro group, have been reported.12b,185,208 The reaction efficiency of their ethers was tested in terms of kinetics of release, quantum yields in solution, or quantum yields in the solid state as a function of the irradiation wavelength for some of them.
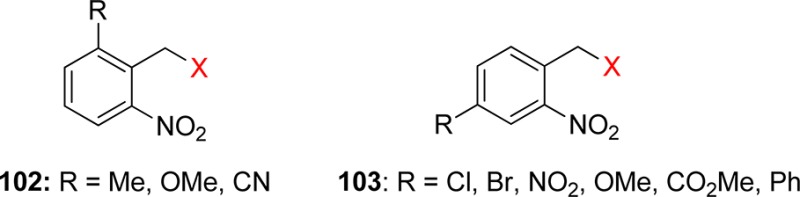
In an effort to increase the absorbance at longer wavelength, Singh and Khade extended the aromatic core as a naphthalene210 or 7-methoxynaphthalene.211 The corresponding esters 104 were able to release aromatic rings containing carboxylic acids at 380 nm, with Φ ranging between 0.08 and 0.16 (in acetonitrile–water).210 The nitrodibenzofuran 105 also had very interesting properties (such as efficient deprotection), which were applied in recent examples.164,165
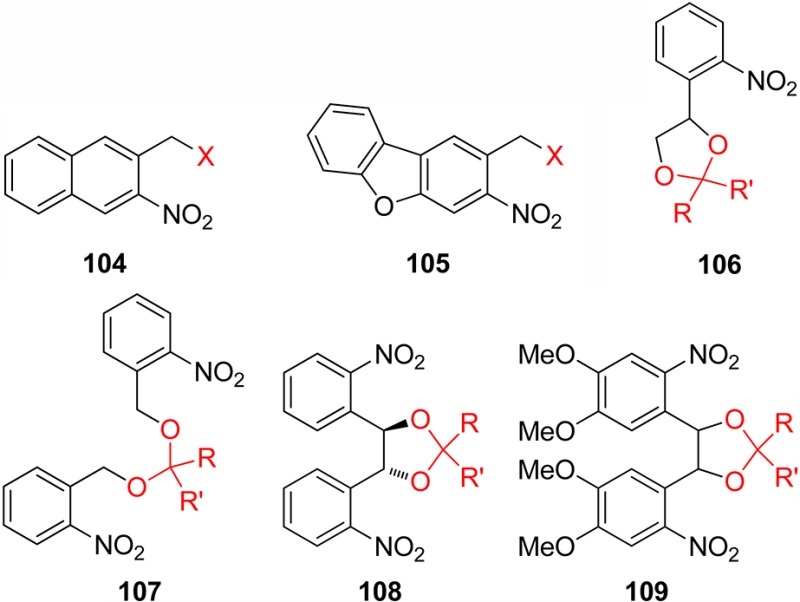
The carbonyl group is among the groups most frequently in need of facile protection–deprotection protocols, so it is surprising that very few PPGs have been developed for it. Gravel and co-workers were among the first to address this issue, by first introducing the ketal 106 in 1974.212 One issue was the generation of a mixture of diastereoisomers if the ketone to be protected was nonsymmetrical. The same group found later that the even simpler ketal 107 could circumvent the problem.213 A solid-support version of 106 was developed.214
Another way of dealing with the issue of chirality was proposed by one of us with the ketal 108, derived from the very easily prepared enantiopure (1R,2R)-bis(o-nitrophenyl)ethanediol,215 which was used by Walsh and co-workers for the preparation of the otherwise too sensitive intermediate.216 The substituted version 109 with a red-shifted absorption was proposed later.217 On the other hand, for the release of simple aldehydes, it is not necessary to consume two aromatic-releasing units, and α-acetyl acetals have been shown to release aldehydes efficiently upon irradiation (Scheme 39).218
Scheme 39. Photolysis of α-Acetyl Acetals218.
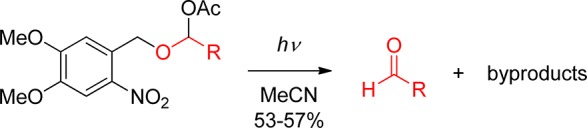
In a similar strategy, the o-nitrobenzaldehyde derived acetals 110 or 111 were used to protect 1,2- or 1,3-diols, principally for protection of saccharides.219 However, in this case, only one alcohol can be deprotected, leading to photostable 2-nitrosobenzoic acid derivatives. Moderate to high regioselectivities were obtained during the deprotection, mostly determined by steric and electronic effects of the nonsymmetrical substrates (Scheme 40).219a The final deprotection of diols could be achieved under basic conditions or by intracellular hydrolysis. A reverse strategy was developed by Iwamura et al. with the treatment of 111 by boron trifluoride in the first step to release an ether of 57, which was subsequently cleaved photochemically.
Scheme 40. Selectivity in the Deprotection of 110-Protected 1,2-Diol219a.

Two-Photon Absorption
The two-photon irradiation technique (see also section 9) enhances the efficient temporal and spatial control of biomolecule delivery. Taking advantage of this for a biological application, Kao and co-workers reported that vanilloid derivatives of oNB (57) and NV (83) can be photoreleased in situ to activate TRPV1 receptors on nociceptive neurons with one-photon quantum efficiencies of 0.13 and 0.041 (Scheme 41).220 In another example, retinoic acid derivatives of 72 and 83 were prepared and photolyzed, first at 365 nm.221 The presence of a long-wavelength tail in the absorption spectrum does not prevent liberation, although isomerization of double bonds did occur. In the two-photon mode (excitation at 750 nm; 25 mGM) retinoic acid was also liberated from cage 57, and dynamic studies on zebrafish embryogenesis were carried out (vide infra).
Scheme 41. Two-Photon Activation of Vanilloid Derivatives220.
An interesting alternative to the simultaneous absorption of two photons is the use of lanthanide-containing upconverting nanoparticles (UCNPs; see section 8.3). In this case, two near-infrared (NIR) photons are absorbed sequentially, and the nanoparticle re-emits one higher-energy photon, in a spectral range that can be exploited by PPGs. Such an example was recently shown by Branda, Zhao, and co-workers, where nitroveratrylester-containing micelles were dissociated upon irradiation with a continuous-wave NIR laser.222 A similar strategy was also used to photolyze benzoin esters.223
Isotopic Substitution
As the transfer of hydrogen from the benzylic site to the nitro group is involved in the primary photochemical step, it was found that the rupture of the stronger C–D bond was less efficient.224 The photolysis of the esters 57 bearing a perdeuterated benzylic position was 3.8–8.3-times less efficient (depending on the irradiation wavelength). The substituents on the aromatic ring in the analogous esters 83 diminished this isotope effect. The origin of these observations has not yet been fully elucidated; so far the phenomenon was utilized in the selective removal of two photolabile (oNB and NV) groups (Scheme 42 a and b depicts two different isotopically labeled derivatives).225 The photochemistry of the oNB derivatives is summarized in Table 8.
Scheme 42. Impact of Isotopic Substitution on the Photoreactivity225.
Table 8. Photolysis Quantum Yields for Nitrobenzylic Derivatives.
| PPG | X (leaving group) | solvent (λirr/nm) | Φ | ref |
|---|---|---|---|---|
| 57 (oNB) | thymidine-OCO2 | MeOH/H2O, 1:1 (365) | 0.033 | (226) |
| 57 (oNB) | t-BuCO2 | MeCN (254) | 0.13 | (185) |
| 61 | CH3CO2 | MeCN (254) | 0.2 | (168b) |
| 66 | t-BuCO2 | polymer (254) | 0.09 | (185) |
| 66 | t-BuCO2 | MeCN (254) | 0.64 | (185) |
| 66 | c-Hex-NHCO2 | polymer (254) | 0.11 | (186) |
| 67 | choline-O | buffer, pH 7 (364) | 0.7 | (189) |
| 67 | sugar-O | buffer, pH 7 (364) | 0.62 | (189) |
| 69 | arsenocholine-O | buffer, pH 7 (364) | 0.43 | (189) |
| 69 | sugar-O | buffer, pH 7 (364) | 0.52 | (189) |
| 70 | coumarin-CO2 | MeCN/buffer, pH 7, 1:1 (325) | 0.013 | (190) |
| 71 | coumarin-CO2 | MeCN/buffer, pH 7, 1:1 (325) | 0.011 | (190) |
| 72 | coumarin-CO2 | MeCN/buffer, pH 7, 1:1 (325) | 0.003 | (190) |
| 73 | PhCO2 | EtOH/H2O, 1:1 (355) | 0.17 | (193) |
| 77 | PhCO2 | CDCl3 (350) | 0.22 | (199) |
| 77 | BnO | CDCl3 (350) | 0.41 | (199) |
| 77 | c-Hex-NHCO2 | CDCl3 (350) | 0.38 | (199) |
| 79 | c-Hex-NHCO2 | THF (254) | 0.62 | (186) |
| 79 | c-Hex-NHCO2 | polymer (254) | 0.13 | (186) |
| 83 (NV) | thymidine-OCO2 | MeOH/H2O, 1:1 (365) | 0.0013 | (204) |
| 83 | coumarin-CO2 | MeCN/buffer, pH 7, 1:1 (325) | 0.006 | (190) |
| 90 (MeNPOC) | thymidine-OCO2 | MeOH/H2O, 1:1 (365) | 0.0075 | (204) |
| 104 | RCO2 | MeCN/H2O, 3:2 (>370) | 0.08–0.16 | (210) |
3.2. o-Nitro-2-phenethyloxycarbonyl Groups
In the late 1990s, Hasan and co-workers designed the oNB one-carbon homologue 112 (1-(2-nitrophenyl)ethyloxycarbonyl, NPEOC; “OC” stands for the −OC(=O)– group) and its α-methylated analogue 113 (NPPOC).226
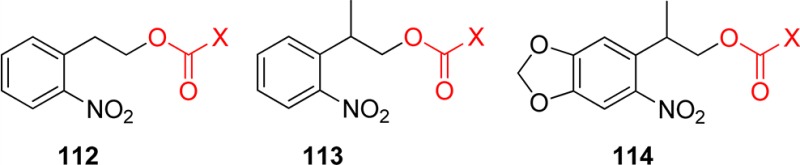
Interestingly, despite the structural similarity between 112/113 and 57, the release mechanism differs markedly, and the observation of a nitrostyrene side product is compatible with a photoinduced elimination (Scheme 43),226 reminiscent to the photoelimination from ketones by photoenolization mentioned earlier (Scheme 11). It turned out that 112 eliminated a 5′-O-nucleoside carbonate (Φ = 0.042) with a higher quantum yield than that of its oNB analogue 57 (Φ = 0.033). The substitution of the benzylic center with a methyl considerably added to the efficiency (Φ = 0.35), which made 113 a candidate of choice for oligonucleotide synthesis.227 This group was further modified to increase the absorbance, such as in the 2-(3,4-methylenedioxy-6-nitrophenyl)propoxycarbonyl (MNPPOC; 114)204,228 or analogous 3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl (DMNPB, section 9)229 derivatives, but 113 was applied in automated light-directed olgonucleotide synthesis (DNA-chips).230 Amino acids231 and carbohydrates were protected with NPPOC at the 6-position, including thiophenyl glycoside donors.232 NPPOC derivatives are themselves relatively inefficient in two-photon deprotections; however, in the presence of sensitizers (section 8) (it has been shown by Wöll and co-workers that NPPOC can be sensitized),166,233 the cross section can reach useful levels for several applications.234 This is discussed in more detail in section 9. A very recent example of sensitization was shown by Winssinger and co-workers, where a thioxanthenone sensitizer is bound to a nucleic acid sequence, which is able to promote the photolysis only when hybridized with a NPPOC-containing sequence with the matching complementary bases.235
Scheme 43. Photolysis of 2-Nitro-2-Phenethyl Derivatives226.
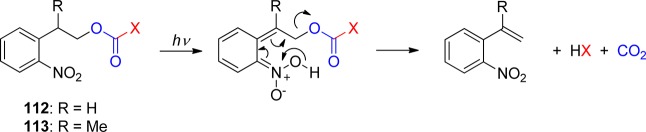
Further modification of the backbone may increase the two-photon absorption cross section (section 9), such as in the biphenyl derivative 115(236) or highly conjugated systems 116 and 117.237 This has also been recently demonstrated by Goeldner and co-workers, who studied the photochemical properties of biphenyl-containing photoremovable protecting groups possessing the o-nitrobenzyl, phenacyl, and 2-(o-nitrophenyl)propyl functionalities.238

Replacement of the methoxy substituent of 115 by an amino group increased significantly the two-photon cross section (section 9).239 The high efficiency of NPPOC was exploited in many applications.166,204,228,230−234,236,240
3.3. o-Nitroanilides
Nitroanilide derivatives, such as 118, were shown in the 1970s to be photolabile and to release carboxylic acids (Scheme 44) and various byproducts depending on the conditions.241 The early works concluded that the reaction is a rearrangement and not a solvolysis, as no isotopic incorporation was observed when the photolysis was carried out in H218O. Likewise, derivatives of 119 are also photolabile, but their cyclic nature prevents a direct H-abstraction on the amide alkyl substituent.242 Failure to provide esters when the reaction is carried out in alcoholic solvents was an additional argument against a simple solvolysis.
Scheme 44. Photoactive o-Nitroanilide Derivatives241.
Neither 118 nor 119 were widely exploited in synthesis, to the exception of the carbamate derivatives of 118 (R = O–alkyl, R1 = Me, R2 = H) that were used as a PPG for alcohols (Scheme 45).243
Scheme 45. Use of o-Nitroanilide Carbamates as PPGs for Alcohols243.
The size of the fused nitrogen heterocycle proved to be crucial for the reactivity, and the 5-membered analogues of 119 (Scheme 44), such as 120 and 121 (R = alkyl), released carboxylic acids very efficiently upon photolysis.244 Most remarkably, when the reaction was carried out in nucleophilic solvents, such as methanol or ammonia-containing dichloromethane, the primary amide was obtained. Corrie, Bochet, Toscano, and co-workers succeeded in providing a comprehensive picture of the reaction mechanism.245 The mechanistic studies concluded that a photoinduced acyl migration from the indoline nitrogen atom of 122 to one of the nitro group oxygens occurs, leading to a highly electrophilic N–O–acyl intermediate 123 (Scheme 46).245a,245b
Scheme 46. Diverging Reaction Pathways in Organic Solvents and in Water245a,245b.
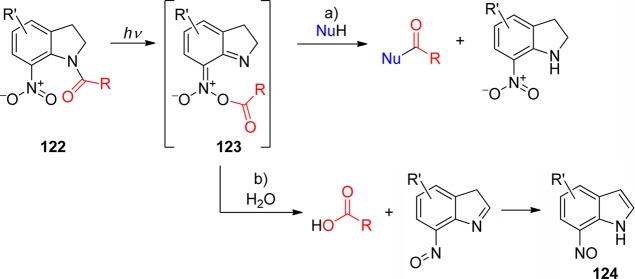
Interestingly, this intermediate further reacts via two distinct pathways depending on the solvent. In water, deprotonation releases a carboxylic acid, with the concomitant formation of a nitrosoindole 124 (pathway b, Scheme 46), whereas in moist organic solvents, the acyl moiety undergoes a nucleophilic attack either by the solvent (water or alcohol) or by an external nucleophile (pathway a). Variation in the substitution pattern on the indoline aromatic ring can to a certain extent influence the ratio of the side-products.246 These 7-nitroindolines release their protected function efficiently,247 and tuning of the aromatic substituents at the C-4 and C-5 position has an impact on their reactivity. Thus, the acetamide 120 (R = Me) releases acetic acid with quantum yields of 0.06 and 0.12 in water and MeCN/H2O 99:1, respectively.245a On the other hand, the reaction of 125 was about 2.5 times more efficient than that of 121 or 126, and 5 times more efficient than that of 127; 128 and 129 were totally photochemically inert, possibly due to a low-lying charge transfer excited state.246,248 Corrie and co-workers also designed a clever combination of a nitroindoline core and an absorbing “antenna”, which will be discussed later (section 8.1).249 The compounds 130(250) and 131(251) have recently been proposed as PPGs.
The bromonitroindoline 120 was used in an early example of peptide synthesis as a protecting group for the C-terminus.252 The solvolysis pathway was used to give the carboxamide by using an ammonia-containing solvent, a feature frequently sought in peptide synthesis. A few years later, a spectacular exploitation of this nucleophilic attack of the reactive intermediate was published by Pass, Amit, and Patchornik, with the use of a N-nucleophilic peptide fragment (Scheme 47).253 Otherwise sensitive glycopeptides were assembled using this strategy.254
Scheme 47. Peptide Coupling Using Acylated 5-Bromo-7-Nitroindolines (Bni)253.

More recently, the derivatives 121 were used in regular organic synthesis for preparing amides from amines,255 but also by using weaker nucleophiles in the formation of thioesters from thiols256 and esters from alcohols.257 The preparation of more complex derivatives of 121 can be difficult, as the nucleophilicity of the indoline nitrogen is greatly reduced by delocalization through the two strong electron-withdrawing groups, but indirect routes were designed and the synthesis of a series of amino acids was recently published.258 The availability of such photoactivable amino acids allowed the all-photochemical synthesis of peptides, by exploiting the different-wavelengths sensitivity of the indoline protecting group on the N-terminus.259 For other application of this chromatic orthogonality, see section 10.
Carbamates can also be prepared by photoinduced transfer of fluorenylmethyloxycarbonyl (Fmoc) 132 or benzyloxycarbonyl (Cbz) 133 derivatives. So far, the Boc analogues were significantly inferior (Scheme 48).260
Scheme 48. Photochemical Introduction of the Fmoc and Cbz Groups260.
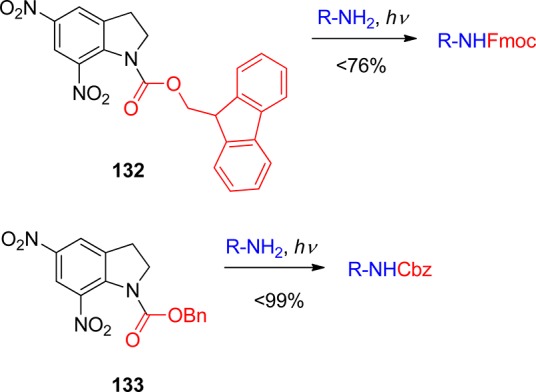
Other functional groups were released from nitroindoline derivatives; Hassner et al. published the release of alcohols from carbamates of 120 (R = OR), or amines from ureas of 120 (R = NR1R2).261 The 7-nitroindolines found the applications in neuronal studies262 and other physiological sciences. For example, derivatives of 125 were found to be active in releasing glutamate or GABA in two-photon absorption mode.249e,263 In some instances, 7-nitroindolines proved to be inferior compared to nitrobenzyl cages.264 In general, photoinduced acyl transfers are relatively rare,255c and recent attempts to design groups with similar properties were only moderately successful.265
4. Coumarin-4-ylmethyl Groups
Photolysis of 7-methoxycoumarinyl-4-methyl derivatives, which established coumarins as a photoactivatable phosphate-releasing group, was first demonstrated by Givens with diethyl phosphate ester 134 (Scheme 49).266
Scheme 49. Release of Phosphates from 7-Methoxycoumarin Derivatives266.
This discovery set the stage for the development of a new class of (coumarin-4-yl)methyl cages (Table 9). The attractive features of the coumarin phototriggers, such as large molar absorption coefficients at longer wavelength, and fast release rates, taken together with improved stability and fluorescent properties that provide a tag for convenient monitoring of the reaction course, have raised researchers’ awareness to this relatively new class of PPGs.9f,9u,267 Clever modifications of the substituents at C6 and C7 in the coumaryl group addressed low water solubility, typical of the first-generation 7-alkoxycoumaryl PPG (types 135 and 136; Table 9), and also helped extend the absorption maximum of the group into the biologically benign range (≥350 nm). Thus, installation of an additional alkoxy group at C6 in the structure of parent 7-alkoxycoumarinylmethanol produced a shift of the absorption maximum by 30 nm. The 6-bromo-derivative was designed to lower the pKa of the 7-hydroxy group by two units to effect a complete deprotonation at physiological pH, thereby enhancing water solubility and causing a λmax shift of 60 nm. Introduction of the second-generation of coumarinylmethyl PPGs with 7-amino substituents (type 137; Table 9) further improved the spectroscopic and photochemical properties of the cage, moving the absorption maxima to 350–400 nm and recording the highest quantum yields (0.21–0.28) among the analogues. The problem of water solubility, which can be an obstacle to biological application, has been addressed by appending polar groups such as carboxylates to the aniline moiety. Polyaromatic analogues were also synthesized to improve fluorophoric properties of the cage for use both as fluorescent tags and as PPGs (type 136; Table 9).
Table 9. Typical Coumarin Chromophores Applied as PPGs.
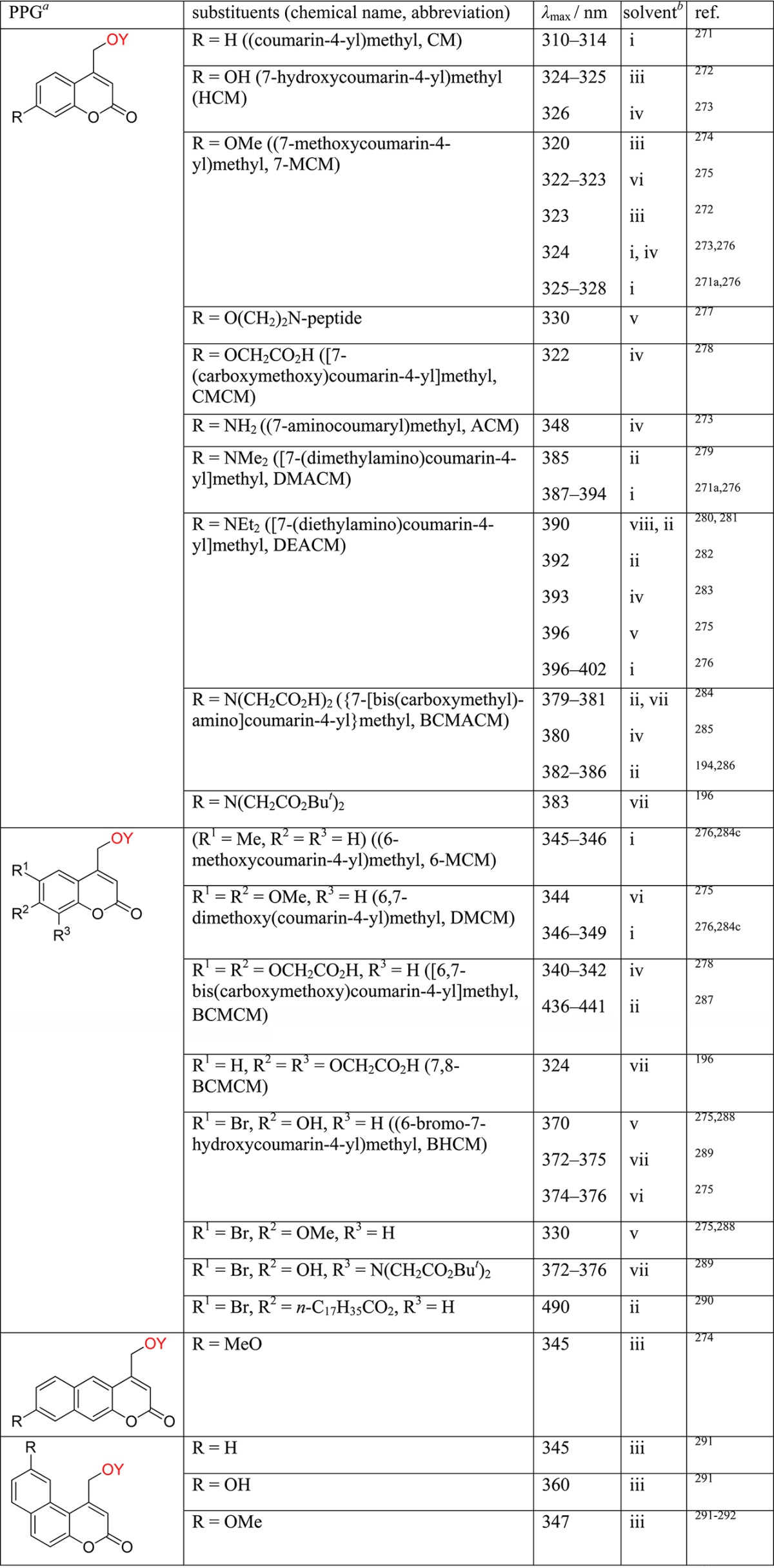
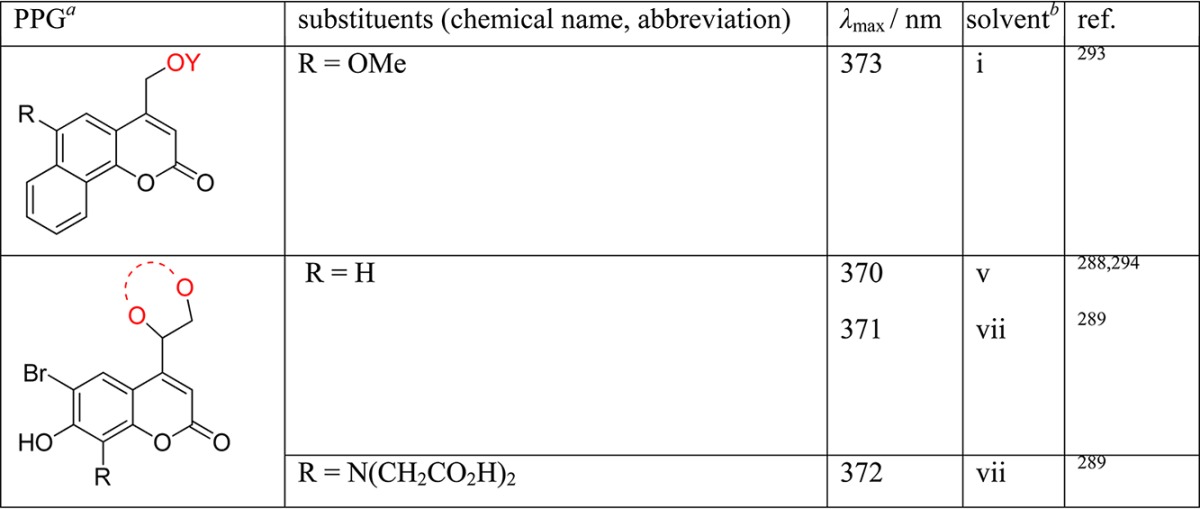
Leaving groups are in red.
Solvent: HEPES/MeOH (i); HEPES buffer (ii); EtOH (iii); PB or PBS buffer (iv); KMOPS buffer (v); KMOPS/MeOH (vi); HEPES/MeCN (vii); and water (viii).
Comprehensive reviews by Furuta,268 Dore,269 and Loudwig and Bayley,270 and more recently one by Givens, Rubina, and Wirz,9u encompass much of the groundwork on the photochemistry of coumarylmethyl PPGs with an emphasis on biological applications. Important mechanistic aspects, synthesis, and several selected recent applications are given below.
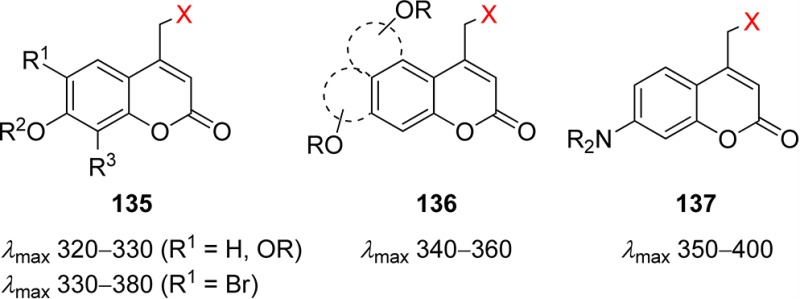
Synthetic Approaches to Coumarin-Caged Compounds
Coumarin-4-ylmethanol 138, easily available from the corresponding halides, is the common precursor of coumarin-caged esters, including phosphates, carboxylates, and sulfonates as well as carbonate, carbamate, and anhydride derivatives (Scheme 50).9a Carboxylic acids, including amino acids, can also be efficiently caged via SN2 substitution of coumarinylmethyl bromide 139. Likewise, coumarin-4-methylamines and -thiols can be directly obtained from bromides 139 by standard alkylation. Coumarin-caged diols, e.g., acetals 140, were prepared in two steps from aldehyde 141, which in turn is available from the corresponding alcohol 138 by oxidation with manganese dioxide (Scheme 50).295 Aldehyde 141 is also a convenient precursor to 4-diazomethylcoumarin 142, which has proven to be effective in protecting complex phosphates, particularly cyclic adenosine nucleotides, when standard approaches employing coumarin-4-ylmethanol 138 or coumarin-4-yl bromide 139 fail to provide the desired product.271a,279a
Scheme 50. Synthetic Approaches to Coumarin-Caged Compounds.

Mechanism of Photorelease and Selected Applications
A general mechanism of photorelease of phosphates, sulfonates, and carboxylic acids, the most successfully applied leaving groups, is summarized in Scheme 51.276,296 After initial light absorption, relaxation takes place to the lowest 1(π,π*) excited singlet state, which partitions between unproductive radiationless decay and fluorescence, and heterolytic C–X bond cleavage. The initially formed tight ion pair (coumarinylmethyl cation and leaving group conjugate base) is the key intermediate. The coumarinylmethyl cation either reacts directly with adventitious nucleophiles or solvent to generate a new stable coumarylmethyl product. Alternatively, the tight ion pair escapes the solvent cage and reacts with available nucleophiles. Although there is evidence of intersystem crossing occurring with 7-aminocoumarins,284c there are no indications of triplet reactivity. Recombination of the tight ion pair regenerates the ground-state caged derivative, an additional nonproductive pathway.
Scheme 51. Mechanism of Photorelease of Coumarin-Caged Compounds297.

Time-resolved absorption studies have demonstrated that the heterolytic bond cleavage is very fast with rate constants reaching 2 × 1010 s–1 (measured for phosphate esters), among the most rapid photorelease rates for any caged compounds.297 However, ion pair recombination dominates the subsequent reactions and is about 10 times faster than nucleophilic trapping of the coumarinylmethyl cation by solvent.276,296 Evidence in support of SN1 cleavage at carbon, vis-à-vis a photosolvolysis pathway, was obtained using 18O-labeled water, which afforded labeled coumarinylmethanol and label-free phosphoric acid (Scheme 51).297 Installation of electron-donating substituents on the coumaryl moiety and choice of a leaving group with low pKa values facilitate the reaction and, at the same time, impede ion pair recombination.276
Poor leaving groups such as alcohols, phenols, and thiols render (coumarin-4-yl)methyl derivatives resistant to heterolysis. Such groups can be more efficiently released when caged through a carbonate linkage with the (6-bromo-7-hydroxy/alkoxycoumarin-4-yl)methyl moiety 143 (X = O, S, Scheme 52). The initially liberated carbonic or thiocarbonic acid is unstable and undergoes decarboxylation to give free alcohol or thiol.196,272,275,289 The rates of these decarboxylation reactions are usually quite slow, k–CO2 = 10–3 s–1,298 and subject to both acid and base catalysis.
Scheme 52. Decarboxylative Photorelease of Alcohols, Thiols, and Amines.
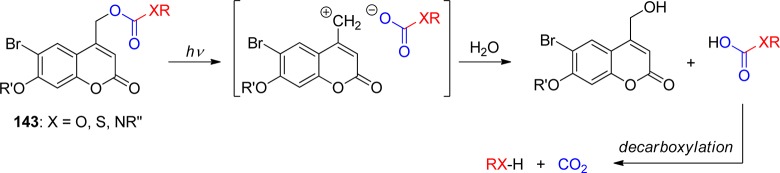
Analogous to alcohols, release of amines from coumarin-4-ylmethyl carbamates 143 (X = NR′′) proceeds uneventfully, although at a slower rate (Scheme 52).272,288,290,299 As in the case of carbonates, the rate-limiting step is decarboxylation of the released carbamate anion, which is more strongly dependent on the pH and on the nature of the released amine or amino acid.62,300 This could become a limitation for applications requiring a very rapid substrate release.
Carbamate-linked coumaryl PPG was used by Hayashi, Kiso, and co-workers to mask the benzylamine moiety and suppress the pharmacophore activity in isotaxel (Scheme 53).283 The resulting photoresponsive prodrug phototaxel (144) was selectively activated using visible light (430 nm) to release isotaxel (145), which converted into paclitaxel (146) by a spontaneous intramolecular O- to N-acyl migration.283 To improve water solubility of the phototaxel prodrug, the N-ethyl groups of the coumarin cage were replaced with N-(2-(dimethylamino)ethyl)acetamide groups.301 It should be mentioned that an alternative O-protected paclitaxel prodrug with a carbonate-linked coumarin was unstable in aqueous solutions.
Scheme 53. Activation of Paclitaxel by Visible Light283.

Among the most significant advances in the photochemistry of coumarin-based protecting groups has been the discovery and development of two-photon activation (see section 9).9h,302 Initially reported for the two-photon release from coumarin-caged substrates by Furuta, Dore, Tsien, and co-workers303 this approach for decaging has developed into a major area of research and discovery.9a,269
Block methylmethacrylate copolymer 147 esterified with the (7-diethylaminocoumarin-4-yl)methyl group exhibiting a large two-photon absorption cross section was used by Morris, Zhao, and co-workers to study light-responsive micelle disruption (Scheme 54).304 UV irradiation of the micelles loaded with the hydrophobic Nile red dye revealed nonradiative energy transfer between the coumarin chromophore and the dye. Micelles without dye showed significant fluorescence self-quenching due to tight packing of the coumarin groups within a confined space; however, photorelease of the chromophore was accompanied by micelle disruption, which caused a dramatic increase in its fluorescence. Experiments with dye-loaded micelles placed in a dialysis cap immersed in water showed efficient diffusion of photoreleased coumarin into water (measured by fluorescence emission), while hydrophobic guest dye molecules remained within the cap. Multiphoton irradiation using near-infrared light was less disruptive for micelles yet sufficient to release loaded dye.304
Scheme 54. Light-Responsive Micelle Disruption304.
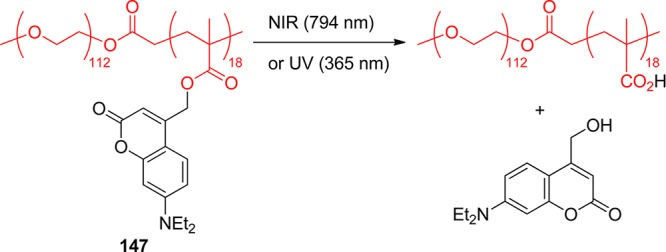
An efficient multiphoton uncaging permitted exposure of the functional groups (primary amines and sulfides) buried within modified aragose gel 148, creating complex 3D sites ready for selective immobilization of biologically relevant entities without affecting the mechanical properties of the patterned material (Scheme 55).299,305
Scheme 55. Selective Photoactivation of Functional Groups within Aragose Gel299,305.

