

Summary
Background
Neoadjuvant chemotherapy is used in the treatment of breast carcinoma because it substantially reduces the size of the primary tumor and lymph node metastases. The present study investigated biomarkers that can predict a pathologic response to the therapy.
Material/Methods
The role of apoptosis in regression of the tumors after neoadjuvant chemotherapy was determined by TUNEL and anti-active caspase 3 assay. The transcriptional profile of 84 key apoptosis genes was evaluated in both pre-therapeutically obtained tumor tissue by core needle biopsy and in specimens removed by final surgery, using a pathway-specific real-time PCR assay. Obtained data were analyzed by hierarchical cluster analysis and correlation analysis. The immunohistochemical profile of each tumor was determined using the standard ABC method.
Results
On the basis of a hierarchical cluster analysis of 13 significantly changed genes, we divided patients into good and poor prognosis groups, which correlate well with progression-free survival. In the good prognosis group, we found a statistically significant down-regulation of the expression of MCL1 and IGF1R genes after neoadjuvant treatment. We also found a statistically significant overexpression of BCL2L10, BCL2AF1, CASP8, CASP10, CASP14, CIDEB, FADD, HRK, TNFRSF25, TNFSF8 and CD70 genes. In contrast, we found up-regulation of IGF1R after the treatment in the group with poor prognosis.
Conclusions
Gene expression profiling using real-time PCR assay is a valuable research tool for the investigation of molecular markers, which reflect tumor biology and treatment response.
Keywords: biomarkers, apoptosis, breast cancer, neoadjuvant chemotherapy
Background
Breast cancer is the most common malignant disease in women, occurring more frequently in developed countries [1]. Neoadjuvant chemotherapy, also called pre-operative chemotherapy, is now widely used in the treatment of breast carcinoma. It has several potential advantages compared with the traditional strategy of surgery followed by adjuvant chemotherapy. Neoadjuvant chemotherapy substantially reduces the size of the primary tumor and lymph node metastasis in more than 80% of cases, and increases the probability that breast-conserving surgery can be performed instead of mastectomy [2]. Neoadjuvant chemotherapy is also a valuable research tool for the discovery of predictive markers of chemotherapy response.
Clinicians have long recognized the heterogeneity of human breast cancers, not only in terms of their diverse natural histories despite identical morphological features, but also in their varied response to treatment [3].
Unfortunately, little progress has been made regarding new prognostic and predictive markers that can assist oncologists in treatment decision-making for breast cancer. Generally accepted prognostic and predictive factors include age, tumor size, lymph node status, histologic tumor type, grade, mitotic rate, Her2 and hormone receptor status [2,4,5]. In general, the poor prognostic features include young age (<35 years), high tumor grade, large size (>2 cm), nodal involvement, lack of hormone receptor expression, and the overexpression of Her2. Other negative features are a high proliferative rate and the presence of lymphovascular invasion [6].
Until the development of microarray techniques in the late 1990s, the main approach used for identifying prognostically significant groups consisted of testing 1 or several markers in a group of patients [7]. However, no single tumor marker has been shown to possess a sufficient predictive value to render it clinically useful [8,9]. To achieve a greater predictive value, multiple markers need to be examined and correlated with the response of tumor cells to the treatment. The development of microarray technology has provided us with just such an opportunity. Molecular profiling has identified at least 5 breast cancer subtypes: luminal-A, luminal-B, Her2-overexpressed, basal-like, and normal breast-like [10,11]. These tumor subtypes have diverse disease biology, behavior, relapse risk, and therapy sensitivity [12,13].
Clinicians also have other tools based on microarray technology to assist in treatment decisions for breast cancer patients. The most recent St. Gallen consensus panel agreed that validated multigene tests (OncotypeDX and Mammaprint) could assist in deciding whether to add chemotherapy in cases where its use was uncertain after the consideration of conventional markers. On the other hand, molecular-based technologies are not regarded by the panel as sufficiently reliable yet to make a definitive contribution to the therapeutic decision [14].
The present study was aimed at the investigation of biomarkers that can predict a pathologic response to therapy, and included 16 patients with invasive breast carcinoma, who had undergone neoadjuvant chemotherapy and subsequent surgery (lumpectomy or mastectomy) at the General University Hospital (Prague, Czech Republic). In all cases, the expression profile of 84 key apoptosis genes was evaluated both in pre-therapeutically obtained tumor tissue from a core needle biopsy and in specimens removed during final surgery, using a real-time PCR assay.
Material and Methods
Patients and treatment
This study included 16 patients, all female, aged 52±11 (mean ±standard deviation) years, neoadjuvantly treated for breast carcinoma at the General University Hospital (Prague, Czech Republic) between 2005 and 2010. Two patients (12.5%) had a stage I tumor, 9 patients (56.2%) stage II, 3 patients (18.8%) stage III, and 2 patients (12.5%) stage IV. The scheme of neoadjuvant chemotherapy is summarized in Table 1.
Table 1.
Clinical, histological and immunohistochemical characteristics of patients before and after neoadjuvant therapy.
| No. | Clinical stage | TNM | Her2 (0–3+) | ER (%) | PR (%) | Ki67 (%) | Casp. 3 (%) | Tunel (%) | Age (years) | Diagnosis | Neoadjuvant treatment | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Her2 positive | ||||||||||||
| 1 | Before | IIA | T2 N0 MX | 3 | >90 | <1 | 35–40 | 10–15 | 20 | 62 62 |
ILC | 4× doxorubicin and cyclophosphamide + 4× docetaxel and trastuzumab |
| After | I | T1 N0 MX | 1 | 30–40 | <1 | 2–3 | 2–3 | 1 | ||||
| 2 | Before | IIIB | T4,NX,MX | 3 | 80 | 80–90 | 70 | <1 | 1 | 61 61 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel + 6× paclitaxel and trastuzumab |
| After | I | T1 N0 MX | 2 | 50–60 | 20–30 | <5 | 0 | 0 | ||||
| 3 | Before | IIA | T2 N0 MX | 3 | 20–25 | <1 | 15–20 | 10–13 | 15 | 45 45 |
IDC | 4× doxorubicin and cyclophosphamide |
| After | IIA | TX N1 MX | 3 | 20 | 0 | <5 | 9 | 10 | ||||
| 4 | Before | IIB | T2 N0 MX | 3 | 0 | 0 | 25–35 | 2–3 | 0 | 54 54 |
IDC | 5× fluorouracil, epirubicin and cyclophosphamide + 12× paclitaxel and trastuzumab |
| After | I | T1 NX MX | 0 | 20 | 10 | N/A | N/A | 1 | ||||
| 5 | Before | IV | T5 N1 MX | 3 | 0 | 0 | 75–80 | 5 | 1 | 49 49 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IV | T4 N1 MX | 3 | 10–15; 70–80 * | <5; 5–10 * | <3; 30–40 * | 2–3 | 5 | ||||
| 6 | Before | IIB | T3 N0 MX | 3 | 0 | 0 | 50–60 | 3–5 | 1 | 38 38 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIIA | T1 N2 MX | 3 | 10 | 10 | 25–30 | 2–3 | 5 | ||||
| Luminal | ||||||||||||
| 7 | Before | I | T1 N0 MX | 1 | 70–80 | 70–80 | 5–10 | <1 | 1 | 48 48 |
ILC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | 0 | T0 N0 M0 | pCR | |||||||||
| 8 | Before | IIIB | T4 N1 MX | 0 | 40–50 | 10 | 15–20 | N/A | 0 | 71 71 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIIC | T2 N3 MX | 0 | 5–10 | 10 | <2 | <1 | 5 | ||||
| 9 | Before | IIB | T2 N1 MX | 1 | 60–70 | 70–80 | 10–15 | 4 | 2 | 47 47 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | I | T1 N0 MX | 0 | >90 | 100 | 3–4 | <1 | 1 | ||||
| 10 | Before | IIA | T2 N0 MX | 0 | >90 | 100 | 5–10 | 0 | 0 | 46 46 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIA | T1 N1 MX | 0 | 70–80 | 10–15 | 0 | 1–2 | 5 | ||||
| 11 | Before | IIIB | T4 N1 MX | 0 | 70–80 | 0 | 10–15 | <1 | 5 | 47 47 |
IDC | 4× doxorubicin, cyclophosphamide and doxorubicin + 16× paclitaxel + letrozole |
| After | IV | TX N3 M1 | 1 | 0 | 0 | 25–30 | 60–70 | 50 | ||||
| 12 | Before | IV | T4 N2 MX | 1 | 80–90 | <1 | 50–60 | N/A | N/A | 75 75 |
ILC | tamoxifen + letrozole + 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIA | T2 N0 MX | 2 | 40 | <1 | <5 | 0 | 1 | ||||
| Basal–like | ||||||||||||
| 13 | Before | I | T1 N0 MX | 1 | 0 | 0 | 75–85 | 5 | 5 | 39 39 |
IDC | 4× doxorubicin and cyclophosphamide |
| After | I | T0 N0 M0 | pCR | |||||||||