A single example of photodeprotection of a coumarinylmethylamine through a C–N bond cleavage has been reported. This reaction proceeded efficiently only in the presence of an excess of a hydrogen-atom donor, such as n-decanethiol or 1,4-cyclohexadiene (Scheme 56).306 A radical mechanism has been proposed that involved electron transfer between the amine and coumaryl moieties forming the intramolecular radical ion pair 149. Subsequent cleavage of the C–N bond generated an aminyl radical and the resonance-stabilized coumarinylmethyl radical 150, both of which were trapped by hydrogen-atom donors.
Scheme 56. Uncaging of Amines via Direct C–N Bond Cleavage306.
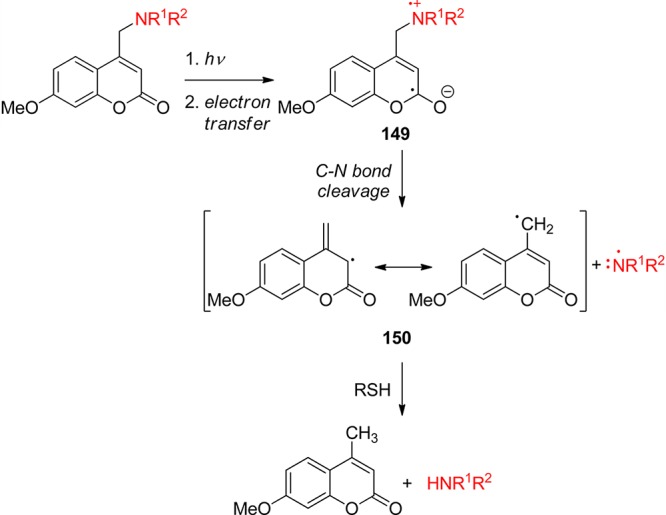
When the released group is the conjugate base of a very strong acid, the caged substrate becomes a “caged proton source” that permits the spatial and temporal control of rapid change in pH. Thus, the release of phosphoric acid, methanesulfonic acid, or sulfuric acid derivatives from their coumarin-caged precursors 151 has been employed as a proton trigger in studies of proton-dependent cellular signal transduction (Scheme 57).307 The release occurred within 2 ns, giving a significant drop in the pH of up to three units. It was also noted that hydrophobic phosphate derivatives penetrate cell membranes whereas charged sulfate was membrane-impermeant.
Scheme 57. Caged Proton Source: Photorelease of Strong Acids307.
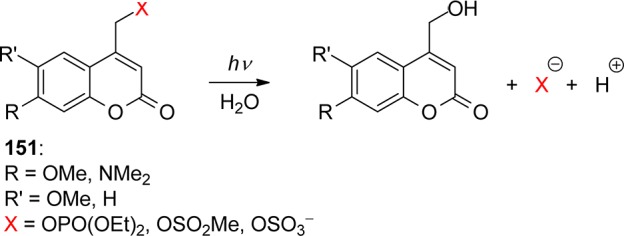
Photorelease of diols from cyclic (coumarin-4-yl)methyl acetals 152 proceeds via an ion-pair intermediate 153, which then reacts with water to produce the hemiacetal 154. The latter decomposes to give free diol and coumaryl aldehyde 155 (Scheme 58).308 Interestingly, the photodegradation of coumaryl-caged diols was found to be dependent on the size of the cyclic acetal. Thus, 1,2- and 1,4-diols were released equally efficiently (Φ = 0.005–0.03), whereas the corresponding 1,3-diols appeared to be completely inert to photolysis. Although different stereoelectronic properties of the three acetals precluded direct comparison of the propensity toward ring-opening,308 it is probable that the divergent quantum yields result from lack of recyclization because of the increasing ring strain for 5- and 7-membered acetals (by ca. 6 kcal mol–1) compared with recyclization to the “strainless” 6-membered analogue.309
Scheme 58. Photorelease of Coumarin-Caged Diols308.
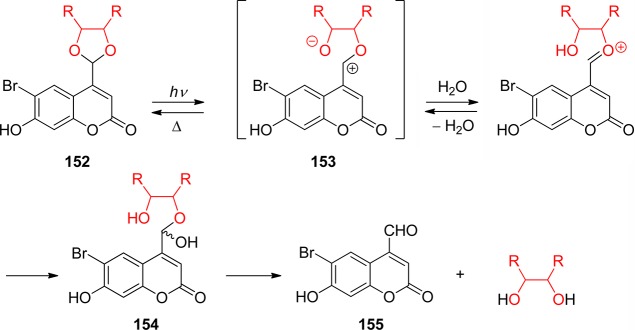
A complementary approach for protecting carbonyl compounds in the form of a photolabile cyclic coumaryl acetal has also been demonstrated (Scheme 59).287,291 Remarkably, both single- and two-photon uncaging (section 9) of 4-coumarin-4-yl-1,3-dioxolanes 156 were successfully achieved under physiological conditions. In line with the above example, the proposed mechanism involved photoinitiated heterolysis and subsequent solvolysis of the zwitterionic species 157.
Scheme 59. Photorelease of Coumarin-Caged Carbonyl Compounds287,291.
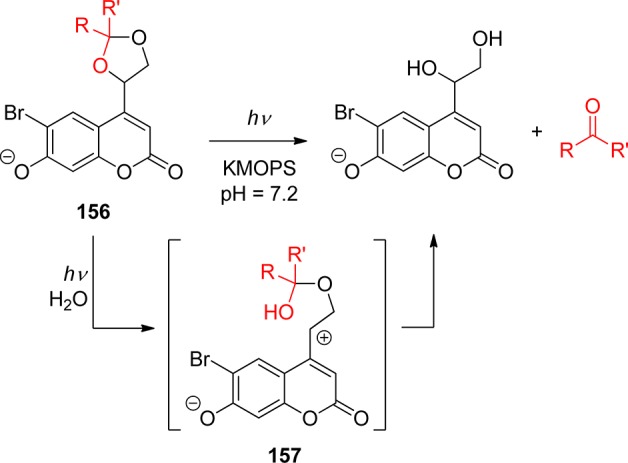
Caged carbonyl compounds 158 were obtained from (coumarin-4-yl)methyl chloride (159) by Wittig olefination followed by dihydroxylation of the resulting alkene with OsO4 and subsequent acetalization or ketalization under standard conditions (Scheme 60).291 Alternatively, caged progesterone 160 was synthesized by Hagen and co-workers via ketalization of the commercially available pregnenolone 161, followed by Oppenauer oxidation of intermediate 162 (Scheme 61).310 Photoactivation of coumaryl-caged progesterone produced 163 in only 30% yield, which, however, was sufficient to initiate progesterone-mediated response in sperm. Two-photon excitation of 160 (755 nm) also caused release of progesterone, albeit with ca. 5 times lower efficiency compared with photolysis of the analogous glutamate derivative.301
Scheme 60. Synthesis of Coumarin-Caged Carbonyl Compounds291.

Scheme 61. Photoactivation of Coumaryl-Caged Progesterone310.
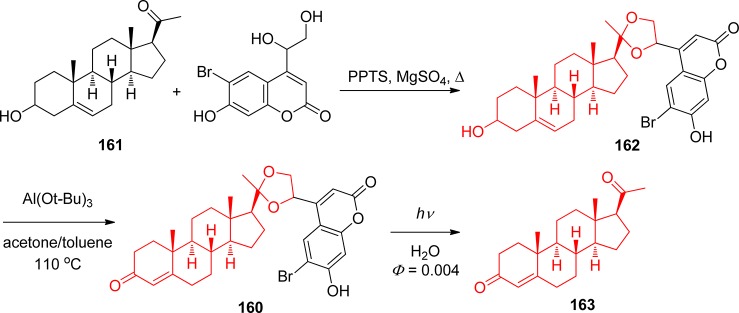
5. Arylmethyl Groups
5.1. Simple Arylmethyl Groups
A large family of photolabile protecting groups is based on photochemically induced hydrolysis (or less commonly alcoholysis) of benzyl or heterobenzyl esters or ethers 164 (Scheme 62).
Scheme 62. Photochemical Cleavage of Benzyl Protection.
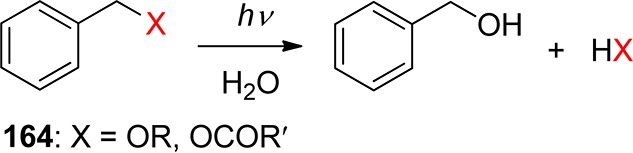
In fact, photochemical release of glycine from its benzyloxycarbonyl derivative was the first application of a “photosensitive protecting group” in organic chemistry.1 Barltrop and Schofield reported that irradiation of N-benzyloxycarbonylglycine 165 with 254 nm light resulted in the release of glycine in 75% yield and a quantum yield of 0.15 (Scheme 63). Photolabile protection of amino acids and dipeptides was further demonstrated by Chamberlin, who used the 3,5-dimethoxybenzyloxycarbonyl protecting group (166).311 Substrates were released in up to 85% yield upon illumination with a high-pressure mercury lamp for 1.5 h.
Scheme 63. Uncaging of Glycine1,311.
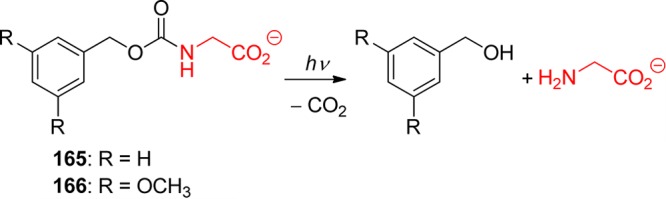
From a mechanistic point of view, this photosolvolysis can proceed via initial light-induced homolysis of the C–O bond, followed by rapid electron transfer to give an ion pair (Scheme 64). The latter is then trapped by solvent to give the final products. Pincock and co-workers have shown that this mechanism is predominant in the photolysis of simple benzyl derivatives.101 In fact, photosolvolysis of benzyl esters is often accompanied by the formation of radical products.312
Scheme 64. Photochemistry of Arylmethyl Ethers.
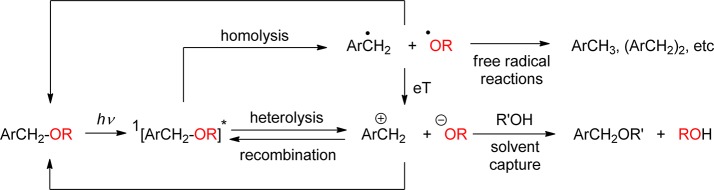
Calculations by Zimmerman and co-workers312a,313 suggested that heterolysis of the C–O bond is energetically preferred over the homolysis, especially in the presence of ortho and meta substituents stabilizing the cation. While studying the photochemical solvolysis of benzyl acetates, Zimmerman observed the now famous “meta-effect”: a higher efficiency of photocleavage for meta-methoxy-substituted benzyl esters.
Structural features that help to delocalize positive charge in the ground state enhance the efficiency of the electron-transfer step (Scheme 64), whereas excited-state cation-stabilizing moieties promote the direct photoheterolysis of the C–O bond. The efficiency of the uncaging reaction depicted in Scheme 64 is defined by the competition between solvent capture of the ion pair to give the liberated hydroxyl compound ROH and recombination leading to the starting material (polar protic solvents enhance both the heterolytic cleavage and electron transfer). Therefore, photodeprotection of carboxylates, carbonates, carbamates, phosphates, and other good leaving groups is usually quite efficient, whereas uncaging of alcohols is more challenging. Enhanced stability of the benzyl cation and increased steric hindrance around the cationic center also help to slow down the ion recombination. Thus, the design of new PPGs of this class was mostly focused on the incorporation of features helping to delocalize positive charge. However, there is a pitfall associated with this approach: a very stable cation makes the PPG susceptible to acid-catalyzed hydrolysis. In fact, the conventional acid-labile trityl protecting group can be removed from caged nucleosides (167) in 71–99% yield upon 254 nm irradiation with ca. 10% quantum efficiency (Scheme 65).314
Scheme 65. Photocleavage of the Trityl PPG314.
Wang and co-workers systematically explored various trityl derivatives and found that the 3-(dimethylamino)trityl group (168, DMATr, Scheme 65) is the most efficient alcohol-caging group in the trityl series (Φ = 0.2).315 Introduction of a strong electron-donating meta-substituent in one of the trityl rings improves the photochemical reactivity due to the meta-effect, stabilizing the PPG derivative to hydrolysis in the dark. This PPG has a relatively long absorption wavelength (λmax = 309 nm), is stable in acidic media, and releases alcohols with 0.12–0.20 quantum yield efficiency. DMATr can be easily installed by simple solvent-free heating of the DMATr acetate with the alcohol. Several alcohols, including sugars and thymidine, have been protected and released upon irradiation with a medium-pressure mercury lamp in methanol with 80–90% yields. DMATr is also orthogonal to conventional methoxytrityl protecting groups, as it can be removed by irradiation in methanol, whereas the methoxytrityl protection is cleaved upon treatment with 80% acetic acid.
A similar α,α-dimethyl-3,5-dimethoxybenzyloxycarbonyl PPG (169, Ddz, Scheme 66) was used by Cameron and Frechet for caging of amines.316 Irradiation of these thermally stable carbamates with a medium-pressure mercury lamp results in liberation of free amines both in THF solution and in the solid state.
Scheme 66. Uncaging of Amines316.
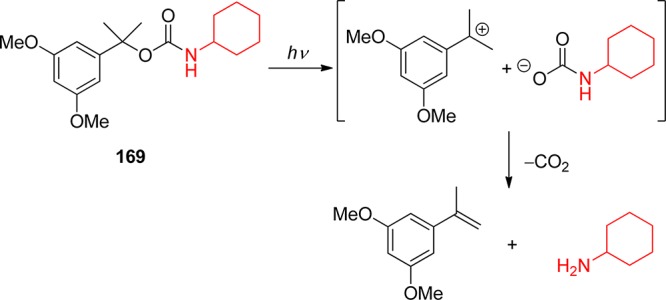
The stability of 9-phenylxanthylium cation prompted the development of the 9-phenylxanthyl (170, pixyl, Px) PPG for primary alcohols (Scheme 67).317
Scheme 67. Protection and Photochemical Release of Primary Alcohols317.
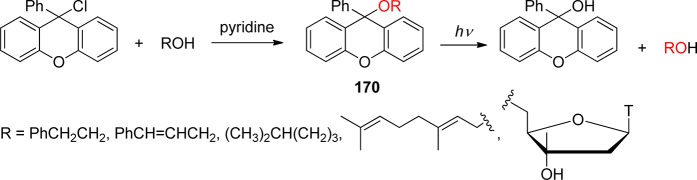
The pixyl PPG is readily introduced in 65–74% yield by treating alcohols with commercially available pixyl chloride in the presence of pyridine. Irradiation of the pixyl ethers at 254 or 300 nm results in the release of the alcohol in 78–97% yield. On the basis of deprotection of the nucleoside thymidine, the authors suggested that Px could be an effective protecting group for the combinatorial synthesis of oligoribonucleotides.
The 9-phenylthioxanthyl group (171, S-pixyl, S-Px, Scheme 68) was found to be an even more efficient cage for primary alcohols, as it absorbs at longer wavelengths and has a higher quantum yield of deprotection.318 It was also shown that i-butyryl and benzoyl protection of amino groups in nucleobases is not affected by the photolysis. The chemical yield of deprotection varied from 75% for cytidine to 97% for thymidine.
Scheme 68. Use of the S-Pixyl PPG for Caging and Release of Nucleosides318.
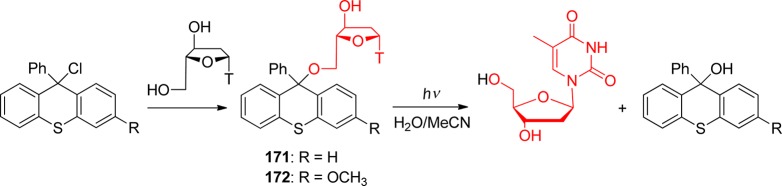
Further studies of the substituent effects on the photochemical release of thymidine from the S-Px cage showed that an electron-donating group in the 3-position enhances the efficiency of the photolysis by a factor of 3.319 Moreover, some of these compounds proved to be suitable for deprotection under irradiation with 350 nm light. For example, 5′-O-(3-methoxy-9-phenylthioxanthyl)-2′-deoxythymidine (172, Scheme 68) quantitatively releases thymidine by irradiation at 300 or 350 nm.
Direct photosolvolysis of ethers, such as 167, 168, and 170–172, has great advantage for alcohol caging over the use of photolabile carbonates. Substrates are released directly from the excited state of the photocage within a few microseconds in the former case, whereas decarboxylation of photochemically generated monocarbonate in the latter case is a much slower process.
Photosolvolysis of arylmethanol derivatives has also been used for the deprotection of carbonyl compound components of photolabile acetals. Wang and co-workers developed a photoremovable protecting group for carbonyl compounds based on trityl photochemistry.320 Ketones and aldehydes are converted into cyclic acetals (173) by treatment with 5-methoxysalicylic alcohol derivatives (Scheme 69). Upon irradiation, the carbonyl compounds are released in 75–90% yield and good quantum efficiency (Φ = 0.11). The authors proposed that this reaction proceeds via a two-step mechanism involving a zwitterionic intermediate. However, it is also possible that initial photocleavage of the benzylic C–O bond produces a hemiacetal, which undergoes further hydrolysis. This cage is remarkably stable to a variety of reagents including metalloorganic (PhLi, LiAlH4, and t-BuOK), strong acids (AcOH, TFA, and concentrated HCl), and mild oxidants (DDQ). Besides, Thevenet and Neier have recently studied photorelease of alcohols from the corresponding naphthalen-2-ylmethylidene and arylethynylmethylidene acetals.321
Scheme 69. Photolabile Acetals for the Protection of Ketones and Aldehydes320.
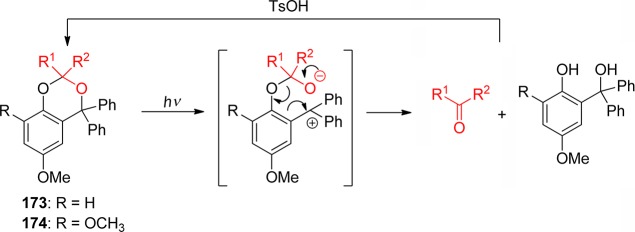
The methoxy substituent in 173 was strategically positioned to exploit Zimmerman’s meta-effect.312a,313 The second methoxy group in 174 makes photoheterolysis of benzylic C–O even more efficient, as noted earlier. A series of carbonyl compounds were protected in the presence of p-toluenesulfonic acid322 or in the neat state and with no added catalysts at 140 °C323 and then released (Φ = 0.17–0.23)320c,320e from the photocage 174 upon irradiation with a 450 W medium-pressure mercury lamp in aqueous acetonitrile. Both protection and uncaging proceeded in 74–99% yields. Introduction of electron-donating or -withdrawing substituents in the para-position of the phenyl substituents in 174 allows for the adjustment of λmax of the chromophore.320c
The majority of trityl- and benzyl-based PPGs require irradiation with light of rather short wavelength to achieve efficient substrate release. To shift the caging chromophore absorbance to longer wavelengths, the photocleavage of several polyaromatic analogues of the benzyl protecting group, including (2-naphthyl)methyl,324 (anthracen-9-yl)methyl,325 (pyren-1-yl)methyl (175, Scheme 70),326 (perylen-3-yl)methyl,327 and (phenanthren-9-yl)methyl,326a−326c was explored.
Scheme 70. Photocleavage of the Fluorescent (Pyren-1-yl)methyl Protecting Group326.
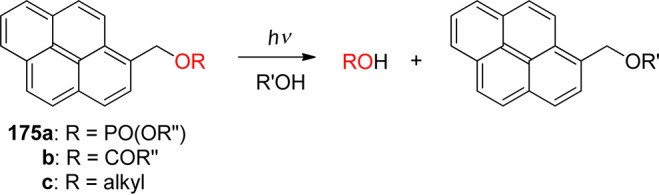
These cages have significant absorbance at 350 nm and show weak to moderate fluorescence (Φfl = 0.01–0.2). Polyaromatic PPGs are well-suited for release of very good leaving groups, such as phosphates (175a). Uncaging of carboxylic acids, on the other hand, is less reliable, and alcohols are released with <0.005 quantum efficiency. The common drawback of polyaromatic PPGs is their poor aqueous solubility. Addressing this problem, Furuta and co-workers have developed the anthraquinon-2-ylmethoxycarbonyl cage (176, Aqmoc),324,326d which undergoes fairly efficient photocleavage at 350 nm (Φ350 nm = 0.10) and has good aqueous solubility (Scheme 71). The utility of Aqmoc group for photoremovable protection of carbohydrates and nucleosides has been demonstrated with 68% and 91% chemical yields, respectively. 1,3-Pentadiene efficiently quenches the photosolvolysis reaction, suggesting that cleavage of Aqmoc protection proceeds via the triplet state of the chromophore.
Scheme 71. Aqmoc Caging of Primary Alcohols324,326d.
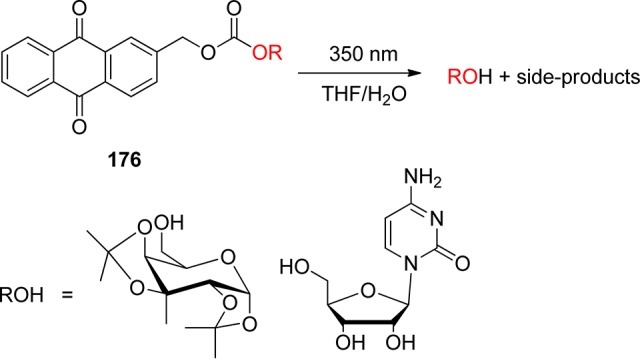
A variation of the anthraquinon-2-ylmethoxy group with a methyl substituent in the benzylic position (177, anthraquinon-2-yleth-2-yl, Aqe) was tested for caging of carboxylic acids (Scheme 72).328
Scheme 72. Aqe Cage for Carboxylic Acids328.
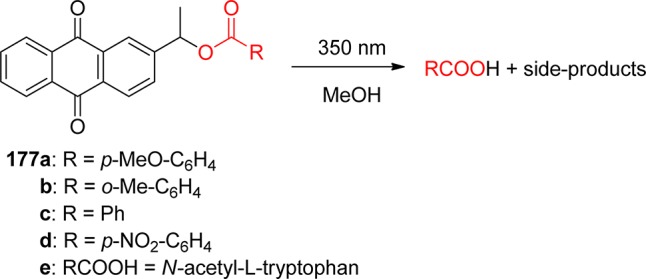
This chromophore has a λmax at 325 nm and significant absorbance at 350 nm. Irradiation of 2-(1′-hydroxyethyl)anthraquinone carboxylates 177a–c at the latter wavelength resulted in relatively efficient (Φ ≈ 0.12) release of the corresponding carboxylic acids in good yields. Uncaging of p-nitrobenzoic acid and N-acetyl-l-tryptophan proceeds with a low quantum (Φ < 0.01) and poor chemical yield, apparently due to intramolecular energy transfer from anthraquinone to the caged acids.328 On the basis of quenching studies and product analysis, the authors proposed a two-step mechanism for the photocleavage of esters 177 (Scheme 73). The initial photoreduction of anthraquinone, which proceeds via the triplet state, produces hydroquinone 178. For this step to work, the reaction media must contain hydrogen donors (e.g., methanol or THF). The second photochemical step resembles the cleavage of o-hydroxybenzyl esters (section 5.2). The (anthraquinon-2-yl)methyl chromophore has been used in the development of photolabile acetals 179 (Scheme 74).329
Scheme 73. Mechanism of Carboxylic Acid Release from the Aqe Cage328.
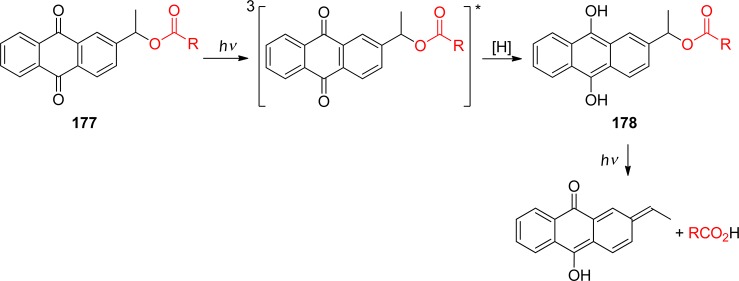
Scheme 74. (Anthraquinon-2-yl)methyl-Based Photolabile Acetals329.
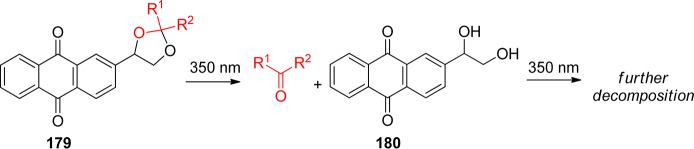
The caged carbonyl compounds 179a–d were prepared in 40–60% yield by the reaction of anthraquinon-2-ylethyl-1′,2′-diol (Aqe-diol, 180) with ketones or aldehydes in the presence pyridinium p-toluenesulfonate (PPTS) and a dehydrating agent. Acetals 179a–d are stable in the dark in neutral solution. Upon 350-nm irradiation in MeCN/aqueous buffer (1:1), carbonyl compounds are liberated with 0.03–0.09 quantum efficiency in 60–90% chemical yield. Under these reaction conditions, Aqe-diol 180 undergoes further decomposition, producing undetermined byproducts.329
Dore and co-workers developed heteroaryl-based PPG for carboxylic acids, (8-bromo-7-hydroxyquinoline-2-yl)methyl (BHQ), which has a strong band with λmax= 370 nm.330 Photodeprotection of acetic acid from the hydroxyquinoline 181a (Scheme 75) was found to be more efficient compared to 4,5-dimethoxy-2-nitrobenzyl (DMNB, section 3.1) and (6-bromo-7-hydroxycoumarin-4-yl)methyl (BHCM, section 4) acetates.331 It is noteworthy that this group can be removed under two-photon excitation conditions332 (see also section 9). BHQ-caged carboxylates (181b), phosphates (181c), and diols (182) are efficiently released under simulated physiological conditions using single-photon and two-photon activation.331 In addition, a (2-phenylquinolin-4-yl)methyl group333 and the corresponding arylmethylsulfonyl analogue334 have been proposed as PPGs for alcohols and amines, respectively.
Scheme 75. (8-Bromo-7-hydroxyquinoline-2-yl)methyl (BHQ)-Based PPGs331.
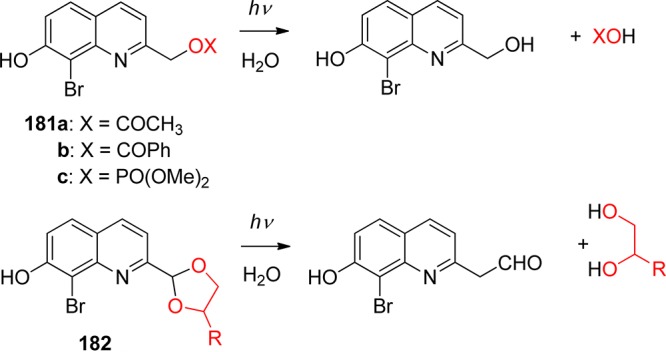
Photochemical properties of the quinoline-based PPGs could be further adjusted by varying the substituent in the aromatic ring.335 For example, replacing the bromine substituent in BHQ with a cyano group ((8-cyano-7-hydroxyquinoline-2-yl)methyl, CyHQ) results in a significant red-shift of the chromophore. However, none of the (quinoline-2-yl)methyl derivatives had higher sensitivity toward two-photon absorption than the parent BHQ group.
Another heterocyclic analogue of benzyl PPG, (benzoxazol-2-yl)methyl, and its derivatives were tested for photolabile protection of the carboxyl group in amino acids.336N-(Phenylmethoxy)carbonyl-d-alanine was released in moderate to quantitative yield upon rather lengthy irradiation at 350 nm.
5.2. o-Hydroxyarylmethyl Groups
Introduction of the hydroxy substituent ortho to the benzylic position dramatically changes the mechanism of C–O bond cleavage and enhances the overall efficiency of the process. Irradiation of o-hydroxybenzyl ethers (183) or their naphthyl analogues (184, (3-hydroxynaphthalen-2-yl)methyl, NQMP) results in the quantitative release of an alcohol and the formation of o-quinonemethide (185) (Scheme 76).337 In the presence of water, the latter undergoes very rapid hydration to give the parent diol or can be trapped with vinyl ether to give photostable chroman.
Scheme 76. Mechanism of Substrate Release from the o-Hydroxybenzyl/Naphthyl Cage337a,337b.
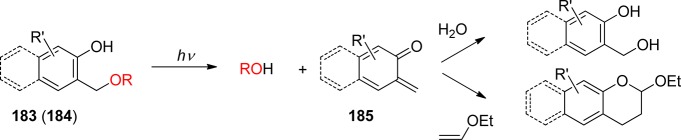
The release of the substrate proceeds within 12 μs after excitation with 0.20–0.40 quantum efficiency and in high to quantitative yield. The quantum and chemical yields of the uncaging reaction show little dependence on the nature of the hydroxy compound. Thus, alcohols, phenols, and carboxylic acids caged with the (3-hydroxy-2-naphthalenyl)methyl group (184) are released in 91–98% yield upon 300 or 350 nm irradiation (Scheme 77).338
Scheme 77. Photochemical Release of Alcohols, Phenols, and Carboxylic Acid from NQMP Cage338.
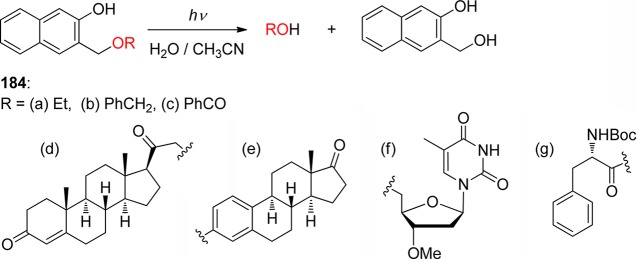
The common drawback of photolabile protecting groups described in this section is that the chromophore of the cage is usually preserved in the photoreaction, reducing the efficiency of the photolysis at higher concentration of caged compounds due to the internal filter effect. The 2,5-dihydroxybenzyl (186, DHB) cage alleviates this problem. The quinone methide 187 formed upon the release of a substrate undergoes tautomerization into methyl-p-quinone 188 (Scheme 78).339 UV spectra of the caged compounds 186a–d show a characteristic absorption band at 297 nm, whereas 188 has no absorbance at this wavelength. The compound 189 incorporates a safety-catch feature: p-quinone precursor is photochemically inert, but mild in situ reduction (sodium dithionite, NADH, etc.) of this compound produces the photoreactive hydroquinone 186.339
Scheme 78. 2,5-Dihydroxybenzyl Cage Incorporating a “Safety-Catch” Feature339.
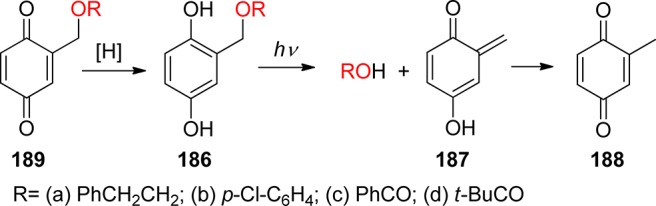
A variation of the DHB group was adapted for caging 1,2- and 1,3-glycols, as well as for photolabile benzylidene protection of carbohydrates. Thus, glucose was released with 0.30 quantum yield and 97% chemical yield upon irradiation of 190 (Scheme 79).340
Scheme 79. Photolabile Benzylidene Protection of Carbohydrates340.
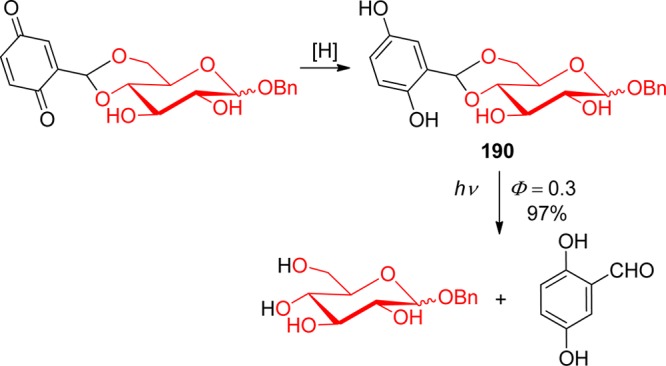
o-Hydroxybenzylidene acetals, such as 190 or 191 (Scheme 80), undergo slow dark hydrolysis in aqueous solutions with a half-life from 2 days to several weeks, whereas p-quinone precursors are stable both in solution and in the solid state. When hydrolytic stability and bleaching of the caging chromophore are not important, glycols can be efficiently caged as photolabile acetals of 5-methoxysalicyl aldehyde (Scheme 80).339
Scheme 80. Photolabile Protection of Glycols339.
A dihydroxybenzyl-based group (192) was also developed for the protection of carbonyl compounds (Scheme 81).341 Ketones and aldehydes are converted into 2-(1,2-dihydroxyethyl)-1,2-benzoquinone acetals 193. The latter compounds are stable in the dark but are quantitatively converted to the corresponding photolabile acetals 192 using mild reducing agents. Irradiation of the latter with 300-nm light results in the efficient photocleavage of the acetals, releasing carbonyl compounds in 88–100% chemical yields.341
Scheme 81. Safety-Catch Photolabile Acetal for Carbonyl Group Protection341.
In addition, a novel bichromophoric fluorescent photolabile protecting group, based on (3-hydroxynaphthalene-2-yl)methyl and dansyl moieties, was recently shown to combine the photochemical release with fluorescent imaging by the caged substrates.342 Falvey and Sundararajan have used an entirely different strategy for the photocleavage of benzyl analogues, such as 4-picolyl and N-methyl-4-picolinium esters using photosensitizers (see section 8.2).8b,343 In addition, visible-light photoredox catalysts have also been used to trigger deprotection of benzyl ethers344 or benzyl amines.345 The properties of the most useful arylmethyl-based PPGs from section 5 are summarized in Table 10.
Table 10. Arylmethyl- and Heteroarylmethyl-Based Photoremovable Protecting Groups.
| PPG | protected group | λmax/nm | λirr/nm | release yield/% | Φ | ref |
|---|---|---|---|---|---|---|
| 164 (benzyl) | amine (as a carbamate) | 254 | 75 | 0.15 | (1) | |
| 166 (DMB) | amine (as a carbamate) | 280 | 263–312 | 85 | (311) | |
| 168 (DMATr) | alcohol | 309 | broadband | ∼85 | 0.12 | (315) |
| 168b | alcohol | 92–100 | 0.23 | (346) | ||
| 169 (Ddz) | amines (as a carbamate) | 276, 282 | broadband | (316) | ||
| 170 (Px) | alcohol | 254 or 300 | ∼90 | (317) | ||
| 171 (S-Px) | alcohol | 300 or 350 | ∼90 | (318,319) | ||
| 174 | carbonyl | 297 | >280 | 92 | 0.17 | (315) |
| 176 (Aqm) | carboxylate, alcohol (as a carbonate), carbonyl | 330 | 350 | ∼80 | ∼0.1 | (324,326d) |
| 181 (BHQ) | carboxylate, phosphate, carbonyl | 369 | 365 | ∼90 | 0.04 | (330a, 331, 335) |
| 184 (NQMP) | alcohol, phenol, carboxylate | 307 | 300 or 350 | 72–100 | 0.1–0.3 | (338) |
| 186 (DHB) | alcohol, phenol, carboxylate, carbonyl, glycol | 297 | 300 | 60–100 | 0.1–0.3 | (339, 340) |
| 194 | carboxylate, alcohol | 438 | >410 | 94–97 | 0.072–0.093 | (327) |
6. Metal-Containing Groups
Metal-containing photoremovable protecting groups upon irradiation have been shown to release coordinated metal ions, gaseous inorganic molecules, such as NO or CO, or organic molecules, often of biological interest. This innovative research topic has recently been reviewed by Haas and Franz,347 Schatzschneider,9o Ciesienski and Franz,9t and Schiller and his co-workers;10b thus, it is not covered comprehensively here.
These cages are metal complexes that undergo a change in the metal coordination environment upon single-photon or multiphoton excitation. Several types of organic ligands, such as amines, nitriles, azides, or thioethers, have been shown to be released. Upon irradiation with visible light above 480 nm, the most common [Ru2+(bpy)2]2+ core (195; an amine is a leaving group in this case) liberates various nitrogen atom-containing ligands (Scheme 82), such as 4-aminopyridine, serotonin, butylamine, tryptamine, tyramine,348 γ-amino butyric acid,349 glutamate,350 or azide.351 One of the amino group-containing ligands has also been replaced by triphenylphosphine to increase the release quantum yield.349b Etchenique and collaborators have reported that ruthenium–bipyridine-based caged nicotine ([Ru(bpy)2(nic)2]2+ where nic = nicotine) is released with the quantum yield Φ = 0.23 upon irradiation with blue (473 nm) or green (532 nm) light.352 Gobetto, Sadler, and co-workers have used spectroscopic and DFT computational methods to study the release mechanism.353 They showed that the photochemical activity is related to the presence of singlet and triplet transitions involving σ-antibonding orbitals and identified and characterized the triplet state responsible for the photodissociation process.353a Excitation of the metal–ligand-to-ligand charge-transfer (MLLCT) band of the [Ru(bpy)(4AP)4]2+ complex (4AP = 4-aminopyridine) was found to provide selective photodissociation of two 4AP moieties in two consecutive steps with quantum yields of approximately 6 × 10–3 and 2 × 10–4, respectively.353c The photorelease of an aliphatic amine from a ruthenium–bipyridine-based PPG attached to silica surfaces has recently been shown to occur upon single-photon (460 nm) and two-photon (900 nm) excitation (section 9).354 A caged glutamate can also be liberated upon single- and two-photon activation from a Ru complex.355 In addition, a photoactivatable fluorescent probe356 as well as caging peptidomimetic inhibitors357 are recent examples in which a ruthenium–bipyridine-based PPG releases a nitrile ligand. Thioether ligands, such as thioether-cholestanol hybrid,358N-acetylmethionine,359 or biotin,359 can also be liberated from the [Ru2+(bpy)2]2+ core upon irradiation with visible light.
Scheme 82. Photorelease from the [Ru2+(bpy)2]2+ Cage.
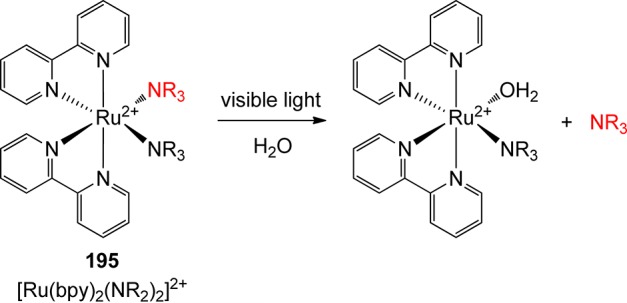
7. Miscellaneous Groups
7.1. Pivaloyl Group
Initially used as part of a photolabile linker for solid-phase synthesis, pivaloyl derivatives such as 196 fragment by a Norrish-type I mechanism equally well in solution, releasing carboxylic acids or alcohols (Scheme 83).360 The carbonyl group might seem to be a nuisance due to cross-reactivity, but these pivaloyl esters show remarkable stability under typical synthetic conditions (such as acids, bases, or transition-metal catalysts). The side-products are all volatile: carbon monoxide, isobutene, and acetone, and the quantum yield of release is quite high (Φ = 0.56). They require, however, the use of a relatively short-wavelength source (280–340 nm), which can be problematic when dealing with biomolecules.
Scheme 83. Photolysis of Pivaloylglycol Derivatives360.
7.2. Esters of Carboxylic Acids
The 2-benzoylbenzoic acid moiety (197) has been utilized as a PPG for primary and secondary alcohols, or thiols (Scheme 84).361 This chromophore exhibits photochemical reactivity typical for benzophenone, such as photoinitiated H-atom or electron transfer to the excited carbonyl group. Alcohol release is accompanied by the formation of a dimeric side-product 198 in the presence of an H-atom donor, whereas the isobenzofuranone derivative 199 is obtained by photoreduction with, for example, amines.
Scheme 84. Photochemistry of 2-Benzoylbenzoic Acid Esters361.
2-Benzoylbenzoic acid derivatives were utilized as photoreversible inhibitors of serine proteases by Jones and Porter.362 Various esters 200 served as efficient chymotrypsin (a digestive enzyme) inhibitors (Scheme 85). The synthesized acyl-chymotrypsins were found to be stable to hydrolysis from hours to months; a sharp increase of enzyme activity was then observed upon irradiation at 366 nm: up to 80% of the preinhibition activity was recovered. The corresponding 2-aroylbenzoic acid was formed as a side-product; Scheme 85 shows one of the proposed reaction mechanisms.
Scheme 85. Chymotrypsin Photorelease362.
A photofragmentation reaction of xanthenoic esters has been shown to give lactones of various ring sizes.363 The initial homolytic O–C bond cleavage of the esters 201 leads to xanthene and formyl radicals, which rearrange to lactones 202 and 203 in moderate to low chemical yields (10–55%; Scheme 86). The corresponding alcohols 204 and formates 205 were isolated as side-products.
Scheme 86. Photofragmentation of Xanthenoic Esters363.
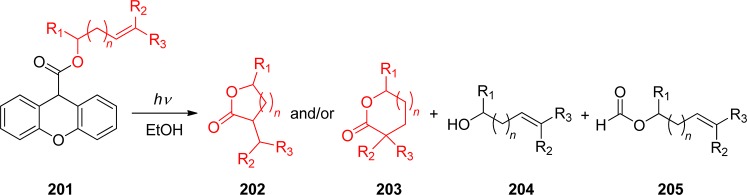
A water-soluble visible-light absorbing (ε400 > 9000 M–1 cm–1) PPG for carboxylic acids based on the aminonitrophenyl chromophore 206 has been demonstrated to photorelease β-alanine (207), which then activates the inhibitory glycine receptor in the mammalian central nervous system (Scheme 87).364 β-Alanine was found to be released in 5 μs with rather low quantum yields of Φ = 0.03 and 0.002, when irradiated at λ = 360 or 450 nm, respectively.
Scheme 87. Liberation of β-Alanine from the Aminonitrophenyl Chromophore364.
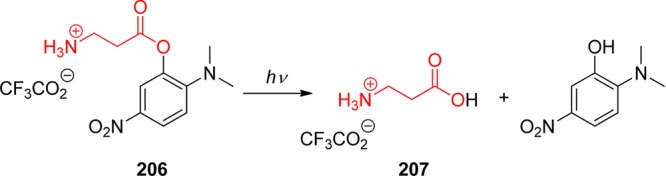
7.3. Arylsulfonyl Group
The photochemical generation of acids (photoacids) is an important strategy in the development of coating and imaging technologies as well as the synthesis of polymers.365 In such a process, the acids are typically photochemically produced from relatively small amounts of an excited initiator to start a chain reaction. However, this topic is beyond the scope of this review; here we only cite a few recent reports366 as examples.
Arylsulfonyl ester (Scheme 88) or amide photoacids are also frequently used as PPGs (e.g., ref (367)). For example, this strategy was utilized for dopamine release from a photocleavable biotin linker368 or for reactivation of a benzenesulfonyl-caged Zn2+ tetraazacyclododecane complex.369 Acid (proton) photorelease has been used to study proton binding in the sarcoplasmic reticulum.370
Scheme 88. Photochemistry of Arylsulfonyl Esters.
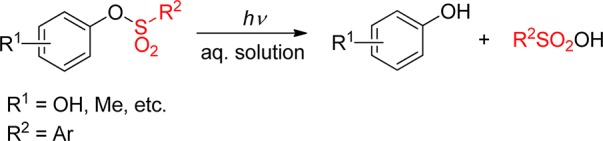
7.4. Ketones: 1,5- and 1,6-Hydrogen Abstraction
A considerable focus has been devoted to the development of photochemically releasable volatile odoriferous compounds—fragrances. Such applications have been the subject of several recent reviews.11,371 Aldehydes and ketones are important classes of fragrance molecules. Herrmann and co-workers have shown that protected carbonyl compounds can be photoreleased from α-keto esters (208) via the Norrish Type II photofragmentation under mild conditions in the presence of air (Scheme 89).372 The initially formed 1,4-biradical reacts with air oxygen followed by the release of the aldehyde or ketone 209, carbon dioxide, and the corresponding carboxylic acid. In the absence of oxygen, 208 can also undergo β-cleavage to give 209 and a ketene intermediate.
Scheme 89. Photochemistry of α-Keto Esters372.
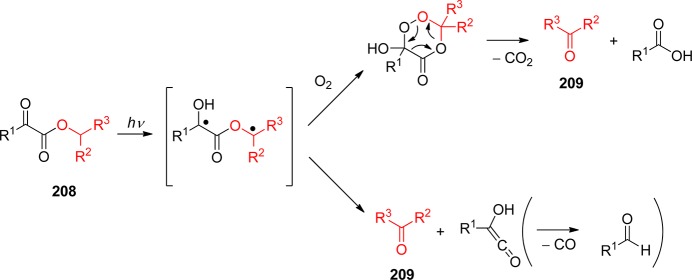
Photochemically triggered hydrolysis of protected volatile aldehydes or ketones has also been demonstrated using 1,5-diketones with abstractable γ-hydrogen373 or 1-alkoxy-9,10-anthraquinones, e.g., 210 (Scheme 90).374 The reaction starts with 1,6-hydrogen atom abstraction followed by electron transfer to form the zwitterion 211, which is trapped by an alcohol. The final fast hydrolytic step liberates the protected compound 212.
Scheme 90. Photochemistry of 1-Alkoxy-9,10-anthraquinones374.
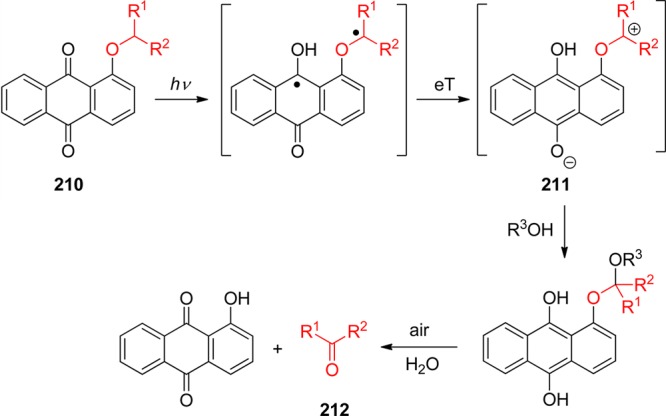
7.5. Carbanion-Mediated Groups
Another type of PPG relies on the photochemical formation of a carbon-centered anion, which drives the subsequent release of a leaving group via β-elimination (Scheme 91). This strategy, developed by Scaiano and co-workers,375 is often based on the photochemical decarboxylation in the side-chain of a chromophore and is compatible with an aqueous environment. Benzophenone (LG = carboxylate; Φ < 0.7),375a xanthone (LG = carboxylate; Φ < 0.7376),377 and phthalimide378 moieties have been used as the chromophores. In this context, a stereodifferentiation process involving the release of the caged molecule has been demonstrated on photoinduced decarboxylation of 2-phthalimido-3-hydroxy-propionate derivatives in which different reaction efficiencies were found for cages with threo or erythro configurations.379 Griesbeck and co-workers have shown that methylthiomethyl esters of ω-phthalimido carboxylic acids can liberate the carboxylic acids upon irradiation.380
Scheme 91. Carbanion-Mediated Photocleavage.
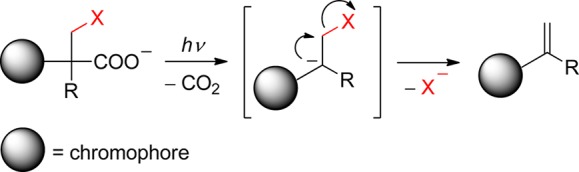
7.6. Sisyl and Other Silicon-Based Groups
Silicon-based protecting groups are often used in organic synthesis to protect alcohols as silyl ethers due to their predictable and selective deprotection and tolerance to many organic reagents. A bulky sisyl (tris(trimethylsilyl)) group has been introduced as a photocleavable protecting group for primary and secondary alcohols.381 It was shown that it is stable to aqueous bases and Grignard and Wittig reagents, as well as resistant to selected fluoride salts. Irradiation of sisyl ethers at 254 nm leads to the deprotection of the alcohols in good chemical yields (62–95%)382 by a radical mechanism.383
The Si–O bond of various trialkylsilyl esters can also be photocleaved by in situ generation of HBr from a catalytic amount of CBr4.384 Such an approach has been used for the deprotection of t-butyldimethylsilyl and β-(trimethylsilyl)ethoxy methyl ethers.385 It was shown that the primary silyl ethers on carbohydrate molecules can be selectively liberated in the presence of secondary silyl ethers. Photochemical protodesilylation of 213 in the presence of hexafluoroisopropanol (HFIP) or isopropanol (IPA) was reported to form an alkoxy intermediate, in which the C–Si bond is oxidized in a subsequent step to release a triol (Scheme 92).386
Scheme 92. Photochemistry of Trialkylsilyl Esters386.
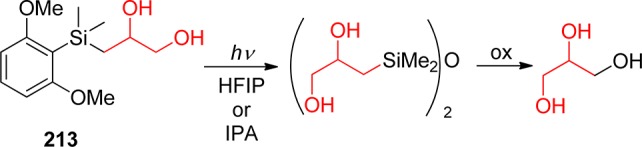
7.7. 2-Hydroxycinnamyl Groups
Porter and co-workers have been first to utilize a 2-hydroxycinnamyl moiety (214, Scheme 93) as a PPG in 1987, in this particular case, for the photochemical activation of thrombin, a serine proteinase.387 In general, this system undergoes initial photoinitiated isomerization followed by cyclization, which facilitates the release of caged substrates such as alcohols. Hiramatsu and co-workers utilized a caged reagent for a fluorimetric assay of peroxidase that led to uniform addition of a reagent without stirring.388 A trihydroxycinnamyl ester-based photocleavable detergent was applied for enhancing the water solubility of cell proteins followed by their MALDI-MS determination.389 The 3,5-dibromo-2,4-dihydroxycinnamic caging group was synthesized and used for 2-photon release of a biologically active substrate in a specific region within the cell.390 The fluorescence emission of the photocyclization byproduct, 6,8-dibromo-7-hydroxycoumarin, then provided the information about the concentration of the released species. Applications of this chemistry to release of a fluorescent reporter and the desired caged substrate391 or mask bioactivity of complex enzymes, such as thrombin, factor Xa, and trypsin, have been demonstrated.392 A small library of the o-hydroxycinnamic derivatives was recently synthesized to study and improve one- and two-photon release.393 Cinnamate esters were also successfully coupled with a CdSe nanocrystal surface.394 The corresponding coumarins were then released upon irradiation of the CdSe nanocrystal by visible light. This protocol also aided the understanding of fundamental nanocrystal–ligand interactions.
Scheme 93. Photochemistry of o-Hydroxycinnamic Derivatives.
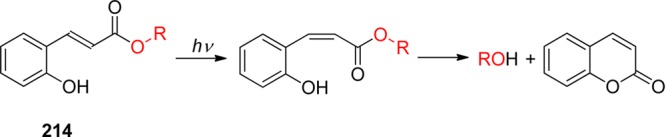
Silyl analogues of 2-hydroxycinnamyl derivatives (215) as PPGs have been introduced by Pirrung and co-workers (Scheme 94).395 A clean and high-yield (<92%) deprotection of primary and secondary alcohols was reported.
Scheme 94. Photochemistry of Silyl Analogues of 2-Hydroxycinnamyl Derivatives395.
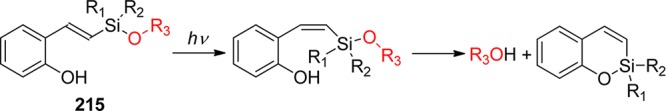
7.8. α-Keto Amides, α,β-Unsaturated Anilides, and Methyl(phenyl)thiocarbamic Acid
Steinmetz and co-workers reported that the carboxylates (including GABA, BocAla, and Glu) attached to the α-carbon of α-ketoamides (216) could be photochemically released, possibly through a zwitterionic intermediate 217, in very good chemical (<93%) and quantum (0.28–0.37) yields, along with the formation of a mixture of two diastereomeric hemiacetals 218 and a small amount of oxazolidinone 219 as byproducts (Scheme 95).396 The yield of 219 was found to depend strongly on the type of alkyl substituent on the carbon adjacent to the amide nitrogen.397 Time-resolved pH-jump experiments showed that the reaction rate is on the microsecond time scale and that carboxylate release occurs in the rate-determining step.398 Phenolates can also be liberated from 216.399 Laser flash photolysis experiments demonstrated that p-substituted phenolic substituents undergo photocleavage to give the corresponding phenol with good quantum yields (0.2–0.3).400 The authors proposed a mechanism that involves H-atom transfer from an N-alkyl group to the carbonyl to produce a zwitterionic intermediate that eliminates the phenolate.
Scheme 95. α-Ketoamides as PPGs396.
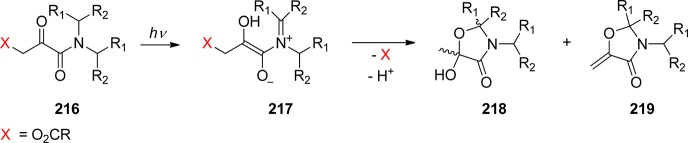
α,β-Unsaturated anilides (220) have been utilized as PPGs for carboxylates or phenolates (Scheme 96).401 The chemical (<71%) and quantum (<0.083) yields of LG release were reported. A zwitterionic intermediate analogous to 217, responsible for LG extrusion, is assumed to form.
Scheme 96. α,β-Unsaturated Anilides as PPGs401.
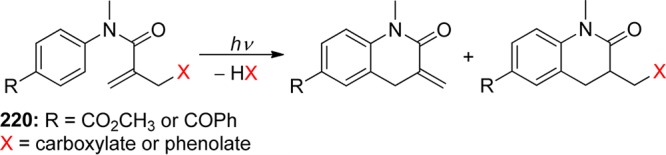
The cysteinyl radical was shown to be photochemically released from the methyl(phenyl)thiocarbamic acid chromophore 221 (Scheme 97).402 The S–S bond cleavage and the cysteinyl radicals’ recombination was studied by time-resolved IR spectroscopic techniques.
Scheme 97. Photochemistry of a Methyl(phenyl)thiocarbamic Acid Chromophore402.
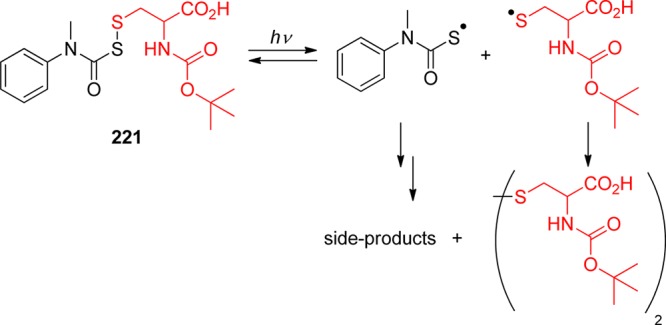
7.9. Thiochromone S,S-Dioxide
Kakiuchi and co-workers recently introduced a novel PPG based on the thiochromone S,S-dioxide chromophore 222 (Scheme 98).403 The alcohols (as carbonates), amines (as carbamates), and carboxylic acids were released in excellent chemical yields (up to 99%) upon irradiation at λ = 280 nm in methanol. The reaction progress was monitored by fluorescence spectroscopy.
Scheme 98. Photochemistry of a Thiochromone S,S-Dioxide Derivative403.
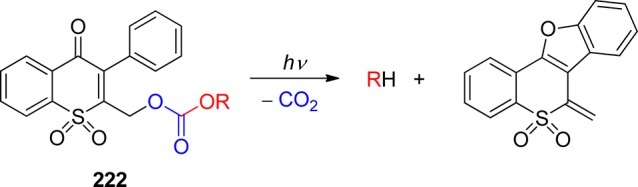
7.10. 2-Pyrrolidino-1,4-Benzoquinone Group
Chen and Steinmetz have demonstrated that 2-pyrrolidino-1,4-benzoquinone 223 gives, upon irradiation with visible light (below ∼700 nm), an unstable benzoxazoline photoproduct, which expels a leaving group, such as carboxylate or phenolate, in a subsequent dark elimination reaction (Scheme 99).404 The reaction proceeds with quantitative chemical but modest quantum (Φ = 0.03–0.10) yields.
Scheme 99. Photochemistry of 2-Pyrrolidino-1,4-Benzoquinone Derivatives404.
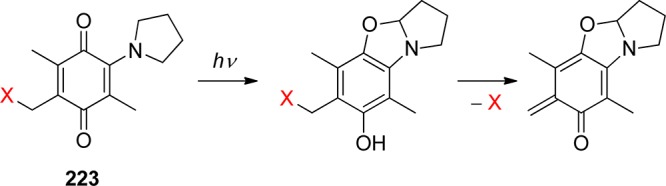
7.11. Triazine and Arylmethyleneimino Groups
A triazine moiety can be used as a (thermally) labile linker in solid-phase synthesis.405 Its applicability as a photocleavable linker has also been reported.406 Laser irradiation of 224 at 355 nm leads to liberation of the protected amines or amino acid derivatives in moderate to high chemical yields (77–100%; Scheme 100). A triazine linker has also been used as part of a homopolymer backbone to yield photoinduced backbone fragmentation.407
Scheme 100. Triazine Moiety as a Photolabile Linker406.
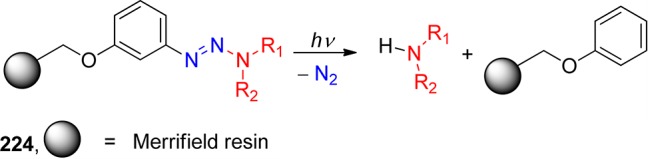
It has been shown that the naphthylmethyleneimino group releases aliphatic or aromatic primary amines as well as α-amino acids (NH2R) in good to excellent chemical yields (51–96%) upon irradiation at 280 or 340 nm. The reaction proceeds via a homolytic cleavage of the N–O(CO) bond. Such strategies are similar to those utilized in the field of photobase generation, which is not covered by this review.12a
7.12. Xanthene and Pyronin Groups
Wirz and co-workers have recently demonstrated that the (6-hydroxy-3-oxo-3H-xanthen-9-yl)methyl derivatives 225 release diethyl phosphate or carboxylic acid upon irradiation with visible light (over 500 nm) with quantum yields of 0.005–0.04 (Scheme 101).408 This reaction is the subject of further investigation.
Scheme 101. Photorelease from the Xanthene Derivatives408.
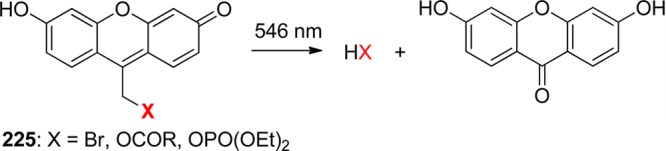
A novel class of pyronin analogues 226, which undergo a photochemical cleavage of the C–C bond in the presence of water both in solution and on a silica gel surface upon direct irradiation with yellow light (Scheme 102), has been reported by Klán and co-workers.409 The final chromophoric photoproduct was shown to be a stable compound absorbing below 430 nm. The course of the reaction was monitored by the characteristic fluorescence emissions of both the starting compound (λmax = 607 nm) and the final product (λmax = 448 nm). As one of the rare examples of visible-light triggered caged systems, it was suggested that the moiety could be used in the field of photoremovable protecting groups or caged fluorophores (section 11).
Scheme 102. Photochemistry of Pyronine409.
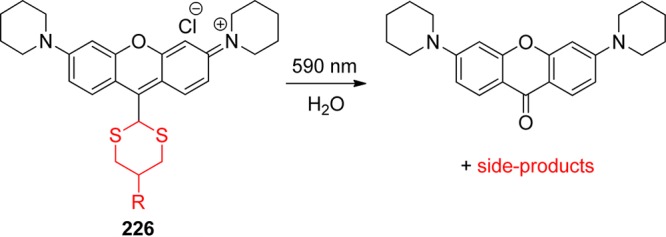
7.13. Retro-Cycloaddition Reactions
A photocleavable linker based on 7-hydroxy-1,1-dimethylnaphthalenone (227) for drug attachment to a polymer support has recently been introduced.410 It undergoes a photochemical [2 + 2]-cycloaddition with 5-fluoro-1-heptanoyl uracil (228), a well-known cytotoxic agent derivative, to form a heterodimer (229; Scheme 103). The linker–drug conjugate is then photochemically polymerized at 470 nm with methyl methacrylate (230), yielding a (hydroxyethyl)methacrylate (HEMA)/methyl methacrylate (MMA) copolymer. Photoinduced release of 228 from the polymer via single- (Φ = 0.01 at λ = 331 nm) or two-photon absorption was observed.
Scheme 103. [2 + 2]-Photocycloaddition of 7-Hydroxy-1,1-dimethylnaphthalenone410.
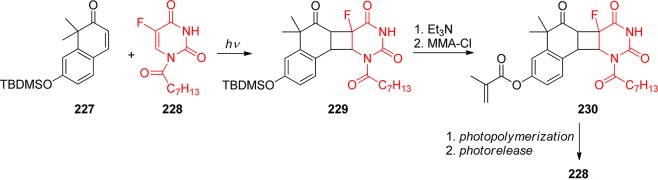
A different strategy—photosensitizer drug delivery via an optical fiber—has recently been designed by Greer and co-workers.411 A singlet oxygen sensitizer (pheophorbide) bound to a porous silica cap of a hollow optical fiber via an alkene spacer (231) was shown to be released in an oxygen stream (Scheme 104). A (Z)-enol ether bridge of 231 reacts with 1O2 to give the dioxetane intermediate 232, which cleaves to liberate the photosensitizer at the probe tip in the proximity of a tumor cell. Such a sensitizer delivery has the potential for photodynamic therapy. In a different study, Wilson and co-workers described the photorelease of o-quinones from pyrene dihydrodioxin (233, Scheme 105).412
Scheme 104. Photosensitizer Drug Delivery (a: Optical Fiber Equipped with Porous Vycor Glass)411.
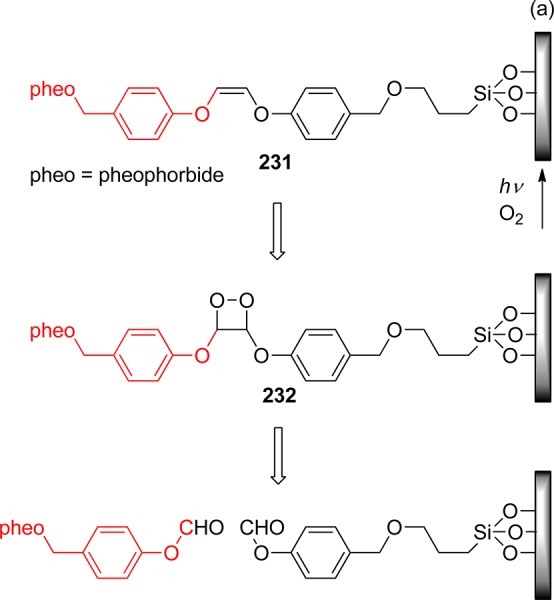
Scheme 105. Photorelease of o-Quinones from Pyrene Dihydrodioxin412a.
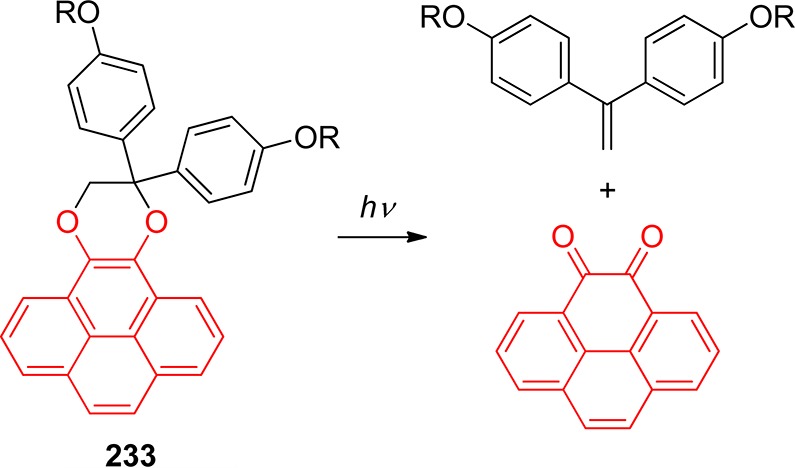
Utilization of a dithienylethene photochemical switch413 for the thermal release of an electron-deficient alkene has been reported.414 The thermally stable derivative 234 undergoes photochemically reversible ring-opening reaction to give 235, which liberates ethylene via reverse Diels–Alder reaction (Scheme 106). The reaction can be controlled by selection of the irradiation wavelength.
Scheme 106. Sequential Photorelease Using a Molecular Switch414.
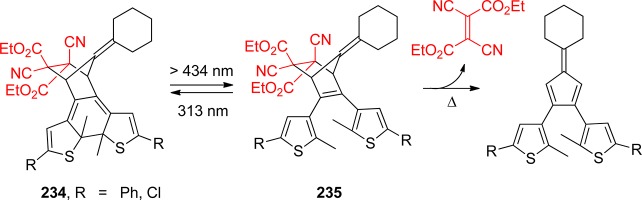
Triggering of a retro Diels–Alder reaction through a photothermal effect was demonstrated by Gates and co-workers for the first time.415 A nanoparticle attached to the caged compound through a linker absorbs the incident light at a specific frequency (of a plasmon resonance) and converts it into heat, which then promotes the chemical reaction to liberate the attached chemical species.415
An alternative approach that utilizes coumarin dimers as photocleavable linkers between the substrate and the polymeric carrier was investigated by Hampp and co-workers.416 The core dimeric coumarin cage 236 was constructed by [2 + 2]-photocycloaddition (Scheme 107). Deprotection of a t-butyldimethylsilyl (TBS) group, followed by esterification, was used to successively install a polymer tether (237) and chlorambucil (238).416b
Scheme 107. Immobilization of Chloroambucil on Polymer Support via a Photolabile Linker416.