| 14 | Before | IIB | T2–3 N1 MX | 0 | 0 | 0 | 70–80 | 25–35 | 20 | 45 45 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIB | T0 N0 M0 | pCR | |||||||||
| 15 | Before | IIB | T2 N1 MX | 0 | 0 | 0 | 35–45 | 60–70 | 50 | 55 55 |
IDC | 3× fluorouracil, epirubicin and cyclophosphamide + 2× docetaxel |
| After | I | T0 N0 MX | pCR | |||||||||
| 16 | Before | IIB | T3 N0 MX | 0 | 0 | 0 | >90 | 5 | 5–10 | 48 48 |
IDC | 4× doxorubicin and cyclophosphamide + 4× docetaxel |
| After | IIA | T2 N0 M0 | 0 | 0 | 0 | 10 | 20–30 | 40 | ||||
inv; DCIS; ILC – Invasive Lobular Carcinoma; IDC – Invasive Ductal Carcinoma; pCR – pathological complete response; N/A – not available.
In all cases, tumor tissue was obtained from both a diagnostic core needle biopsy and from specimens removed during final surgery (lumpectomy or mastectomy).
Histological evaluation
The specimens from the core needle biopsy and surgery were fixed in 10% formalin and embedded in paraffin wax. The histological evaluation was done on slides routinely stained with hematoxylin and eosin (HE). Tumor stage was determined according to WHO guidelines [15] regarding the usage of clinical data. The molecular classification of the tumors was based on the immunohistochemical profile of Her2, ER, PR, CK5,6, CK8 and CK18 [16]. After the histological evaluation, areas of eventual tumor necrosis were macro-dissected using a scalpel.
Immunohistochemistry
Immunohistochemistry was performed using the avidin-biotin complex (ABC) method as was described in our previous work [17]. Primary antibodies were applied against c-erbB-2 protein (1:100, clone 5A2, Novocastra); ER (1:20, clone ER-6F11, Novocastra); PR (1:100, clone PGR-312, Novocastra); Ki-67 (1:50, clone MIB-1, DakoCytomation); CK5,6 (1:50, clone D5/16B4, DakoCytomation), CK8 (ready to use, clone 35bH11, DakoCytomation), CK18 (1:50, clone DC10, DakoCytomation) and cleaved caspase-3 (1:250, Asp175, Cell Signaling Technology, Beverly, MA, USA). In the case of active caspase-3, antigen retrieval was performed with a 10 mM sodium citrate buffer (pH 6.0) in a water bath for 40 minutes.
TUNEL assay
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) was performed with a fluorometric TUNEL system (Promega, Madison, WI, USA) according to the manufacturer’s protocol. In brief, the tissue sections were treated with 20 μg/ml of proteinase K for 20 minutes and then incubated in a nucleotide mixture containing fluorescein-12-dUTP and TdT (terminal deoxynucleotidyl transferase) for 1 hour at 37°C. Positive controls were pretreated with 1 U/ml of DNase, and negative controls were incubated without the TdT enzyme. The fluorescein-12-dUTP-labeled DNA was observed using a Provis AX70 (Olympus, Japan) fluorescence microscope. TUNEL-positive nuclei were expressed as a percent of the total nuclei (DAPI-positive) per slide.
RNA isolation and cDNA preparation
Deparaffinized slides of formalin-fixed, paraffin-embedded tissues, the isolation of total RNA and the synthesis of cDNA were performed by standard procedures described in our previous work [18].
In brief, total RNA was extracted using an RNeasy Micro Kit (Qiagen); reverse transcription was performed by a RevertAid – First Strand cDNA Synthesis Kit (Fermentas), which employs a random hexamer primer and Moloney-Murine Leukemia Virus (MMLV) reverse transcriptase.
Real-Time PCR assay (Gene expression profiling)
The gene expression of 84 key apoptosis genes was profiled by the RT2 Profiler Apoptosis PCR Array (PAHS-012, SABioscience, Frederick, MD, USA). RT-PCRs were performed in a 96-well plate format using a LightCycler 480 (Roche Applied Science) according to the manufacturer’s instructions (SuperArray Bioscience). In brief, approximately 500 pg RNA was first converted to cDNA by using an RT2 First Strand Kit (C-03, SuperArray Bioscience), then the cDNA was pre-amplified using an RT2 Nano PreAMP Kit (C-06, SuperArray Bioscience) and a Nano PreAMP Human Apoptosis Primer Mix (PBH-7012, SuperArray Bioscience). Finally, PCR reactions were performed using real-time PCR with an RT2 Real-Time SYBR Green PCR master mix PA-010; SuperArray Bioscience). The total volume of the PCR reaction was 25 μL. An equivalent of 5 pg of RNA was applied to the PCR reaction. The thermocycler parameters were 95°C for 10 minutes, followed by 45 cycles of 95°C for 15 seconds, and then 60°C for 1 minute. All data was analyzed by an RT2 Profiler PCR Array Data Analysis Template v3.0 (SABioscience) and relative changes in gene expression were calculated using the ΔΔCt (cycle threshold) method [19]. An average of the number of cycles of the 5 housekeeping genes – β-2-microglobulin (B2M), hypoxanthine phosphoribosyltransferase 1 (HPRT1), ribosomal protein L13a (RPL13A), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and β-actin (ACTB) – was used to normalize the expression between samples, and a 2.0-fold change in gene expression was used as the cut-off threshold to determine up- or down-regulation, as previously described by Li et al. [20]. In each case, the difference in gene expression between the tumor before and after treatment was calculated.
Statistical analysis
Statistical analysis was performed using programs RT2 Profiler PCR Array Data Analysis Template v3.0 (SABioscience) and Statistica v9.1 (StatSoft, Czech Republic).
The Student’s t test was used to evaluate the statistical significance of the results. Differences with P values <0.05 were considered significant.
Results
Histologic findings, tumor staging
Out of the 16 patients, 13 (81%) had infiltrating ductal carcinoma, and 3 (19%) had infiltrating lobular carcinoma. Tumor stage was determined both before therapy and after therapy (Table 1). In the pre-therapeutical investigation, 2 patients (12.5%) had tumors of stage I, 9 patients (56.2%) stage II, 3 patients (18.8%) stage III, and 2 patients (12.5%) stage IV. In post-therapeutical investigation, 6 (37.5%) patients had tumor stage I, 5 (31.25%) patients stage II, 2 (12.5%) patients stage III, 2 (12.5%) patients stage IV, and 1 (6.25%) patient stage 0. These results indicate tumor reduction and a decrease of tumor stages after therapy in 8 patients (50%). No significant change was observed in 4 patients (25%). A progression of disease was found in 4 patients (25%). In 4 patients (25%) a pathologically complete response (pCR) was observed. Surprisingly, not only tumors differing in the stage of the disease, but also tumors of the same histology and stage (cases No. 4 vs. 6 and No. 14 vs. 16; see Table 1) reacted differently to neoadjuvant therapy. Disease progressed early in cases No. 6 and 16 (data not shown).
Immunohistochemistry
The molecular classification of the tumors was determined on the basis of the immunohistochemical profile of Her2, ER, PR, CK5,6, CK8 and CK18. Subsequently, we investigated Ki-67 proliferation index. The status of all markers was determined both before and after the treatment, which allowed us to compare the differences among patients in each group and also the changes in expression after the treatment. The detailed outcome is shown in Table 1 (for CK5,6, CK8 and CK18 data not shown) along with the results of TUNEL and anti-active caspase 3 assay.
Cleaved Caspase-3 Immunohistochemistry and TUNEL assay
The role of apoptosis in the regression of tumors after neoadjuvant chemotherapy was determined by 2 independent methods: anti-active caspase 3 assay and TUNEL. The activated caspase-3 antibody selectively labeled the cytoplasm of cells (Figure 1A, B), whereas the TUNEL assay showed specific nuclear staining (Figure 1C, D). The cells had morphology consistent with apoptosis (Figure 1B, D).