This type of caging presumes linking through the 7-hydroxy group of one coumarin moiety to the active substance while the other coumarin is attached to a polymer support. The cleavage of the dimer occurs via [2 + 2]-cycloreversion, which can be triggered both by single- and two-photon absorption (Scheme 108).417
Scheme 108. Photorelease via [2 + 2]-Cycloreversion417.

8. Sensitized Release
In most cases, a PPG is the chromophore that is responsible for both the light absorption and the primary photochemical step, resulting in a specific bond-breaking process. There have been more or less successful attempts to modify the structure of a PPG in order to enhance its absorption properties and chemical reactivity. Recent research efforts have focused on the development of the systems composed of two, separated or linked, molecular components, in which the photorelease occurs from the moiety that is indirectly activated by energy transfer from or electron transfer from/to the excited auxiliary chromophore (photosensitizer).27 Such a sensitizer moiety should absorb strongly in the region of interest.
8.1. Sensitized Release: Photoinduced Energy Transfer
Energy transfer is a process by which the excitation energy of an excited molecule (donor; sensitizer, S) is transferred (kET) to a neighboring molecule (acceptor; quencher, Q, Scheme 109).27 Photosensitization via energy transfer is one of the most common and practical ways to generate excited states, particularly triplet states, especially when direct excitation at a desired wavelength cannot be achieved or when it does not lead to the desired excited state. For efficient triplet–triplet transfer, the process should be exergonic to prevent reverse transfer (kd); the donor molecule has to undergo efficient intersystem crossing and have a high molar absorption coefficient at the irradiation wavelength as well as a sufficiently long triplet lifetime to enable quantitative energy transfer.27,418 If the acceptor molecule is a PPG, the reaction (kr) then leads to release of a leaving group. Bimolecular sensitization is achieved through diffusive encounters. When the donor is covalently bound to the acceptor molecule, intramolecular energy transfer enables a more specific control of the transfer;419 in such a case, only “equimolar” amounts of a sensitizer are needed. This strategy, benefiting from an increased “uncaging cross section” (Φrelε(λirr); see ref (22) for discussion) of release using triplet sensitization, has already been demonstrated on several examples.
Scheme 109. Photoinduced Energy Transfer (Blue Color Depicts an Electronic Excitation).
In 2004, Steiner, Green, and co-workers demonstrated that triplet sensitization of a known photoremovable protecting 2-(2-nitrophenyl)propyl moiety (239, Scheme 110; see also section 3.2) in homogeneous solutions or on glass substrates (microarray chips) enhances its uncaging cross section.418 The leaving group (alcohol), attached as a carbonate, is liberated in the course of the reaction as a carbonate monoester that disintegrates thermally into the corresponding alcohol and CO2. Acridin-9(10H)-one (240) and 9H-thioxanthen-9-one (241) were used as sensitizers (sens) that were excited by both continuous illumination and laser flash photolysis to study the reaction mechanism. Transient absorption observed at ∼400 nm was assigned to the aci-nitro intermediate 242 (see section 3.1). The sensitization kinetics of this bimolecular process were found to be nearly diffusion-controlled both in solution (kq = 1–4 × 109 M–1 s–1; in acetonitrile or methanol) and on the chip. Four photolabile phosphoramidites were synthesized for testing high-density oligonucleotide microarrays; the average chemical yield for release of the bases by using 241 was 97%. The kinetic analysis of the photosensitized cleavage reaction of surface-bound photolabile chromophores with free diffusion of sensitizer molecules from the bulk of a solution to the surface has recently been reported.167
Scheme 110. Intermolecular Triplet Sensitization of the 2-(2-Nitrophenyl)propyl Chromophore418.

In addition, Pirrung, Dore, and co-workers have recently demonstrated that, while the 2-(2-nitrophenyl)propyl group has a low sensitivity to two-photon excitation (see also section 9), the presence of a sensitizer with a large two-photon absorption cross section, such as 241, in the solution improves it considerably.234 Steiner and co-workers have also designed and studied covalently linked 9H-thioxanthen-9-one (the sensitizer) and 2-(2-nitrophenyl)propoxycarbonyl chromophores.166,233,240b,240g Scheme 111 shows an example of such a compound (243), in which the sensitizer is attached to the chromophore via a saturated four-bond aliphatic tether. After the sensitizer is excited and the intersystem crosses to the triplet state, the energy is transferred through space to the 2-(2-nitrophenyl)propyl group, which liberates thymidine.
Scheme 111. Intramolecular Triplet Sensitization of the 2-(2-Nitrophenyl)propyl Chromophore.
A variety of other covalently linked 9H-thioxanthen-9-one derivatives, including 244 (Scheme 111), in which the sensitizer is directly attached to the photoactive chromophore, were also prepared and studied to compare their photophysical and photochemical properties using stationary fluorescence and nanosecond and femtosecond time-resolved laser spectroscopy.166 The authors proposed a dual mechanism of triplet–triplet energy transfer. It was suggested that a slower transfer involves the lowest triplet (T1; π,π*) state of 9H-thioxanthen-9-one in the case of long-tether bichromophores. When bichromophores are connected through a short linker, the energy is believed to be transferred from the T2 (n,π*) state. These protecting groups were tested under conditions for lithographic DNA-chip synthesis.233 Their speed of release was found to be 10 times higher than that of the nonmodified 2-(2-nitrophenyl)propyl moiety.
Corrie and co-workers have designed the benzophenone antenna-sensitized 1-acyl-7-nitroindolines 245,249a−249d which display a significantly enhanced extent of photochemical cleavage in solution compared to their nonsensitized analogues247,248 (Scheme 112; see also section 3.3). The 4,4′-dialkoxybenzophenone chromophore (sens), which has substantially higher molar absorption coefficients in the region 270–320 nm than those of the nitroindoline moiety,249d serves as a light-harvesting antenna that transfers its triplet energy to form the reactive triplet state of the acceptor (PPG). It subsequently undergoes rapid photocleavage to liberate the 1-acyl substituent as the carboxylate in aqueous media. The triplet energy of 4,4′-dialkoxybenzophenone (ET = 70 kcal mol–1) is about 10 kcal mol–1 higher than the estimated ET of nitroindoline, which is one of the key conditions for efficient transfer.249d The authors then evaluated biological effects of released l-glutamate from 245 (R = CH2CH2CH(NH3+)COO–) using hippocampal neurons in primary culture and cerebellar granule cells in an acute brain slice preparation.249c The data suggested that glutamate was completely released in the irradiated samples. Recently, a more water-soluble analogue of 245 was synthesized and tested.249a
Scheme 112. Antenna-Sensitized 1-Acyl-7-nitroindoline System247,248.
8.2. Sensitized Release: Photoinduced Electron Transfer
The release of a leaving group can also be induced by photoinduced electron transfer (PET). In general, the excited state of a sensitizer (S) is quenched due to electron transfer to a quencher (Q; keT; Scheme 113). S and Q can be either separated or interconnected via a tether. As a result, a radical ion pair is formed provided that both S and Q are neutral prior the reaction. The radical ion pair can undergo a chemical reaction420 to give the products (here, the leaving group is released from one of the radical ions, kr) or a reverse electron transfer to regenerate S and Q (k–eT). This represents an important fork in the reaction sequence that determines the overall release quantum yields. The strategy allows the light-absorption step to be studied separately from the release step; therefore, both processes can be independently optimized. In principle, two major mechanistic strategies can be designed because either of the S or Q moieties can represent a PPG bearing the leaving group. As a result, the PPG can either be oxidized by loss of an electron or reduced by accepting an electron in a direct (the protecting group is a chromophore) or sensitized manner. Obviously, only the latter method has the benefits of a tunable chromophore. Some of the PET-based PPGs and their applications have been reviewed by Falvey and Sundararajan.8b
Scheme 113. Photoinduced Electron Transfer (Blue Color Depicts a Sensitizer).