Figure 1.

Detection of apoptotic cells. Apoptosis was determined by two methods: anti-active caspase 3 immunohistochemistry (A, B) and TUNEL assay (C, D), respectively. In each case, apoptotic cell death assay was performed before (A, C) and after (B, D) the treatment, respectively. The antibody specific for activated caspase-3 selectively labelled the cytoplasm (brown) of apoptotic cells (A, B). Fluorescein-12-dUTP incorporation results in localized green fluorescence within the nucleus of apoptotic cells only (B, D).
The apoptotic index (AI), exactly caspase-3 labeling index and TUNEL labeling index were used as quantitative measures of apoptosis in histologic sections as previously reported by Duan et al. [21]. The AIs calculated from these methods showed a similar pattern (Table 1) with good correlation between the TUNEL AI and caspase-3 AI (R=0.95), suggesting that these 2 method are highly comparable for the detection of apoptotic cells.
In 1 patient (6.3%) a dramatic decrease of AI was observed after the treatment. In 2 cases (12.5%) we found considerable increased AI after the treatment (Table 1). In 8 cases (50%), minor changes (up to 5%) of the AI were observed after the treatment. For technical reasons, 1 TUNEL and 3 activated caspase-3 assays were not evaluated.
Gene expression profiling
In each case, the transcriptional profile of 84 key apoptosis genes was evaluated by using real-time PCR assay.
Firstly, we analyzed all pretreatment samples compared with the control group of 4 cases with pCR, which we selected as a standard of good responders. From the pool of 84 genes, only 13 were significant (p<0.05) across all samples. We analyzed the significant data by hierarchical cluster analysis, which divided patients into 2 groups (Figure 2). One group had a transcriptional profile more similar to patients with pCR than the rest of the cases that we included in the second group. These 2 groups of patients correlate well with progression-free survival (Figure 3) and therefore these groups of patients could be identified as good and poor prognosis groups.
Figure 2.

Dendrogram of 16 breast cancer patients analyzed by hierarchical clustering analysis. Gene expression profiling was performed by qRT-PCR assay. Expression levels above the mean for the gene are shown in red squares and expression levels below the mean for the gene are shown in green squares. Branches representing good responders are shown in blue and those representing bad responders in red.
Figure 3.

Progression-free survival. Progression free survival (PFS) analysis of patients based on the gene expression analysis of 13 apoptosis associated genes. There is a significant PFS difference between the good and poor prognosis group (P<0.05).
Finally, we analyzed changes in expression of individual genes before and after the treatment. In the good prognosis group we found a statistically significant down-regulation of the expression of MCL1 and IGF1R genes after neoadjuvant treatment. We also found a statistically significant overexpression of BCL2L10, BCL2AF1, CASP8, CASP10, CASP14, CIDEB, FADD, HRK, TNFRSF25, TNFSF8 and CD70 genes. In contrast, we found an up-regulation of IGF1R after the treatment in the poor prognosis group, while the expression of all the other genes remained almost unchanged (Figure 4).
Figure 4.
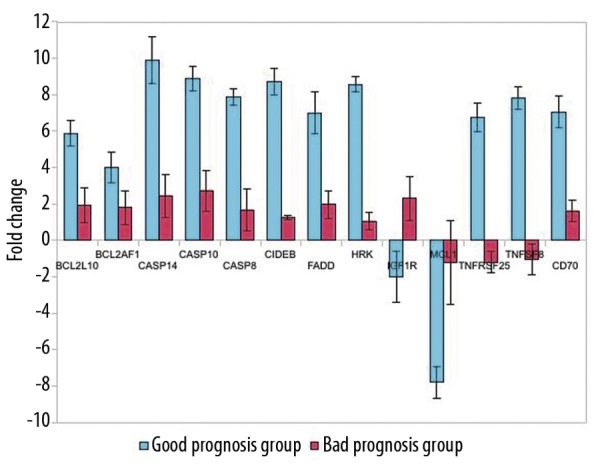
Changes in gene expression after the treatment. Up or down regulation (comparing tumors before treatment) of individual genes is expressed as normalized ratios with standard deviation. A 2-fold change in gene expression was used as the cut-off threshold to determine up- or down-regulation. A positive value indicates gene up-regulation and a negative value indicates gene down regulation. There are significant differences between the tumors before and after treatment (P<0.05).
Discussion
Breast cancer is a clinically heterogeneous disease, and existing histopathological classifications do not fully capture its varied clinical course. Current breast cancer TNM staging is based on the anatomical extent (size, lymph node status, distant metastases), but this classification gives only a limited insight into breast cancer biology and clinical outcome. The recent St. Gallen consensus panel [14] established three risk categories: minimal, intermediate and high risk. Hormone receptors, tumor size, tumor grade and age still dominate as key discriminating factors. Newly accepted prognostic factors are Her2 status, lymphatic and vascular invasion in the primary tumor.
Surprisingly, none of these criteria significantly correlated with the clinical outcome in our study. When the St. Gallen risk categories were analyzed, no significant difference in overall survival and progression-free survival was seen (data not shown). Moreover, the high-risk group identified by the St. Gallen criteria included patients with a pathologically complete response and a good outcome. Similar findings were described by Bauer et al. [22], who found that there was very little difference between the low risk and intermediate risk categories when using a 5-year relative survival rate as the single principal criterion. In addition, other indicators, namely proliferation and the apoptotic index, did not correlate with the longer clinical outcome in our study. On the other hand, they correlated well with the regression of the tumor after primary chemotherapy.
Her2, ER and other molecular markers have become important predictive factors with the development of tailored therapies targeting specific molecules. For example, Her2 positivity predicts a response to trastuzumab, and estrogen receptor positivity predicts a response to hormonal therapy [23]. On the other hand, there are no clinically useful predictive markers of response to conventional chemotherapy [24].
Because many cancer therapies induce classic apoptosis [25], we assume that the regression of tumors after neoadjuvant treatment is executed predominantly by programmed cell death, despite different chemotherapy agents. However, programmed cell death takes many forms distinct from classical apoptosis. Apoptosis-like programmed cell death refers to a cell death process that has similar hallmarks of apoptosis, such as chromatin condensation and the appearance of phosphatidylserine on the outer leaflet of the cell membrane, but not necessarily requiring caspase activity [26]. Necrosis-like programmed cell death describes programmed cell death that does not include chromatin condensation and has varying degrees of other apoptotic features. Caspase-1 and caspase-8 have been implicated in some cases of this type of programmed cell death [26]. Paraptosis describes a cell death that requires gene expression but morphologically does not resemble either apoptosis or necrosis [27]. Unlike necrosis, all of these forms of cell death are programmed because they require gene expression.
In this study, we focused on the end-point of therapy, that is, the death of malignant cells. However, the complexity of apoptosis regulation and the large number of molecular players in the apoptotic signaling pathways result in a crossover of death pathways [26], which may lead eventually to a different phenotypic pattern. Moreover, caspase-independent death or time from death stimulus might explain the low apoptotic index in the good prognosis group.
In spite of the above, or because of it, we determined the expression profile of 84 key apoptosis-associated genes in all tumors, and on this basis we have developed the 13 apoptosis- associated genes expression assay which may be helpful for the stratification of the patients into groups with different responses to chemotherapy. Among these genes are MCL1, BCL2L10, BCL2AF1, CASP8, CASP10, CASP14, CIDEB, FADD, HRK, IGF1R, TNFRSF25, TNFSF8 and CD70.
An understanding of how these molecules interact gives us an insight into breast cancer biology and allows the development of therapeutics able to induce or modulate the pathway leading to the death of malignant cells.