The Gibbs free energy of photoinduced electron transfer ΔeTG° in an excited encounter complex (D··A)* can be calculated from eq 1, where E°(D•+/D) is the standard electrode potential of the donor radical cation, E°(A/A•–) is that of the acceptor A, ΔE0–0 is the 0–0′ excitation energy of the corresponding excited molecule (D* or A*), and w are the electrostatic work terms.27,421
| 1 |
Photofragmentation of tosylamides (246) in the presence of an electron donor (sensitizer), such as 1,4-dimethoxybenzene, and a reducing agent, such as ascorbic acid or sodium borohydride, resulting in the release of a free amine, was demonstrated by Yonemitsu and co-workers (Scheme 114) as the first example of a PET-based deprotection.422 The reductant acts as the source of the hydrogen atom and an electron (thus restoring the sensitizer). The release of simple amines occurs in high chemical (75–91%) and quantum (<0.65) yields. A covalently linked sensitizer approach was also explored.423
Scheme 114. Photosensitized Fragmentation of Tosylamides422.
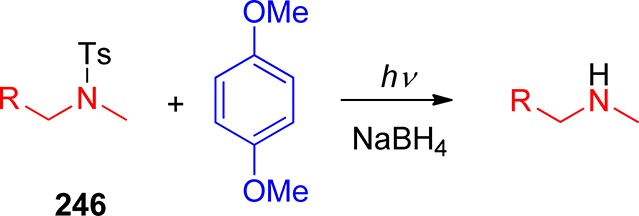
More recently, Corrie and Papageorgiou reported on the photochemistry of sulfonamide derivatives of amino acids.424 Glycine was released from the parent compound in rather low chemical yields unless a large excess of ascorbate was present in the solution. The best yields for the amino acid derivatives were found to be below 30%.425 The concurrent decarboxylation, triggered by intramolecular hydrogen-atom or electron transfer from the peptide bond, was believed to represent the major competing reaction.426 The tosyl group has also been used as a protecting group for near-quantitative thymidine liberation in the presence of 1,5-dimethoxynaphthalene as an electron donor in the synthesis of 5′-amino analogues of 3′-azido-3′-deoxythymidine (AZT).427 In contrast, methanesulfonyl and pentafluorosulfonyl esters were shown to be less suitable PET-based protection moieties.428
In addition to their intrinsic photoreactivity, phenacyl derivatives (section 2.1) can also release the leaving groups from the α-position upon one-electron reduction to the corresponding phenacyl anion radicals (Scheme 115). The carboxylates, for example, are liberated with a rate constant of ∼108 s–1.429 Falvey and co-workers demonstrated that these leaving groups are readily released from phenacyl esters (247) upon photosensitized electron reduction by light-absorbing amines (sens), provided they possess excited-state oxidation potentials below or equal to −2.2 V.430 Quantitative yields were reported for various derivatives. This approach was later extended for sensitizers that absorb at wavelengths in the near-UV region (<400 nm).430b Similarly, irradiation of phenacyl esters in micelles,57a or 1-oxoindan-2-yl and 1,3-dioxoindan-2-yl esters in acetonitrile solutions,60 was shown to liberate the carboxylic acids in high chemical yields. The release of other leaving groups has also been examined. A high-yielding (68–100%) liberation of phosphates, diacids, and alcohols (protected via carbonate linker) was reported.431
Scheme 115. Photosensitized Release of Carboxylic Acids from Phenacyl Esters430.
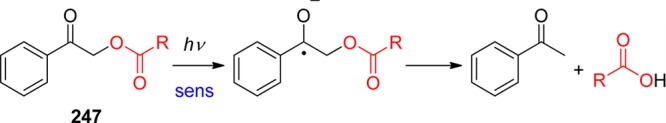
The electron-donor sensitizers can also be covalently attached to the phenacyl moiety. Laser flash photolysis studies revealed that a charge-transfer state (τ ≈ 500 ns) between N,N-dimethylaniline and the phenacyl chromophores of 248 is formed upon irradiation (Scheme 116).343a The phenacyl anion radical subsequently releases the leaving group (carboxylate) or regenerates the ground state of the parent compound.
Scheme 116. Bimolecular Sensitization of Phenacyl Esters343a.
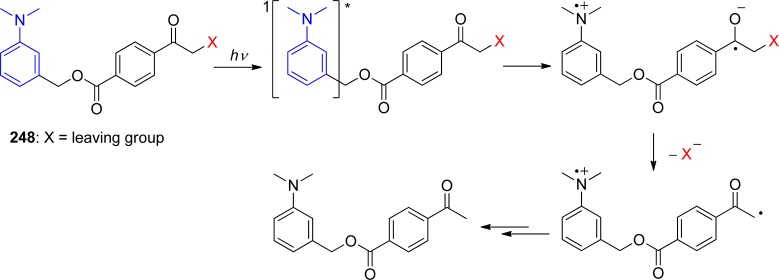
Falvey and co-workers demonstrated that the 4-pyridylmethyl (picolyl) group, previously used as a conventional protecting group for carboxylates in peptide synthesis, could be photochemically reduced in the presence of N,N,N′,N′-tetramethylbenzidine.343b The low reduction potential of the picolyl group (E[pyridine/pyridine–•]= −2.62 V vs SCE)38 precludes this group from being reduced by most sensitizers; therefore, alkylation on the nitrogen (249, Scheme 117) was chosen to increase its reduction potential. Irradiation of a perchlorate salt of 249, in the presence of, for example, the carbazole 250 as an electron donor, liberates a leaving group, such as a carboxylate.343b The addition of 1,4-cyclohexadiene as a hydrogen atom donor (to suppress back electron transfer) was found to result in increased chemical (86%) and quantum (0.39) yields of carboxylic acid release. The N-methylpicolinium esters can also serve as protecting moieties in the absence of any external photosensitizer when the iodide counterion (an electron donor) is exchanged for perchlorate.343b
Scheme 117. 4-Pyridylmethyl Group343b.
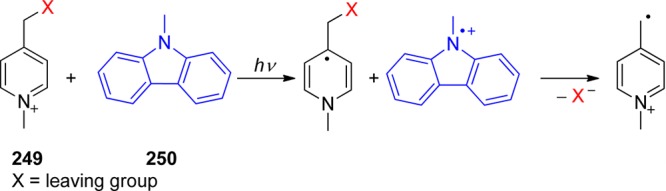
Deprotection of carboxylic acids, amino acids, and phosphates from 249 in the presence of visible-light absorbing photosensitizers and 1,4-cyclohexadiene has been reported to take place in high chemical yields (87–100%).432 The pyrromethene photosensitizers (PM 546 and 597) employed in this work are dyes originally developed for use in lasers. Mediated electron transfer with benzophenone or xanthone tethered to the picolinium moiety in the presence of a photosensitizer was used to increase the quantum yield of carboxylic or amino acids release to Φ = 0.72 at λ = 380 (Scheme 118).343c,433 Gold nanoparticles absorbing the visible light (>500 nm) were shown to mediate electron transfer between dithiothreitol, a good electron donor, and an N-methylpicolinium ester in aqueous solution with quantum yields in the range between 0.5 and 4.5, suggesting involvement of a radical chain mechanism.434 Ketocoumarin dyes were also used in a similar application.435 Tris(bipyridyl)ruthenium(II) as both a photosensitizer and a mediator for electron transfer between a good electron donor and the newly synthesized water-soluble 2-cyanopicolinium protecting group was recently demonstrated to result in the release of various carboxylic acids.436
Scheme 118. Photorelease via Mediated Electron Transfer.
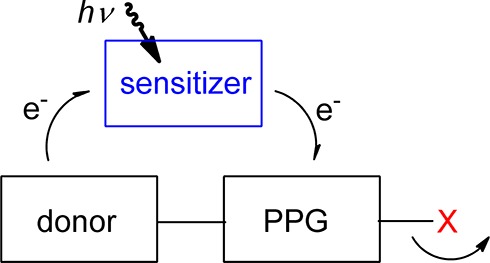
Falvey and co-workers then exercised the same principle to control the viscosity of aqueous solutions of 251 and cetyl trimethylammonium bromide by using visible light to initiate sensitized photorelease from the picolinium group and UV light to control photoisomerization of the attached cinnamic acid (Rubpy = tris(bipyridyl)ruthenium(II) as a sensitizer; Scheme 119).437 The irradiation protocols are thus conveniently orthogonal to one another.
Scheme 119. Orthogonal Photorelease and Photoisomerization437.
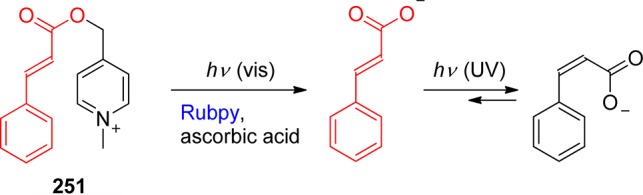
Visible-light-absorbing tris(bipyridyl)ruthenium(II) has also been recently used to mediate electron transfer to N-methylpicolinium carbamates (252) to give free amines in several steps (Scheme 120).438 Another application of a visible-light photoredox catalyst, such as Ir[dF(CF3)ppy]2(dtbbpy)PF6, in the oxidation of electron-rich arenes resulting in the selective deprotection of p-methoxybenzyl ethers in 69–91% yields has been demonstrated by Stephenson and collaborators.344a 2,3-Dichloro-5,6-dicyano-p-benzoquinone as an electron acceptor has been used for deprotection of various benzyl ethers by Toshima and collaborators.344b In addition, Lechner and König have shown that flavin photocatalysis can be used as visible-light-absorbing sensitizers in the deprotection of benzyl amines.345
Scheme 120. Visible-Light Deprotection of N-Methylpicolinium Carbamates438.
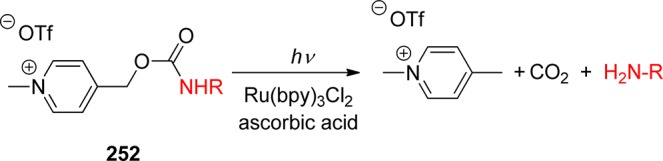
Benzyl ethers can release alcohols through oxidatively sensitized irradiation;439 however, no additional reports have been published in the past decade. In a different strategy, Tu and Floreancig demonstrated the protection of carbonyl and carboxyl compounds, which can be released by intramolecular photoinduced electron transfer from a phenethyl alcohol/ether group to covalently tethered anthraquinone (as a sensitizer) in 253 according to Scheme 121.440 The reported chemical yields of the deprotection were between 6 and 97%.
Scheme 121. Intramolecular Sensitization of the Phenethyl Alcohol/Ether Group440.
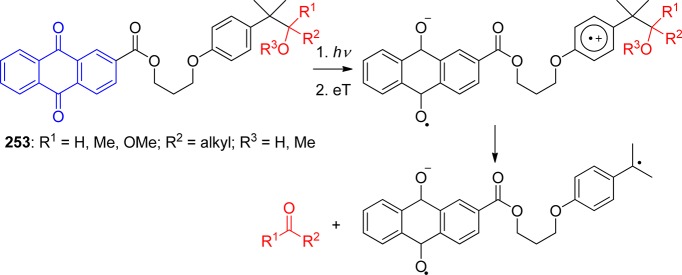
Cossy and Rakotoarisoa have discovered that the 2-acetoxyethyl group can be used to protect secondary amines.441 The deprotection is facilitated by photoinduced electron transfer from the acetoxyethyl derivative 254 to the dimethoxybenzophenone sensitizer 255 via a cation radical intermediate and the iminium salt 256, which subsequently hydrolyzes to release the secondary amine 257 in 60–80% chemical yields (Scheme 122).
Scheme 122. Sensitization of Acetoxyethyl Derivatives441.
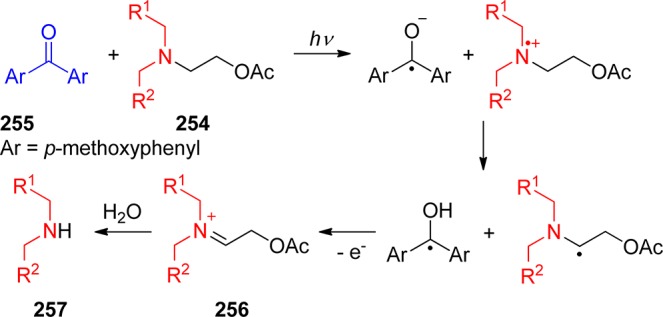
A blue-light triggered photorelease system based on the riboflavin-binding protein dodecin (a dodecameric hollow-spherical flavoprotein with six flavin binding sites) has recently been developed by Noll and co-workers.442 The release of the reduced flavin derivatives is reported to be triggered by irradiation with blue light of the dodecin-bound flavin anchor.
Kutateladze and co-workers have developed a novel strategy of aldehyde and ketone (258) protection using the dithiane moiety, which can be efficiently removed via oxidative photoinduced electron transfer in the presence of a sensitizer (sens), such as benzophenone (Scheme 123).443 The starting compounds 259 are easily synthesized from the lithium salts of 1,3-dithianes and the corresponding carbonyl compounds. Upon irradiation, the carbonyl compounds are released in excellent to mediocre chemical yields (e.g., 35–97%)443 and with reasonable efficiency (Φ = 0.06–0.17).444
Scheme 123. Photochemistry of Dithiane Moiety443.
A simplified mechanism of the oxidative deprotection is shown in Scheme 124. The reaction of 260 is facilitated by electron transfer to the excited state of benzophenone, resulting in formation of the cation radical 261 and the benzophenone radical anion 262. Depending on the solvent polarity, 262 deprotonates 261 in the solvent cage, or both species escape the solvent cage and form the same compounds through the reaction with water. It was evident from LFP studies that in-cage deprotonation occurs in dry acetonitrile whereas the latter process is dominant in more polar acetonitrile/water mixtures.445 Deprotonation of the radical cation of an adduct of dithiane and, for example, a t-butyl-substituted carbonyl compound gives the zwitterionic intermediate 263, which may, following intramolecular electron transfer, lead to an alternative C–C bond scission to give a stable t-butyl radical.446 However, the detailed mechanism is still not completely clear.447 Adducts of trithianes and trithiabicyclo[2.2.2]octanes, analogous to those of 1,3-dithianes, have also been studied as protection moieties.448 In addition, the dithiane moiety was successfully coupled with amino acids to form novel dithiazane photocleavable linkers.449 9H-Thioxanthen-9-one has also been introduced as a sensitizer in the dithiane-, trithiane-, and dithiazane-based photolabile scaffolds.450
Scheme 124. Mechanism of Oxidative Deprotection of Dithiane Moiety445,446.
The dithiane-based linkers were explored for various applications, such as photolabile phospholipids and amphiphiles,451 calixarene-based rosette,452 or barbiturate receptors.453 Dithiane-spiro-crown ethers (e.g., 264; Scheme 125) were used as photolabile moieties, which allowed photochemical interruption of transport through liquid membranes,448a or as photolabile linkers.454
Scheme 125. Photochemistry of Dithiane-spiro-crown Ethers448a.
Triggering of two-photon fluorescence (see also section 9) as a reporting function has been developed.455 Kutateladze and co-workers used dithianes to mask the sensitizer molecules immobilized on beads, dendrimers, or peptides to demonstrate that excitation of a single free sensitizer leads to liberation of its own copy, thus leading to signal amplification.456 Events, which occur on an attomolar concentration scale, can now be detected via molecular recognition-triggered photoamplified fluorescence quenching.457 Dithiane-based photolabile tags have also been used for encoding and direct screening of solution-phase combinatorial libraries.458
Dalko and co-workers have recently reported on a very interesting bichromophoric system composed from hydroxymethyl quinolene-derived probes, which are activated through interactions of X-ray or γ-irradiation with gadolinium(III) complexes (Scheme 126).459 These metal complexes act as intramolecular antennae and convert part of the energy resulting in electron transfer and subsequent fragmentation of the quinolines.
Scheme 126. Photo- and Radiolysis of Caged Hydroxymethyl Quinolone Derivatives459.
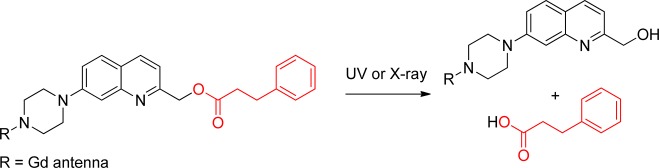
8.3. Sensitized Release: Light Upconverting Nanoparticles
In contrast to multiphoton excitation, which comprises the simultaneous absorption of two or more photons using pulses with femtosecond duration (section 9), photon upconversion is a sequential absorption of two or more photons.460 Light upconverting nanoparticles (UCNPs) have attracted considerable attention as visible or near-IR (NIR) harvesting light antennae that, upon energy or heat transfer (photothermal effect), can trigger a release of species, such as drugs.461
Branda and co-workers used a dimethoxybenzoin PPG coupled to monodispersed core–shell nanoparticles (NPs) composed of NaYF4 nanocrystals doped with lanthanides to release carboxylates upon NIR light activation (980 nm) followed by NP fluorescence, exciting the benzoin chromophore.223 The wavelength of emission may be controlled by the power density of lasers used for NPs excitation.462 The same principle has also been demonstrated on photoswitches.463
Remotely triggered release was achieved by heat transfer from gold nanoparticles, which can be tethered to, encapsulated within, or suspended freely outside liposomes or micelles that can serve as drug carriers.222,464 In a similar study, gold NPs were enclosed in biodegradable and biocompatible microspheres (1–15 μm) containing the antitumor drug paclitaxel.465 The NIR-activated photothermal effect leads to efficient paclitaxel release.
A variety of target compounds, such as oligonucleotides or si-RNA, can also be adsorbed on the modified surface of a gold NP466 or coupled to the NP surface via gold–thiol conjugation.467 For example, a high payload of doxorubicin was coated and successfully released from both the outer and inner shells of polyethylene glycol (PEG) hollow gold nanoparticles.468 Chromatic orthogonality (see also section 10) to release two different DNA oligonucleotides has been demostrated on gold nanorods differing in their aspect ratio,469 which is promising for future applications in cancer theranostics.470
9. Two-Photon Excitation-Induced Photorelease
The removal of the photolabile protecting groups discussed in this review usually requires UV irradiation. These conditions, however, are not compatible with many biomedical applications, as UV–vis light is efficiently absorbed by the tissue.471 The major absorbing species in vertebrate tissues is oxyhemoglobin, which filters practically all the light of wavelengths shorter than 650 nm.471 On the other hand, water becomes increasingly absorbant at wavelengths longer than 950 nm. These two factors define the “phototherapeutic window”, the region of relative tissue transparency between 650 and 950 nm.472 The increased depth of tissue penetration of radiation in this region is accompanied by additional advantages: lower scattering and reduced phototoxic effects. However, red and NIR photons have relatively low energy, limiting the range of processes they can initiate. In addition, the majority of chromophores with useful single-photon absorbance in the “phototherapeutic window” are also sensitive to visible light, complicating the handling of the caged substrates. One of the approaches that overcomes most of these problems is to use nonresonant two-photon excitation (2PE). At high light intensity, chromophores may simultaneously absorb two red/NIR photons, producing higher-energy excited states, the same as or similar to those accessible by direct excitation with UV photons of about twice the frequency.473 Additionally, focusing the irradiation of 2PE on UV chromophores within UV-absorbing materials provides an opportunity to control substrate release in three dimensions. The probability of 2PE is proportional to radiant intensity squared, which is many orders of magnitude higher in the λ3 volume around the focal point than in other areas of the laser beam.474 This phenomenon has made possible the development of 3-D applications in fluorescent imaging,473a,474b,475 microscopy,474b,476 photonics,477 3-D fabrication,478 and potentially even drug delivery304,479 and photodynamic therapy.480 Substantially less data are available on two-photon induced photochemical reactions473c,481 and their use in biochemistry.164,476b,482 The 3-D capabilities of two-photon-induced uncaging were successfully used in neuron mapping,10d,483 investigation of intracellular processes,484 and regulation of protein activity in vitro221,485 and in vivo,221,485b as well as in the development of 2PE-uncaging microscopy.486
2PE requires very high light intensities, which can be achieved only using ultrafast pulsed lasers. Thus, a commercially available mode-locked Ti:sapphire laser can provide a photon irradiance (Ep) of 1025 photons cm–2 s–1 (3 mm beam at 100 fs pulse duration, and repetition frequency ν = 90 MHz) or more. The principal emission of the most common Ti:sapphire lasers is at 800 nm, which almost perfectly corresponds to the minimum of mammalian tissues absorbance. Lasers that can be tuned to shorter wavelengths are usually more expensive. The differential form of the Beer’s law for two-photon excitation487 is shown below (eq 2), where Ep is the photon irradiance (photons cm–2 s–1), δ is the two-photon cross section (cm4 s photon–1 molecule–1), N is the concentration (molecules cm–3), and x is the sample thickness (cm).
| 2 |
| 3 |
As light intensity virtually does not change when passing through the sample due to low two-photon absorption, we can write eq 2 in a simplified form (eq 3). The rate of photoreaction with quantum yield Φ (molecule photon–1) is proportional to photon irradiance absorbed by the sample, which is in turn proportional to squared photon irradiance (eq 4).481f The coefficient 1/2 in eq 4 reflects the two-photon nature of the process.
| 4 |
| 5 |
Integration of eq 4 and taking into account the pulsed nature of the irradiation gives eq 5.481fN0 and Nt represent starting and current concentration of a substrate; Ep represents the averaged photon irradiance during the pulse, tpulse is pulse duration, ν is laser repetition rate, and tirr is the time of exposure. Strictly speaking, Ep2 should be integrated for the duration of the pulse, but this approximation introduces insignificant error. For low-conversion experiments that use the Ti:sapphire laser discussed above, we can write a simplified eq 6, where δ is expressed in Goeppert-Mayer units (1 GM = 10–50 cm4 s photon–1 molecule–1).
| 6 |
Thus, to achieve 1% release of a substrate within the focal volume of a laser beam using a PPG with a quantum yield of uncaging Φ = 0.5 and a 2PE cross-section of δ = 1 GM, the sample has to be irradiated for about 30 min. One should also take into account the background one-photon absorbance of tissue materials, which is about 10–3 per 1 cm at 750 nm,476b,488 and the broadening of ultrashort pulses in dense media. For an efficient two-photon photochemistry in tissues, the product δEp should be at least 10–25 cm2 molecule–1.487 This means that, at light intensities produced by a Ti:sapphire laser, the two-photon absorption cross section δ of the PPG should be around 1 GM or higher.
The cross section of two-photon absorption for most organic chromophores is rather low (δ < 1 GM); however, some recently synthesized fluorophores, which usually contain D−π–A−π–D (D = donor, A = acceptor) or a similar structural motif, have δ over 1000 GM.477b,477c Unfortunately, these advances in the design of two-photon chromophores have not yet been applied to photolabile protecting groups. The suitability of several conventional (single-photon) PPGs for 2PE-induced substrate release has been explored instead (Table 11). The efficiency of this process is better characterized by the two-photon cross section of uncaging (δunc), which is a product of the two-photon absorption cross section (δ) and the 2PE quantum efficiency (Φ2PE),489 δunc = Φ2PEδ. It should be noted that the value of δunc is strongly dependent on the wavelength of irradiation. Many cages that show decent efficiency of substrate release at 740–750 nm have very low δunc at 800 nm.
Table 11. Single-Photon and Two-Photon Uncaging Quantum Efficiencies of Photolabile Protecting Groups.
| PPG | Φa | λ/nm | δunc/GMb | λ/nm | ref |
|---|---|---|---|---|---|
| 83 (4,5-dimethoxy-2-nitrobenzyl, NV) | 0.16 | 305 | 0.035c | 730 | (490) |
| 0.006 | 365 | 0.03 | 740 | (303) | |
| 0.08 | >365 | 0.01 | 800 | (496) | |
| 0.23c | 740 | (221, 485b) | |||
| 0.004c–0.025 | 750 | (221, 485b) | |||
| 57 (o-nitrobenzyl, oNB; R = Cl, OH, MeO, NH2, p-MeOC6H4, etc.) | 0.001–0.1 | 325 | 0.015–0.065 | 750 | (190) |
| 265 (3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl, DMNPB) | 0.26 | 365 | 0.17 | 720 | (492) |
| 115 ((4′-methoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl, PMNB) | 0.1 | 315 | 0.45 | 800 | (236) |
| 3.1 | 740 | (237) | |||
| 266 ((4′-tris-ethoxymethoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl, PENB) | 0.1 | 315 | 3.7 | 740 | (493) |
| 117 (2,7-bis-{4-nitro-8-[3-(2-propyl)-styryl]}-9,9-bis-[1-(3,6-dioxaheptyl)]-fluorene, BNSF) | 0.25 | 354 | 5.0 | 800 | (237) |
| 267 ((7-hydroxycoumarin-4-yl)methyl) | 0.025 | 365 | 1.07 | 740 | (331) |
| 0.13 | 800 | (303) | |||
| 268 ((6-chloro-7-hydroxycoumarin-4-yl)methyl) | 0.01 | 365 | 1.07 | 740 | (303) |
| 0.34 | 800 | (303) | |||
| 143 ((6-bromo-7-hydroxycoumarin-4-yl)methyl, BHCM) | 0.084 | 350 | 0.35 | 740 | (288) |
| 0.019–0.037 | 365 | 0.51–1.99 | 740 | (294, 303) | |
| 0.21 | 780 | (330a) | |||
| 0.37–0.42 | 800 | (303) | |||
| 269 ({7-[bis(carboxymethyl)-amino]coumarin-4-yl}methyl, BCMACM) | 0.29 | 330–430 | ca. 1c | 800 | (286) |
| 270 ({7-[4-(dimethylamino)styryl]coumarin-4-yl}methoxycarbonyl) | 0.83 × 10–3 | 407 | 0.26 | 800 | (497) |
| 271 ((3,6,8-tribromo-7-hydroxycoumarin-4-yl)methyl) | 0.065 | 365 | 0.96 | 740 | (303) |
| 3.1 | 800 | (303) | |||
| 181 ((8-bromo-7-hydroxyquinoline-2-yl)methyl, BHQ) | 0.29–0.39 | 365 | 0.59–0.90 | 740 | (331) |
| 0.087 | 780 | (330a) | |||
| 272 ((8-cyano-7-hydroxyquinoline-2-yl)methyl, CyHQ) | 0.31 | 365 | 0.32 | 740 | (335a) |
| 273 ((8-chloro-7-hydroxyquinoline-2-yl)methyl, CHQ) | 0.10 | 365 | 0.12 | 740 | (335a) |
| 274 ([7-(dimethylamino)quinoline-2-yl]methyl, DMAQ) | 0.046 | 365 | 0.13 | 740 | (335a) |
| 275 ([7-(dimethylamino)-4-chloroquinoline-2-yl]methyl, DMAQ-Cl) | 0.09 | 365 | 0.47 | 740 | (335a) |
| 276 ((7-mercaptoquinoline-2-yl)methyl, TQ) | 0.063 | 365 | 0.42 | 740 | (335a) |
| 277 ((Z)-butyl 3-(4-(diethylamino)-2-hydroxyphenyl)-2-acetamidoacrylate) | 0.05 | 300–400 | 0.3 | 750 | (393b) |
| 278 (3,5-dibromo-2,4-dihydroxycinnamate) | 0.05 | 369 | 1.6 | 750 | (390) |
| 0.05 | 300–400 | 0.6 | 750 | (393a) | |
| 279 ((E)-3-(6-hydroxy-benzo(1,3)dioxo-5-yl)acrylate) | 0.03 | 300–400 | 3.8 | 750 | (393a) |
| 280 ((E)-3-(4-diethylamino-2-hydroxy-phenyl)acrylate) | 0.02 | 300–400 | 2.0 | 750 | (393a) |
| 281 ((Z)-butyl 2-acetamido-3-(5-hydroxybenzo[d][1,3]dioxol-6-yl)acrylate) | 0.07 | 300–400 | 2.0 | 750 | (393b) |
| 282 ((E)-3-(8-hydroxy-2,3,6,7-tetrahydro-1H,5H-pyrido(3,2,1-ij)quinolin-9-yl)acrylate) | 0.03 | 300–400 | 4.7 | 750 | (393a) |
| 125 (4-methoxy-7-nitroindolinyl, MNI) | 0.085 | 350 | 0.006 | 730 | (10d, 483) |
| 127 (4-methoxy-5,7-dinitroindolinyl, MDNI) | 0.47 | 350 | 0.06c | 730 | (494) |
| 283 (4-carboxymethoxy-5,7-dinitroindolinyl, CDNI) | 0.5c | 334–364 | 0.06 | 720 | (483a) |
| 130 (5-methoxy-8-nitro-1,2-dihydroquinolinyl, MNDQ) | 0.05 | 365 | <0.06c | 720 | (250) |
The single-photon quantum yield of substrate release.
The two-photon cross section of uncaging.
By comparison with the literature data.
The most widely used o-nitrobenzyl-based PPGs have a rather low two-photon uncaging cross section, which varies, depending on the substitution in the ring and the wavelength of excitation, from 0.01 to 0.035 GM (Table 11). The NV cage (83) shows the highest sensitivity to 2PE in this family, with δunc = 0.035 GM at 730 nm.490 When o-nitrobenzyl (oNB) PPGs are used for caging of fluorescent dyes (section 11), the quantum yield and two-photon cross section of uncaging is often improved apparently due to the energy transfer from the dye to the caging chromophore. Thus, coumarin is released from the oNB cage (57) with 53% quantum efficiency in single-photon excitation at 365 nm and with 2PE δ = 0.37–0.68 GM at 740 nm.491 The analogous 2-(2-nitrophenyl)prop-1-yl-based PPGs, such as NPPOC, NPEOC (section 3.2), or DMNPB (265), possess the same o-nitrophenyl chromophore but have 5–12 times higher δunc due to more efficient photochemistry.492 The two-photon uncaging cross section of these groups can be also improved by sensitization. Thus, in the presence of a triplet sensitizer with a large 2PE cross section (i.e., thioxanthone), the two-photon uncaging action cross section of NPEOC (section 3.2) is enhanced to 0.86 from 0.12 GM.234 Extension of the conjugated π-system of the chromophore [(4′-methoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl, PMNB, 115; (4′-tris-ethoxymethoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl, PENB, 266],236,237,493 especially when combined with a symmetrical structure (BNSF, 117),237 brings the two-photon uncaging cross sections into a respectable 3–5 GM range. However, these groups are rather bulky and suffer from poor solubility.
(Coumarin-4-yl)methyl-based (section 4) PPGs 267, 268, 269, 270, and 143 are usually more efficient two-photon cages than simple 1-(2-nitrophenyl)ethyl (NPE) and 2-(2-nitrophenyl)prop-1-yl (NPP) (section 3.2) analogues (Table 11). The uncaging cross sections of the popular BHCM group (143, section 4) range from 0.35 to 2 GM at 740 nm depending on the caged substrate.288,294 Incorporation of two additional bromine atoms in the benzene ring of BHCM (271) results in a red-shift of the 2PE maximum and further enhancement of the two-photon uncaging efficiency (Table 11).303
Dore and co-workers have systematically studied the effects of electron-donating and electron-withdrawing substituents on the two-photon uncaging cross sections of (quinoline-4-yl)methyl PPGs (section 5).331 The most efficient two-photon-induced substrate release in this family was reported for the BHQ cage (181, δunc = 0.6–0.9 GM at 740 nm).330a Replacement of the bromo substituent for the cyano- (272) or chloro- (273) groups resulted in a reduced δunc, nitro group largely suppressed the two-photon sensitivity.335a The 7-dimethylamino- (274 and 275) and mercapto- (276) (quinoline-4-yl)methyl derivatives are also presented in the table.
The 2-hydroxycinnamyl cage (section 7.7) developed by Porter’s group387a was also found to be amenable to 2PE-triggered substrate release. Jullien and co-workers have demonstrated that, with the appropriate choice of substituents in the aromatic ring (277, 278, 279, and 280), the photochemical step of the uncaging process, trans–cis isomerization of the (E)-cinnamate moiety, can be induced under two-photon photolysis conditions.393b 4-Amino substituted (E)-(o-hydroxyphenyl)acrylates possess the highest two-photon activation cross section (281, δunc = 2.0 GM; 282, δunc = 4.7 GM at 740 nm).393a Although the substrate release from these cages is relatively slow at a biological pH (section 7.7), the 2PE-sensitive (E)-cinnamates are remarkably easy to synthesize. Despite the rather low two-photon cross sections of nitroindolyl cages (125, 127, 283, and 130, Table 11; see also section 3.3), they have been successfully employed in several biochemical applications.10d,250,483,494
Ruthenium complexes that contain amino compounds in their coordination sphere can release these substrates (section 6) under two-photon irradiation in aqueous solution. Thus, 800-nm irradiation of Ru(bipy)2(4-aminopyridine)22+ or Ru(bpy)2(PMe3)(GluH2)2+ was found to result in the release of 4-aminopyridine or glutamine with 0.01 to 0.1 GM cross sections.495
10. Chromatic Orthogonality
The diversity of different PPGs, operating by different mechanisms and bearing different types of chromophores, opens the door for wavelength-selective deprotection. Indeed, the possibility of releasing on demand various types of molecules (e.g., in a cell, or wherever access by conventional injection techniques is not possible) is a very appealing prospect. A first-order approximation would suggest that absorption maxima sufficiently distant could work; however, fast energy transfer may thwart such a strategy, in particular for cases where both chromophores are part of the same molecular entity. Careful choice of PPG pairs made such a wavelength discrimination possible, allowing for the selective release of two different carboxylic acids, in what was an early example of chromatic orthogonality (Scheme 127).208,498
Scheme 127. Example of Intermolecular Chromatic Orthogonality498.
The same benzoin (section 2.4)/nitroveratryl (NV, section 3.3) pair was then shown to be equally orthogonal in the intramolecular case, where spatial proximity is more critical than in separate molecules (Scheme 128).499 Reactivity tuning by way of a kinetic isotope effect was also exploited to make two derivatives of o-nitrobenzylic esters (section 3.1) chromatically orthogonal.225
Scheme 128. Example of Intramolecular Chromatic Orthogonality499.
Examples of this principle were shown soon after in the solid-phase synthesis of peptides360c and in the selective release from resins by using the nitroveratry/pivaloylglycol pair.500 del Campo and co-workers investigated a more complex version of the wavelength-selective cleavage by putting seven photolabile protecting groups on various functionalities immobilized through an organosilane tether on a glass surface against each other.240e,501 Systematic analysis of photolytic characteristics of common photolabile groups helped establish several protecting strategies that permit the simultaneous use of up to four orthogonal photoactivated groups, such as benzoin (λex = 255 nm, section 2.4), p-hydroxyphenacyl (pHP, λex = 275 nm, section 2.3), 5,7-dinitroindolinyl (DNI, λex = 360 nm, section 3.3), and [7-(diethylamino)coumarin-4-yl]methyl (DEACM, λex = 435 nm, section 4), that can be cleaved sequentially from the same solid support under different irradiation conditions.240e It was also noted that two-photon excitation can potentially expand the spectral window and increase the number of possible functional levels for selective spatiotemporal activation.
Applications in biochemistry are appearing more and more frequently, such as the protection of cysteines (using variously substituted coumarins),196 nucleotides (using a coumarin/nitrophenethyl pair),502 or both,503 and the release of both substrates and inhibitors (also using a coumarin/nitrophenethyl pair),504 or the selective release of glutamate or GABA by two-colors/two-photon excitation.263b The latter strategy was also applied very recently with a NV/p-MeO-phenacyl pair by Emptage, Conway, and co-workers.106c The same concept was shown to be valid in the solid phase, where the wavelength-selective modification of surfaces was performed with orthogonal pairs135a,501 or quartets.240e
11. Photoactivatable Fluorescent Dyes
Photoactivatable (caged) fluorophores13b are fluorogenic and release a fluorescent molecule upon irradiation. They are obtained by coupling a fluorescent dye to a PPG that prevents it from displaying fluorescence. Caged fluorescent dyes provide a highly sensitive tool to monitor the flow of liquids (rheology) and to follow the movements and the distribution of particular species of interest at the single molecule level and with a spatial resolution at the nanometer scale, i.e., beyond the diffraction limit of optical microscopy (∼250 nm). Fluorescence imaging microscopy is among the most powerful techniques for observing dynamic processes in living cells. To monitor the movement of target molecules or species in real time, it is necessary to create a local region within the cell where the fluorescence intensity is higher than in the bulk. Imaging with high target-to-background contrast requires a high activation ratio, i.e., a high ratio of postactivation to preactivation signal intensity. Thus, the signal intensity arising from the caged fluorophore should be close to zero.
The classic method for creating a differentially labeled region has been fluorescence photobleaching, i.e, reducing fluorescence intensity by irradiation, usually by reaction of a fluorescent dye with singlet oxygen. Alternatively, photochemically labile fluorophores such as the aminophthalimide–serine shown in Scheme 129 can be used.378 Disadvantages of photobleaching through singlet oxygen are that it may cause local damage to proteins and membranes and that it is difficult to track a region of reduced fluorescence within a background of higher fluorescence. The use of a nonbleachable reference fluorophore has been proposed to track the distribution of the bleached molecules by image differencing.505
Scheme 129. “Armed” Phthalimide (λmax = 340 nm, Exhibits Strong Fluorescence, λmax = 513 nm, in Aqueous Media at pH = 7; Irradiation Liberates Acetate and CO2, and the Fluorescence Decreases)378.
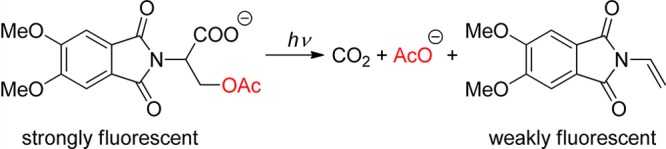
To overcome these limitations, numerous photoactivatable fluorophores have been developed. The design and use of caged fluorophores have been reviewed.9n,13a,13b,506 Desiderata for an effective caged fluorescent probe are biostability and -compatibility; a high affinity to or, preferentially, specificity for the target (e.g., cancer cells);507 rapid and efficient photoactivation providing a high ratio of pre- and postirradiation fluorescence; a high fluorescence quantum yield; photostability of the uncaged fluorophore; and practicable synthesis. Up to 1998,506 caged fluorophores have all used variants of the oNB (section 3) caging group (e.g., 284).
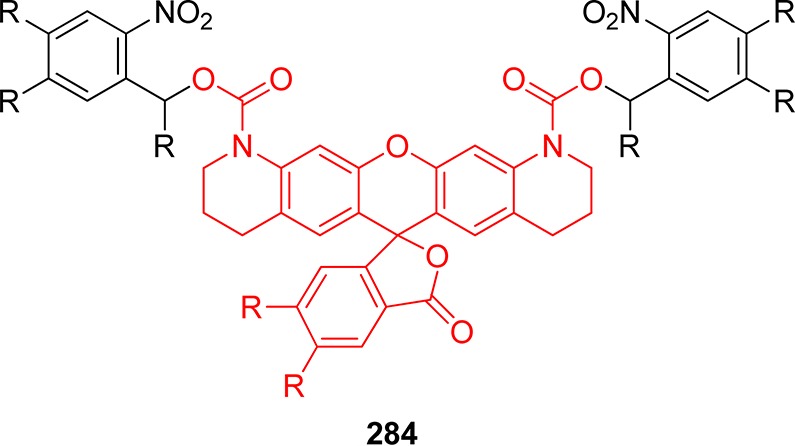
The full activation of such bis-caged lactone (leuco) forms of rhodamine or fluorescein requires the removal of both PPGs. Removal of one PPG is sufficient to restore the chromophore, but the remaining nitroaromatic group often largely quenches the fluorescence emission of the monocaged dye. Full deprotection therefore requires high doses of UV irradiation. On the other hand, the non-negligible fluorescence quantum yield of monoprotected fluorescein hampers its use as a caged fluorophore, because the activation ratio upon deprotection is usually insufficient. A monoprotected variant of fluorescein (TokyoGreen, 285, Scheme 130) has, however, been developed that exhibits very little fluorescence.508 The fluorescence quantum yield of singly 2-(2-nitrophenyl)prop-1-yl (NPP) (section 3.1) protected TokyoGreen (286) is less than 1/100th that of protected fluorescein. It liberates the highly fluorescent free form (Φfl = 0.96) by removal of the PPG with a quantum yield of 0.03.509
Scheme 130. Deprotection of Mono-Caged Tokyo Green (Its Fluorescence Is Quenched by Intramolecular Electron Transfer in the Excited Singlet State)508.
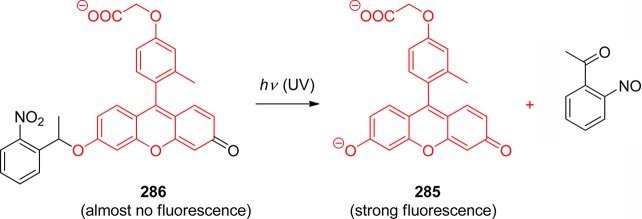
Several other examples of fluorescence activation by photochemical removal of a quencher moiety attached near the fluorophore have been reported. Other than by electron transfer, fluorescent singlet states can be quenched by Förster resonance energy transfer (FRET, somewhat inadequately called “fluorescence resonance energy transfer” by most biochemists)510 to an adjacent, nonfluorescent, but photoremovable chromophore. For example, a synthetic route to incorporate a photocleavable 4-dimethylaminoazobenzene-4′-sulfonyl (287, dabsyl)511 moiety and fluorescein at adjacent cytidines in the middle of a 25-mer oligodeoxynucleotide has been reported.512 UV irradiation removed the dabsyl moiety, which increased the fluorescence intensity 51-fold and restored the melting temperature of the nucleotide. Such caged fluorescent oligodeoxynucleotides will allow many DNA processes to be controlled with light.