Apoptosis can be induced in response to many external stimuli, including the activation of cell surface receptors such as Fas, TNFR family members and others [27]. These “death receptors” have 2 distinct signaling motifs – death domains and death effector domains – that allow them to interact with other proteins involved in the apoptosis cascade. The Fas-associated death domain protein (FADD) binds to the ligand-bound receptor through its C-terminal death domain, which leads to the activation of procaspase 8. Caspase 8 is involved in initiating apoptosis induced by Fas and various apoptotic stimuli [28] and is known as an “initiator caspase”. It can activate the effector caspases, including caspase 3, 10 and 14, by proteolytic processing. Once caspase 3 is activated, it cleaves downstream targets and irreversibly directs the cell to the apoptotic death. The changes in expression of Bcl2 family members (MCL1, BCL2L10 and BCL2AF1) suggest that not only are the death receptors pathways activated, but the mitochondrial apoptosis pathway also play a role in the chemotherapy treatment. A product of the Harakiri (Hrk) gene is a regulator of cell death that physically interacts with death-repressor proteins Bcl2 and BclX(L), and forms inactive complexes of Bcl2-Hrk and/or BclX(L)-Hrk [29]. In breast cancer, the overexpression of IGF1R and the activation of its downstream signaling molecules have been linked to an inhibition of apoptosis, disease progression, increased resistance to cytotoxic chemotherapeutic drugs or radiotherapy, and to poor prognosis [30,31]. CIDEB is a member of the CIDE (cell death-inducing DFF45 [DNA fragmentation factor 45]-like effector) family of apoptosis-inducing factors [32]. The N-terminal region of CIDEs is homologous to the CIDE-N domains in DFF40/CAD (caspase-activated nuclease) and its inhibitor (DFF45/ICAD [inhibitor of CAD]), which are 2 subunits of the DFF complex [33,34]. Cleavage of DFF45/ICAD by caspase 3 releases DFF40/CAD from the complex, which leads to DNA fragmentation and nuclear condensation [35–37].
Although the precise pathway leading to the chemotherapy-induced cell death is still unclear, it is not surprising that various kinds of molecules – from receptors through caspases to molecules that provide DNA fragmentation – play a significant role. Changes in the expression of these molecules inform us about the nature and sequence of intracellular events leading to programed cell death, show potential therapeutic targets, and reveal the strategies of cancer cells to escape apoptosis.
Conclusions
In order to devise effective, rational treatments, we need a reliable tool for predicting response to the treatment. As we have shown, gene expression profiling after neoadjuvant chemotherapy is a valuable research tool for investigating molecular markers, which may better reflect tumor biology and treatment response than standard prognostic and predictive factors. Moreover, a pathway-focused real-time PCR assay provides a cheaper alternative to microarrays, which allows easier implementation in clinical practice. However, larger groups of patients need to be studied to verify our findings.
Acknowledgments
We wish to thank Štěpánka Melčáková for expert assistance with TUNEL assay.
Abbreviations
- PCR
polymerase chain reaction
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- ABC
avidin-biotin complex
- pCR
pathologically complete response
Footnotes
Source of support: This work was supported by Grant IGA MZ ČR NS/10575-3
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Buchholz TA, Hunt KK, Whitman GJ, et al. Neoadjuvant chemotherapy for Brest carcinoma. Cancer. 2003;98:1150–60. doi: 10.1002/cncr.11603. [DOI] [PubMed] [Google Scholar]
- 3.Jones C, Ford E, Gillett C, et al. Molecular cytogenetic identification of subgroups of grade III invasive ductal breast carcinomas with different clinical outcomes. Clin Cancer Res. 2004;10:5988–97. doi: 10.1158/1078-0432.CCR-03-0731. [DOI] [PubMed] [Google Scholar]
- 4.Goldhirsch A, Glick JH, Gelber RD, et al. Meeting Highlights: Intemationa1 Expert Consensus on the Primary Therapy of Early Breast Cancer 2005. Ann Oncol. 2005;16:1569–83. doi: 10.1093/annonc/mdi326. [DOI] [PubMed] [Google Scholar]
- 5.Eifel P, Axelson JA, Costa J, et al. NIH Consensus Development Panel: Adjuvant Therapy for Breast Cancer. J Natl Cancer Inst. 2001;93:979–89. doi: 10.1093/jnci/93.13.979. [DOI] [PubMed] [Google Scholar]
- 6.Oakman C, Santarpia L, Di Leo A. Breast cancer assessment tools and optimizing adjuvant therapy. Nat Rev Clin Oncol. 2010;7:725–32. doi: 10.1038/nrclinonc.2010.170. [DOI] [PubMed] [Google Scholar]