The same principle was used to regulate the DNA polymerase reaction by the Klenow fragment of polymerase I with UV light. A 25-mer caged fluorescent oligodeoxynucleotide as the template was functionalized with a fluorescein reporter and the photoremovable dabsyl quencher moiety (287). With this template, the Klenow fragment was blocked from extending a complementary 12-mer primer. Removal of the quencher by short UV photolysis partly restored the activity of the Klenow fragment, and the reactivation could be monitored by fluorescence.511
A related approach was used to provide for real-time monitoring of Smad2, a key protein involved in the transforming growth factor β (TGF-β) signaling pathway, spatial and temporal control of its activity, and differentiation between its active and inactive state.513 The protein was labeled at neighboring sites with a fluorescein chromophore and with a 4-dimethylaminoazobenzene-4′-carboxylic (dabcyl) acid quencher through a photocleavable 4-[4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy] butanoic acid linker. This both suppressed the protein activity and quenched its fluorescence. Photocleavage of the caging group resulted in the simultaneous restoration of protein activity and luminescence of the fluorescent tag.
Numerous photocleavable (coumarin-4-yl)methyl esters with fast release have been described (section 4).276,289,296 NPP-caged coumarins that display more than 200-fold fluorescence enhancement upon UV irradiation were developed (Scheme 131).491 The combined advantages of robust fluorescence contrast enhancement, high uncaging efficiency, noninvasive cellular delivery, and flexible chemistry for bioconjugations are promising for the use of these caged coumarins in biochemical and biological research. The high uncaging efficiency is attributed to the antenna function of the coumarin, i.e., light absorbed by the coumarin chromophore is utilized to cleave the NPP cage. It should, however, be noted that NPE-caged fluorophores are subject to the known disadvantages of o-nitrobenzyl photochemistry (see section 3.1).
Scheme 131. Caged Coumarin491.
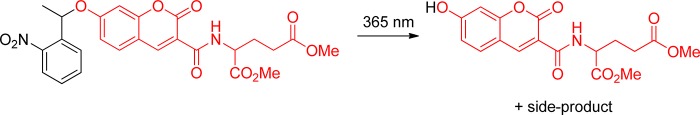
To prepare a caged coumarin emitting a different color, the o-nitrobenzyl-caged coumarin was linked to a water-soluble fluorescein dye emitting at 520 nm.514 The caged dye can be localized in the sample prior to photolysis by directly exciting the fluorescein moiety at 490 nm. After uncaging the coumarin chromophore, the green emission can be excited at 410 nm due to efficient FRET from the coumarin moiety to the fluorescein dye. This is especially desirable for experiments demanding highly localized photoactivation by two-photon uncaging, which requires knowing the distribution of the label in three dimensions. These probes thus offer new opportunities to image molecular and cellular dynamics.
A light-activated fluorescent reporter of intracellular protein kinase activity has been designed (Scheme 132) that furnishes a fluorescent readout.515 The photolytically labile appendage affords control over both the timing and the amount of active sensor release. The quantum yield for photolytic conversion is 0.06. The caged fluorescent substrate was introduced into HeLa cells via microinjection. Following in situ illumination of the caged peptide, time-dependent changes of the fluorescence intensity due to phosphorylation of the released hydroxyl function provided a measure of protein kinase activity in the cells.
Scheme 132. Fluorescent Reporter of Protein Kinase Activity515.
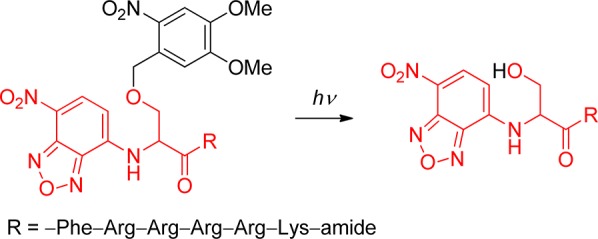
Azidomethyl-caged fluorescein derivatives (288 and 289) were utilized to monitor the dynamics of oligonucleotides in living human cells. Both compounds were rapidly activated upon brief irradiation and showed a strong increase in fluorescence intensity.516

The addition of lithiated dithianes to 2-amidothioxanthones disrupts conjugation, resulting in a blue-shift of the absorption and a dramatic decrease in fluorescence intensity. The product represents a new caging system capable of fast quantification of released payloads such as TentaGel beads by single- or two-photon fluorescence.455 Irradiation induces homolytic C–C fragmentation followed by disproportionation of the two radicals (Scheme 133). The half-life of the radical intermediates was 1.7 μs.
Scheme 133. Caged Thioxanthone455.
Rhodamine dyes having a 2-diazo caging group incorporated into a spiro-9H-xanthene fragment have been synthesized by reaction of diazomethane with the acid chloride of rhodamine B.517 The yellow crystalline diazoketone 290 was obtained in high yield (Scheme 134). Caged rhodamines with an additional carboxy group in the benzoyl fragment of the diazoketone that may be linked to an amino or a thiol reactive site were also made. Application of a related system featuring a high contrast ratio (1:140) and activation by visible light (λ < 420 nm) to high-resolution microscopy was recently reported.518
Scheme 134. Photochemistry of Caged Rhodamine (a Wolff Rearrangement)517.

A ruthenium-rhodamine complex 291 (see Section 6) has been evaluated as an activatable fluorescent probe (Scheme 135).356 Upon irradiation, it releases rhodamine in a fast and clean reaction, increasing its fluorescence nearly 6-fold.
Scheme 135. Photochemistry of Caged Rhodamine356.
A photoactivatable nitrobenzyl-caged (section 3.1) green fluorescent protein (292, GFP) that is practically nonfluorescent prior to irradiation was synthesized recently (Scheme 136).519 The relative brightness at the wavelength of maximum emission (486 nm) increased by at least 4 orders of magnitude during photoactivation at 365 nm.
Scheme 136. o-Nitrobenzyl-Caged GFP519.
Photoactivatable fluorescent proteins have become an important addition to the set of molecular probes used to understand cellular function. Wild-type GFP520 is in fact already a caged fluorophore of sorts. The protein can adopt two conformations with different absorption maxima, and irradiation shifts the molecules toward the longer-wavelength absorbing form. Photoconversion in wild-type GFP is thought to involve rotation of threonine 203 and decarboxylation of glutamic acid 222. A mutant of wild-type GFP, in which the threonine 203 position is replaced by histidine, was shown to increase its fluorescence intensity (λex = 488 nm, λem = 520 nm) 100 times upon intense irradiation at 413 nm.521 The activated protein remains stable for days under aerobic conditions. This photoactivatable variant of the Aequorea victoria GFP was used both as a free protein to measure protein diffusion across the nuclear envelope and as a chimera with a lysosomal membrane protein to demonstrate rapid interlysosomal membrane exchange.
Optical microscopy provides spatial resolution down to the diffraction limit of ∼250 nm, but single fluorescent particles can now be localized down to ∼1 nm precision, and spectroscopic techniques, such as FRET, can offer yet smaller distance changes. Super-resolution (subdiffraction) imaging techniques based on sequential imaging of sparse subsets of single molecules have used fluorophores whose emission can be photoactivated or photoswitched.522 The common principle is that only one pointlike light source is active in a diffraction-limited area at any time. Imaging with a sensitive camera localizes this fluorophore. Its exact location can then be determined to well below the diffraction limit by fitting a point-spread function. In the beginning of data acquisition, all except a few fluorophores are prepared in the off-state and the number of active fluorophores is kept constant by applying an activating light source that compensates for the loss of fluorophores by photobleaching or switching off. A super-resolution image of a labeled complex structure can then be reconstructed from many successive rounds of weak photoactivation and fitting. This method has been used to image intracellular proteins at nanometer spatial resolution. As an alternative to caged fluorophores for super-resolution microscopy, on- and off-states have been engineered by controlling the photophysics of fluorophores by electron transfer reactions.523
Because good organic fluorophores are more photochemically stable than most fluorescent proteins, organic fluorophores have a potential benefit in super-resolution imaging schemes, but targeting to specific cellular proteins must be provided. The design and application of target-specific an azido derivative of 2-dicyanomethylene-3-cyano-2,5-dihydrofuran, a photoactivatable push–pull fluorogen that produces bright fluorescent labels suitable for single-molecule super-resolution imaging in live bacterial and fixed mammalian cells, were reported (293).524

The same authors subsequently demonstrated that the azide-to-amine photoactivation process is generally applicable to a variety of push–pull chromophores, which provide a new class of photoactivatable single-molecule probes for fluorescent labeling and super-resolution microscopy.525 Moreover, these photoactivated push–pull dyes can insert into bonds of nearby biomolecules, simultaneously forming a covalent bond and becoming fluorescent (fluorogenic photoaffinity labeling).
The design, synthesis, and photophysical characterization of a class of ligand-directed, photoactivatable, turn-on fluorescent probes for the spatially controlled imaging of microtubules in live mammalian cells was reported.526 A series of taxoid–tetrazoles was reported in which 7-β-alanyltaxol L was attached to a water-soluble tetrazole bearing an o-allyloxy group at the N-phenyl ring via a flexible linker. Upon irradiation at 302, 365, or even 405 nm, the tetrazoles 294 undergo deazotization generating reactive nitrile imine dipoles that spontaneously react with the prealigned allyl group to form pyrazoline fluorophores (Scheme 137). Whereas none of the taxoid–tetrazoles was fluorescent, the taxoid–pyrazolines exhibited strong fluorescence emission in the range of 500–800 nm. With this photoactivatable fluorescent probe, it should be possible to label microtubules asymmetrically within a single cell and identify factors that break cellular symmetry during cell division.
Scheme 137. Intramolecular Photoclick Reaction526.

12. Conclusion
Photoremovable protecting groups (PPGs) release molecules such as enzymes, neurotransmitters, signaling molecules, fluorophores, insecticides, pheromones, and fragrances that thereby exhibit desirable physical, chemical, or biological qualities upon photoactivation. They are synthetically malleable and accessible, offering a wide range of structures for designed applications. They offer excellent spatial and temporal control for the substrate release. Their applications span many scientific fields, from DNA chip technology, drug delivery, and photoregulation of proteins, to rheology, solid-phase synthesis, surface chemistry, and nanotechnology. PPGs are excellent, versatile tools for time-resolved studies of chemical processes in living cells. Multiphoton excitation can provide superior spatial resolution, and the new chromophores included herein offer more precise temporal control for addressing the dynamics of in vivo events in living organisms.
There has been an upsurge in interest in PPGs over the past decade that has resulted in a dramatic increase in the number of new designs and the development of both new and known PPGs to fulfill the demands for better sensitivity, faster kinetics, and more demanding bioanalytical applications. The many new discoveries have brought substantial improvements and versatility to the synthesis, thermal stability, solubility, and absorption properties of PPGs, as well as improved efficiencies and rates of release. Nonetheless, there still remain many important research goals, such as extending the wavelength coverage for activation of PPGs with visible and infrared light and improving the absorption properties for wavelength-selective, orthogonal activation, and melding these features with the inherent advantages of photoactivation that PPG possess as outlined at the beginning of this review. Our goal has been to aid and assist researchers in applying these tools and to attract and encourage their participation in this rapidly developing field.
Acknowledgments
Support for this work was provided by the Grant Agency of the Czech Republic (203/09/0748), the Ministry of Education, Youth and Sports of the Czech Republic (ME09021, KONTAKT/AMVIS), and the project CETOCOEN (CZ.1.05/2.1.00/01.0001) granted by the European Regional Development Fund, (P.K. and T.S.). T.S. profited from the Brno Ph.D. Talent Program funded by Brno City Municipality. C.G.B. thanks the Swiss National Science Foundation (200020-129617) for the support. A.B. thanks the CNRS, the French Ministry of Research, and the Agence Nationale de la Recherche (ANR-11-JS07-001-01) for financial support. R.G. and M.R. thank the NIH, grant RO1 GM72910, for financial support.
Glossary
List of Abbreviations
- 2PE
two-photon excitation
- Ac
acetyl
- ACM
(7-aminocoumarin-4-yl)methyl
- α-CNB
α-carboxynitrobenzyl
- α-CNV
α-carboxy-6-nitroveratryl
- ADP
adenosine diphosphate
- Ala
alanine
- ANB
4-acetyl-2-nitrobenzyl
- Aqe
anthraquinon-2-yleth-2-yl
- Aqm
anthraquinon-2-ylmethoxy
- Aqmoc
anthraquinon-2-ylmethoxycarbonyl
- ATP
adenosine-5′-triphosphate
- BCMACM
{7-[bis(carboxymethyl)amino]coumarin-4-yl}methyl
- BCMCM
[6,7-bis(carboxymethoxy)coumarin-4-yl]methyl
- BDP
benzoin diethyl phosphate
- BHCM
(6-bromo-7-hydroxycoumarin-4-yl)methyl
- BHQ
(8-bromo-7-hydroxyquinoline-2-yl)methyl
- BNSF
2,7-bis-{4-nitro-8-[3-(2-propyl)-styryl]}-9,9-bis-[1-(3,6-dioxaheptyl)]fluorene
- Bni
5-bromo-7-nitroindolinyl
- Boc
N-(t-butoxycarbonyl)
- cAMP
cyclic adenosine monophosphate
- Cbz
benzyloxycarbonyl
- CDNI
4-carboxymethoxy-5,7-dinitroindolinyl
- CM
(coumarin-4-yl)methyl
- CMCM
[7-(carboxymethoxy)coumarin-4-yl]methyl
- CT
charge transfer
- CyHQ
(8-cyano-7-hydroxyquinoline-2-yl)methyl
- dabsyl
dimethylaminoazobenzene-4′-sulfonyl
- dabcyl
4-dimethylaminoazobenzene-4′-carboxylic
- DDQ
2,3-dichloro-5,6-dicyanobenzoquinone
- Ddz
α,α-dimethyl-3,5-dimethoxybenzyloxycarbonyl
- DEACM
[7-(diethylamino)coumarin-4-yl]methyl
- DEP
diethyl phosphate
- DFT
density functional theory
- DHB
2,5-dihydroxybenzyl
- DMACM
[7-(dimethylamino)coumarin-4-yl]methyl
- DMAQ
[7-(dimethylamino)quinoline-2-yl]methyl
- DMAQ-Cl
[7-(dimethylamino)-4-chloroquinoline-2-yl]methyl
- DMATr
3-(dimethylamino)trityl
- DMB
3′,5′-dimethoxybenzoin
- DMBF
2-phenyl-5,7-dimethoxybenzofuran
- DMCM
6,7-dimethoxy(coumarin-4-yl)methyl
- DMNB
4,5-dimethoxy-2-nitrobenzyl
- DMNPB
3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl
- DMP
2,5-dimethylphenacyl
- DNI
5,7-dinitroindolinyl
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ES
energy of the lowest excited singlet state
- ET
energy of the lowest excited triplet state
- Fmoc
fluorenylmethyloxycarbonyl
- FRET
Förster resonance energy transfer
- GABA
γ-aminobutyric acid
- GAP
GTPase activating protein
- GDP
guanosine diphosphate
- GFP
green fluorescent protein
- GTP
guanosine triphosphate
- HCM
(7-hydroxycoumarin-4-yl)methyl
- HEMA
(hydroxyethyl)methacrylate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HFIP
hexafluoroisopropanol
- CHQ
(8-chloro-7-hydroxyquinoline-2-yl)methyl
- IPA
isopropanol
- ISC
intersystem crossing
- KMOPS
potassium 3-(N-morpholino)propanesulfonate
- KTRF
Kerr-gated time-resolved fluorescence
- LFP
laser flash spectroscopy
- LG
leaving group
- LTC4
Leukotriene C4
- MCM
(methoxycoumarin-4-yl)methyl
- MDNI
4-methoxy-5,7-dinitroindolinyl
- MeNPOC
3,4-(methylenedioxy)-6-nitrophenylethoxycarbonyl
- MeNV
α-methyl-6-nitroveratryl
- MeNVOC
α-methyl-6-nitroveratryloxycarbonyl
- MeNPOM
α-methyl-(6-nitropiperonyloxymethyl)
- MLLCT
metal–ligand-to-ligand charge transfer
- MMA
methyl methacrylate
- MNDQ
5-methoxy-8-nitro-1,2-dihydroquinolinyl
- MNI
4-methoxy-7-nitroindolinyl
- MNPPOC
2-(3,4-methylenedioxy-6-nitrophenyl)propoxycarbonyl
- NDBF
3-nitrodibenzofuran-2-yl
- NIR
near-infrared
- NOPA
noncollinear optical parametric amplifier
- NP
nanoparticle
- NPE
1-(2-nitrophenyl)ethyl
- NPEOC
1-(2-nitrophenyl)ethyloxycarbonyl
- NPP
2-(2-nitrophenyl)prop-1-yl
- NPPOC
2-(2-nitrophenyl)prop-1-oxycarbonyl
- NQMP
(3-hydroxynaphthalen-2-yl)methyl
- NV
6-nitroveratryl; (3,4-dimethoxy-6-nitrophenyl)methyl
- NVOC
6-nitroveratryloxycarbonyl
- oNB
2-nitrobenzyl
- oNT
2-nitrotoluene
- PAK
protein kinase A
- PB
phosphate buffer
- PBS
phosphate-buffered saline
- PCA
photoremovable chiral auxiliary
- PEG
polyethylene glycol
- PENB
(4′-trisethoxymethoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl
- PET
photoinduced electron transfer
- pHA
4-hydroxyacetophenone
- pHP
4-hydroxyphenacyl
- PMNB
(4′-methoxy-4-nitrobiphenyl-3-yleth-2-yl)methyl
- PPG
photoremovable protecting group
- PPTS
pyridinium p-toluenesulfonate
- PTK
protein tyrosine phosphatase
- Px
9-phenylxanthyl, pixyl
- SCE
saturated calomel electrode
- S-Px
9-phenylthioxanthyl, S-pixyl
- TBS
t-butyldimethylsilyl
- TFA
trifluoroacetic acid
- TFE
2,2,2-trifluoroethanol
- TGF
transforming growth factor
- TMS
trimethylsilyl
- TQ
(7-mercaptoquinoline-2-yl)methyl
- TR-FTIR
time-resolved Fourier transform infrared
- TR-RR
time-resolved resonance Raman
- TR-TA
time-resolved transient absorption
- Ts
tosyl
- UCNP
upconverting nanoparticle
- X
leaving group
Biographies

Petr Klán received an M.S. degree from Masaryk University, Brno, Czech Republic, in 1986. After working in industry for 5 years, he pursued his Ph.D. at Michigan State University under the tutelage of Prof. Peter J. Wagner. After receiving his Ph.D. in chemistry in 1998, he joined the faculty at Masaryk University, where he is now a full professor. His current research focuses on photochemistry, mechanisms of organic reactions, kinetic studies by flash photolysis, spectroscopy, photoremovable protecting groups, and environmental photochemistry. He is a co-author of “Photochemistry of Organic Compounds: From Concepts to Practice” (Wiley, 2009) with Prof. Jakob Wirz.

Tomáš Šolomek received his M.S. degree from Masaryk University, Brno, Czech Republic, in 2009. He is currently a Ph.D. candidate in Prof. Petr Klán’s group at Masaryk University, conducting research on the study and development of new photoremovable protecting groups. His interests encompass experimental and theoretical photochemistry and the chemistry of reactive intermediates, especially open-shell biradicals.

Christian Bochet received his B.Sc. degree in chemistry in 1990 and an M.Sc. in 1991 in inorganic chemistry under the guidance of Prof. Alan Williams at the University of Geneva. He then joined the group of the late Prof. Wolfgang Oppolzer, receiving his Ph.D. in organic chemistry in 1996, focusing on total synthesis of alkaloids. From 1996 to 1998, he was a postdoctoral fellow at Stanford University with Prof. Barry M. Trost, where he worked on transition metal catalysis applied to the synthesis of natural products. In 1998, he returned to the University of Geneva where he began his independent research program in organic photochemistry. In 2002, he moved to the University of Fribourg as a SNF associate professor. He is currently professor of chemistry (2006). His interests include reagent-controlled selective reactions, total synthesis of natural products, and organic photochemistry. When not doing chemistry, he enjoys playing the violin as a member of the Geneva Symphony Orchestra.

Aurélien Blanc graduated from the University of Geneva in 2004 with a Ph.D. in organic photochemistry under the direction of Prof. C. Bochet. He then joined Prof. F. D. Toste (University of California, Berkeley) as a postdoctoral fellow of the Swiss National Science Foundation working on asymmetric synthesis of oxygenated rings. In 2006, he moved to the University J. Fourier in Grenoble as a teaching assistant working on natural product synthesis with Dr. A. Greene. In 2007, he joined Prof. Pale (University of Strasbourg) as a CNRS fellow. His main interests are in photochemistry toward organic synthesis and on the development of new methods in organic synthesis based on coinage-metal catalysis, especially gold- and silver-catalyzed heterocyclization reactions applied to natural product synthesis.

Richard S. Givens is Emeritus Professor of Chemistry (2010) at the University of Kansas. He earned his BA degree in Chemistry (H.-G. Gilde) at Marietta College (1962) and his Ph.D. with H. E. Zimmerman at the University of Wisconsin, Madison (1967). After a year as an NIH postdoctoral associate with the late G. A. Russell at Iowa State University, he began his independent research career at the University of Kansas (1967). His work centers on physical organic chemistry, photochemistry, bioanalytical chemistry, and their applications in chemistry and biology. He is a co-editor with Maurice Goeldner of “Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules” (Wiley-VCH, 2005).

Marina Rubina received her B.S. degree in chemistry and chemical education from Syktyvkar State University (Russia) in 1996. She was a research associate at the Moscow State University (Russia) before earning her Ph.D. degree with Vladimir Gevorgyan (2004) at the University of Illinois at Chicago. She has been a postdoctoral associate at the University of Kansas with Michael Rubin and Richard Givens. She now holds the position of Laboratory Director for the Department of Chemistry and serves as a Research Associate in the Center for Environmentally Beneficial Catalysis at the University of Kansas.

Vladimir Popik received his Ph.D. degree from St. Petersburg State University in 1990 and continued as a Research Scientist until 1992 when he joined Jerry Kresge at the University of Toronto as a postdoctoral fellow and Research Professor. In 1999 he joined the faculty of the Center for Photochemical Sciences at the Bowling Green State University and in 2006 moved to the University of Georgia, where he is currently Professor of Chemistry. Vladimir’s recent awards include NSF Career Award and Georgia Cancer Coalition Distinguished Cancer Scholar. His current research involves design of photoswitchable antibiotics, development of novel photolabile protecting groups and linkers, and biochemical and material science applications of photo-click chemistry.

Dr. Alexey P. Kostikov received his M.S. degree from the Saint-Petersburg State University in Saint-Petersburg, Russia. He began his Ph.D. studies with Prof. Vladimir V. Popik at Bowling Green State University and transferred with him to the University of Georgia to complete his Ph.D. (2007). His research included developing novel photoremovable protecting groups based on the photochemistry of 2-hydroxybenzyl alcohol derivatives. After graduation, he joined Prof. John Baldwin’s group at Syracuse University as a postdoctoral fellow. In 2009 he became Research Associate in Prof. Ralf Schirrmacher’s group at McGill University, Montreal, Canada. He currently specializes in radiochemistry, synthesizing radiotracers for positron emission tomography (PET). His independent research project is focused on the application of recently developed copper-free click chemistry for the quick and efficient radiolabeling of biologically active macromolecules such as peptides with the widely used PET radioisotope fluorine-18.