- 7.Reis-Filho JS, Westbury C, Pierga JY. The impact of expression profiling on prognostic and predictive testing in breast cancer. J Clin Pathol. 2006;59:225–31. doi: 10.1136/jcp.2005.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotiriou C, Powles TJ, Dowsett M, et al. Gene expression profiles derived from fine needle aspiration correlate with response to systemic chemotherapy in breast cancer. Breast Cancer Res. 2002;4:R3. doi: 10.1186/bcr433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pluciennik E, Krol M, Nowakowska M, et al. Breast cancer relapse prediction based on multi-gene RT-PCR algorithm. Med Sci Monit. 2010;16(3):CR132–36. [PubMed] [Google Scholar]
- 10.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 11.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sotiriou C, Neo SY, McShane LM, et al. Breast cancer c1assification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–98. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profi1ing predicts c1inical outcome of breast cancer. Nature. 2002;415:530–36. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 14.Goldhirsch A, Ingle JN, Gelber RD, et al. Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2009. Ann Oncol. 2009;20:1319–29. doi: 10.1093/annonc/mdp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavassoli FA, Devilee P. WHO Classification of tumours Pathology and genetics tumours of the breast and female genital organs. Lyon: IARC Press; 2004. [Google Scholar]
- 16.Normanno N, De Luca A, Carotenuto P, et al. Prognostic Applications of Gene Expression Signatures in Breast Cancer. Oncology. 2009;77(Suppl 1):2–8. doi: 10.1159/000258489. [DOI] [PubMed] [Google Scholar]
- 17.Skálová H, Dundr P, Povýšil C, et al. Altered expression of Her2/neu after neoadjuvant treatment of breast cancer. Folia Biologica. 2011;57(5):191–99. doi: 10.14712/fb2011057050191. [DOI] [PubMed] [Google Scholar]
- 18.Tvrdík D, Svatošová J, Dundr P, Povýšil C. Molecular diagnosis of synovial sarcoma: detection of SYT-SSX1/2 fusion transcripts by RT-PCR in paraffin-embedded tissue. Med Sci Monit. 2005;11(3):MT1–7. [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Liu B, Maminishkis A, et al. Gene expression profiling in autoimmune noninfectious uveitis disease. J Immunol. 2008;181:5147–57. doi: 10.4049/jimmunol.181.7.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan WR, Garner DS, Williams SD, et al. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol. 2003;199:221–28. doi: 10.1002/path.1289. [DOI] [PubMed] [Google Scholar]
- 22.Bauer K, Parise C, Caggiano V. Use of ER/PR/HER2 subtypes in conjunction with the 2007 St Gallen Consensus Statement for early breast cancer. BMC Cancer. 2010;10:228–40. doi: 10.1186/1471-2407-10-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van de Vijver M. Gene-Expression Profiling and the Future of Adjuvant Therapy. The Oncologist. 2005;10(Suppl 2):30–34. doi: 10.1634/theoncologist.10-90002-30. [DOI] [PubMed] [Google Scholar]
- 24.Chang JC, Wooten EC, Tsimelzon A, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–69. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- 25.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–26. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 26.Leist M, Jäättelä M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–98. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 27.Sperandio S, de Belle I, Bredesen DE. An alternative, non-apoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–81. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wajant H, Gerspach J, Pfizenmaier K. Engineering death receptor ligands for cancer therapy. Cancer Lett. 2011 [Google Scholar]
- 29.Daniel PT, Wieder T, Sturm I, Schulze-Osthoff K. The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia. 2001;15:1022–32. doi: 10.1038/sj.leu.2402169. [DOI] [PubMed] [Google Scholar]
- 30.Inohara N, Ding L, Chen S, Núńez G. Harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L) EMBO J. 1997;16:1686–94. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha RL, Hilsenbeck SG, Jackson JG, et al. Insulin-like growth factor binding protein 3 and insulin receptor substrate 1 in breast cancer: correlation with clinical parameters and disease-free survival. Clinical Cancer Research. 1997;3:103–9. [PubMed] [Google Scholar]
- 32.Turner BC, Haffty BG, Narayanan L, et al. IGF-I receptor and cyclin D1 expression influence cellular radiosensitivity and local breast cancer recurrence after lumpectomy and radiation. Cancer Research. 1997;57:3079–83. [PubMed] [Google Scholar]
- 33.Inohara N, Koseki T, Chen S, et al. CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J. 1998;17:2526–33. doi: 10.1093/emboj/17.9.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enari M, Sakahira H, Yokoyama H, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–84. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Li P, Widlak P, et al. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci USA. 1998;95:8461–66. doi: 10.1073/pnas.95.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]