Jakob Wirz is Emeritus Professor of Chemistry at the University of Basel. He studied at the ETH Zürich with Prof. E. Heilbronner and, after postdoctoral studies in London with Profs. G. Porter and D. H. R. Barton, habilitated at the University of Basel, where he led a research group until 2007. He co-authored the book “Photochemistry of Organic Compounds: From Concepts to Practice” (Wiley, 2009) with Prof. P. Klán. His research interests include photochemistry and spectroscopy, laser flash photolysis, reaction mechanisms, and photochemical protecting groups.
The authors declare no competing financial interest.
Dedication
This review is dedicated to the memory of Professor Howard E. Zimmerman, University of Wisconsin, Madison (July 5, 1926−February 11, 2012).
Funding Statement
National Institutes of Health, United States
References
- Barltrop J. A.; Schofield P. Tetrahedron Lett. 1962, 16, 697. [Google Scholar]
- a Barton D. H. R.; Chow Y. L.; Cox A.; Kirby G. W. Tetrahedron Lett. 1962, 23, 1055. [Google Scholar]; b Barton D. H. R.; Chow Y. L.; Cox A.; Kirby G. W. J. Chem. Soc. 1965, 3571. [Google Scholar]
- Patchornik A.; Amit B.; Woodward R. B. J. Am. Chem. Soc. 1970, 92, 6333. [Google Scholar]
- Sheehan J. C.; Wilson R. M. J. Am. Chem. Soc. 1964, 86, 5277. [Google Scholar]
- Engels J.; Schlaeger E. J. J. Med. Chem. 1977, 20, 907. [DOI] [PubMed] [Google Scholar]
- Kaplan J. H.; Forbush B. III; Hoffman J. F. Biochemistry 1978, 17, 1929. [DOI] [PubMed] [Google Scholar]
- a Pelliccioli A. P.; Wirz J. Photochem. Photobiol. Sci. 2002, 1, 441. [DOI] [PubMed] [Google Scholar]; b Givens R. S.; Conrad P. G. I.; Yousef A. L.; Lee J.-I.. Photoremovable Protecting Groups. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, 2004. [Google Scholar]
- a Bochet C. G. J. Chem. Soc., Perkin Trans. 1 2002, 2, 125. [Google Scholar]; b Falvey D. E.; Sundararajan C. Photochem. Photobiol. Sci. 2004, 3, 831. [DOI] [PubMed] [Google Scholar]; c Hoffmann N. Chem. Rev. 2008, 108, 1052. [DOI] [PubMed] [Google Scholar]; d Sankaranarayanan J.; Muthukrishnan S.; Gudmundsdottir A. D. Adv. Phys. Org. Chem. 2009, 43, 39. [Google Scholar]; e Bochet C. G.; Blanc A.. Photolabile Protecting Groups in Organic Synthesis. In Handbook of Synthetic Photochemistry; Albini A., Fagnoni M., Eds.; Wiley: Weinheim, Germany, 2010; Chapter 13, p 417. [Google Scholar]
- a Goeldner M.; Givens R. S.. Dynamic Studies in Biology; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]; b Giovannardi S.; Landò L.; Peres A. News Physiol. Sci. 1998, 13, 251. [DOI] [PubMed] [Google Scholar]; c Shigeri Y.; Tatsu Y.; Yumoto N. Pharmacol. Ther. 2001, 91, 85. [DOI] [PubMed] [Google Scholar]; d Lawrence D. S. Curr. Opin. Chem. Biol. 2005, 9, 570. [DOI] [PubMed] [Google Scholar]; e Kramer R. H.; Chambers J. J.; Trauner D. Nature Chem. Biol. 2005, 1, 360. [DOI] [PubMed] [Google Scholar]; f Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2006, 45, 4900. [DOI] [PubMed] [Google Scholar]; g Gorostiza P.; Isacoff E. Mol. Biosyst. 2007, 3, 686. [DOI] [PubMed] [Google Scholar]; h Ellis-Davies G. C. R. Nat. Methods 2007, 4, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Tang X.; Dmochowski I. J. Mol. Biosyst. 2007, 3, 100. [DOI] [PubMed] [Google Scholar]; j Xiangming M.; Xiaoyun C.; Yao F.; Qingxiang G. Progr. Chem. 2008, 20, 2034. [Google Scholar]; k Sjulson L.; Miesenböck G. Chem. Rev. 2008, 108, 1588. [DOI] [PubMed] [Google Scholar]; l Lee H.-M.; Larson D. R.; Lawrence D. S. ACS Chem. Biol. 2009, 4, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Yu H.; Li J.; Wu D.; Qiu Z.; Zhang Y. Chem. Soc. Rev. 2010, 39, 464. [DOI] [PubMed] [Google Scholar]; n Specht A.; Bolze F.; Omran Z.; Nicoud J.-F.; Goeldner M. HFSP J. 2009, 3, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Schatzschneider U. Eur. J. Inorg. Chem. 2010, 10, 1451. [Google Scholar]; p Katz J. S.; Burdick J. A. Macromol. Biosci. 2010, 10, 339. [DOI] [PubMed] [Google Scholar]; q Priestman M. A.; Lawrence D. S. Biochim. Biophys. Acta 2010, 1804, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Riggsbee C. W.; Deiters A. Trends Biotechnol. 2010, 28, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]; s Krauss U.; Drepper T.; Jaeger K.-E. Chem.—Eur. J. 2011, 17, 2552. [DOI] [PubMed] [Google Scholar]; t Ciesienski K. L.; Franz K. J. Angew. Chem., Int. Ed. 2011, 50, 814. [DOI] [PubMed] [Google Scholar]; u Givens R. S.; Rubina M.; Wirz J. Photochem. Photobiol. Sci. 2012, 11, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]; v Fehrentz T.; Schonberger M.; Trauner D. Angew. Chem., Int. Ed. 2011, 50, 12156. [DOI] [PubMed] [Google Scholar]; w Photosensitive Molecules for Controlling Biological Function; Chambers J. J., Kramer R. H., Eds.; Humana Press: New York, 2011. [Google Scholar]; x Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446. [DOI] [PubMed] [Google Scholar]
- a Lin C.-C.; Anseth K. S. Pharm. Res. 2009, 26, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crespy D.; Landfester K.; Schubert U. S.; Schiller A. Chem. Commun. 2010, 46, 6651. [DOI] [PubMed] [Google Scholar]; c Lovell J. F.; Liu T. W. B.; Chen J.; Zheng G. Chem. Rev. 2010, 110, 2839. [DOI] [PubMed] [Google Scholar]; d Warther D.; Gug S.; Specht A.; Bolze F.; Nicoud J. F.; Mourot A.; Goeldner M. Bioorg. Med. Chem. 2010, 18, 7753. [DOI] [PubMed] [Google Scholar]
- a Herrmann A. Angew. Chem., Int. Ed. 2007, 46, 5836. [DOI] [PubMed] [Google Scholar]; b Herrmann A. Photochem. Photobiol. Sci. 2012, 11, 446. [DOI] [PubMed] [Google Scholar]
- a Suyama K.; Shirai M. Prog. Polym. Sci. 2009, 34, 194. [Google Scholar]; b Zhao H.; Sterner E. S.; Coughlin E. B.; Theato P. Macromolecules 2012, 45, 1723. [Google Scholar]
- a Puliti D.; Warther D.; Orange C.; Specht A.; Goeldner M. Bioorg. Med. Chem. 2011, 19, 1023. [DOI] [PubMed] [Google Scholar]; b Li W.-H.; Zheng G. Photochem. Photobiol. Sci. 2012, 11, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fukaminato T. J. Photochem. Photobiol., C 2011, 12, 177. [Google Scholar]
- A Special Issue of Photochem. Photobiol. Sci. on Photoremovable Protecting Groups: Developments and Applications; Wirz J., Ed.; Royal Society of Chemistry: Cambridge, U.K., 2012; Vol. 11, pp 433–600. [DOI] [PubMed] [Google Scholar]
- a Rose M. J.; Mascharak P. K. Coord. Chem. Rev. 2008, 252, 2093. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hishikawa K.; Nakagawa H.; Miyata N. Yakugaku Zasshi 2011, 131, 317. [DOI] [PubMed] [Google Scholar]; c Tfouni E.; Truzzi D. R.; Tavares A.; Gomes A. J.; Figueiredo L. E.; Franco D. W. Nitric Oxide–Biol. Ch. 2012, 26, 38. [DOI] [PubMed] [Google Scholar]
- a Schatzschneider U. Eur. J. Inorg. Chem. 2010, 1451. [Google Scholar]; b Rimmer R. D.; Richter H.; Ford P. C. Inorg. Chem. 2010, 49, 1180. [DOI] [PubMed] [Google Scholar]; c Zhang W. Q.; Atkin A. J.; Fairlamb I. J. S.; Whitwood A. C.; Lynam J. M. Organometallics 2011, 30, 4643. [Google Scholar]
- a Gomez T. M.; Spitzer N. C. Nature 1999, 397, 350. [DOI] [PubMed] [Google Scholar]; b Zucker R. Methods Cell Biol. 1994, 40, 31. [DOI] [PubMed] [Google Scholar]; c Zucker R.Photorelease Techniques for Raising or Lowering Intracellular Ca(2+). In Calcium in Living Cells; Whitaker M., Ed.; Academic Press: Waltham, MA, 2010; Vol. 99; p 27. [DOI] [PubMed] [Google Scholar]; d Cui J. X.; Gropeanu R. A.; Stevens D. R.; Rettig J.; Campo A. J. Am. Chem. Soc. 2012, 134, 7733. [DOI] [PubMed] [Google Scholar]
- a Bandara H. M. D.; Walsh T. P.; Burdette S. C. Chem.—Eur. J. 2011, 17, 3932. [DOI] [PubMed] [Google Scholar]; b Stephens M. R.; Geary C. D.; Weber S. G. Photochem. Photobiol. 2002, 75, 211. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Chen Y. Eur. J. Org. Chem. 2011, 7, 1346. [Google Scholar]
- Mbatia H. W.; Bandara H. M. D.; Burdette S. C. Chem. Commun. 2012, 48, 5331. [DOI] [PubMed] [Google Scholar]
- Low molar absorption coefficients may be advantageous to achieve deep penetration when high concentrations are required to release a sufficient amount of the reactant.
- The quantity Φrelε(λirr), the units of which are M–1 cm–1 = dm3 mol–1 cm–1 = 1000 cm2 mol–1, is sometimes called uncaging cross section, although the latter term should have the dimension of an area. Strictly, the net absorption cross section is defined as σnet(λ) = κ/NA, where κ is the molar napierian absorption coefficient and NA is the Avogadro constant. The relation between the net absorption cross section and the molar decadic absorption coefficient is σnet(λ)/cm2 = (3.8236 × 10–21/mol) × [ε(λ)/(M–1 cm–1)].
- The spectra were measured by one of us (T.S.) unless noted otherwise.
- Perkampus H.-H.UV–VIS Atlas of Organic Compounds, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1992. [Google Scholar]
- The spectrum was measured by one of us (C.G.B.).
- Fuh R.-C. A. In http://omlc.ogi.edu/spectra/PhotochemCAD/html/085.html, 1997.
- Klan P.; Wirz J.. Photochemistry of organic compounds: From concepts to practice; Wiley: Chichester, U.K., 2009. [Google Scholar]
- a Warren J. A.; Bernstein E. R. J. Chem. Phys. 1986, 85, 2365. [Google Scholar]; b Park S. T.; Feenstra J. S.; Zewail A. H. J. Chem. Phys. 2006, 124, 174707. [DOI] [PubMed] [Google Scholar]
- Carsey T. P.; Findley G. L.; McGlynn S. P. J. Am. Chem. Soc. 1979, 101, 4502. [Google Scholar]
- a Itoh T.; Baba H.; Takemura T. Bull. Chem. Soc. Jpn. 1978, 51, 2841. [Google Scholar]; b Kanda Y.; Kaseda H.; Matumura T. Spectrochim. Acta 1964, 20, 1387. [Google Scholar]; c Silva C. R.; Reilly J. P. J. Phys. Chem. 1996, 100, 17111. [Google Scholar]
- Fang W. H.; Phillips D. L. ChemPhysChem 2002, 3, 889. [Google Scholar]
- Wagner P. J.; Klan P.. Norrish Type II Photoelimination of Ketones: Cleavage of 1,4-Biradicals Formed by α-Hydrogen Abstraction. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool W. M., Lenci F., Eds.; CRC Press LLC: Boca Raton, FL, 2003; Chapter 52, p 1. [Google Scholar]
- McGarry P. F.; Doubleday C. E.; Wu C. H.; Staab H. A.; Turro N. J. J. Photochem. Photobiol., A 1994, 77, 109. [Google Scholar]
- Merkel P. B.; Kearns D. R. J. Chem. Phys. 1973, 58, 398. [Google Scholar]
- Chattopadhyay S. K.; Kumar C. V.; Das P. K. J. Photochem. 1985, 30, 81. [Google Scholar]
- Ghoshal S. K.; Sarkar S. K.; Kastha G. S. Bull. Chem. Soc. Jpn. 1981, 54, 3556. [Google Scholar]
- Clark W. D. K.; Litt A. D.; Steel C. J. Am. Chem. Soc. 1969, 91, 5413. [Google Scholar]
- Montalti M.; Credi A.; Prodi L.; Gandolfi M. T.. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, 2006. [Google Scholar]
- Sheehan J. C.; Umezawa K. J. Org. Chem. 1973, 38, 3771. [Google Scholar]
- Banerjee A.; Falvey D. E. J. Am. Chem. Soc. 1998, 120, 2965. [Google Scholar]
- Literak J.; Dostalova A.; Klan P. J. Org. Chem. 2006, 71, 713. [DOI] [PubMed] [Google Scholar]
- Khade P. K.; Singh A. K. Tetrahedron Lett. 2007, 48, 6920. [Google Scholar]
- a Jana A.; Atta S.; Sarkar S. K.; Singh N. D. P. Tetrahedron 2010, 66, 9798. [Google Scholar]; b Okada S.; Yamashita S.; Furuta T.; Iwamura M. Photochem. Photobiol. 1995, 61, 431. [Google Scholar]
- a Jovanovic S. V.; Renaud J.; Berinstain A. B.; Scaiano J. C. Can. J. Chem. 1995, 73, 223. [Google Scholar]; b Renaud J.; Scaiano J. C. Res. Chem. Intermed. 1995, 21, 457. [Google Scholar]
- Singh P. N. D.; Mandel S. M.; Robinson R. M.; Zhu Z. D.; Franz R.; Ault B. S.; Gudmundsdottir A. D. J. Org. Chem. 2003, 68, 7951. [DOI] [PubMed] [Google Scholar]
- a Kasapoglu F.; Onen A.; Bicak N.; Yagci Y. Polymer 2002, 43, 2575. [Google Scholar]; b Takahashi E.; Sanda F.; Endo T. J. Appl. Polym. Sci. 2004, 91, 3470. [Google Scholar]; c Yagci Y.; Durmaz Y. Y.; Aydogan B. Chem. Rec. 2007, 7, 78. [DOI] [PubMed] [Google Scholar]
- Tazhe Veetil A.; Solomek T.; Ngoy B. P.; Pavlíkova N.; Heger D.; Klan P. J. Org. Chem. 2011, 76, 8232. [DOI] [PubMed] [Google Scholar]
- Kammari L.; Solomek T.; Ngoy B. P.; Heger D.; Klan P. J. Am. Chem. Soc. 2010, 132, 11431. [DOI] [PubMed] [Google Scholar]
- a Haag R.; Wirz J.; Wagner P. J. Helv. Chim. Acta 1977, 60, 2595. [Google Scholar]; b Das P. K.; Encinas M. V.; Small R. D.; Scaiano J. C. J. Am. Chem. Soc. 1979, 101, 6965. [Google Scholar]; c Small R. D.; Scaiano J. C. J. Am. Chem. Soc. 1977, 99, 7713. [Google Scholar]
- Weedon A. C.Photochemical reactions involving enols. In The chemistry of enols; Rappoport Z., Ed.; Wiley: New York, 1990; p 591. [Google Scholar]
- Sammes P. G. Tetrahedron 1976, 32, 405. [Google Scholar]
- Klan P.; Wirz J.; Gudmundsdottir A.. Photoenolization and its Applications. In CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; Griesbeck A., Oelgemoeller M., Ghetti F., Eds.; CRC Press: Boca Raton, FL, 2012. [Google Scholar]
- a Michalak J.; Gebicki J. Wiad. Chem 1993, 47, 407. [Google Scholar]; b Segura J. L.; Martin N. Chem. Rev. 1999, 99, 3199. [DOI] [PubMed] [Google Scholar]; c Mehta G.; Kotha S. Tetrahedron 2001, 57, 625. [Google Scholar]; d Yoshioka M.; Saito M. J. Synth. Org. Chem. Jpn 2001, 59, 689. [Google Scholar]
- a Klan P.; Pelliccioli A. P.; Pospisil T.; Wirz J. Photochem. Photobiol. Sci. 2002, 1, 920. [DOI] [PubMed] [Google Scholar]; b Klan P.; Zabadal M.; Heger D. Org. Lett. 2000, 2, 1569. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Ryu H. J. Tetrahedron Lett. 2010, 51, 1512. [Google Scholar]
- a Bergmark W. R. J. Chem. Soc., Chem. Commun. 1978, 2, 61. [Google Scholar]; b Bergmark W. R.; Barnes C.; Clark J.; Paparian S.; Marynowski S. J. Org. Chem. 1985, 50, 5612. [Google Scholar]
- a Ruzicka R.; Zabadal M.; Klan P. Synth. Commun. 2002, 32, 2581. [Google Scholar]; b Zabadal M.; Pelliccioli A. P.; Klan P.; Wirz J. J. Phys. Chem. A 2001, 105, 10329. [DOI] [PubMed] [Google Scholar]; c Literak J.; Relich S.; Kulhanek P.; Klan P. Mol. Divers. 2003, 7, 265. [DOI] [PubMed] [Google Scholar]
- Literak J.; Wirz J.; Klan P. Photochem. Photobiol. Sci. 2005, 4, 43. [DOI] [PubMed] [Google Scholar]
- Kammari L.; Plistil L.; Wirz J.; Klan P. Photochem. Photobiol. Sci. 2007, 6, 50. [DOI] [PubMed] [Google Scholar]
- Literak J.; Hroudna L.; Klan P. J. Photochem. Photobiol., A 2008, 194, 59. [Google Scholar]
- Pelliccioli A. P.; Klan P.; Zabadal M.; Wirz J. J. Am. Chem. Soc. 2001, 123, 7931. [DOI] [PubMed] [Google Scholar]
- Papageorgiou G.; Barth A.; Corrie J. E. T. Photochem. Photobiol. Sci. 2005, 4, 216. [DOI] [PubMed] [Google Scholar]
- Du L. H.; Zhang S. H.; Wang Y. G. Tetrahedron Lett. 2005, 46, 3399. [Google Scholar]
- Park B. S.; Lee H. M. Bull. Korean Chem. Soc. 2008, 29, 2054. [Google Scholar]
- Wessig P.; Glombitza C.; Muller G.; Teubner J. J. Org. Chem. 2004, 69, 7582. [DOI] [PubMed] [Google Scholar]
- Wessig P.; Teubner J. Synlett 2006, 10, 1543. [Google Scholar]
- Pospisil T.; Veetil A. T.; Lovely Angel P. A.; Klan P. Photochem. Photobiol. Sci. 2008, 7, 625. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Jeong S. Bull. Korean Chem. Soc. 2009, 30, 3053. [Google Scholar]
- Lehmann T. E.; Muller G.; Berkessel A. J. Org. Chem. 2000, 65, 2508. [DOI] [PubMed] [Google Scholar]
- Plistil L.; Solomek T.; Wirz J.; Heger D.; Klan P. J. Org. Chem. 2006, 71, 8050. [DOI] [PubMed] [Google Scholar]
- Solomek T.; Stacko P.; Veetil A. T.; Pospisil T.; Klan P. J. Org. Chem. 2010, 75, 7300. [DOI] [PubMed] [Google Scholar]
- Stacko P.; Solomek T.; Klan P. Org. Lett. 2011, 13, 6556. [DOI] [PubMed] [Google Scholar]
- a Sebej P.; Lim B. H.; Park B. S.; Givens R. S.; Klan P. Org. Lett. 2011, 13, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ngoy B. P.; Sebej P.; Solomek T.; Lim B. H.; Pastierik T.; Park B. S.; Givens R. S.; Heger D.; Klan P. Photochem. Photobiol. Sci. 2012, 11, 1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng S.; Ullman E. F. J. Am. Chem. Soc. 1976, 98, 541. [Google Scholar]
- Pirrung M. C.; Roy B. G.; Gadamsetty S. Tetrahedron 2010, 66, 3147. [Google Scholar]
- Atemnkeng W. N.; Louisiana L. D.; Yong P. K.; Vottero B.; Banerjee A. Org. Lett. 2003, 5, 4469. [DOI] [PubMed] [Google Scholar]
- Sobczak M.; Wagner P. J. Org. Lett. 2002, 4, 379. [DOI] [PubMed] [Google Scholar]
- Kamdzhilov Y.; Wirz J. Photochem. Photobiol. Sci. 2007, 6, 865. [DOI] [PubMed] [Google Scholar]
- Chiang Y.; Kresge A. J.; Hellrung B.; Schunemann P.; Wirz J. Helv. Chim. Acta 1997, 80, 1106. [Google Scholar]
- a Konosonoks A.; Wright P. J.; Tsao M. L.; Pika J.; Novak K.; Mandel S. M.; Bauer J. A. K.; Bohne C.; Gudmundsdottir A. D. J. Org. Chem. 2005, 70, 2763. [DOI] [PubMed] [Google Scholar]; b Muthukrishnan S.; Pace T. C. S.; Li Q. A.; Seok B.; de Jong G.; Bohne C.; Gudmundsdottir A. D. Can. J. Chem. 2011, 89, 331. [Google Scholar]
- Muthukrishnan S.; Sankaranarayanan J.; Pace T. C. S.; Konosonoks A.; DeMichiei M. E.; Meese M. J.; Bohne C.; Gudmundsdottir A. J. Org. Chem. 2010, 75, 1393. [DOI] [PubMed] [Google Scholar]
- a Givens R. S.; Park C. H. Tetrahedron Lett. 1996, 37, 6259. [Google Scholar]; b Park C. H.; Givens R. S. J. Am. Chem. Soc. 1997, 119, 2453. [Google Scholar]
- a Givens R. S.; Yousef A. L.. p-Hydroxyphenacyl: A photoremovable protecting group for caging bioactive substrates. In Dynamic Studies in Biology; Goeldner M., Givens R. S., Eds.; Wiley-VCH: Weinheim, Germany, 2006; p 55. [Google Scholar]; b Givens R. S.; Lee J.-I. J. Photosci 2003, 10, 37. [Google Scholar]; c Givens R. S.; Weber J. F. W.; Jung A. H.; Park C.-H.. New photoprotecting groups: Desyl and p-hydroxyphenacyl phosphate and carboxylate esters. In Methods Enzymol.; Marriott G., Ed.; Academic Press: New York, 1998; Vol. 291, p 1. [DOI] [PubMed] [Google Scholar]
- Greene T. W.; Wutts P. G. M.. Protective Groups in Organic Synthesis; Wiley-Interscience: New York, 2006. [Google Scholar]
- a Ma C. S.; Zuo P.; Kwok W. M.; Chan W. S.; Kan J. T. W.; Toy P. H.; Phillips D. L. J. Org. Chem. 2004, 69, 6641. [DOI] [PubMed] [Google Scholar]; b Ma C. S.; Kwok W. M.; Chan W. S.; Du Y.; Kan J. T. W.; Toy P. H.; Phillips D. L. J. Am. Chem. Soc. 2006, 128, 2558. [DOI] [PubMed] [Google Scholar]; c Chen X. B.; Ma C. S.; Kwok W. M.; Guan X. G.; Du Y.; Phillips D. L. J. Phys. Chem. A 2006, 110, 12406. [DOI] [PubMed] [Google Scholar]; d Givens R. S.; Heger D.; Hellrung B.; Kamdzhilov Y.; Mac M.; Conrad P. G.; Cope E.; Lee J. I.; Mata-Segreda J. F.; Schowen R. L.; Wirz J. J. Am. Chem. Soc. 2008, 130, 3307. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Stensrud K. F.; Heger D.; Sebej P.; Wirz J.; Givens R. S. Photochem. Photobiol. Sci. 2008, 7, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favorskii A. E. J. Russ. Phys. Chem. Soc 1894, 26, 590. [Google Scholar]
- Anderson J. C.; Reese C. B. Tetrahedron Lett. 1962, 1, 1. [Google Scholar]
- The spectra were measured by Sanjeewa Senadheera, University of Kansas. [Google Scholar]
- Klicova L.; Sebej P.; Solomek T.; Hellrung B.; Slavicek P.; Klan P.; Heger D.; Wirz J. J. Phys. Chem. A 2012, 116, 2935. [DOI] [PubMed] [Google Scholar]
- Geibel S.; Barth A.; Amslinger S.; Jung A. H.; Burzik C.; Clarke R. J.; Givens R. S.; Fendler K. Biophys. J. 2000, 79, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Du X. L.; Frei H.; Kim S. H. J. Biol. Chem. 2000, 275, 8492. [DOI] [PubMed] [Google Scholar]; b Du X. L.; Frei H.; Kim S. H. Biopolymers 2001, 62, 147. [DOI] [PubMed] [Google Scholar]
- a Kötting C.; Gerwert K. Chem. Phys. 2004, 307, 227. [Google Scholar]; b Kötting C.; Kallenbach A.; Suveyzclis Y.; Wittinghofer A.; Gerwert K. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remes M.; Roithova J.; Schroder D.; Cope E. D.; Perera C.; Senadheera S. N.; Stensrud K.; Ma C. C.; Givens R. S. J. Org. Chem. 2011, 76, 2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens R. S.; Stensrud K.; Conrad P. G.; Yousef A. L.; Perera C.; Senadheera S. N.; Heger D.; Wirz J. Can. J. Chem. 2011, 89, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Corrie J. E. T.; Munasinghe V. R. N.; Wan P. J. Am. Chem. Soc. 1999, 121, 5625. [Google Scholar]
- a Ma C. S.; Kwok W. M.; Chan W. S.; Zuo P.; Kan J. T. W.; Toy P. H.; Phillips D. L. J. Am. Chem. Soc. 2005, 127, 1463. [DOI] [PubMed] [Google Scholar]; b Zuo P.; Ma C. S.; Kwok W. M.; Chan W. S.; Phillips D. L. J. Org. Chem. 2005, 70, 8661. [DOI] [PubMed] [Google Scholar]; c Ma C. S.; Kwok W. M.; Chan W. S.; Du Y.; Zuo P.; Kan J. T. W.; Toy P. H.; Phillips D. L. Curr. Sci. 2009, 97, 202. [Google Scholar]; d Ma C. S.; Chan W. S.; Kwok W. M.; Zuo P.; Phillips D. L. J. Chem. Phys. B 2004, 108, 9264. [Google Scholar]
- a Conrad P. G.; Givens R. S.; Hellrung B.; Rajesh C. S.; Ramseier M.; Wirz J. J. Am. Chem. Soc. 2000, 122, 9346. [Google Scholar]; b Stensrud K.; Noh J.; Kandler K.; Wirz J.; Heger D.; Givens R. S. J. Org. Chem. 2009, 74, 5219. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Conrad P. G.; Givens R. S.; Weber J. F. W.; Kandler K. Org. Lett. 2000, 2, 1545. [DOI] [PubMed] [Google Scholar]
- Balachandran Kammath V.; Šolomek T.; Ngoy B. P.; Heger D.; Klan P.; Rubina M.; Givens R. S.. J. Org. Chem. 2013, DOI: 10.1021/jo300850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. B.; Ma C. S.; Kwok W. M.; Guan X. G.; Du Y.; Phillips D. L. J. Chem. Phys. B 2007, 111, 11832. [DOI] [PubMed] [Google Scholar]
- Cao Q.; Guan X. G.; George M. W.; Phillips D. L.; Ma C. S.; Kwok W. M.; Li M. D.; Du Y.; Sun X. Z.; Xue J. D. Faraday Discuss. 2010, 145, 171. [Google Scholar]
- Pincock J. A. Acc. Chem. Res. 1997, 30, 43. [Google Scholar]
- a Aston J. G.; Newkirk J. D. J. Am. Chem. Soc. 1951, 73, 3900. [Google Scholar]; b Bordwell F. G.; Scamehorn R. G.; Springer W. R. J. Am. Chem. Soc. 1969, 91, 2087. [Google Scholar]; c Burr J. G.; Dewar M. J. S. J. Chem. Soc. 1954, 1201. [Google Scholar]; d Loftfield R. B. J. Am. Chem. Soc. 1950, 72, 632. [Google Scholar]; e Loftfield R. B. J. Am. Chem. Soc. 1951, 73, 4707. [Google Scholar]
- Chiang Y.; Kresge A. J.; Zhu Y. J. Am. Chem. Soc. 2002, 124, 6349. [DOI] [PubMed] [Google Scholar]
- a The mechanism of the photo-Favorskii rearrangement has challenged researchers for over a decade. See, for example, Anslyn E. V.; Dougherty D. A.. Question 14 in Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2004; p 994. [Google Scholar]; b A recent study provides evidence for the oxyallyl-phenoxy intermediate:Givens R. S.; Rubina M.; Stensrud K. L.. J. Org. Chem., 2013, 10.1021/jo301640q [DOI] [PMC free article] [PubMed]
- Lukeman M.; Veale D.; Wan P.; Munasinghe V. R. N.; Corrie J. E. T. Can. J. Chem. 2004, 82, 240. [Google Scholar]
- a Baldwin J. E.; McConnaughie A. W.; Moloney M. G.; Pratt A. J.; Shim S. B. Tetrahedron 1990, 46, 6879. [Google Scholar]; b Epstein W. W.; Garrossian M. J. Chem. Soc., Chem. Commun. 1987, 8, 532. [Google Scholar]; c Stanton-Humphreys M. N.; Taylor R. D. T.; McDougall C.; Hart M. L.; Brown C. T. A.; Emptage N. J.; Conway S. J. Chem. Commun. 2012, 48, 657. [DOI] [PubMed] [Google Scholar]
- a Givens R. S.; Athey P. S.; Kueper L. W.; Matuszewski B.; Xue J. Y. J. Am. Chem. Soc. 1992, 114, 8708. [Google Scholar]; b Givens R. S.; Kueper L. W. Chem. Rev. 1993, 93, 55. [Google Scholar]; c Givens R. S.; Athey P. S.; Matuszewski B.; Kueper L. W. III; Xue J. Y.; Fister T. J. Am. Chem. Soc. 1993, 115, 6001. [Google Scholar]
- An H. Y.; Kwok W. M.; Ma C. S.; Guan X. G.; Kan J. T. W.; Toy P. H.; Phillips D. L. J. Org. Chem. 2010, 75, 5837. [DOI] [PubMed] [Google Scholar]
- Fischer N. O.; Paulini R.; Drechsler U.; Rotello V. M. Chem. Commun. 2004, 24, 2866. [DOI] [PubMed] [Google Scholar]
- a Shaginian A.; Patel M.; Li M. H.; Flickinger S. T.; Kim C. H.; Cerrina F.; Belshaw P. J. J. Am. Chem. Soc. 2004, 126, 16704. [DOI] [PubMed] [Google Scholar]; b Flickinger S. T.; Patel M.; Binkowski B. F.; Lowe A. M.; Li M. H.; Kim C.; Cerrina F.; Belshaw P. J. Org. Lett. 2006, 8, 2357. [DOI] [PubMed] [Google Scholar]; c Ueda S.; Fujita M.; Tamamura H.; Fujii N.; Otaka A. ChemBioChem 2005, 6, 1983. [DOI] [PubMed] [Google Scholar]
- a Zou K. Y.; Cheley S.; Givens R. S.; Bayley H. J. Am. Chem. Soc. 2002, 124, 8220. [DOI] [PubMed] [Google Scholar]; b Zou K. Y.; Miller W. T.; Givens R. S.; Bayley H. Angew. Chem., Int. Ed. 2001, 40, 3049. [DOI] [PubMed] [Google Scholar]
- a Givens R. S.; Weber J. F. W.; Conrad P. G.; Orosz G.; Donahue S. L.; Thayer S. A. J. Am. Chem. Soc. 2000, 122, 2687. [Google Scholar]; b Sul J. Y.; Orosz G.; Givens R. S.; Haydon P. G. Neuron Glia Biol. 2004, 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabaci G.; Guo X. C.; Beebe K. D.; Coggeshall K. M.; Pei D. J. Am. Chem. Soc. 1999, 121, 5085. [Google Scholar]
- Cope E. D. Ph.D. thesis, University of Kansas, http://kuscholarworks.ku.edu/dspace/bitstream/1808/4256/1/umi-ku-2581_1.pdf, 2008. [Google Scholar]
- Specht A.; Loudwig S.; Peng L.; Goeldner M. Tetrahedron Lett. 2002, 43, 8947. [Google Scholar]
- Kötting C.; Kallenbach A.; Suveyzdis Y.; Eichholz C.; Gerwert K. ChemBioChem 2007, 8, 781. [DOI] [PubMed] [Google Scholar]
- a Kim G.; Kandler K. Nat. Neurosci. 2003, 6, 282. [DOI] [PubMed] [Google Scholar]; b Noh J.; Seal R. P.; Garver J. A.; Edwards R. H.; Kandler K. Nat. Neurosci. 2010, 13, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kandler K.; Givens R. S.; Katz L. C.. Photostimulation with caged glutamate. In Imaging Neurons; Yuste R., Lanni F., Konnerth A., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 2000; p 27/1 . [Google Scholar]; d Kim G.; Kandler K. J. Neurosci. Methods 2011, 200, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan J. C.; Wilson R. M.; Oxford A. W. J. Am. Chem. Soc. 1971, 93, 7222. [Google Scholar]
- Corrie J. E. T.; Trentham D. R. J. Chem. Soc., Perkin Trans. 1 1992, 2409. [Google Scholar]
- Shi Y.; Corrie J. E. T.; Wan P. J. Org. Chem. 1997, 62, 8278. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Ye T.; Zhou Z.; Simon J. D. Photochem. Photobiol. 2006, 82, 1258. [DOI] [PubMed] [Google Scholar]
- Boudebous H.; Kosmrlj B.; Sket B.; Wirz J. J. Phys. Chem. A 2007, 111, 2811. [DOI] [PubMed] [Google Scholar]
- a Chan W. S.; Ma C. S.; Kwok W. M.; Zuo P.; Phillips D. L. J. Phys. Chem. A 2004, 108, 4047. [Google Scholar]; b Ma C.; Kwok W. M.; An H.-Y.; Guan X.; Fu M. Y.; Toy P. H.; Phillips D. L. Chem.—Eur. J. 2010, 16, 5102. [DOI] [PubMed] [Google Scholar]
- a Chen R. P. Y.; Huang J. J. T.; Chen H.-L.; Jan H.; Velusamy M.; Lee C.-T.; Fann W.; Larsen R. W.; Chan S. I. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7305. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rock R. S.; Chan S. I. J. Org. Chem. 1996, 61, 1526. [Google Scholar]; c Rock R. S.; Hansen K. C.; Larsen R. W.; Chan S. I. Chem. Phys. 2004, 307, 201. [Google Scholar]
- Rock R. S.; Chan S. I. J. Am. Chem. Soc. 1998, 120, 10766. [Google Scholar]
- Boudebous H., Ph.D. thesis, University of Basel, Basel, Switzerland, 2006. [Google Scholar]
- Chen X.; Ma C.; Phillips D. L.; Fang W.-H. Org. Lett. 2010, 12, 5108. [DOI] [PubMed] [Google Scholar]
- Givens R. S.; Matuszewski B. J. Am. Chem. Soc. 1984, 106, 6860. [Google Scholar]
- Rajesh C. S.; Givens R. S.; Wirz J. J. Am. Chem. Soc. 2000, 122, 611. [Google Scholar]
- Ma C.; Du Y.; Kwok W. M.; Phillips D. L. Chem.—Eur. J. 2007, 13, 2290. [DOI] [PubMed] [Google Scholar]
- Chan W. S.; Ma C. S.; Kwok W. M.; Phillips D. L. J. Phys. Chem. A 2005, 109, 3454. [DOI] [PubMed] [Google Scholar]
- Sheehan J. C.; Daves G. D. Jr. J. Org. Chem. 1964, 29, 2006. [Google Scholar]
- Pirrung M. C.; Fallon L.; Lever D. C.; Shuey S. W. J. Org. Chem. 1996, 61, 2129. [Google Scholar]
- McCoy C. P.; Rooney C.; Edwards C. R.; Jones D. S.; Gorman S. P. J. Am. Chem. Soc. 2007, 129, 9572. [DOI] [PubMed] [Google Scholar]
- a del Campo A.; Boos D.; Spiess H. W.; Jonas U. Angew. Chem., Int. Ed. 2005, 44, 4707. [DOI] [PubMed] [Google Scholar]; b Pirrung M. C.; Bradley J.-C. J. Org. Chem. 1995, 60, 6270. [Google Scholar]; c Pirrung M. C.; Fallon L.; McGall G. J. Org. Chem. 1998, 63, 241. [Google Scholar]
- Pirrung M. C.; Bradley J.-C. J. Org. Chem. 1995, 60, 1116. [Google Scholar]
- Wu T.; Tang H.; Bohne C.; Branda N. R. Angew. Chem., Int. Ed. 2012, 51, 2741. [DOI] [PubMed] [Google Scholar]
- Givens R. S.; Athey P. S.; Kueper L. W. III; Matuszewski B.; Xue J. Y. J. Am. Chem. Soc. 1992, 114, 8708. [Google Scholar]
- Gee K. R.; Kueper L. W. III; Barnes J.; Dudley G.; Givens R. S. J. Org. Chem. 1996, 61, 1228. [Google Scholar]
- a Papageorgiou G.; Corrie J. E. T. Tetrahedron 1997, 53, 3917. [Google Scholar]; b Pirrung M. C.; Huang C.-Y. Tetrahedron Lett. 1995, 36, 5883. [Google Scholar]; c Cameron J. F.; Willson C. G.; Fréchet J. M. J. J. Chem. Soc., Chem. Commun. 1995, 9, 923. [Google Scholar]
- Cameron J. F.; Willson C. G.; Fréchet J. M. J. J. Chem. Soc., Perkin Trans. 1 1997, 16, 2429. [Google Scholar]
- Cameron J. F.; Willson C. G.; Fréchet J. M. J. J. Am. Chem. Soc. 1996, 118, 12925. [Google Scholar]
- Pirrung M. C.; Shuey S. W. J. Org. Chem. 1994, 59, 3890. [Google Scholar]
- Stowell M. H. B.; Rock R. S.; Rees D. C.; Chan S. I. Tetrahedron Lett. 1996, 37, 307. [Google Scholar]
- Stowell M. H. B.; Wang G.; Day M. W.; Chan S. I. J. Am. Chem. Soc. 1998, 120, 1657. [Google Scholar]
- a Cano M.; Ladlow M.; Balasubramanian S. J. Org. Chem. 2002, 67, 129. [DOI] [PubMed] [Google Scholar]; b Lee H. B.; Balasubramanian S. J. Org. Chem. 1999, 64, 3454. [DOI] [PubMed] [Google Scholar]; c Routledge A.; Abell C.; Balasubramanian S. Tetrahedron Lett. 1997, 38, 1227. [Google Scholar]
- Chumachenko N.; Novikov Y.; Shoemaker R. K.; Copley S. D. J. Org. Chem. 2011, 76, 9409. [DOI] [PubMed] [Google Scholar]
- Peach J. M.; Pratt A. J.; Snaith J. S. Tetrahedron 1995, 51, 10013. [Google Scholar]
- Ashraf M. A.; Russell A. G.; Wharton C. W.; Snaith J. S. Tetrahedron 2007, 63, 586. [Google Scholar]
- Balachandran Kammath V.; Sebej P.; Slanina T.; Kriz Z.; Klan P. Photochem. Photobiol. Sci. 2012, 11, 500. [DOI] [PubMed] [Google Scholar]
- Barltrop J. A.; Plant P. J.; Schofield P. J. Chem. Soc., Chem. Commun. 1966, 22, 822. [Google Scholar]
- Ciamician G.; Silber P. Chem. Ber. 1901, 34, 2040. [Google Scholar]
- a Yip R. W.; Wen Y. X.; Gravel D.; Giasson R.; Sharma D. K. J. Phys. Chem. 1991, 95, 6078. [Google Scholar]; b Wettermark G. J. Phys. Chem. 1962, 66, 2560. [Google Scholar]; c Yip R. W.; Sharma D. K.; Giasson R.; Gravel D. J. Phys. Chem. 1985, 89, 5328. [Google Scholar]; d Gravel D.; Giasson R.; Blanchet D.; Yip R. W.; Sharma D. K. Can. J. Chem. 1991, 69, 1193. [Google Scholar]; e Takezaki M.; Hirota N.; Terazima M. J. Phys. Chem. A 1997, 101, 3443. [Google Scholar]; f Schwörer M.; Wirz J. Helv. Chim. Acta 2001, 84, 1441. [Google Scholar]
- a Schmierer T.; Laimgruber S.; Haiser K.; Kiewisch K.; Neugebauer J.; Gilch P. Phys. Chem. Chem. Phys. 2010, 12, 15653. [DOI] [PubMed] [Google Scholar]; b Heinz B.; Schmierer T.; Laimgruber S.; Gilch P. J. Photochem. Photobiol., A 2008, 199, 274. [Google Scholar]; c Laimgruber S.; Schachenmayr H.; Schmidt B.; Zinth W.; Gilch P. Appl. Phys. B: Lasers Opt. 2006, 85, 557. [Google Scholar]; d Laimgruber S.; Schmierer T.; Gilch P.; Kiewisch K.; Neugebauer J. Phys. Chem. Chem. Phys. 2008, 10, 3872. [DOI] [PubMed] [Google Scholar]; e Laimgruber S.; Schreier W. J.; Schrader T.; Koller F.; Zinth W.; Gilch P. Angew. Chem., Int. Ed. 2005, 44, 7901. [DOI] [PubMed] [Google Scholar]; f Leyva V.; Corral I.; Schmierer T.; Gilch P.; Gonzalez L. Phys. Chem. Chem. Phys. 2011, 13, 4269. [DOI] [PubMed] [Google Scholar]; g Leyva V.; Corral I.; Schmierer T.; Heinz B.; Feixas F.; Migani A.; Blancafort L.; Gilch P.; Gonzalez L. J. Phys. Chem. A 2008, 112, 5046. [DOI] [PubMed] [Google Scholar]; h Migani A.; Leyva V.; Feixas F.; Schmierer T.; Gilch P.; Corral I.; Gonzalez L.; Blancafort L. Chem. Commun. 2011, 47, 6383. [DOI] [PubMed] [Google Scholar]; i Schmierer T.; Schreier W. J.; Koller F. O.; Schrader T. E.; Gilch P. Phys. Chem. Chem. Phys. 2009, 11, 11596. [DOI] [PubMed] [Google Scholar]; j Schmierer T.; Ryseck G.; Villnow T.; Regner N.; Gilch P. Photochem. Photobiol. Sci. 2012, 11, 1313. [DOI] [PubMed] [Google Scholar]
- Margerum J. D.; Petrusis C. T. J. Am. Chem. Soc. 1969, 91, 2467. [Google Scholar]
- Walker J. W.; Reid G. P.; McCray J. A.; Trentham D. R. J. Am. Chem. Soc. 1988, 110, 7170. [Google Scholar]
- a Barth A.; Hauser K.; Maentele W.; Corrie J. E. T.; Trentham D. R. J. Am. Chem. Soc. 1995, 117, 10311. [Google Scholar]; b Barth A.; Corrie J. E. T.; Gradwell M. J.; Maeda Y.; Maentele W.; Meier T.; Trentham D. R. J. Am. Chem. Soc. 1997, 119, 4149. [Google Scholar]; c Cepus V.; Ulbrich C.; Allin C.; Troullier A.; Gerwert K. Methods Enzymol. 1998, 291, 223. [DOI] [PubMed] [Google Scholar]
- Il’ichev Y. V.; Schwoerer M. A.; Wirz J. J. Am. Chem. Soc. 2004, 126, 4581. [DOI] [PubMed] [Google Scholar]
- Corrie J. E. T.; Barth A.; Munasinghe V. R. N.; Trentham D. R.; Hutter M. C. J. Am. Chem. Soc. 2003, 125, 8546. [DOI] [PubMed] [Google Scholar]
- Il’ichev Y. V.; Wirz J. J. Phys. Chem. A 2000, 104, 7856. [Google Scholar]
- a Dunkin I. R.; Gebicki J.; Kiszka M.; Sanin-Leira D. Spectrochim. Acta, Part A 1997, 53, 2553. [Google Scholar]; b Dunkin I. R.; Gebicki J.; Kiszka M.; Sanin-Leira D. J. Chem. Soc., Perkin Trans. 2 2001, 8, 1414. [Google Scholar]
- Hellrung B.; Kamdzhilov Y.; Schwoerer M.; Wirz J. J. Am. Chem. Soc. 2005, 127, 8934. [DOI] [PubMed] [Google Scholar]
- Ellis-Davies G. C. R.; Barsotti R. J. Cell Calcium 2006, 39, 75. [DOI] [PubMed] [Google Scholar]
- Momotake A.; Lindegger N.; Niggli E.; Barsotti R. J.; Ellis-Davies G. C. R. Nat. Methods 2006, 3, 35. [DOI] [PubMed] [Google Scholar]
- Kantevari S.; Buskila Y.; Ellis-Davies G. C. R. Photochem. Photobiol. Sci. 2012, 11, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöll D.; Laimgruber S.; Galetskaya M.; Smirnova J.; Pfleiderer W.; Heinz B.; Gilch P.; Steiner U. E. J. Am. Chem. Soc. 2007, 129, 12148. [DOI] [PubMed] [Google Scholar]
- Wöll D.; Lukzen N.; Steiner U. E. Photochem. Photobiol. Sci. 2012, 11, 533. [DOI] [PubMed] [Google Scholar]
- a Görner H. Photochem. Photobiol. Sci. 2005, 4, 822. [DOI] [PubMed] [Google Scholar]; b Bley F.; Schaper K.; Goerner H. Photochem. Photobiol. 2008, 84, 162. [DOI] [PubMed] [Google Scholar]
- a Breitinger H.-G. A.; Wieboldt R.; Ramesh D.; Carpenter B. K.; Hess G. P. Biochemistry 2000, 39, 5500. [DOI] [PubMed] [Google Scholar]; b Milburn T.; Matsubara N.; Billington A. P.; Udgaonkar J. B.; Walker J. W.; Carpenter B. K.; Webb W. W.; Marque J.; Denk W.; McCray J. A.; Hess G. P. Biochemistry 1989, 28, 49. [DOI] [PubMed] [Google Scholar]; c Wieboldt R.; Ramesh D.; Jabri E.; Karplus P. A.; Carpenter B. K.; Hess G. P. J. Org. Chem. 2002, 67, 8827. [DOI] [PubMed] [Google Scholar]
- Barth A.; Martin S. R.; Corrie J. E. T. Photochem. Photobiol. Sci. 2006, 5, 107. [DOI] [PubMed] [Google Scholar]
- Solomek T.; Mercier S.; Bally T.; Bochet C. G. Photochem. Photobiol. Sci. 2012, 11, 548. [DOI] [PubMed] [Google Scholar]
- Gaplovsky M.; Il’ichev Y. V.; Kamdzhilov Y.; Kombarova S. V.; Mac M.; Schwoerer M. A.; Wirz J. Photochem. Photobiol. Sci. 2005, 4, 33. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Y.Kao J. P.; Grynkiewicz G.; Minta A.; Tsien R. Y. J. Am. Chem. Soc. 1998, 110, 3212. [Google Scholar]
- Bayley H.; Chang C.-Y.; Miller W. T.; Niblack B.; Pan P. Methods Enzymol. 1998, 291, 117. [DOI] [PubMed] [Google Scholar]
- Kalbag S. M.; Roeske R. W. J. Am. Chem. Soc. 1975, 97, 440. [DOI] [PubMed] [Google Scholar]
- a Deiters A. Curr. Opin. Chem. Biol. 2009, 13, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ellis-Davies G. C. R. Chem. Rev. 2008, 108, 1603. [DOI] [PubMed] [Google Scholar]; c Loudwig S.; Specht A.; Goeldner M. Actual. Chim. 2002, 1, 7. [Google Scholar]; d Dorman G.; Prestwich G. D. Trends Biotechnol. 2000, 18, 64. [DOI] [PubMed] [Google Scholar]; e Curley K.; Lawrence D. S. Pharmacol. Ther. 1999, 82, 347. [DOI] [PubMed] [Google Scholar]
- Stutz A.; Pitsch S. Synlett 1999, SI, 930. [Google Scholar]
- Chalmers S.; Caldwell S. T.; Quin C.; Prime T. A.; James A. M.; Cairns A. G.; Murphy M. P.; McCarron J. G.; Hartley R. C. J. Am. Chem. Soc. 2012, 134, 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Zeng Y.; Xun Z.; Han Y.; Chen J.; Li Y.-Y.; Li Y. Langmuir 2012, 28, 1733. [DOI] [PubMed] [Google Scholar]
- Deshpande R. K.; Waterhouse G. I. N.; Jameson G. B.; Telfer S. G. Chem. Commun. 2012, 48, 1574. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Hummel C. W.; Nakada M.; Shibayama K.; Pitsinos E. N.; Saimoto H.; Mizuno Y.; Baldenius K. U.; Smith A. L. J. Am. Chem. Soc. 1993, 115, 7625. [Google Scholar]
- Gareau Y.; Zamboni R.; Wong A. W. J. Org. Chem. 1993, 58, 1582. [Google Scholar]
- Kevwitch R. M.; McGrath D. V. New J. Chem. 2007, 31, 1332. [Google Scholar]
- Ajayaghosh A.; Pillai V. N. R. Tetrahedron Lett. 1995, 36, 777. [Google Scholar]
- a Reichmanis E.; Smith B. C.; Gooden R. J. Polym. Sci. 1985, 23, 1. [Google Scholar]; b Reichmanis E.; Gooden R.; Wilkins C. W. Jr.; Schonhorn H. J. Polym. Sci. 1983, 21, 1075. [Google Scholar]
- Cameron J. F.; Frechet J. M. J. J. Am. Chem. Soc. 1991, 113, 4303. [Google Scholar]
- Allan A. C.; Ward J. L.; Beale M. H.; Trewavas A. J. Methods Enzymol. 1998, 291, 474. [Google Scholar]
- Ajayaghosh A.; Pillai V. N. R. Tetrahedron 1988, 44, 6661. [Google Scholar]
- Specht A.; Goeldner M. Angew. Chem., Int. Ed. 2004, 43, 2008. [DOI] [PubMed] [Google Scholar]
- Aujard I.; Benbrahim C.; Gouget M.; Ruel O.; Baudin J. B.; Neveu P.; Jullien L. Chem.—Eur. J. 2006, 12, 6865. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Pieper W. H.; Kaliappan K. P.; Dhananjeyan M. R. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A. G.; Sadler M. J.; Laidlaw H. J.; Gutiérrez-Loriente A.; Wharton C. W.; Carteau D.; Bassani D. M.; Snaith J. S. Photochem. Photobiol. Sci. 2012, 11, 556. [DOI] [PubMed] [Google Scholar]
- Russell A. G.; Ragoussi M.-E.; Ramalho R.; Wharton C. W.; Carteau D.; Bassani D. M.; Snaith J. S. J. Org. Chem. 2010, 75, 4648. [DOI] [PubMed] [Google Scholar]
- Gilbert D.; Funk K.; Dekowski B.; Lechler R.; Keller S.; Moehrlen F.; Frings S.; Hagen V. ChemBioChem 2007, 8, 89. [DOI] [PubMed] [Google Scholar]
- Schaper K.; Mobarekeh S. A. M.; Grewer C. Eur. J. Org. Chem. 2002, 6, 1037. [Google Scholar]
- Kotzur N.; Briand B.; Beyermann M.; Hagen V. J. Am. Chem. Soc. 2009, 131, 16927. [DOI] [PubMed] [Google Scholar]
- Corrie J. E. T.; Munasinghe V. R. N.; Trentham D. R.; Barth A. Photochem. Photobiol. Sci. 2008, 7, 84. [DOI] [PubMed] [Google Scholar]
- Schaper K.; Etinski M.; Fleig T. Photochem. Photobiol. 2009, 85, 1075. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Lee Y. R.; Park K.; Springer J. B. J. Org. Chem. 1999, 64, 5042. [DOI] [PubMed] [Google Scholar]
- Bochet C.; Mercier S.. Photolabile linker units. In Linker Strategies in Solid-Phase Organic Synthesis; Scott. P., Ed.; J. Wiley and Sons: Chichester, U.K., 2009; p 151. [Google Scholar]
- Mitchison T. J.; Sawin K. E.; Theriot J. A.; Gee T. K.; Mallavarapu A. Methods Enzymol. 1998, 291, 63. [DOI] [PubMed] [Google Scholar]
- Alvarez M.; Alonso J. M.; Filevich O.; Bhagawati M.; Etchenique R.; Piehler J.; del Campo A. Langmuir 2011, 27, 2789. [DOI] [PubMed] [Google Scholar]
- a Holmes C. P. J. Org. Chem. 1997, 62, 2370. [DOI] [PubMed] [Google Scholar]; b Pease A. C.; Solas D.; Sullivan E. J.; Cronin M. T.; Holmes C. P.; Fodor S. P. A. Proc. Natl. Acad. Sci. U.S.A 1994, 91, 5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berroy P.; Viriot M. L.; Carre M. C. Sens. Actuators, B 2001, B74, 186. [Google Scholar]
- a Lusic H.; Deiters A. Synthesis 2006, 2147. [Google Scholar]; b Lusic H.; Young D. D.; Lively M. O.; Deiters A. Org. Lett. 2007, 9, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Deiters A. Bioorg. Med. Chem. Lett. 2006, 16, 2658. [DOI] [PubMed] [Google Scholar]
- Wysocki L. M.; Grimm J. B.; Tkachuk A. N.; Brown T. A.; Betzig E.; Lavis L. D. Angew. Chem., Int. Ed. 2011, 50, 11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochet C. G. Tetrahedron Lett. 2000, 41, 6341. [Google Scholar]
- Riguet E.; Bochet C. G. Org. Lett. 2007, 9, 5453. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Khade P. K. Tetrahedron 2005, 61, 10007. [Google Scholar]
- Singh A. K.; Khade P. K. Tetrahedron Lett. 2011, 52, 4899. [Google Scholar]
- a Gravel D.; Hébert J.; Thoraval D. Can. J. Chem. 1983, 61, 400. [Google Scholar]; b Hébert J.; Gravel D. Can. J. Chem. 1974, 52, 187. [Google Scholar]
- Gravel D.; Murray S.; Ladouceur G. J. Chem. Soc., Chem. Commun. 1985, 24, 1828. [Google Scholar]
- Aurell M. J.; Boix C.; Ceita M. L.; Llopis C.; Tortajada A.; Mestres R. J. Chem. Res. (S) 1995, 11, 452. [Google Scholar]
- Blanc A.; Bochet C. G. J. Org. Chem. 2003, 68, 1138. [DOI] [PubMed] [Google Scholar]
- Strieter E. R.; Koglin A.; Aron Z. D.; Walsh C. T. J. Am. Chem. Soc. 2009, 131, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantevari S.; Narasimhaji C. V.; Mereyala H. B. Tetrahedron 2005, 61, 5849. [Google Scholar]
- Lage Robles J.; Bochet C. G. Org. Lett. 2005, 7, 3545. [DOI] [PubMed] [Google Scholar]
- a Sebej P.; Solomek T.; Hroudna L.; Brancova P.; Klan P. J. Org. Chem. 2009, 74, 8647. [DOI] [PubMed] [Google Scholar]; b Young D. D.; Deiters A. Angew. Chem., Int. Ed. 2007, 46, 4290. [DOI] [PubMed] [Google Scholar]; c Watanabe S.; Sueyoshi T.; Ichihara M.; Uehara C.; Iwamura M. Org. Lett. 2001, 3, 255. [DOI] [PubMed] [Google Scholar]; d Collins P. M.; Munasinghe V. R. N. J. Chem. Soc., Perkin Trans. 1 1983, 8, 1879. [Google Scholar]; e Collins P. M.; Munasinghe V. R. N. J. Chem. Soc., Perkin Trans. 1 1983, 5, 921. [Google Scholar]; f Collins P. M.; Eder H. J. Chem. Soc., Perkin Trans. 1 1983, 5, 927. [Google Scholar]
- Zhao J.; Gover T. D.; Muralidharan S.; Auston D. A.; Weinreich D.; Kao J. P. Y. Biochemistry 2006, 45, 4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu P.; Aujard I.; Benbrahim C.; Le Saux T.; Allemand J. F.; Vriz S.; Bensimon D.; Jullien L. Angew. Chem., Int. Ed. 2008, 47, 3744. [DOI] [PubMed] [Google Scholar]
- Yan B.; Boyer J.-C.; Branda N. R.; Zhao Y. J. Am. Chem. Soc. 2011, 133, 19714. [DOI] [PubMed] [Google Scholar]
- Carling C. J.; Nourmohammadian F.; Boyer J. C.; Branda N. R. Angew. Chem., Int. Ed. 2010, 49, 3782. [DOI] [PubMed] [Google Scholar]
- Blanc A.; Bochet C. G. J. Am. Chem. Soc. 2004, 126, 7174. [DOI] [PubMed] [Google Scholar]
- Blanc A.; Bochet C. G. Org. Lett. 2007, 9, 2649. [DOI] [PubMed] [Google Scholar]
- Hasan A.; Stengele K.-P.; Giegrich H.; Cornwell P.; Isham K. R.; Sachleben R. A.; Pfleiderer W.; Foote R. S. Tetrahedron 1997, 53, 4247. [Google Scholar]
- Beier M.; Hoheisel J. D. Nucleic Acids Res. 2000, 28, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan K. R. Org. Biomol. Chem. 2006, 4, 1857. [DOI] [PubMed] [Google Scholar]
- Petersen S.; Alonso J. M.; Specht A.; Duodu P.; Goeldner M.; del Campo A. Angew. Chem., Int. Ed. 2008, 47, 3192. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Wang L. X.; Montague-Smith M. P. Org. Lett. 2001, 3, 1105. [DOI] [PubMed] [Google Scholar]
- Bhushan K. R.; DeLisi C.; Laursen R. A. Tetrahedron Lett. 2003, 44, 8585. [Google Scholar]
- Yi H.; Maisonneuve S.; Xie J. Org. Biomol. Chem. 2009, 7, 3847. [DOI] [PubMed] [Google Scholar]
- Wöll D.; Smirnova J.; Galetskaya M.; Prykota T.; Buhler J.; Stengele K. P.; Pfleiderer W.; Steiner U. E. Chem.—Eur. J. 2008, 14, 6490. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Dore T. M.; Zhu Y.; Rana V. S. Chem. Commun. 2010, 46, 5313. [DOI] [PubMed] [Google Scholar]
- Roethlingshoefer M.; Gorska K.; Winssinger N. J. Am. Chem. Soc. 2011, 133, 18110. [DOI] [PubMed] [Google Scholar]
- Gug S.; Charon S.; Specht A.; Alarcon K.; Ogden D.; Zietz B.; Leonard J.; Haacke S.; Bolze F.; Nicoud J. F.; Goeldner M. ChemBioChem 2008, 9, 1303. [DOI] [PubMed] [Google Scholar]
- Gug S.; Bolze F.; Specht A.; Bourgogne C.; Goeldner M.; Nicoud J. F. Angew. Chem., Int. Ed. 2008, 47, 9525. [DOI] [PubMed] [Google Scholar]
- Specht A.; Bolze F.; Donato L.; Herbivo C.; Charon S.; Warther D.; Gug S.; Nicoud J.-F.; Goeldner M. Photochem. Photobiol. Sci. 2012, 11, 578. [DOI] [PubMed] [Google Scholar]
- Donato L.; Mourot A.; Davenport C. M.; Herbivo C.; Warther D.; Léonard J.; Bolze F.; Nicoud J.-F.; Kramer R. H.; Goeldner M.; Specht A. Angew. Chem., Int. Ed. 2012, 51, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Buehler S.; Lagoja I.; Giegrich H.; Stengele K.-P.; Pfleiderer W. Helv. Chim. Acta 2004, 87, 620. [Google Scholar]; b Smirnova J.; Woll D.; Pfleiderer W.; Steiner U. E. Helv. Chim. Acta 2005, 88, 891. [Google Scholar]; c Bhushan K. R. Synlett 2006, 13, 2130. [Google Scholar]; d Drexler K.; Smirnova J.; Galetskaya M.; Voss S.; Fonin M.; Boneberg J.; Rudiger U.; Leiderer P.; Steiner U. E. Langmuir 2009, 25, 10794. [DOI] [PubMed] [Google Scholar]; e San Miguel V.; Bochet C. G.; del Campo A. J. Am. Chem. Soc. 2011, 133, 5380. [DOI] [PubMed] [Google Scholar]; f Wirkner M.; Weis S.; San Miguel V.; Álvarez M.; Gropeanu R. A.; Salierno M.; Sartoris A.; Unger R. E.; Kirkpatrick C. J.; del Campo A. ChemBioChem 2011, 12, 2623. [DOI] [PubMed] [Google Scholar]; g Wöll D.; Smirnova J.; Pfleiderer W.; Steiner U. E. Angew. Chem., Int. Ed. 2006, 45, 2975. [DOI] [PubMed] [Google Scholar]
- Amit B.; Patchornik A. Tetrahedron Lett. 1973, 14, 2205. [Google Scholar]
- Amit B.; Ben-Efraim D. A.; Patchornik A. J. Chem. Soc., Perkin Trans. 1 1976, 1, 57. [Google Scholar]
- Loudwig S.; Goeldner M. Tetrahedron Lett. 2001, 42, 7957. [Google Scholar]
- Amit B.; Ben-Efraim D. A.; Patchornik A. J. Am. Chem. Soc. 1976, 98, 843. [Google Scholar]
- a Morrison J.; Wan P.; Corrie J. E. T.; Papageorgiou G. Photochem. Photobiol. Sci. 2002, 1, 960. [PubMed] [Google Scholar]; b Cohen A. D.; Helgen C.; Bochet C. G.; Toscano J. P. Org. Lett. 2005, 7, 2845. [DOI] [PubMed] [Google Scholar]; c Corrie J. E. T.; Barth A.; Papageorgiou G. J. Labelled Compd. Radiopharm. 2001, 44, 619. [Google Scholar]
- Papageorgiou G.; Ogden D.; Kelly G.; Corrie J. E. T. Photochem. Photobiol. Sci. 2005, 4, 887. [DOI] [PubMed] [Google Scholar]
- Papageorgiou G.; Ogden D. C.; Barth A.; Corrie J. E. T. J. Am. Chem. Soc. 1999, 121, 6503. [Google Scholar]
- Papageorgiou G.; Corrie J. E. T. Tetrahedron 2000, 56, 8197. [Google Scholar]
- a Papageorgiou G.; Ogden D.; Corrie J. E. T. Photochem. Photobiol. Sci. 2008, 7, 423. [DOI] [PubMed] [Google Scholar]; b Papageorgiou G.; Corrie J. E. T. Tetrahedron 2005, 61, 609. [Google Scholar]; c Papageorgiou G.; Ogden D.; Corrie J. E. T. J. Org. Chem. 2004, 69, 7228. [DOI] [PubMed] [Google Scholar]; d Papageorgiou G.; Lukeman M.; Wan P.; Corrie J. E. T. Photochem. Photobiol. Sci. 2004, 3, 366. [DOI] [PubMed] [Google Scholar]; e Matsuzaki M.; Hayama T.; Kasai H.; Ellis-Davies G. C. R. Nat. Chem. Biol. 2010, 6, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obi N.; Momotake A.; Kanemoto Y.; Matsuzaki M.; Kasai H.; Arai T. Tetrahedron Lett. 2010, 51, 1642. [Google Scholar]
- Honda T.; Momotake A.; Arai T. Photochem. Photobiol. Sci. 2012, 11, 493. [DOI] [PubMed] [Google Scholar]
- Goissis G.; Erickson B. W.; Merrifield R. B. Proc. Am. Pept. Symp. 1977, 5, 559. [Google Scholar]
- Pass S.; Amit B.; Patchornik A. J. Am. Chem. Soc. 1981, 103, 7674. [Google Scholar]
- a Kaneshiro C. M.; Michael K. Angew. Chem., Int. Ed. 2006, 45, 1077. [DOI] [PubMed] [Google Scholar]; b Simo O.; Lee V. P.; Davis A. S.; Kreutz C.; Gross P. H.; Jones P. R.; Michael K. Carbohydr. Res. 2005, 340, 557. [DOI] [PubMed] [Google Scholar]; c Vizvardi K.; Kreutz C.; Davis A. S.; Lee V. P.; Philmus B. J.; Simo O.; Michael K. Chem. Lett. 2003, 32, 348. [Google Scholar]
- a Helgen C.; Bochet C. G. Synlett 2001, 12, 1968. [Google Scholar]; b Nicolaou K. C.; Safina B. S.; Winssinger N. Synlett 2001, SI, 900. [Google Scholar]; c Débieux J.-L.; Bochet C. G. J. Phys. Org. Chem. 2010, 23, 272. [Google Scholar]
- Hogenauer T. J.; Wang Q.; Sanki A. K.; Gammon A. J.; Chu C. H. L.; Kaneshiro C. M.; Kajihara Y.; Michael K. Org. Biomol. Chem. 2007, 5, 759. [DOI] [PubMed] [Google Scholar]
- Débieux J.-L.; Cosandey A.; Helgen C.; Bochet C. G. Eur. J. Org. Chem. 2007, 13, 2073. [Google Scholar]
- Débieux J.-L.; Bochet C. G. J. Org. Chem. 2009, 74, 4519. [DOI] [PubMed] [Google Scholar]
- Débieux J.-L.; Bochet C. Chem. Sci. 2012, 3, 405. [Google Scholar]
- Helgen C.; Bochet C. G. J. Org. Chem. 2003, 68, 2483. [DOI] [PubMed] [Google Scholar]
- Hassner A.; Yagudayev D.; Pradhan T. K.; Nudelman A.; Amit B. Synlett 2007, 15, 2405. [Google Scholar]
- a Canepari M.; Nelson L.; Papageorgiou G.; Corrie J. E. T.; Ogden D. J. Neurosci. Methods 2001, 112, 29. [DOI] [PubMed] [Google Scholar]; b Papageorgiou G.; Corrie J. E. T. Tetrahedron 2007, 63, 9668. [Google Scholar]; c Zhang Z.; Papageorgiou G.; Corrie J. E. T.; Grewer C. Biochemistry 2007, 46, 3872. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Trigo F. F.; Papageorgiou G.; Corrie J. E. T.; Ogden D. J. Neurosci. Methods 2009, 181, 159. [DOI] [PubMed] [Google Scholar]; e Trigo F. F.; Bouhours B.; Rostaing P.; Papageorgiou G.; Corrie J. E. T.; Triller A.; Ogden D.; Marty A. Neuron 2010, 66, 235. [DOI] [PubMed] [Google Scholar]
- a Tanaka J. I.; Horiike Y.; Matsuzaki M.; Miyazaki T.; Ellis-Davies G. C. R.; Kasai H. Science 2008, 319, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kantevari S.; Matsuzaki M.; Kanemoto Y.; Kasai H.; Ellis-Davies G. C. R. Nat. Methods 2010, 7, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noguchi J.; Nagaoka A.; Watanabe S.; Ellis-Davies G. C. R.; Kitamura K.; Kano M.; Matsuzaki M.; Kasai H. J. Physiol. (London) 2011, 589, 2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier W.; Corrie J. E. T.; Papageorgiou G.; Laube B.; Grewer C. J. Neurosci. Methods 2005, 142, 1. [DOI] [PubMed] [Google Scholar]
- Helgen C.; Bochet C. G. Heterocycles 2006, 67, 797. [Google Scholar]
- Givens R. S.; Matuszewski B. J. Am. Chem. Soc. 1984, 106, 6860. [Google Scholar]
- Schultz C. HFSP J. 2007, 1, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T.Coumarin-4-ylmethyl phototriggers. In Dynamic Studies in Biology; Goeldner M., Givens R. S., Eds.; Wiley-VCH: Weinheim, Germany, 2006; p 29. [Google Scholar]
- Dore T. M.Multiphoton phototriggers for exploring cell physiology. In Dynamic Studies in Biology; Goeldner M., Givens R. S., Eds.; Wiley-VCH: Weinheim, Germany, 2006; p 435. [Google Scholar]
- Loudwig S.; Bayley H.. Light-activated proteins: An overview. In Dynamic Studies in Biology; Goeldner M., Givens R. S., Eds.; Wiley-VCH: Weinheim, Germany, 2006; p 253. [Google Scholar]
- a Eckardt T.; Hagen V.; Schade B.; Schmidt R.; Schweitzer C.; Bendig J. J. Org. Chem. 2002, 67, 703. [DOI] [PubMed] [Google Scholar]; b Fonseca A. S. C.; Goncalves M. S. T.; Costa S. P. G. Amino Acids 2010, 39, 699. [DOI] [PubMed] [Google Scholar]
- Fonseca A. S. C.; Goncalves M. S. T.; Costa S. P. G. Tetrahedron 2007, 63, 1353. [Google Scholar]
- Curten B.; Kullmann P. H. M.; Bier M. E.; Kandler K.; Schmidt B. F. Photochem. Photobiol. 2005, 81, 641. [DOI] [PubMed] [Google Scholar]
- Fernandes M. J. G.; Goncalves M. S. T.; Costa S. P. G. Tetrahedron 2008, 64, 3032. [Google Scholar]
- Suzuki A. Z.; Watanabe T.; Kawamoto M.; Nishiyama K.; Yamashita H.; Ishii M.; Iwamura M.; Furuta T. Org. Lett. 2003, 5, 4867. [DOI] [PubMed] [Google Scholar]
- Schmidt R.; Geissler D.; Hagen V.; Bendig J. J. Phys. Chem. A 2007, 111, 5768. [DOI] [PubMed] [Google Scholar]
- Katayama K.; Tsukiji S.; Furuta T.; Nagamune T. Chem. Commun. 2008, 42, 5399. [DOI] [PubMed] [Google Scholar]
- Takaoka K.; Tatsu Y.; Yumoto N.; Nakajima T.; Shimamoto K. Bioorgan. Med. Chem. 2004, 12, 3687. [DOI] [PubMed] [Google Scholar]
- a Geissler D.; Kresse W.; Wiesner B.; Bendig J.; Kettnemann H.; Hagen V. ChemBioChem 2003, 4, 162. [DOI] [PubMed] [Google Scholar]; b Hagen V.; Frings S.; Wiesner B.; Helm S.; Kaupp U. B.; Bendig J. ChemBioChem 2003, 4, 434. [DOI] [PubMed] [Google Scholar]
- Pinheiro A. V.; Baptista P.; Lima J. C. Nucleic Acids Res. 2008, 36, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shembekar V. R.; Chen Y. L.; Carpenter B. K.; Hess G. P. Biochemistry 2005, 44, 7107. [DOI] [PubMed] [Google Scholar]; b Shembekar V. R.; Chen Y. L.; Carpenter B. K.; Hess G. P. Biochemistry 2007, 46, 5479. [DOI] [PubMed] [Google Scholar]
- Schonleber R. O.; Bendig J.; Hagen V.; Giese B. Bioorg. Med. Chem. 2002, 10, 97. [DOI] [PubMed] [Google Scholar]
- Skwarczynski M.; Noguchi M.; Hirota S.; Sohma Y.; Kimura T.; Hayashi Y.; Kiso Y. Bioorg. Med. Chem. Lett. 2006, 16, 4492. [DOI] [PubMed] [Google Scholar]
- a Senda N.; Momotake A.; Arai T. Bull. Chem. Soc. Jpn. 2007, 80, 2384. [Google Scholar]; b Hagen V.; Dekowski B.; Kotzur N.; Lechler R.; Wiesner B.; Briand B.; Beyermann M. Chem.—Eur. J. 2008, 14, 1621. [DOI] [PubMed] [Google Scholar]; c Senda N.; Momotake A.; Nishimura Y.; Arai T. Bull. Chem. Soc. Jpn. 2006, 79, 1753. [Google Scholar]
- Taniguchi A.; Skwarczynski M.; Sohma Y.; Okada T.; Ikeda K.; Prakash H.; Mukai H.; Hayashi Y.; Kimura T.; Hirota S.; Matsuzaki K.; Kiso Y. ChemBioChem 2008, 9, 3055. [DOI] [PubMed] [Google Scholar]
- Hagen V.; Dekowski B.; Nache V.; Schmidt R.; Geissler D.; Lorenz D.; Eichhorst J.; Keller S.; Kaneko H.; Benndorf K.; Wiesner B. Angew. Chem., Int. Ed. 2005, 44, 7887. [DOI] [PubMed] [Google Scholar]
- Hagen V.; Frings S.; Bendig J.; Lorenz D.; Wiesner B.; Kaupp U. B. Angew. Chem., Int. Ed. 2002, 41, 3625. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Watanabe T.; Tanabe S.; Sakyo J.; Matsuba C. Org. Lett. 2007, 9, 4717. [DOI] [PubMed] [Google Scholar]
- Hagen V.; Kilic F.; Schaal J.; Dekowski B.; Schmidt R.; Kotzur N. J. Org. Chem. 2010, 75, 2790. [DOI] [PubMed] [Google Scholar]
- Subramaniam R.; Xioa Y.; Li Y. J.; Qian S. Y.; Sun W. F.; Mallik S. Tetrahedron Lett. 2010, 51, 529. [Google Scholar]
- Piloto A. M.; Rovira D.; Costa S. P. G.; Goncalves M. S. T. Tetrahedron 2006, 62, 11955. [Google Scholar]
- a Fernandes M. J. G.; Goncalves M. S. T.; Costa S. P. G. Tetrahedron 2008, 64, 11175. [Google Scholar]; b Fernandes M. J. G.; Costa S. P. G.; Goncalves M. S. T. Tetrahedron 2011, 67, 2422. [Google Scholar]; c Fernandes M. J.; Goncalves M. S. T.; Costa S. P. G. J. Pept. Sci. 2010, 16, 54. [Google Scholar]
- Soares A. M. S.; Costa S. P. G.; Goncalves M. S. T. Amino Acids 2010, 39, 121. [DOI] [PubMed] [Google Scholar]
- Lu M.; Fedoryak O. D.; Moister B. R.; Dore T. M. Org. Lett. 2003, 5, 2119. [DOI] [PubMed] [Google Scholar]
- Lin W. Y.; Lawrence D. S. J. Org. Chem. 2002, 67, 2723. [DOI] [PubMed] [Google Scholar]
- Schmidt R.; Geissler D.; Hagen V.; Bendig J. J. Phys. Chem. A 2005, 109, 5000. [DOI] [PubMed] [Google Scholar]
- Schade B.; Hagen V.; Schmidt R.; Herbrich R.; Krause E.; Eckardt T.; Bendig J. J. Org. Chem. 1999, 64, 9109. [Google Scholar]
- a Rossi F. M.; Kao J. P. Y. J. Biol. Chem. 1997, 272, 3266. [DOI] [PubMed] [Google Scholar]; b Pocker Y.; Davison B. L.; Deits T. L. J. Am. Chem. Soc. 1978, 100, 3564. [Google Scholar]; c Rossi F. M.; Margulis M.; Tang C. M.; Kao J. P. Y. J. Biol. Chem. 1997, 272, 32933. [DOI] [PubMed] [Google Scholar]
- Wylie R. G.; Shoichet M. S. J. Mater. Chem. 2008, 18, 2716. [Google Scholar]
- Johnson S. L.; Morrison D. L. J. Am. Chem. Soc. 1972, 94, 1323. [DOI] [PubMed] [Google Scholar]
- Noguchi M.; Skwarczynski M.; Prakash H.; Hirota S.; Kimura T.; Hayashi Y.; Kiso Y. Bioorg. Med. Chem. 2008, 16, 5389. [DOI] [PubMed] [Google Scholar]
- a Denk W.; Strickler J. H.; Webb W. W. Science 1990, 248, 73. [DOI] [PubMed] [Google Scholar]; b Denk W. Proc. Natl. Acad. Sci. U.S.A 1994, 91, 6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T.; Wang S. S. H.; Dantzker J. L.; Dore T. M.; Bybee W. J.; Callaway E. M.; Denk W.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin J.; Pelletier M.; Lepage M.; Allard J. F.; Morris D.; Zhao Y. Angew. Chem., Int. Ed. 2009, 48, 3329. [DOI] [PubMed] [Google Scholar]
- Wosnick J. H.; Shoichet M. S. Chem. Mater. 2008, 20, 55. [Google Scholar]
- Schoenleber R. O.; Giese B. Synlett 2003, 4, 501. [Google Scholar]
- Geissler D.; Antonenko Y. N.; Schmidt R.; Keller S.; Krylova O. O.; Wiesner B.; Bendig J.; Pohl P.; Hagen V. Angew. Chem., Int. Ed. 2005, 44, 1195. [DOI] [PubMed] [Google Scholar]
- Oshima T.; Ueno S. Y.; Nagai T. Heterocycles 1995, 40, 607. [Google Scholar]
- Wiberg K. B. Angew. Chem., Int. Ed. 1986, 25, 312. [Google Scholar]
- Kilic F.; Kashikar N. D.; Schmidt R.; Alvarez L.; Dai L.; Weyand I.; Wiesner B.; Goodwin N.; Hagen V.; Kaupp U. B. J. Am. Chem. Soc. 2009, 131, 4027. [DOI] [PubMed] [Google Scholar]
- Chamberlin J. W. J. Org. Chem. 1966, 31, 1658. [Google Scholar]
- a Zimmerman H. E.; Sandel V. R. J. Am. Chem. Soc. 1963, 85, 915. [Google Scholar]; b Givens R. S.; Oettle W. F. J. Am. Chem. Soc. 1971, 93, 3301. [Google Scholar]; c Givens R. S.; Oettle W. F. J. Org. Chem. 1972, 37, 4325. [Google Scholar]
- a Zimmerman H. E. J. Am. Chem. Soc. 1995, 117, 8988. [Google Scholar]; b Zimmerman H. E. J. Phys. Chem. A 1998, 102, 5616. [Google Scholar]
- Kulikov A. Diss. Abstr. Int., B 2006, 67, 2008. [Google Scholar]
- a Wang P. F.; Zhou L.; Zhang X.; Liang X. Chem. Commun. 2010, 46, 1514. [DOI] [PubMed] [Google Scholar]; b Zhou L.; Yang H. S.; Wang P. F. J. Org. Chem. 2011, 76, 5873. [DOI] [PubMed] [Google Scholar]
- Cameron J. F.; Frechet J. M. J. J. Org. Chem. 1990, 55, 5919. [Google Scholar]
- Misetic A.; Boyd M. K. Tetrahedron Lett. 1998, 39, 1653. [Google Scholar]
- Coleman M. P.; Boyd M. K. Tetrahedron Lett. 1999, 40, 7911. [Google Scholar]
- Coleman M. P.; Boyd M. K. J. Org. Chem. 2002, 67, 7641. [DOI] [PubMed] [Google Scholar]
- a Wang P. F.; Hu H. Y.; Wang Y. Org. Lett. 2007, 9, 1533. [DOI] [PubMed] [Google Scholar]; b Wang P. F.; Mondal M.; Wang Y. Eur. J. Org. Chem. 2009, 13, 2055. [Google Scholar]; c Yang H. S.; Zhang X.; Zhou L.; Wang P. F. J. Org. Chem. 2011, 76, 2040. [DOI] [PubMed] [Google Scholar]; d Yang H. S.; Mu F.; Wang P. F. J. Org. Chem. 2011, 76, 8955. [DOI] [PubMed] [Google Scholar]; e Wang P. F.; Wang Y.; Hu H. Y.; Spencer C.; Liang X.; Pan L. R. J. Org. Chem. 2008, 73, 6152. [DOI] [PubMed] [Google Scholar]
- Thevenet D.; Neier R. Helv. Chim. Acta 2011, 94, 331. [Google Scholar]
- Wang P. F.; Hu H. Y.; Wang Y. Org. Lett. 2007, 9, 2831. [DOI] [PubMed] [Google Scholar]
- Wang P. F.; Wang Y.; Hu H. Y.; Liang X. Eur. J. Org. Chem. 2009, 2, 208. [Google Scholar]
- Furuta T.; Torigai H.; Sugimoto M.; Iwamura M. J. Org. Chem. 1995, 60, 3953. [Google Scholar]
- Singh A. K.; Khade P. K. Tetrahedron Lett. 2005, 46, 5563. [Google Scholar]
- a Iwamura M.; Hodota C.; Ishibashi M. Synlett 1991, 1, 35. [Google Scholar]; b Furuta T.; Torigai H.; Osawa T.; Iwamura M. Chem. Lett. 1993, 7, 1179. [Google Scholar]; c Fernandes M. J. G.; Sameiro M.; Goncalves T.; Costa S. P. G. Tetrahedron 2007, 63, 10133. [Google Scholar]; d Furuta T.; Hirayama Y.; Iwamura M. Org. Lett. 2001, 3, 1809. [DOI] [PubMed] [Google Scholar]
- Jana A.; Ikbal M.; Singh N. D. P. Tetrahedron 2012, 68, 1128. [Google Scholar]
- Ren M. G.; Bi N. M.; Mao M.; Song Q. H. J. Photochem. Photobiol., A 2009, 204, 13. [Google Scholar]
- Yu J. Y.; Tang W. J.; Wang H. B.; Song Q. H. J. Photochem. Photobiol., A 2007, 185, 101. [Google Scholar]
- a Fedoryak O. D.; Dore T. M. Org. Lett. 2002, 4, 3419. [DOI] [PubMed] [Google Scholar]; b An H. Y.; Ma C. S.; Li W.; Harris K. T.; Dore T. M.; Phillips D. L. J. Phys. Chem. A 2010, 114, 2498. [DOI] [PubMed] [Google Scholar]; c Ma J. N.; Cheng S. C.; An H. Y.; Li M. D.; Ma C. S.; Rea A. C.; Zhu Y.; Nganga J. L.; Dore T. M.; Phillips D. L. J. Phys. Chem. A 2011, 115, 11632. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Pavlos C. M.; Toscano J. P.; Dore T. M. J. Am. Chem. Soc. 2006, 128, 4267. [DOI] [PubMed] [Google Scholar]
- Ma J. N.; Rea A. C.; An H. Y.; Ma C. S.; Guan X. G.; Li M. D.; Su T.; Yeung C. S.; Harris K. T.; Zhu Y.; Nganga J. L.; Fedoryak O. D.; Dore T. M.; Phillips D. L. Chem.—Eur. J. 2012, 18, 6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epling G. A.; Provatas A. A. Chem. Commun. 2002, 10, 1036. [DOI] [PubMed] [Google Scholar]
- Epling G. A.; Walker M. E. Tetrahedron Lett. 1982, 23, 3843. [Google Scholar]
- a Davis M. J.; Kragor C. H.; Reddie K. G.; Wilson H. C.; Zhu Y.; Dore T. M. J. Org. Chem. 2009, 74, 1721. [DOI] [PubMed] [Google Scholar]; b Li Y. M.; Shi J.; Cai R.; Chen X. Y.; Guo Q. X.; Liu L. Tetrahedron Lett. 2010, 51, 1609. [Google Scholar]
- Soares A. M. S.; Costa S. P. G.; Goncalves M. S. T. Tetrahedron 2010, 66, 8189. [Google Scholar]
- a Arumugam S.; Popik V. V. J. Am. Chem. Soc. 2009, 131, 11892. [DOI] [PubMed] [Google Scholar]; b Arumugam S.; Popik V. V. J. Org. Chem. 2010, 75, 7338. [DOI] [PubMed] [Google Scholar]; c Arumugam S.; Popik V. V. J. Am. Chem. Soc. 2012, 134, 8408. [DOI] [PubMed] [Google Scholar]
- Kulikov A.; Arumugam S.; Popik V. V. J. Org. Chem. 2008, 73, 7611. [DOI] [PubMed] [Google Scholar]
- Kostikov A. P.; Popik V. V. J. Org. Chem. 2007, 72, 9190. [DOI] [PubMed] [Google Scholar]
- Kostikov A. P.; Popik V. V. Org. Lett. 2008, 10, 5277. [DOI] [PubMed] [Google Scholar]
- Kostikov A. P.; Malashikhina N.; Popik V. V. J. Org. Chem. 2009, 74, 1802. [DOI] [PubMed] [Google Scholar]
- Arumugam S.; Popik V. V. Photochem. Photobiol. Sci. 2012, 11, 518. [DOI] [PubMed] [Google Scholar]
- a Lee K.; Falvey D. E. J. Am. Chem. Soc. 2000, 122, 9361. [Google Scholar]; b Sundararajan C.; Falvey D. E. J. Org. Chem. 2004, 69, 5547. [DOI] [PubMed] [Google Scholar]; c Sundararajan C.; Falvey D. E. Org. Lett. 2005, 7, 2631. [DOI] [PubMed] [Google Scholar]
- a Tucker J. W.; Narayanam J. M. R.; Shah P. S.; Stephenson C. R. J. Chem. Commun. 2011, 47, 5040. [DOI] [PubMed] [Google Scholar]; b Rahim M. A.; Matsumura S.; Toshima K. Tetrahedron Lett. 2005, 46, 7307. [Google Scholar]
- Lechner R.; König B. Synthesis 2010, 10, 1712. [Google Scholar]
- Yang H.; Zhou L.; Wang P. F. Photochem. Photobiol. Sci. 2012, 11, 514. [DOI] [PubMed] [Google Scholar]
- Haas K. L.; Franz K. J. Chem. Rev. 2009, 109, 4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zayat L.; Calero C.; Albores P.; Baraldo L.; Etchenique R. J. Am. Chem. Soc. 2003, 125, 882. [DOI] [PubMed] [Google Scholar]; b Zayat L.; Salierno M.; Etchenique R. Inorg. Chem. 2006, 45, 1728. [DOI] [PubMed] [Google Scholar]
- a Verde E. M. R.; Zayat L.; Etchenique R.; Yuste R. Front. Neural Circuits 2008, 2, Art. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zayat L.; Noval M. G.; Campi J.; Calero C. I.; Calvo D. J.; Etchenique R. ChemBioChem 2007, 8, 2035. [DOI] [PubMed] [Google Scholar]
- Salierno M.; Fameli C.; Etchenique R. Eur. J. Inorg. Chem. 2008, 7, 1125. [Google Scholar]
- a Farrer N. J.; Woods J. A.; Salassa L.; Zhao Y.; Robinson K. S.; Clarkson G.; Mackay F. S.; Sadler P. J. Angew. Chem., Int. Ed. 2010, 49, 8905. [DOI] [PubMed] [Google Scholar]; b Sokolov A. Y.; Schaefer H. F. Dalton Trans. 2011, 40, 7571. [DOI] [PubMed] [Google Scholar]
- Filevich O.; Salierno M.; Etchenique R. J. Inorg. Biochem. 2010, 104, 1248. [DOI] [PubMed] [Google Scholar]
- a Salassa L.; Garino C.; Salassa G.; Gobetto R.; Nervi C. J. Am. Chem. Soc. 2008, 130, 9590. [DOI] [PubMed] [Google Scholar]; b Ruiu T.; Garino C.; Salassa L.; Pizarro A. M.; Nervi C.; Gobetto R.; Sadler P. J. Eur. J. Inorg. Chem. 2010, 8, 1186. [Google Scholar]; c Salassa L.; Garino C.; Salassa G.; Nervi C.; Gobetto R.; Lamberti C.; Gianolio D.; Bizzarri R.; Sadler P. J. Inorg. Chem. 2009, 48, 1469. [DOI] [PubMed] [Google Scholar]
- San Miguel V.; Álvarez M.; Filevich O.; Etchenique R.; del Campo A. Langmuir 2012, 28, 1217. [DOI] [PubMed] [Google Scholar]
- Salierno M.; Marceca E.; Peterka D. S.; Yuste R.; Etchenique R. J. Inorg. Biochem. 2010, 104, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Marmol J.; Filevich O.; Etchenique R. Anal. Chem. 2010, 82, 6259. [DOI] [PubMed] [Google Scholar]
- Respondek T.; Garner R. N.; Herroon M. K.; Podgorski I.; Turro C.; Kodanko J. J. J. Am. Chem. Soc. 2011, 133, 17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S.; Limburg B.; Meeldijk J. D.; Gebbink R.; Killian J. A. J. Am. Chem. Soc. 2011, 133, 252. [DOI] [PubMed] [Google Scholar]
- Goldbach R. E.; Rodriguez-Garcia I.; van Lenthe J. H.; Siegler M. A.; Bonnet S. Chem.—Eur. J. 2011, 17, 9924. [DOI] [PubMed] [Google Scholar]
- a Peukert S.; Giese B. J. Org. Chem. 1998, 63, 9045. [Google Scholar]; b Glatthar R.; Giese B. Org. Lett. 2000, 2, 2315. [DOI] [PubMed] [Google Scholar]; c Kessler M.; Glatthar R.; Giese B.; Bochet C. G. Org. Lett. 2003, 5, 1179. [DOI] [PubMed] [Google Scholar]
- Jones P. B.; Pollastri M. P.; Porter N. A. J. Org. Chem. 1996, 61, 9455. [Google Scholar]
- Jones P. B.; Porter N. A. J. Am. Chem. Soc. 1999, 121, 2753. [Google Scholar]
- Plessis C.; Derrer S. Tetrahedron Lett. 2001, 42, 6519. [Google Scholar]
- Banerjee A.; Grewer C.; Ramakrishnan L.; Jager J.; Gameiro A.; Breitinger H. G. A.; Gee K. R.; Carpenter B. K.; Hess G. P. J. Org. Chem. 2003, 68, 8361. [DOI] [PubMed] [Google Scholar]
- a Shirai M.; Tsunooka M. Bull. Chem. Soc. Jpn. 1998, 71, 2483. [Google Scholar]; b Shirai M.; Tsunooka M. Prog. Polym. Sci. 1996, 21, 1. [Google Scholar]
- a Andraos J.; Barclay G. G.; Medeiros D. R.; Baldovi M. V.; Scaiano J. C.; Sinta R. Chem. Mater. 1998, 10, 1694. [Google Scholar]; b Ortica F.; Coenjarts C.; Scaiano J. C.; Liu H.; Pohlers G.; Cameron J. F. Chem. Mater. 2001, 13, 2297. [Google Scholar]; c Malval J. P.; Suzuki S.; Morlet-Savary F.; Allonas X.; Fouassier J. P.; Takahara S.; Yamaoka T. J. Phys. Chem. A 2008, 112, 3879. [DOI] [PubMed] [Google Scholar]; d Malval J. P.; Morlet-Savary F.; Allonas X.; Fouassier J. P.; Suzuki S.; Takahara S.; Yamaoka T. Chem. Phys. Lett. 2007, 443, 323. [DOI] [PubMed] [Google Scholar]; e Arnold P. A.; Fratesi L. E.; Bejan E.; Cameron J.; Pohlers G.; Liu H.; Scaiano J. C. Photochem. Photobiol. Sci. 2004, 3, 864. [DOI] [PubMed] [Google Scholar]; f Steidl L.; Jhaveri S. J.; Ayothi R.; Sha J.; McMullen J. D.; Ng S. Y. C.; Zipfel W. R.; Zentel R.; Ober C. K. J. Mater. Chem. 2009, 19, 505. [Google Scholar]; g Shirai M.; Okamura H. Prog. Org. Coat. 2009, 64, 175. [Google Scholar]
- Kageyama Y.; Ohshima R.; Sakurama K.; Fujiwara Y.; Tanimoto Y.; Yamada Y.; Aoki S. Chem. Pharm. Bull. 2009, 57, 1257. [DOI] [PubMed] [Google Scholar]
- Aoki S.; Matsuo N.; Hanaya K.; Yamada Y.; Kageyama Y. Bioorg. Med. Chem. 2009, 17, 3405. [DOI] [PubMed] [Google Scholar]
- Ohshima R.; Kitamura M.; Morita A.; Shiro M.; Yamada Y.; Ikekita M.; Kimura E.; Aoki S. Inorg. Chem. 2010, 49, 888. [DOI] [PubMed] [Google Scholar]
- Fibich A.; Janko K.; Apell H. J. R. Biophys. J. 2007, 93, 3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrer S.; Flachsmann F.; Plessis C.; Stang M. Chimia 2007, 61, 665. [Google Scholar]
- a Rochat S.; Minardi C.; de saint Laumer J. Y.; Herrmann A. Helv. Chim. Acta 2000, 83, 1645. [Google Scholar]; b Herrmann A.; Debonneville C.; Laubscher V.; Aymard L. Flavour Frag. J. 2000, 15, 415. [Google Scholar]; c Levrand B.; Herrmann A. Chimia 2007, 61, 661. [Google Scholar]; d Levrand B.; Herrmann A. Photochem. Photobiol. Sci. 2002, 1, 907. [DOI] [PubMed] [Google Scholar]
- Griesbeck A. G.; Hinze O.; Görner H.; Huchel U.; Kropf C.; Sundermeierc U.; Gerkec T. Photochem. Photobiol. Sci. 2012, 11, 587. [DOI] [PubMed] [Google Scholar]
- a Levrand B.; Herrmann A. Flavour Frag. J. 2006, 21, 400. [Google Scholar]; b Jones P. B.; Brinson R. G.; Sarma S. J.; Elkazaz S. Org. Biomol. Chem. 2008, 6, 4204. [DOI] [PubMed] [Google Scholar]; c Brinson R. G.; Jones P. B. Org. Lett. 2004, 6, 3767. [DOI] [PubMed] [Google Scholar]; d Brinson R. G.; Hubbard S. C.; Zuidema D. R.; Jones P. B. J. Photochem. Photobiol., A 2005, 175, 118. [Google Scholar]; e Blankespoor R. L.; DeVries T.; Hansen E.; Kallemeyn J. M.; Klooster A. M.; Mulder J. A.; Smart R. P.; Griend D. A. V. J. Org. Chem. 2002, 67, 2677. [DOI] [PubMed] [Google Scholar]
- a Lukeman M.; Scaiano J. C. J. Am. Chem. Soc. 2005, 127, 7698. [DOI] [PubMed] [Google Scholar]; b Cosa G.; Lukeman M.; Scaiano J. C. Acc. Chem. Res. 2009, 42, 599. [DOI] [PubMed] [Google Scholar]; c Blake J. A.; Gagnon E.; Lukeman M.; Scaiano J. C. Org. Lett. 2006, 8, 1057. [DOI] [PubMed] [Google Scholar]
- Blake J. A.; Lukeman M.; Scaiano J. C. J. Am. Chem. Soc. 2009, 131, 4127. [DOI] [PubMed] [Google Scholar]
- Blake J. A.; Bareiss B.; Jimenez L.; Griffith M.; Scaiano J. C. Photochem. Photobiol. Sci. 2012, 11, 539. [DOI] [PubMed] [Google Scholar]
- Soldevilla A.; Perez-Ruiz R.; Miara Y. D.; Griesbeck A. G. Chem. Commun. 2010, 46, 3747. [DOI] [PubMed] [Google Scholar]
- Soldevilla A.; Griesbeck A. G. J. Am. Chem. Soc. 2006, 128, 16472. [DOI] [PubMed] [Google Scholar]
- Griesbeck A. G.; Oelgemoller M.; Lex J. J. Org. Chem. 2000, 65, 9028. [DOI] [PubMed] [Google Scholar]
- Brook M. A.; Gottardo C.; Balduzzi S.; Mohamed M. Tetrahedron Lett. 1997, 38, 6997. [Google Scholar]
- Brook M. A.; Balduzzi S.; Mohamed M.; Gottardo C. Tetrahedron 1999, 55, 10027. [Google Scholar]
- Mohamed M.; Brook M. A. Can. J. Chem. 2000, 78, 1357. [Google Scholar]
- Chen M. Y.; Lee A. S. Y. J. Org. Chem. 2002, 67, 1384. [DOI] [PubMed] [Google Scholar]
- a Chen M. Y.; Patkar L. N.; Jan M. D.; Lee A. S. Y.; Lin C. C. Tetrahedron Lett. 2004, 45, 635. [Google Scholar]; b Chen M. Y.; Lu K. C.; Lee A. S. Y.; Lin C. C. Tetrahedron Lett. 2002, 43, 2777. [Google Scholar]; c Chen M. Y.; Patkar L. N.; Lu K. C.; Lee A. S. Y.; Lin C. C. Tetrahedron 2004, 60, 11465. [Google Scholar]
- Werle S.; Robert F.; Bouas-Laurent H.; Landais Y. Tetrahedron Lett. 2007, 48, 8909. [Google Scholar]
- a Turner A. D.; Pizzo S. V.; Rozakis G.; Porter N. A. J. Am. Chem. Soc. 1988, 110, 244. [Google Scholar]; b Turner A. D.; Pizzo S. V.; Rozakis G. W.; Porter N. A. J. Am. Chem. Soc. 1987, 109, 1274. [Google Scholar]
- Shiono H.; Nohta H.; Utsuyama C.; Hiramatsu M. Anal. Chim. Acta 2000, 405, 17. [Google Scholar]
- Norris J. L.; Hangauer M. J.; Porter N. A.; Caprioli R. M. J. Mass. Spectrom. 2005, 40, 1319. [DOI] [PubMed] [Google Scholar]
- Gagey N.; Neveu P.; Jullien L. Angew. Chem., Int. Ed. 2007, 46, 2467. [DOI] [PubMed] [Google Scholar]
- Duan X.-Y.; Zhai B.-C.; Song Q.-H. Photochem. Photobiol. Sci. 2012, 11, 593. [DOI] [PubMed] [Google Scholar]
- a Thuring J. W.; Li H.; Porter N. A. Biochemistry 2002, 41, 2002. [DOI] [PubMed] [Google Scholar]; b Porter N. A.; Bruhnke J. D. J. Am. Chem. Soc. 1989, 111, 7616. [Google Scholar]; c Porter N. A.; Bruhnke J. D. Photochem. Photobiol. 1990, 51, 37. [DOI] [PubMed] [Google Scholar]; d Porter N. A.; Bush K. A.; Kinter K. S. J. Photochem. Photobiol., B 1997, 38, 61. [DOI] [PubMed] [Google Scholar]; e Stoddard B. L.; Bruhnke J.; Koenigs P.; Porter N.; Ringe D.; Petsko G. A. Biochemistry 1990, 29, 8042. [DOI] [PubMed] [Google Scholar]; f Stoddard B. L.; Bruhnke J.; Porter N.; Ringe D.; Petsko G. A. Biochemistry 1990, 29, 4871. [DOI] [PubMed] [Google Scholar]
- a Gagey N.; Neveu P.; Benbrahim C.; Goetz B.; Aujard I.; Baudin J. B.; Jullien L. J. Am. Chem. Soc. 2007, 129, 9986. [DOI] [PubMed] [Google Scholar]; b Gagey N.; Emond M.; Neveu P.; Benbrahim C.; Goetz B.; Aujard I.; Baudin J. B.; Jullien L. Org. Lett. 2008, 10, 2341. [DOI] [PubMed] [Google Scholar]
- Wijtmans M.; Rosenthal S. J.; Zwanenburg B.; Porter N. A. J. Am. Chem. Soc. 2006, 128, 11720. [DOI] [PubMed] [Google Scholar]
- Pirrung M. C.; Fallon L.; Zhu J.; Lee Y. R. J. Am. Chem. Soc. 2001, 123, 3638. [DOI] [PubMed] [Google Scholar]
- Ma C. C.; Steinmetz M. G.; Cheng Q.; Jayaraman V. Org. Lett. 2003, 5, 71. [DOI] [PubMed] [Google Scholar]
- Ma C. C.; Steinmetz M. G.; Kopatz E. J.; Rathore R. Tetrahedron Lett. 2005, 46, 1045. [Google Scholar]
- Ma C. C.; Steinmetz M. G.; Kopatz E. J.; Rathore R. J. Org. Chem. 2005, 70, 4431. [DOI] [PubMed] [Google Scholar]
- Ma C. C.; Steinmetz M. G. Org. Lett. 2004, 6, 629. [DOI] [PubMed] [Google Scholar]
- Ma C. C.; Chen Y. G.; Steinmetz M. G. J. Org. Chem. 2006, 71, 4206. [DOI] [PubMed] [Google Scholar]
- a Jia J. L.; Sarker M.; Steinmetz M. G.; Shukla R.; Rathore R. J. Org. Chem. 2008, 73, 8867. [DOI] [PubMed] [Google Scholar]; b Jia J. L.; Steinmetz M. G.; Shukla R.; Rathore R. Tetrahedron Lett. 2008, 49, 4621. [Google Scholar]
- Kolano C.; Sander W. Eur. J. Org. Chem. 2003, 6, 1074. [Google Scholar]
- Kitani S.; Sugawara K.; Tsutsumi K.; Morimoto T.; Kakiuchi K. Chem. Commun. 2008, 18, 2103. [DOI] [PubMed] [Google Scholar]
- a Chen Y. G.; Steinmetz M. G. Org. Lett. 2005, 7, 3729. [DOI] [PubMed] [Google Scholar]; b Chen Y. G.; Steinmetz M. G. J. Org. Chem. 2006, 71, 6053. [DOI] [PubMed] [Google Scholar]
- Bräse S. Acc. Chem. Res. 2004, 37, 805. [DOI] [PubMed] [Google Scholar]
- Enders D.; Rijksen C.; Bremus-Kobberling E.; Gillner A.; Kobberling J. Tetrahedron Lett. 2004, 45, 2839. [Google Scholar]
- Nagel M.; Hany R.; Lippert T.; Molberg M.; Nuesch F. A.; Rentsch D. Macromol. Chem. Phys. 2007, 208, 277. [Google Scholar]
- Sebej P.; Slanina T.; Al Anshori J.; Lovely Angel P. A.; Klan P.; Müller P.; Wintner J.; Wirz J.. J. Org. Chem. 2013, DOI: 10.1021/jo301455n. [DOI] [PubMed] [Google Scholar]
- Stacko P.; Sebej P.; Klan P.. Org. Lett. 2012, 14, 4918. [DOI] [PubMed] [Google Scholar]
- Liese J.; Hampp N. A. J. Photochem. Photobiol., A 2011, 219, 228. [Google Scholar]
- Zamadar M.; Ghosh G.; Mahendran A.; Minnis M.; Kruft B. I.; Ghogare A.; Aebisher D.; Greer A. J. Am. Chem. Soc. 2011, 133, 7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mack E. T.; Carle A. B.; Liang J. T. M.; Coyle W.; Wilson R. M. J. Am. Chem. Soc. 2004, 126, 15324. [DOI] [PubMed] [Google Scholar]; b Mack E. T.; Birzniece D.; Veach D. R.; Coyle W.; Wilson R. M. Bioorg. Med. Chem. Lett. 2005, 15, 2173. [DOI] [PubMed] [Google Scholar]
- Natali M.; Giordani S. Chem. Soc. Rev. 2012, 41, 4010. [DOI] [PubMed] [Google Scholar]
- Lemieux V.; Gauthier S.; Branda N. R. Angew. Chem., Int. Ed. 2006, 45, 6820. [DOI] [PubMed] [Google Scholar]
- Bakhtiari A. B. S.; Hsiao D.; Jin G. X.; Gates B. D.; Branda N. R. Angew. Chem., Int. Ed. 2009, 48, 4166. [DOI] [PubMed] [Google Scholar]
- a Kim H. C.; Hartner S.; Hampp N. J. Photochem. Photobiol., A 2008, 197, 239. [Google Scholar]; b Hartner S.; Kim H. C.; Hampp N. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 2443. [Google Scholar]; c Hartner S.; Kim H. C.; Hampp N.. Phototriggered multifunctional drug delivery device. In Optical Methods for Tumor Treatment and Detection: Mechanisms and Techniques in Photodynamic Therapy Xv; Kessel D., Ed.; SPIE: San Jose, CA, 2006; Vol. 6139; p 260. [Google Scholar]
- Liese J.; Hampp N. A. J. Photochem. Photobiol., A 2010, 209, 128. [Google Scholar]
- Wöll D.; Walbert S.; Stengele K. P.; Albert T. J.; Richmond T.; Norton J.; Singer M.; Green R. D.; Pfleiderer W.; Steiner U. E. Helv. Chim. Acta 2004, 87, 28. [Google Scholar]
- a Dietliker K.; Broillet S.; Heltrung B.; Rzadek P.; Rist G.; Wirz J.; Neshchadin D.; Gescheidt G. Helv. Chim. Acta 2006, 89, 2211. [Google Scholar]; b Wagner P. J.; Klan P. J. Am. Chem. Soc. 1999, 121, 9626. [Google Scholar]; c Speiser S. Chem. Rev. 1996, 96, 1953. [DOI] [PubMed] [Google Scholar]
- Houmam A. Chem. Rev. 2008, 108, 2180. [DOI] [PubMed] [Google Scholar]
- Rehm D.; Weller A. Ber. Bunsen-Ges. Phys. Chem 1969, 73, 834. [Google Scholar]
- Hamada T.; Nishida A.; Yonemitsu O. J. Am. Chem. Soc. 1986, 108, 140. [Google Scholar]
- Hamada T.; Nishida A.; Yonemitsu O. Tetrahedron Lett. 1989, 30, 4241. [Google Scholar]
- Corrie J. E. T.; Papageorgiou G. J. Chem. Soc., Perkin Trans. 1 1996, 13, 1583. [Google Scholar]
- Papageorgiou G.; Corrie J. E. T. Tetrahedron 1999, 55, 237. [Google Scholar]
- a Hill R. R.; Jeffs G. E.; Roberts D. R.; Wood S. A. Chem. Commun. 1999, 17, 1735. [Google Scholar]; b Hill R. R.; Moore S. A.; Roberts D. R. Chem. Commun. 2003, 22, 2838. [DOI] [PubMed] [Google Scholar]; c Hill R. R.; Moore S. A.; Roberts D. R. Photochem. Photobiol. 2005, 81, 1439. [DOI] [PubMed] [Google Scholar]
- Urjasz W.; Celewicz L. J. Phys. Org. Chem. 1998, 11, 618. [Google Scholar]
- a Binkley R. W.; Liu X. G. J. Carbohydr. Chem 1992, 11, 183. [Google Scholar]; b Liu X. G.; Binkley R. W. J. Carbohydr. Chem 1993, 12, 779. [Google Scholar]
- Tanner D. D.; Chen J. J.; Chen L.; Luelo C. J. Am. Chem. Soc. 1991, 113, 8074. [Google Scholar]
- a Banerjee A.; Falvey D. E. J. Org. Chem. 1997, 62, 6245. [Google Scholar]; b Banerjee A.; Lee K.; Yu Q.; Fang A. G.; Falvey D. E. Tetrahedron Lett. 1998, 39, 4635. [Google Scholar]
- Banerjee A.; Lee K.; Falvey D. E. Tetrahedron 1999, 55, 12699. [Google Scholar]
- Sundararajan C.; Falvey D. E. J. Am. Chem. Soc. 2005, 127, 8000. [DOI] [PubMed] [Google Scholar]
- Sundararajan C.; Falvey D. E. Photochem. Photobiol. Sci. 2006, 5, 116. [DOI] [PubMed] [Google Scholar]
- Borak J. B.; Lopez-Sola S.; Falvey D. E. Org. Lett. 2008, 10, 457. [DOI] [PubMed] [Google Scholar]
- Borak J. B.; Falvey D. E. Photochem. Photobiol. Sci. 2010, 9, 854. [DOI] [PubMed] [Google Scholar]
- Borak J. B.; Falvey D. E. J. Org. Chem. 2009, 74, 3894. [DOI] [PubMed] [Google Scholar]
- Borak J. B.; Lee H. Y.; Raghavan S. R.; Falvey D. E. Chem. Commun. 2010, 46, 8983. [DOI] [PubMed] [Google Scholar]
- Edson J. B.; Spencer L. P.; Boncella J. M. Org. Lett. 2011, 13, 6156. [DOI] [PubMed] [Google Scholar]
- Pandey G.; Krishna A. Synth. Commun. 1988, 18, 2309. [Google Scholar]
- Tu W. Y.; Floreancig P. E. Org. Lett. 2007, 9, 2389. [DOI] [PubMed] [Google Scholar]
- Cossy J.; Rakotoarisoa H. Tetrahedron Lett. 2000, 41, 2097. [Google Scholar]
- Noll G.; Trawoeger S.; von Sanden-Flohe M.; Dick B.; Grininger M. ChemBioChem 2009, 10, 834. [DOI] [PubMed] [Google Scholar]
- McHale W. A.; Kutateladze A. G. J. Org. Chem. 1998, 63, 9924. [Google Scholar]
- Mitkin O. D.; Kurchan A. N.; Wan Y. Q.; Schiwal B. F.; Kutateladze A. G. Org. Lett. 2001, 3, 1841. [DOI] [PubMed] [Google Scholar]
- Vath P.; Falvey D. E. J. Org. Chem. 2001, 66, 2887. [DOI] [PubMed] [Google Scholar]
- a Li Z. G.; Kutateladze A. G. J. Org. Chem. 2003, 68, 8236. [DOI] [PubMed] [Google Scholar]; b Gustafson T. P.; Kurchan A. N.; Kutateladze A. G. Tetrahedron 2006, 62, 6574. [Google Scholar]
- a Valiulin R. V.; Kutateladze A. G. J. Org. Chem. 2008, 73, 6393. [DOI] [PubMed] [Google Scholar]; b Valiulin R. A.; Lakkakula S.; Kutateladze A. G. J. Photochem. Photobiol., A 2009, 206, 80. [Google Scholar]
- a Barnhurst L. A.; Kutateladze A. G. Org. Lett. 2001, 3, 2633. [DOI] [PubMed] [Google Scholar]; b Valiulin R. A.; Kutateladze A. G. J. Org. Chem. 2008, 73, 335. [DOI] [PubMed] [Google Scholar]
- Kurchan A. N.; Kutateladze A. G. Org. Lett. 2002, 4, 4129. [DOI] [PubMed] [Google Scholar]
- Kutateladze A. G.; Kottani R.; Kurchan A. N.; Majjigapu J. R. R.; Shirk S. M. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 1379. [Google Scholar]
- a Wan Y. Q.; Angleson J. K.; Kutateladze A. G. J. Am. Chem. Soc. 2002, 124, 5610. [DOI] [PubMed] [Google Scholar]; b Li Z. G.; Wan Y. Q.; Kutateladze A. G. Langmuir 2003, 19, 6381. [Google Scholar]; c Ezhov R. N.; Rozhkov V. V.; Majjigapu J. R. R.; Kutateladze A. G. J. Sulfur Chem. 2008, 29, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. G.; Chiu H.; Kutateladze A. G. Can. J. Chem. 2003, 81, 807. [Google Scholar]
- Lakkakula S.; Mitkin O. D.; Valiulin R. A.; Kutateladze A. G. Org. Lett. 2007, 9, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mitkin O. D.; Wan Y. Q.; Kurchan A. N.; Kutateladze A. G. Synthesis 2001, 8, 1133. [Google Scholar]; b Wan Y. Q.; Mitkin O.; Barnhurst L.; Kurchan A.; Kutateladze A. G. Org. Lett. 2000, 2, 3817. [DOI] [PubMed] [Google Scholar]
- Majjigapu J. R. R.; Kurchan A. N.; Kottani R.; Gustafson T. P.; Kutateladze A. G. J. Am. Chem. Soc. 2005, 127, 12458. [DOI] [PubMed] [Google Scholar]
- a Kottani R.; Majjigapu J. R. R.; Kurchan A.; Majjigapu K.; Gustafson T. P.; Kutateladze A. G. J. Am. Chem. Soc. 2006, 128, 14794. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Majjigapu K.; Majjigapu J. R. R.; Kutateladze A. G. Angew. Chem., Int. Ed. 2007, 46, 6137. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gustafson T. P.; Metzel G. A.; Kutateladze A. G. Photochem. Photobiol. Sci. 2012, 11, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T. P.; Metzel G. A.; Kutateladze A. G. Org. Biomol. Chem. 2011, 9, 4752. [DOI] [PubMed] [Google Scholar]
- Kottani R.; Valiulin R. A.; Kutateladze A. G. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit M.; Bort G.; Doan B. T.; Sicard C.; Ogden D.; Scherman D.; Ferroud C.; Dalko P. I. Angew. Chem., Int. Ed. 2011, 50, 9708. [DOI] [PubMed] [Google Scholar]
- Auzel F. Chem. Rev. 2004, 104, 139. [DOI] [PubMed] [Google Scholar]
- a Haase M.; Schafer H. Angew. Chem., Int. Ed. 2011, 50, 5808. [DOI] [PubMed] [Google Scholar]; b Doane T. L.; Burda C. Chem. Soc. Rev. 2012, 41, 2885. [DOI] [PubMed] [Google Scholar]; c Kato M. Yakugaku Zasshi 2012, 132, 201. [DOI] [PubMed] [Google Scholar]; d Zandberg W. F.; Bakhtiari A. B. S.; Erno Z.; Hsiao D.; Gates B. D.; Claydon T.; Branda N. R. Nanomed. Nanotechnol. 2012, 8, 908. [DOI] [PubMed] [Google Scholar]
- Boyer J. C.; Carling C. J.; Gates B. D.; Branda N. R. J. Am. Chem. Soc. 2010, 132, 15766. [DOI] [PubMed] [Google Scholar]
- a Carling C.-J.; Boyer J.-C.; Branda N. R. J. Am. Chem. Soc. 2009, 131, 10838. [DOI] [PubMed] [Google Scholar]; b Yang Y.; Shao Q.; Deng R.; Wang C.; Teng X.; Cheng K.; Cheng Z.; Huang L.; Liu Z.; Liu X.; Xing B. Angew. Chem., Int. Ed. 2012, 51, 3125. [DOI] [PubMed] [Google Scholar]
- Wu G. H.; Milkhailovsky A.; Khant H. A.; Fu C.; Chiu W.; Zasadzinski J. A. J. Am. Chem. Soc. 2008, 130, 8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Shao R.; Wei X.; Gupta S.; Li C. Small 2010, 6, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Barhoumi A.; Huschka R.; Bardhan R.; Knight M. W.; Halas N. J. Chem. Phys. Lett. 2009, 482, 171. [Google Scholar]; b Huschka R.; Neumann O.; Barhoumi A.; Halas N. J. Nano Lett. 2010, 10, 4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Braun G. B.; Pallaoro A.; Wu G. H.; Missirlis D.; Zasadzinski J. A.; Tirrell M.; Reich N. O. ACS Nano 2009, 3, 2007. [DOI] [PubMed] [Google Scholar]; b Lu W.; Zhang G. D.; Zhang R.; Flores L. G.; Huang Q.; Gelovani J. G.; Li C. Cancer Res. 2010, 70, 3177. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huschka R.; Zuloaga J.; Knight M. W.; Brown L. V.; Nordlander P.; Halas N. J. J. Am. Chem. Soc. 2011, 133, 12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Zhang G. D.; Li C. ACS Nano 2010, 4, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijaya A.; Schaffer S. B.; Pallares I. G.; Hamad-Schifferli K. ACS Nano 2009, 3, 80. [DOI] [PubMed] [Google Scholar]
- Melancon M. P.; Zhou M.; Li C. Acc. Chem. Res. 2011, 44, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dougherty T. J.Photochemistry in the Treatment of Cancer. In Adv. Photochem.; Volman D. H., Hammond G. S., Neckers D. C., Eds.; Wiley-VCH: Weinheim, Germany, 1992; Vol. 17; p 275. [Google Scholar]; b Szeimies R. M.; Calzavara-Pinton P.; Karrer S.; Ortel B.; Landthaler M. J. Photochem. Photobiol., B 1996, 36, 213. [DOI] [PubMed] [Google Scholar]
- König K. J. Microsc. (Oxford, U.K.) 2000, 200, 83. [DOI] [PubMed] [Google Scholar]
- a Becker A.; Hessenius C.; Licha K.; Ebert B.; Sukowski U.; Semmler W.; Wiedenmann B.; Grotzinger C. Nat. Biotechnol. 2001, 19, 327. [DOI] [PubMed] [Google Scholar]; b Göppert-Mayer M. Ann. Phys. 1931, 9, 273. [Google Scholar]; c Kauffman J. F.; Turner J. M.; Alabugin I. V.; Breiner B.; Kovalenko S. V.; Badaeva E. A.; Masunov A.; Tretiak S. J. Phys. Chem. A 2006, 110, 241. [DOI] [PubMed] [Google Scholar]; d Lakowicz J. R.Two-Photon And Non-Linear Induced Fluorescence; Plenum Press: New York, 1991. [Google Scholar]; e Rubart M. Circ. Res. 2004, 95, 1154. [DOI] [PubMed] [Google Scholar]
- a Brown E. B.; Shear J. B.; Adams S. R.; Tsien R. Y.; Webb W. W. Biophys. J. 1999, 76, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weissleder R. Nat. Biotechnol. 2001, 19, 316. [DOI] [PubMed] [Google Scholar]
- a Dakin K.; Li W. H. Nat. Methods 2006, 3, 959. [DOI] [PubMed] [Google Scholar]; b Kim H. M.; Kim B. R.; Hong J. H.; Park J. S.; Lee K. J.; Cho B. R. Angew. Chem., Int. Ed. 2007, 46, 7445. [DOI] [PubMed] [Google Scholar]; c Pond S. J. K.; Tsutsumi O.; Rumi M.; Kwon O.; Zojer E.; Bredas J. L.; Marder S. R.; Perry J. W. J. Am. Chem. Soc. 2004, 126, 9291. [DOI] [PubMed] [Google Scholar]
- a Croix C. M. S.; Leelavanichkul K.; Watkins S. C. Adv. Drug Delivery Rev. 2006, 58, 834. [DOI] [PubMed] [Google Scholar]; b Denk W.; Piston D. W.; Webb W. W.. Two-Photon Molecular Excitation in Laser-Scanning Microscopy. In Handbook of Biological Confocal Microscopy; Pawley J. B., Ed.; Plenum Press: New York, 1995. [Google Scholar]; c Jenei A.; Kirsch A. K.; Subramaniam V.; Arndt-Jovin D. J.; Jovin T. M. Biophys. J. 1999, 76, 1092. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kielar F.; Congreve A.; Law G. L.; New E. J.; Parker D.; Wong K. L.; Castreno P.; de Mendoza J. Chem. Commun. 2008, 21, 2435. [DOI] [PubMed] [Google Scholar]; e Xu C.; Zipfel W.; Shear J. B.; Williams R. M.; Webb W. W. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 10763. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zoumi A.; Yeh A.; Tromberg B. J. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Belfield K. D.; Schafer K. J. Chem. Mater. 2002, 14, 3656. [Google Scholar]; b Cumpston B. H.; Ananthavel S. P.; Barlow S.; Dyer D. L.; Ehrlich J. E.; Erskine L. L.; Heikal A. A.; Kuebler S. M.; Lee I. Y. S.; McCord-Maughon D.; Qin J. Q.; Rockel H.; Rumi M.; Wu X. L.; Marder S. R.; Perry J. W. Nature 1999, 398, 51. [Google Scholar]; c Kawata S.; Sun H. B. J. Photopolym. Sci. Technol. 2002, 15, 471. [Google Scholar]; d Kawata S.; Sun H. B.; Tanaka T.; Takada K. Nature 2001, 412, 697. [DOI] [PubMed] [Google Scholar]
- La Fratta C. N.; Fourkas J. T.; Baldacchini T.; Farrer R. A. Angew. Chem., Int. Ed. 2007, 46, 6238. [DOI] [PubMed] [Google Scholar]
- a Kim H. C.; Hartner S.; Behe M.; Behr T. M.; Hampp N. A. J. Biochem. Opt. 2006, 11, 034024. [DOI] [PubMed] [Google Scholar]; b Kloxin A. M.; Kasko A. M.; Salinas C. N.; Anseth K. S. Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wong D. Y.; Griffin D. R.; Reed J.; Kasko A. M. Macromolecules 2010, 43, 2824. [Google Scholar]
- a Balaz M.; Collins H. A.; Dahlstedt E.; Anderson H. L. Org. Biomol. Chem. 2009, 7, 874. [DOI] [PubMed] [Google Scholar]; b Bhawalkar J. D.; Kumar N. D.; Zhao C. F.; Prasad P. N. J. Clin. Laser Med. Surg. 1997, 15, 201. [DOI] [PubMed] [Google Scholar]; c Frederiksen P. K.; Jorgensen M.; Ogilby P. R. J. Am. Chem. Soc. 2001, 123, 1215. [DOI] [PubMed] [Google Scholar]; d Goyan R. L.; Cramb D. T. Photochem. Photobiol. 2000, 72, 821. [DOI] [PubMed] [Google Scholar]; e Wachter E. A.; Partridge W. P.; Fisher W. G.; Dees H. C.; Petersen M. G.. Simultaneous two-photon excitation of photodynamic therapy agents. In Commercial Applications of Ultrafast Lasers; Reed M. K., Ed.; SPIE: San Jose, CA, 1998; Vol. 3269, p 68. [Google Scholar]; f Wachter E. A.; Petersen M. G.; Dees H. C.. Photodynamic therapy with ultrafast lasers. In Commercial and Biomedical Applications of Ultrafast Lasers; Reed M. K., Neev J., Eds.; SPIE: San Jose, CA, 1999; Vol. 3616; p 66. [Google Scholar]
- a Belfield K. D. Spectrum 2001, Spring, 1. [Google Scholar]; b Belfield K. D.; Bondar M. V.; Liu Y.; Przhonska O. V. J. Phys. Org. Chem. 2003, 16, 69. [Google Scholar]; c Corredor C. C.; Belfield K. D.; Bondar M. V.; Przhonska O. V.; Hernandez F. E.; Kachkovsky O. D. J. Photochem. Photobiol., A 2006, 184, 177. [Google Scholar]; d Dvornikov A. S.; Bouas-Laurent H.; Desvergne J. P.; Rentzepis P. M. J. Mater. Chem. 1999, 9, 1081. [Google Scholar]; e Kim H. C.; Kreiling S.; Greiner A.; Hampp N. Chem. Phys. Lett. 2003, 372, 899. [Google Scholar]; f Urdabayev N. K.; Popik V. V. J. Am. Chem. Soc. 2004, 126, 4058. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Lev-Ram V.; Tsien R. Y. Chem. Biol. 1997, 4, 867. [DOI] [PubMed] [Google Scholar]
- a Ellis-Davies G. C. R.; Matsuzaki M.; Paukert M.; Kasai H.; Bergles D. E. J. Neurosci. 2007, 27, 6601. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matsuzaki M.; Ellis-Davies G. C. R.; Nemoto T.; Miyashita Y.; Iino M.; Kasai H. Nat. Neurosci. 2001, 4, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Smith M. A.; Ellis-Davies G. C. R.; Magee J. C. J. Physiol. (London) 2003, 548, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T. Bio. Ind. 2006, 23, 58. [Google Scholar]
- a McCray J. A. Methods Enzymol. 1998, 291, 175. [Google Scholar]; b Sinha D. K.; Neveu P.; Gagey N.; Aujard I.; Benbrahim-Bouzidi C.; Le Saux T.; Rampon C.; Gauron C.; Goetz B.; Dubruille S.; Baaden M.; Volovitch M.; Bensimon D.; Vriz S.; Jullien L. ChemBioChem 2010, 11, 653. [DOI] [PubMed] [Google Scholar]
- Kasai H.; Matsuzaki M.; Ellis-Davies G. C. R.. Two-Photon Uncaging Microscopy; Cold Spring Harbor Laboratory Press: Cold Spring, New York, 2005. [DOI] [PubMed] [Google Scholar]
- Friedrich D. M. J. Chem. Educ. 1982, 59, 472. [Google Scholar]
- Cheong W. F.; Prahl S. A.; Welch A. J. IEEE J. Quantum Electron. 1990, 26, 2166. [Google Scholar]
- The quantum efficiency for two-photon induced reactions is defined as the fraction of excited states that undergo chemical transformation.
- Kantevari S.; Hoang C. J.; Ogrodnik J.; Egger M.; Niggli E.; Ellis-Davies G. C. R. ChemBioChem 2006, 7, 174. [DOI] [PubMed] [Google Scholar]
- Zhao Y. R.; Zheng Q.; Dakin K.; Xu K.; Martinez M. L.; Li W. H. J. Am. Chem. Soc. 2004, 126, 4653. [DOI] [PubMed] [Google Scholar]
- Specht A.; Thomann J. S.; Alarcon K.; Wittayanan W.; Ogden D.; Furuta T.; Kurakawa Y.; Goeldner M. ChemBioChem 2006, 7, 1690. [DOI] [PubMed] [Google Scholar]
- Warther D.; Bolze F.; Leonard J.; Gug S.; Specht A.; Puliti D.; Sun X. H.; Kessler P.; Lutz Y.; Vonesch J. L.; Winsor B.; Nicoud J. F.; Goeldner M. J. Am. Chem. Soc. 2010, 132, 2585. [DOI] [PubMed] [Google Scholar]
- Fedoryak O. D.; Sul J. Y.; Haydon P. G.; Ellis-Davies G. C. R. Chem. Commun. 2005, 29, 3664. [DOI] [PubMed] [Google Scholar]
- Nikolenko V.; Yuste R.; Zayat L.; Baraldo L. M.; Etchenique R. Chem. Commun. 2005, 13, 1752. [DOI] [PubMed] [Google Scholar]
- Shigenaga A.; Yamamoto J.; Sumikawa Y.; Furuta T.; Otaka A. Tetrahedron Lett. 2010, 51, 2868. [Google Scholar]
- Bao C. Y.; Fan G. S.; Lin Q. N.; Li B.; Cheng S. Y.; Huang Q.; Zhu L. Y. Org. Lett. 2012, 14, 572. [DOI] [PubMed] [Google Scholar]
- Bochet C. G. Angew. Chem., Int. Ed. 2001, 40, 2071. [DOI] [PubMed] [Google Scholar]
- a Blanc A.; Bochet C. G. J. Org. Chem. 2002, 67, 5567. [DOI] [PubMed] [Google Scholar]; b Bochet C. G. Synlett 2004, 13, 2268. [Google Scholar]
- Ladlow M.; Legge C. H.; Neudeck T.; Pipe A. J.; Sheppard T.; Yang L. Q. L. Chem. Commun. 2003, 16, 2048. [DOI] [PubMed] [Google Scholar]
- Stegmaier P.; Alonso J. M.; del Campo A. Langmuir 2008, 24, 11872. [DOI] [PubMed] [Google Scholar]
- Schafer F.; Joshi K. B.; Fichte M. A. H.; Mack T.; Wachtveitl J.; Heckel A. Org. Lett. 2011, 13, 1450. [DOI] [PubMed] [Google Scholar]
- Priestman M. A.; Sun L.; Lawrence D. S. ACS Chem. Biol. 2011, 6, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiali B.; Goguen B. N.; Aemissegger A. J. Am. Chem. Soc. 2011, 133, 11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn G. A.; Dobbie I. M.; Monypenny J.; Holt M. R.; Zicha D. J. Microsc. (Oxford, U. K.) 2002, 205, 109. [DOI] [PubMed] [Google Scholar]
- Mitchison T. J.; Sawin K. E.; Theriot J. A.; Gee K.; Mallavarapu A. Methods Enzymol. 1998, 291, 63. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Choyke P. L. Acc. Chem. Res. 2011, 44, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano Y.; Kamiya M.; Kanda M.; Ueno T.; Hirose K.; Nagano T. J. Am. Chem. Soc. 2005, 127, 4888. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Urano Y.; Kamiya M.; Ueno T.; Kojima H.; Nagano T. J. Am. Chem. Soc. 2007, 129, 6696. [DOI] [PubMed] [Google Scholar]
- Braslavsky S. E.; Fron E.; Rodriguez H. B.; Roman E. San; Scholes G. D.; Schweitzer G.; Valeur B.; Wirz J. Photochem. Photobiol. Sci. 2008, 7, 1444. [DOI] [PubMed] [Google Scholar]
- Tang X.; Richards J. L.; Peritz A. E.; Dmochowski I. J. Bioorg. Med. Chem. Lett. 2005, 15, 5303. [DOI] [PubMed] [Google Scholar]
- Tang X.; Dmochowski I. J. Org. Lett. 2005, 7, 279. [DOI] [PubMed] [Google Scholar]
- Pellois J.-P.; Hahn M. E.; Muir T. W. J. Am. Chem. Soc. 2004, 126, 7170. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Guo Y.-M.; Li W.-H. J. Am. Chem. Soc. 2007, 129, 10616. [DOI] [PubMed] [Google Scholar]
- a Yeh R.-H.; Yan X.; Cammer M.; Bresnick A. R.; Lawrence D. S. J. Biol. Chem. 2002, 277, 11527. [DOI] [PubMed] [Google Scholar]; b Veldhuyzen W. F.; Nguyen Q.; McMaster G.; Lawrence D. S. J. Am. Chem. Soc. 2003, 125, 13358. [DOI] [PubMed] [Google Scholar]
- Furukawa K.; Abe H.; Tsuneda S.; Ito Y. Org. Biomol. Chem. 2010, 8, 2309. [DOI] [PubMed] [Google Scholar]
- Belov V. N. W.; Christian A.; Boyarskiy V. P.; Jakobs S.; Hell S. W. Angew. Chem., Int. Ed. 2010, 49, 3520. [DOI] [PubMed] [Google Scholar]
- Kolmakov K.; Wurm C.; Sednev M. V.; Bossi M. L.; Belov V. N.; Hell S. W. Photochem. Photobiol. Sci. 2012, 11, 522. [DOI] [PubMed] [Google Scholar]
- Groff D.; Wang F.; Jockusch S.; Turro N. J.; Schultz P. G. Angew. Chem., Int. Ed. 2010, 49, 7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien R. Y. Angew. Chem., Int. Ed. 2009, 48, 5612. [DOI] [PubMed] [Google Scholar]
- Patterson G. H.; Lippincott-Schwartz J. Science 2002, 297, 1873. [DOI] [PubMed] [Google Scholar]
- a Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. Science 2006, 313, 1642. [DOI] [PubMed] [Google Scholar]; b de Schryver F.; Nonell S. Photochem. Photobiol. Sci. 2009, 8, 441.19337655 [Google Scholar]
- Steinhauer C.; Forthmann C.; Vogelsang J.; Tinnefeld P. J. Am. Chem. Soc. 2008, 130, 16840. [DOI] [PubMed] [Google Scholar]
- Lee H.-l. D.; Lord S. J.; Iwanaga S.; Zhan K.; Xie H.; Williams J. C.; Wang H.; Bowman G. R.; Goley E. D.; Shapiro L.; Twieg R. J.; Rao J.; Moerner W. E. J. Am. Chem. Soc. 2010, 132, 15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord S. J.; Lee H.-l. D.; Samuel R.; Weber R.; Liu N.; Conley N. R.; Thompson M. A.; Twieg R. J.; Moerner W. E. J. Phys. Chem. B 2010, 114, 14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Ho L. Y.; Lin Q. J. Am. Chem. Soc. 2011, 133, 11912. [DOI] [PMC free article] [PubMed] [Google Scholar]